

Abstract
A whole-genome scan for identifying selection acting on pairs of linked loci is proposed and implemented. The scan is based on 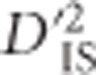 , one of Ohta's 1982 measures of between-population linkage disequilibrium (LD). An approximate empirical null distribution for the statistic is suggested. Although the partitioning of LD into between-population components was originally used to investigate epistatic selection, we demonstrate that values of
, one of Ohta's 1982 measures of between-population linkage disequilibrium (LD). An approximate empirical null distribution for the statistic is suggested. Although the partitioning of LD into between-population components was originally used to investigate epistatic selection, we demonstrate that values of 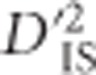 may also be influenced by single-locus selective sweeps with linkage but no epistasis. The proposed scan is implemented in a diverse panel of chickens including 72 distinct breeds genotyped at 538 298 single-nucleotide polymorphisms. In all, 1723 locus pairs are identified as putatively corresponding to a selective sweep or epistatic selection. These pairs of loci generally cluster to form overlapping or neighboring signals of selection. Known variants that were expected to have been under selection in the panel are identified, as well as an assortment of novel regions that have putatively been under selection in chickens. Notably, a promising pair of genes located 8 MB apart on chromosome 9 are identified based on
may also be influenced by single-locus selective sweeps with linkage but no epistasis. The proposed scan is implemented in a diverse panel of chickens including 72 distinct breeds genotyped at 538 298 single-nucleotide polymorphisms. In all, 1723 locus pairs are identified as putatively corresponding to a selective sweep or epistatic selection. These pairs of loci generally cluster to form overlapping or neighboring signals of selection. Known variants that were expected to have been under selection in the panel are identified, as well as an assortment of novel regions that have putatively been under selection in chickens. Notably, a promising pair of genes located 8 MB apart on chromosome 9 are identified based on 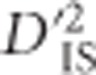 as demonstrating strong evidence of dispersive epistatic selection between populations.
as demonstrating strong evidence of dispersive epistatic selection between populations.
Introduction
A variety of patterns are generated in the genomes of organisms undergoing selection. Such patterns depend on a multitude of factors including the demographic history of the populations in question, the type of selection that is taking place and the relative importance of the variant or variants that are being selected. For many of these factors, appropriate tests for selection have been developed and are in wide use. For instance, in cases of directed evolution with experimental populations, especially for those with biological replication, changes in allele frequency may be directly measured to identify single-locus selection (Wisser et al., 2008; Turner et al., 2011; Parts et al., 2011; Hirsch et al., 2014). In a related test, which is particularly useful if samples from pre-selection populations are not available, patterns of nucleotide variability between vs within populations may be leveraged to identify selection at a single locus (Lewontin and Krakauer, 1973; Akey et al., 2002; Beissinger et al., 2013). This type of test may theoretically be able to distinguish between directional and balancing selection, since the former is expected to drive alleles toward fixation while the latter will maintain an elevated level of variability (Akey et al., 2002). In addition, selection on an individual locus is known to reduce genetic variability at linked sites (Maynard Smith and Haigh, 1974), which has led to an assortment of tests for selection based on data that are observed either within a single population (Sabeti et al., 2002; Voight et al., 2006) or between populations (Sabeti et al., 2007; Tang et al., 2007). This class of tests, however, is most powerful for detecting recent strong selection since the length of any haplotypes showing reduced variability, which are the basis of these tests, will decay over time.
An alternative type of selection that is also expected to produce unique genomic patterns is epistatic selection. Here, the favored phenotype depends on an interacting set of alleles at more than one locus. Therefore, the non-random association, or linkage disequilibrium (LD), between alleles at such interacting loci is expected to increase. If these loci are genetically linked, LD between them may grow over generations (Kimura, 1965). Ohta (1982a, b), developed a set of statistics to partition LD into between and within subpopulation components, in a manner analogous to Wright's F-statistics (Wright, 1949), which are frequently used to identify single-locus selection based on subpopulation variability. According to Ohta, comparisons between these statistics, which are denoted D2IT, D2IS, D2ST, D′2IS, and D′2ST, may suggest whether epistatic selection or random genetic drift is the driving force behind observed levels of LD between pairs of markers. Although this type of test was originally implemented in a purely theoretical setting, software has been developed and implemented to apply these statistics to experimental data (Black and Krafsur, 1985; Garnier-Gere and Dillmann, 1992). Interesting examples applying these statistics to evaluate loci for evidence of epistatic selection depict an array of findings. For instance, Song et al. (2009) found that overall drift was more important in shaping LD patterns than was epistatic selection in Boechera stricta, as did Vitalis et al. (2002) in Marsilea strigosa. Alternatively, Miyashita et al. (1993), investigated patterns of LD across a specific region of Drosophila melanogaster and concluded that epistatic selection was likely to be involved. Extensions of Ohta's theory can be found as well. Storz and Kelly (2008), for example, used a similar approach to measure between-population components of the ZnS statistic (Kelly, 1997), and a related statistic was employed by Ma et al. (2010). Both of these methods are based on expected haplotype frequencies but do not incorporate information regarding gametic phase. Therefore, they represent summaries of the covariance in allele frequencies between loci.
Interestingly, Ohta's D-statistics have not been extended to or implemented in a framework that involves dense genome-wide marker or sequence data on the scale that is commonly employed today. One limitation of the statistics is that although a point estimate of the relative contributions of random processes, such as stochastic sampling and drift, compared with epistatic selection for a particular pair of loci can be obtained, previous research has not addressed the uncertainty of these contributions for the purpose of assigning significance. Second, although D-statistics are analogous to Wright's F-statistics, Ohta's original estimators assume that the effects of epistatic selection will be similar in all subpopulations, which is contrary to the form in which FST tests are typically employed. Modifications have been suggested to address this discrepancy and to allow high subpopulation differentiation to serve as a signature of selection as well (Black and Krafsur, 1985); however these modifications do not address significance. A third and final impediment to applying these statistics in whole-genome scans involves complications related to distinguishing a signature generated by epistatic selection from that due to large sweeps resulting from hitchhiking (Maynard Smith and Haigh, 1974; Sabeti et al., 2002).
Herein we extend the subdivision of LD into its between and within subpopulation components to a framework that may be applied in a whole-genome scan for selection. We follow the approach of Black and Krafsur (1985) in employing Δij, the Burrows composite measure of LD (Cockerham and Weir, 1977), to compute LD when gametic phase is ambiguous. We identify one of Ohta's statistics, 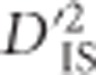 , as most informative for testing pairs of linked loci that are jointly impacted by selection at either the single-locus or epistatic level. An empirically based null distribution for these statistics is proposed that captures much of the variability expected due to random processes alone, such as sampling error and drift, while excluding that which may result from linked selection. Using this null distribution to define a lower limit for significance thresholds, our proposed scan is employed to infer genetically linked pairs of loci that demonstrate extreme divergence in their gametic frequencies between subpopulations relative to the total population. We highlight that such pairs of loci can be generated by either extreme drift, epistatic selection or through an ongoing selection event at a single locus that is in LD with its neighbors, an important limitation of D-statistics that, to our knowledge, has not been noted before. However, by evaluating pairwise patterns of
, as most informative for testing pairs of linked loci that are jointly impacted by selection at either the single-locus or epistatic level. An empirically based null distribution for these statistics is proposed that captures much of the variability expected due to random processes alone, such as sampling error and drift, while excluding that which may result from linked selection. Using this null distribution to define a lower limit for significance thresholds, our proposed scan is employed to infer genetically linked pairs of loci that demonstrate extreme divergence in their gametic frequencies between subpopulations relative to the total population. We highlight that such pairs of loci can be generated by either extreme drift, epistatic selection or through an ongoing selection event at a single locus that is in LD with its neighbors, an important limitation of D-statistics that, to our knowledge, has not been noted before. However, by evaluating pairwise patterns of 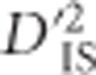 over regions, inferences about the type of selection that is taking place may be possible.
over regions, inferences about the type of selection that is taking place may be possible.
This genome scan was employed using a highly diverse panel of chickens, consisting of 72 breeds with at least 18 individuals per breed genotyped at nearly 600 000 single-nucleotide polymorphism (SNP) markers. Multiple previously identified regions known to impact important traits are shown to contain significant locus pairs, as well as novel, putatively selected genomic regions. Regions that suggest patterns consistent with single-locus sweeps vs epistatic selection are explored.
Background and theory
Variance components of LD in subdivided populations
When a population of individuals is divided into subpopulations with limited migration, the variance of LD for that population may be divided into subcomponents as well. Ohta (1982a, b), denoted these variance components as D2IT, D2IS, D2ST, D′2IS, and D′2ST. Consider two loci, A and B. Let xi,k and yj,k be the frequencies of the ith and jth alleles at loci A and B, respectively, in the kth subpopulation, and define gij,k as the frequency of gametes Ai Bj in the kth subpopulation. Let 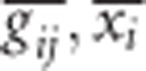 and
and 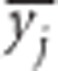 denote their averages over subpopulations. Therefore, if the total population is balanced,
denote their averages over subpopulations. Therefore, if the total population is balanced, 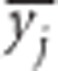 and
and 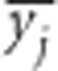 correspond to the gametic and allele frequencies in the total population. According to Ohta (1982a, b), the variance components of LD may be defined according to:
correspond to the gametic and allele frequencies in the total population. According to Ohta (1982a, b), the variance components of LD may be defined according to:
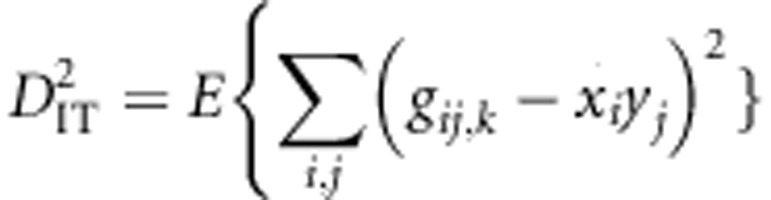 |
 |
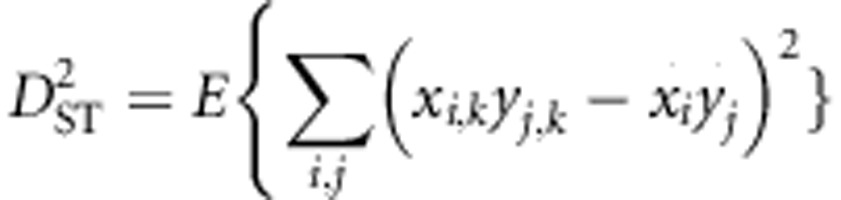 |
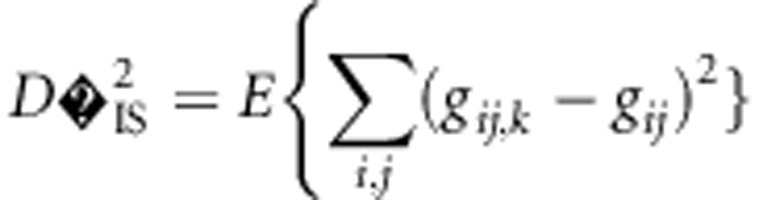 |
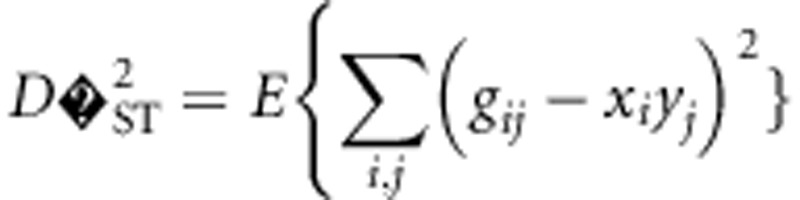 |
where the expectation is taken with respect to the distribution over alleles and subpopulations. These variance components are based on treating xi, k, yj, k, and gij, k as independent and identically distributed random variables each corresponding to a distribution. The complicated nature of drift makes specifying the precise distribution difficult or impossible for all but the simplest scenarios.
Based on these definitions, 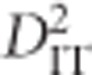 is a measure of the correlation of Ai and Bj on the same gametes of a subpopulation relative to the expectation according to allele frequencies in the total population,
is a measure of the correlation of Ai and Bj on the same gametes of a subpopulation relative to the expectation according to allele frequencies in the total population, 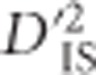 measures the expected variance of LD for subpopulations,
measures the expected variance of LD for subpopulations, 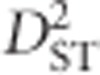 is the expected correlation of Ai and Bj in a subpopulation relative to their expected correlation in the total population,
is the expected correlation of Ai and Bj in a subpopulation relative to their expected correlation in the total population, 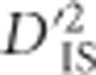 is the correlation of Ai and Bj on the same gamete in a subpopulation relative to that of the total population and
is the correlation of Ai and Bj on the same gamete in a subpopulation relative to that of the total population and 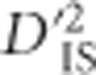 is the variance, computed over alleles only, of the LD of the total population. Observe that although the subscripts are the same (IT, IS and ST), Ohta's use of subscripts differs substantially from Wright's (Wright, 1949). While in Wright's notation I represents individuals, S subpopulations and T the total population, in Ohta's usage the subscript I specifies gametic frequencies within subpopulations, T indicates expected haplotype frequencies in the total population and S may specify either expected haplotype frequencies based on population-specific allele frequencies or population-specific gametic frequencies (coupled with D′ in this latter case).
is the variance, computed over alleles only, of the LD of the total population. Observe that although the subscripts are the same (IT, IS and ST), Ohta's use of subscripts differs substantially from Wright's (Wright, 1949). While in Wright's notation I represents individuals, S subpopulations and T the total population, in Ohta's usage the subscript I specifies gametic frequencies within subpopulations, T indicates expected haplotype frequencies in the total population and S may specify either expected haplotype frequencies based on population-specific allele frequencies or population-specific gametic frequencies (coupled with D′ in this latter case).
If interest lies in identifying pairs of loci that display highly variable LD between subpopulations,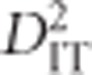 ,
, 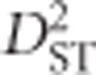 , and
, and 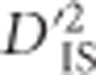 may be appropriate since each of these components compares a population-specific measure with a total-population measure. However, Ohta (1982a, b), has shown that
may be appropriate since each of these components compares a population-specific measure with a total-population measure. However, Ohta (1982a, b), has shown that 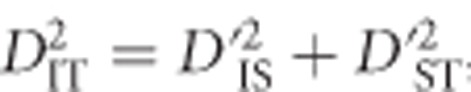 , so the information contained in
, so the information contained in 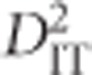 that is relevant to this goal fully resides in
that is relevant to this goal fully resides in 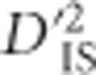 . Notice also that
. Notice also that 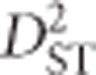 depends only on expected haplotype frequencies and does not incorporate actual gametic information (there is no
depends only on expected haplotype frequencies and does not incorporate actual gametic information (there is no 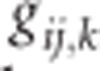 or
or 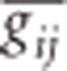 term), precluding its relevance as a test of non-random association between populations. This leaves
term), precluding its relevance as a test of non-random association between populations. This leaves 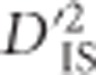 as the most relevant statistic. Its relevance becomes even more pertinent by noting that
as the most relevant statistic. Its relevance becomes even more pertinent by noting that 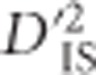 can be manipulated in the following manner, by simply adding and subtracting a
can be manipulated in the following manner, by simply adding and subtracting a 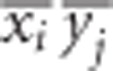 term:
term:
 |
Notice that 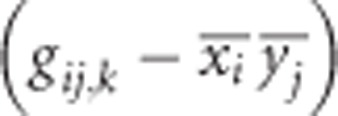 measures the covariance of alleles at loci A and B, or coefficient of LD, in subpopulations relative to that in the total population, and
measures the covariance of alleles at loci A and B, or coefficient of LD, in subpopulations relative to that in the total population, and  measures the coefficient of LD in the total population. Therefore, by measuring the variability of gametic frequencies in subpopulations relative to the total population,
measures the coefficient of LD in the total population. Therefore, by measuring the variability of gametic frequencies in subpopulations relative to the total population, 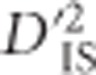 can be considered a measure of the variability of LD measured at two scales. For these reasons,
can be considered a measure of the variability of LD measured at two scales. For these reasons, 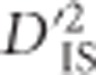 is the preferred statistic to use for identifying pairs of loci jointly undergoing selection.
is the preferred statistic to use for identifying pairs of loci jointly undergoing selection.
Ohta (1982a, b), suggested that, for a pair of loci, if 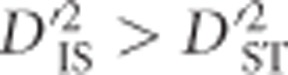 and
and 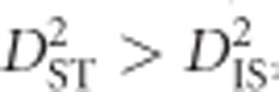 , drift is expected to be more important than epistatic selection in generating LD. Conversely,
, drift is expected to be more important than epistatic selection in generating LD. Conversely, 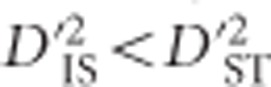 and
and 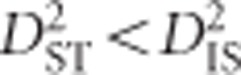 was said to imply that the same allelic combinations are favorable across subpopulations, so epistatic selection is responsible for observed levels of LD. However, these two conditions are based on the assumption of identical combinations of alleles being favored in all subpopulations, leaving out the possibility that selection may vary between subpopulations. Later, Black and Krafsur (1985) proposed that under dispersive epistatic selection between populations it will hold that
was said to imply that the same allelic combinations are favorable across subpopulations, so epistatic selection is responsible for observed levels of LD. However, these two conditions are based on the assumption of identical combinations of alleles being favored in all subpopulations, leaving out the possibility that selection may vary between subpopulations. Later, Black and Krafsur (1985) proposed that under dispersive epistatic selection between populations it will hold that 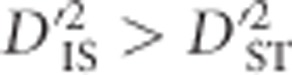 and D2ST<D2IS. Neither Ohta's nor Black and Krafsur's (1985) conditions involve theoretical or empirical distributions for calculating strict or approximate significance thresholds for testing epistatic selection, nor do they incorporate the possibility that selection on an individual locus, with some amount of linkage across a region, may be responsible for the observed values.
and D2ST<D2IS. Neither Ohta's nor Black and Krafsur's (1985) conditions involve theoretical or empirical distributions for calculating strict or approximate significance thresholds for testing epistatic selection, nor do they incorporate the possibility that selection on an individual locus, with some amount of linkage across a region, may be responsible for the observed values.
Genome scan and null distribution
To build on Ohta's (1982a, b), and Black and Krafsur's (1985) tests, an approach designed for a whole-genome scan was developed. It is capable of identifying either epistatic selection on pairs of markers, or sweeps impacting pairs of loci due to linkage. 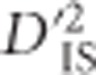 is used as the basis for this scan, and because it is a quadratic function, estimates are known to have large sampling variances and a distribution dependent on allele frequencies (Hill and Weir, 1988). An approximate null distribution, depicting the expected variability of
is used as the basis for this scan, and because it is a quadratic function, estimates are known to have large sampling variances and a distribution dependent on allele frequencies (Hill and Weir, 1988). An approximate null distribution, depicting the expected variability of 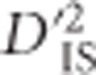 resulting from sampling in a scenario without selection, was identified. To derive this null distribution, notice that when loci A and B are unlinked, the expected value of
resulting from sampling in a scenario without selection, was identified. To derive this null distribution, notice that when loci A and B are unlinked, the expected value of 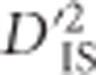 will not include effects of joint selection on the pair. This is because, first, single-locus selection on either locus will not systematically impact the correlation between alleles at loci A and B. Moreover, for unlinked loci epistasis will not indefinitely increase LD between the loci unless the selection coefficient is extremely large (rapidly leading to fixation), a phenomenon termed quasi-linkage equilibrium by Kimura (1965), and later studied in more depth by others (Nagylaki, 1993). Hence, even when there is epistasis between unlinked loci,
will not include effects of joint selection on the pair. This is because, first, single-locus selection on either locus will not systematically impact the correlation between alleles at loci A and B. Moreover, for unlinked loci epistasis will not indefinitely increase LD between the loci unless the selection coefficient is extremely large (rapidly leading to fixation), a phenomenon termed quasi-linkage equilibrium by Kimura (1965), and later studied in more depth by others (Nagylaki, 1993). Hence, even when there is epistasis between unlinked loci, 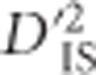 will not portray excessively high values as a result of selection. However, when A and B are linked to some extent, dispersive single-locus selection with linkage will elevate levels of
will not portray excessively high values as a result of selection. However, when A and B are linked to some extent, dispersive single-locus selection with linkage will elevate levels of 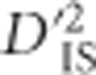 , as will dispersive epistatic selection for a favorable allele combination. In the case of single-locus selection, this signature is due to hitchhiking (Maynard Smith and Haigh, 1974) and will be temporary because, over generations, recombination will break apart the correlation between Ai and Bj. In the case of epistatic selection the signature will last indefinitely because the correlation of favorable allele pairs will increase over time (Kimura, 1965).
, as will dispersive epistatic selection for a favorable allele combination. In the case of single-locus selection, this signature is due to hitchhiking (Maynard Smith and Haigh, 1974) and will be temporary because, over generations, recombination will break apart the correlation between Ai and Bj. In the case of epistatic selection the signature will last indefinitely because the correlation of favorable allele pairs will increase over time (Kimura, 1965).
Therefore, patterns of 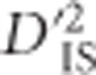 observed for unlinked loci do not reflect selective sweeps or epistatic selection, while they do depend on factors such as population size, mutation rate and migration rate. Only one potential contributor to
observed for unlinked loci do not reflect selective sweeps or epistatic selection, while they do depend on factors such as population size, mutation rate and migration rate. Only one potential contributor to 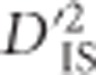 under a model of drift may be missed for unlinked locus pairs compared with linked pairs: for a period of time after a variant appears, but before recombination breaks down its association with the background in which it arose, the pattern of drift shown by that mutation will be correlated to those of its neighbors. Still, the distribution of
under a model of drift may be missed for unlinked locus pairs compared with linked pairs: for a period of time after a variant appears, but before recombination breaks down its association with the background in which it arose, the pattern of drift shown by that mutation will be correlated to those of its neighbors. Still, the distribution of 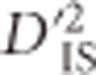 values from unlinked loci is useful to set a boundary that excludes much of the variability expected by chance alone. This boundary corresponds to a lower limit for significance. The implication is that an empirical, population-specific null distribution that accounts for random processes including mutation, migration, sampling error, genotyping error and most drift, but that excludes selection, may be developed for
values from unlinked loci is useful to set a boundary that excludes much of the variability expected by chance alone. This boundary corresponds to a lower limit for significance. The implication is that an empirical, population-specific null distribution that accounts for random processes including mutation, migration, sampling error, genotyping error and most drift, but that excludes selection, may be developed for 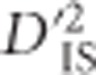 by computing the statistic over pairs of unlinked loci. Using this null distribution, critical thresholds depicting the most outlying values expected for all but the most extreme case of drift are identified. To identify pairs of loci that have been subjected to selection,
by computing the statistic over pairs of unlinked loci. Using this null distribution, critical thresholds depicting the most outlying values expected for all but the most extreme case of drift are identified. To identify pairs of loci that have been subjected to selection, 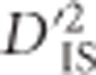 values are computed for pairs of linked loci and compared with the identified critical thresholds. In a practical setting,
values are computed for pairs of linked loci and compared with the identified critical thresholds. In a practical setting, 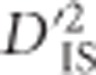 depends on the number of subpopulations included in its calculation, because fewer populations increase sampling error, so this must be accounted for when determining the critical thresholds of drift (see Materials and methods).
depends on the number of subpopulations included in its calculation, because fewer populations increase sampling error, so this must be accounted for when determining the critical thresholds of drift (see Materials and methods).
Distinguishing patterns generated due to a large selective sweep from epistatic selection
Although 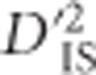 can identify linked epistatic selection and single-locus selection with linkage, the value of this statistic for an isolated pair of loci cannot alone be used to distinguish the type of selection taking place. However, in certain cases the overall pattern of
can identify linked epistatic selection and single-locus selection with linkage, the value of this statistic for an isolated pair of loci cannot alone be used to distinguish the type of selection taking place. However, in certain cases the overall pattern of 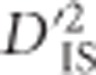 across a region, which depends on gametic frequencies, may indicate whether epistasis is likely to be at play. This results from the fact that, over generations, the extent of increased LD for pairs of loci surrounding an individual locus that is undergoing selection will diminish, since it is not being systematically maintained. When linked epistatic selection is taking place, however, increased LD between the two linked and epistatic loci will be preserved for as long as the advantage of the variant persists (Kimura, 1965). Even though the recombination rate between the loci must be smaller than the epistatic selection coefficient for this pattern to appear at all (Ohta, 1982a), over generations the probability of single and double recombination events between loci increases. Therefore, when epistatic selection is strong it is expected that a specific pair of loci may be held in tight LD due to selection, leading to elevated values of
across a region, which depends on gametic frequencies, may indicate whether epistasis is likely to be at play. This results from the fact that, over generations, the extent of increased LD for pairs of loci surrounding an individual locus that is undergoing selection will diminish, since it is not being systematically maintained. When linked epistatic selection is taking place, however, increased LD between the two linked and epistatic loci will be preserved for as long as the advantage of the variant persists (Kimura, 1965). Even though the recombination rate between the loci must be smaller than the epistatic selection coefficient for this pattern to appear at all (Ohta, 1982a), over generations the probability of single and double recombination events between loci increases. Therefore, when epistatic selection is strong it is expected that a specific pair of loci may be held in tight LD due to selection, leading to elevated values of 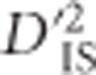 , although pairs of loci between the selected pair will display more neutral values. In other words, epistatic selection is most likely to be taking place if an elevated
, although pairs of loci between the selected pair will display more neutral values. In other words, epistatic selection is most likely to be taking place if an elevated 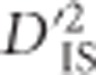 is observed for one or a few pairs of loci but not for others in the same region. Such a pattern is extremely unlikely when selection operates on a single locus with alleles linked to their background, since in this case elevated values of
is observed for one or a few pairs of loci but not for others in the same region. Such a pattern is extremely unlikely when selection operates on a single locus with alleles linked to their background, since in this case elevated values of 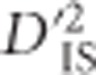 should exist across the entire region.
should exist across the entire region.
Materials and methods
Chicken data
Data were taken from the Synbreed Chicken Diversity Panel (Weigend et al., 2014), which represents a wide range of populations of individually genotyped and phenotyped chickens. The panel encompassed wild populations and domesticated breeds of various origins and histories. The panel has been shown to be capable of reconstituting phylogenetic relationships between breeds based on marker data. Chickens were genotyped using an Affymetrix Axiom HD genotyping array (Affymetrix, Santa Clara, CA, USA) (Kranis et al., 2013), for which SNPs were mapped to the Gallus_gallus_4.0 reference genome. The SNPs in this array were selected to have an approximately uniform distribution across the genome in terms of SNPs per cM, leading to a higher physical density (SNPs per kb) on microchromosomes than on macrochromosomes. Markers that were observed in fewer than 95% of individuals were removed from the data set. Next, individuals with lower than a 95% call rate for SNPs were removed. After markers and individuals were filtered, only breeds with at least 18 individuals represented were included. This left a total of 72 breeds comprised of 32 European breeds, 29 Asian breeds, 8 game breeds (from both Asia and Africa), 2 commercial broiler lines and a sample of the wild red jungle fowl (Gallus gallus gallus). The set reflected variation in major phenotype categories, such as normal sized vs dwarfs, feathering type and color, skin color, comb and crest, among others. These 72 breeds were treated as distinct populations throughout our analysis. After the quality-filtering steps were completed, 1417 individuals genotyped at 538 298 SNPs remained. The average marker spacing across the entire data set was one SNP approximately every 1700 bp.
Computing 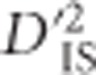
With most genotyping strategies, gametic phase cannot be assigned to double heterozygotes, even for loci on the same chromosome. However, a strategy for computing Ohta's variance components of LD that utilizes the Burrows composite measure of LD, Δij (Cockerham and Weir, 1977), has been previously derived (Black and Krafsur, 1985). The estimation of 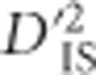 from data is particularly relevant, so we reproduce the formula here. Letting Tij measure the approximate frequency with which Ai and Bjappear in the same gamete, as done when calculating Burrows' composite measure of LD (Schaid, 2004), estimates of
from data is particularly relevant, so we reproduce the formula here. Letting Tij measure the approximate frequency with which Ai and Bjappear in the same gamete, as done when calculating Burrows' composite measure of LD (Schaid, 2004), estimates of 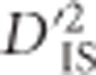 may be computed as
may be computed as
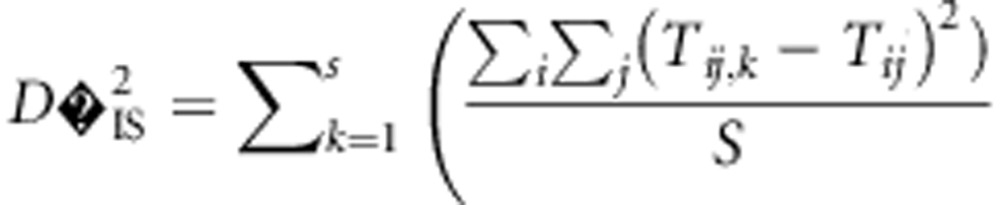 |
where s is the number of subpopulations. Tij is computed on a population basis, so although it is an expectation per population it may be considered a random variable over populations. Notice that Tij is an approximation of the random variable 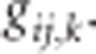 . We employed R for computation (R. Core Team, 2013) and used the resources of the University of Wisconsin, Madison Center for High Throughput Computing.
. We employed R for computation (R. Core Team, 2013) and used the resources of the University of Wisconsin, Madison Center for High Throughput Computing.
To develop the null distribution, a random sample of 2 × 109 pairs of SNPs was chosen, with the requirement that each pair consisted of two SNPs on different chromosomes. For every pair consisting of SNPs each with minor allele frequency ⩾0.1, statistics were calculated. This relatively strict exclusion of loci with low minor allele frequency was imposed to mitigate the high dependency of LD on allele frequencies. Each population was included in the computation only if the minor allele frequency of both SNPs in the pair within that population was ⩾0.05. Pairs that included SNPs on sex chromosomes were excluded from the null distribution. Since 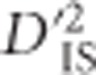 is computed as the mean of a distribution, there is a relationship between the variability of the estimated
is computed as the mean of a distribution, there is a relationship between the variability of the estimated 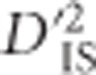 and the number of populations included in its computation. Specifically, including more populations in the computation of
and the number of populations included in its computation. Specifically, including more populations in the computation of 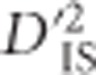 is akin to utilizing a larger sample size, in which case the sampling error of the estimator will decrease. Therefore, separate critical values were identified for each number of included populations from 15 to 60 (providing 46 distinct critical values). Situations where <15 or >60 populations were included in the comparison were too rare for critical values to be reliably drawn and were therefore removed. Critical thresholds were set as an extreme quantile of
is akin to utilizing a larger sample size, in which case the sampling error of the estimator will decrease. Therefore, separate critical values were identified for each number of included populations from 15 to 60 (providing 46 distinct critical values). Situations where <15 or >60 populations were included in the comparison were too rare for critical values to be reliably drawn and were therefore removed. Critical thresholds were set as an extreme quantile of 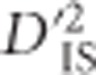 observed in the null distribution. As described in the next paragraph, there were, on average, 1 595 951 tests per threshold, so the quantile utilized was set at
observed in the null distribution. As described in the next paragraph, there were, on average, 1 595 951 tests per threshold, so the quantile utilized was set at 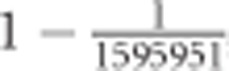 , corresponding to an expectation of one false positive per critical threshold (or 46 across the experiment). After removing locus pairs that did not conform to the filtering criteria, 1 232 649 348 pairs remained from which the critical thresholds were identified.
, corresponding to an expectation of one false positive per critical threshold (or 46 across the experiment). After removing locus pairs that did not conform to the filtering criteria, 1 232 649 348 pairs remained from which the critical thresholds were identified.
The experimental calculations for linked pairs of loci were developed similarly, but the pairs of loci used were not random. Instead, statistics were computed for every pair of markers with no more than 199 markers between them. This 200-marker distance limit was imposed due to computational limitations, as ideally all markers with any non-zero level of linkage would be compared. This restriction means that on average, pairwise comparisons between markers up 341 kb distal from one another were computed. The same allele frequency filtering used for the null distribution above was imposed for the inclusion of markers and populations when statistics was computed. To identify significant pairs of loci, 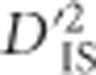 values for each pair of SNPs were compared with the corresponding critical thresholds. After locus pairs that did not conform to the filtering criteria were removed, 73 413 740 pairs were tested, computed from 447 538 unique SNPs. This provides an average of 1 595 951 locus pairs per critical threshold. As mentioned previously, one false positive is expected per threshold, providing a total of 46 over the entire experiment. This is ~2.7% of the number of significant locus pairs that were observed, demonstrating that most identified locus pairs cannot be explained by chance alone.
values for each pair of SNPs were compared with the corresponding critical thresholds. After locus pairs that did not conform to the filtering criteria were removed, 73 413 740 pairs were tested, computed from 447 538 unique SNPs. This provides an average of 1 595 951 locus pairs per critical threshold. As mentioned previously, one false positive is expected per threshold, providing a total of 46 over the entire experiment. This is ~2.7% of the number of significant locus pairs that were observed, demonstrating that most identified locus pairs cannot be explained by chance alone.
Using pathway information to investigate potential cases of linked epistatic selection
It may be the case that epistatic genes are sometimes found in close proximity to one another on chromosomes due to epistatic selection influencing genome architecture through, for example, the arrangement of genes (Schaeffer et al., 2003). Along these lines, Ohta used the variance components of LD between and within subpopulations to investigate the major histocompatibility complex cluster of genes and the possibility that similarities between species are a result of linked epistatic selection (Ohta, 1982a, b). In the spirit of this hypothesis, pathway information was utilized here to evaluate whether evidence of linked genes that are potentially epistatic could be found. This question was addressed in two ways. In the first, all available pathway information for the chicken genome was downloaded from the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa et al., 2014). Groups of genes that are mapped in close proximity to one another and predicted to belong to the same pathway were identified. The criteria for forming these groups were that, for a gene to be included, its start position had to be within 500 kb of the start position of another gene in the group, and the genes were required to belong to the same pathway. The ‘Metabolic pathways' pathway, gga01100, was excluded because it contains a large number of sub-pathways, each of which is included on its own. When a gene was predicted to belong to multiple pathways, it was permitted to belong to separate groups for each. After such gene groups were identified, they were evaluated for whether or not there was any evidence of significance according to 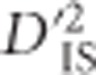 values computed in the whole-genome scan. Significant pairs of loci where one or both SNPs fell within the physical boundaries of the group were identified. This was taken as evidence in support of the hypothesis that epistatic selection on two or more of the grouped genes has taken place in the vicinity of the group of genes.
values computed in the whole-genome scan. Significant pairs of loci where one or both SNPs fell within the physical boundaries of the group were identified. This was taken as evidence in support of the hypothesis that epistatic selection on two or more of the grouped genes has taken place in the vicinity of the group of genes.
Because the whole-genome scan was limited to markers no more than 200 markers distal from one another, it failed to test potentially epistatic markers corresponding to genes further apart than this. Therefore, another analysis was performed for pairs of markers within genes predicted to belong to the same pathway and reside on the same chromosome. Again, gga01100 was excluded. 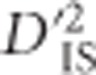 was computed for all pairwise combinations of markers located within these genes, and the computed values were compared with the critical thresholds calculated from the whole-genome scan.
was computed for all pairwise combinations of markers located within these genes, and the computed values were compared with the critical thresholds calculated from the whole-genome scan.
Results
Most 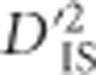 values can be explained by chance
values can be explained by chance
The null distribution of 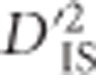 , generated by computing the statistic for pairwise combinations of loci that are unlinked, sets a lower boundary for
, generated by computing the statistic for pairwise combinations of loci that are unlinked, sets a lower boundary for 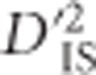 significance by estimating the degree of extremity that is explainable by chance alone. In practice, the threshold set by this null distribution appears to discriminate putatively selected regions well; among the 73 413 740 values that were computed, ~0.0023% were deemed as significant deviations from the null distribution. As mentioned previously,
significance by estimating the degree of extremity that is explainable by chance alone. In practice, the threshold set by this null distribution appears to discriminate putatively selected regions well; among the 73 413 740 values that were computed, ~0.0023% were deemed as significant deviations from the null distribution. As mentioned previously, 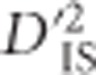 values were dependent on the number of populations included in each locus comparison and, therefore, separate significance thresholds were set for each number of populations. A decreasing trend was observed, such that the more populations included in a comparison, the lower the
values were dependent on the number of populations included in each locus comparison and, therefore, separate significance thresholds were set for each number of populations. A decreasing trend was observed, such that the more populations included in a comparison, the lower the 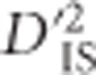 significance threshold tended to be (Supplementary Figure S1). When considering all locus pairs regardless of the number of populations used to compute
significance threshold tended to be (Supplementary Figure S1). When considering all locus pairs regardless of the number of populations used to compute 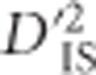 , however, it was observed that the null and experimental distributions of the statistic were quite similar, with the experimental distribution showing a small amount of inflation (Figure 1). For the null distribution, the first quantile, median and third quantile of the distribution are 0.466, 0.530, and 0.592, respectively, while for the experimental distribution the values are 0.495, 0.566, and 0.634, respectively.
, however, it was observed that the null and experimental distributions of the statistic were quite similar, with the experimental distribution showing a small amount of inflation (Figure 1). For the null distribution, the first quantile, median and third quantile of the distribution are 0.466, 0.530, and 0.592, respectively, while for the experimental distribution the values are 0.495, 0.566, and 0.634, respectively.
Figure 1.

Null vs experimental distributions. Histograms comparing the null and experimental distribution for  . Observe that the distributions are quite similar, but the experimental distribution is slightly inflated.
. Observe that the distributions are quite similar, but the experimental distribution is slightly inflated.
Evidence of selection
At the whole-genome level, 1723 locus pairs, encompassing 1802 unique loci, display values of 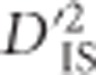 that exceed the significance thresholds, suggesting that they may have been under dispersive selection. Most of these pairs overlap with one another or cluster tightly around potentially interesting regions of the genome. Figure 2 depicts the genome-wide distribution of locus pairs that were identified as significant. Such pairs exist on 25 of the 28 chicken chromosomes studied (heterosomes and chromosomes beyond 28 were not tested), with only chromosomes 16, 25 and 26 displaying no significant pairs.
that exceed the significance thresholds, suggesting that they may have been under dispersive selection. Most of these pairs overlap with one another or cluster tightly around potentially interesting regions of the genome. Figure 2 depicts the genome-wide distribution of locus pairs that were identified as significant. Such pairs exist on 25 of the 28 chicken chromosomes studied (heterosomes and chromosomes beyond 28 were not tested), with only chromosomes 16, 25 and 26 displaying no significant pairs.
Figure 2.

Location of significant locus pairs. A whole-genome map of loci where significant values of 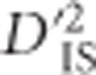 were identified in the chicken genome. The position of every marker that was part of a significant pair is represented. Positions are plotted according to the galGal4 assembly. These loci are those putatively under dispersive selective sweeps or dispersive cis-acting epistatic selection.
were identified in the chicken genome. The position of every marker that was part of a significant pair is represented. Positions are plotted according to the galGal4 assembly. These loci are those putatively under dispersive selective sweeps or dispersive cis-acting epistatic selection.
Among the pairs of loci identified as significant, a subset fell into regions that have already been shown to influence traits known to have been under selection in chickens. This supports the potential usefulness of using 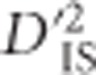 and the corresponding null distribution as a genome-wide scan for selection. One of the best-studied of these examples involves the BCDO2 gene, found on chromosome 24. This region has been shown to control yellow vs white skin in domestic chickens (Eriksson et al., 2008; Lobo et al., 2012). The chicken panel studied here included breeds fixed for yellow, white and black skin, so a sensible hypothesis would be that evidence of selection differentiating these breeds based on skin color would be apparent. This seemed to hold: on chromosome 24, an array of significant
and the corresponding null distribution as a genome-wide scan for selection. One of the best-studied of these examples involves the BCDO2 gene, found on chromosome 24. This region has been shown to control yellow vs white skin in domestic chickens (Eriksson et al., 2008; Lobo et al., 2012). The chicken panel studied here included breeds fixed for yellow, white and black skin, so a sensible hypothesis would be that evidence of selection differentiating these breeds based on skin color would be apparent. This seemed to hold: on chromosome 24, an array of significant 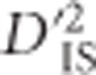 values was observed in the immediate vicinity of BCDO2 (Figure 3,Supplementary Table S1). The lack of many significant SNPs within the BCDO2 gene itself may result from breeds being fixed for one form of the gene or another, since our test was only computed over populations for which both SNPs in a pair were segregating with a minor allele frequency of at least 0.05.
values was observed in the immediate vicinity of BCDO2 (Figure 3,Supplementary Table S1). The lack of many significant SNPs within the BCDO2 gene itself may result from breeds being fixed for one form of the gene or another, since our test was only computed over populations for which both SNPs in a pair were segregating with a minor allele frequency of at least 0.05.
Figure 3.

The BCDO2 locus. Depiction of significant locus pairs identified in the BCDO2 region of chromosome 24, and the 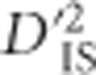 value observed for each significant pair. The height of each dashed line depicts the value of
value observed for each significant pair. The height of each dashed line depicts the value of 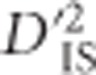 observed for that pair.
observed for that pair.
Another interesting region identified via 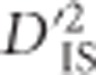 spanned a large portion of chromosome 7. Chickens harbor a segregating inversion on this chromosome from ~14.5 to 21.3 Mb (originally reported as 16.5–23.88 Mb in the galGal3 assembly) (Imsland et al., 2012). Variants within the inverted region code for comb morphology, among other phenotypes.
spanned a large portion of chromosome 7. Chickens harbor a segregating inversion on this chromosome from ~14.5 to 21.3 Mb (originally reported as 16.5–23.88 Mb in the galGal3 assembly) (Imsland et al., 2012). Variants within the inverted region code for comb morphology, among other phenotypes. 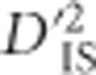 values indicate an abundance of significant locus pairs across this region (Figure 2). Similarly to the case involving BCDO2, this panel includes several breeds with large phenotypic variability for comb morphology, so it is not surprising that a high divergence between these breeds, measured using
values indicate an abundance of significant locus pairs across this region (Figure 2). Similarly to the case involving BCDO2, this panel includes several breeds with large phenotypic variability for comb morphology, so it is not surprising that a high divergence between these breeds, measured using 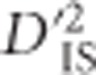 , was observed. In addition, since inversions have large impacts on recombination in a region, therefore affecting LD which the
, was observed. In addition, since inversions have large impacts on recombination in a region, therefore affecting LD which the 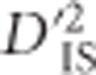 statistic is based on, this suggests that
statistic is based on, this suggests that 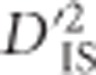 may be useful for detecting selection on structural variation. The possibility that epistatic selection acts on chromosome inversions has been demonstrated previously (Schaeffer et al., 2003).
may be useful for detecting selection on structural variation. The possibility that epistatic selection acts on chromosome inversions has been demonstrated previously (Schaeffer et al., 2003).
Patterns of epistasis and large selective sweeps
Often, regions of the genome harboring pairs of loci with significant values of 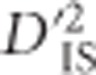 displayed generally elevated values of the statistic across the entire region. This is illustrated in Figure 4a, where significant values of
displayed generally elevated values of the statistic across the entire region. This is illustrated in Figure 4a, where significant values of 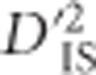 are plotted for a region on chromosome 5 that contained several significant pairs. Figure 4b shows the complete breakdown of
are plotted for a region on chromosome 5 that contained several significant pairs. Figure 4b shows the complete breakdown of 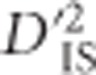 for all locus pairs in this region. Patterns such as this one are consistent with the expectation under a dispersive, large selective sweep between populations, since such an instance is expected to alter gametic frequencies and generate LD across an entire region. However, it cannot be ruled out that epistatic loci on either end of this region and others like it are resulting in the maintenance of LD between locus pairs.
for all locus pairs in this region. Patterns such as this one are consistent with the expectation under a dispersive, large selective sweep between populations, since such an instance is expected to alter gametic frequencies and generate LD across an entire region. However, it cannot be ruled out that epistatic loci on either end of this region and others like it are resulting in the maintenance of LD between locus pairs.
Figure 4.

Sweeps vs potential epistatic selection. 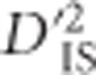 values for two example regions. a and b demonstrate region on chromosome 5 showing patterns consistent with a large selective sweep. c and d are of a region on chromosome 6, where significance was not clear except for specific locus pairs, suggestive of epistasis. (a) Significant locus pairs identified across a region of chromosome 5; (b)
values for two example regions. a and b demonstrate region on chromosome 5 showing patterns consistent with a large selective sweep. c and d are of a region on chromosome 6, where significance was not clear except for specific locus pairs, suggestive of epistasis. (a) Significant locus pairs identified across a region of chromosome 5; (b) 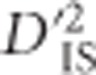 for every locus pair across the same region of chromosome 5. Values are plotted at the midpoint of the locus pair. Significant pairs are highlighted in red; (c) Significant locus pairs identified across a region of chromosome 6; (d)
for every locus pair across the same region of chromosome 5. Values are plotted at the midpoint of the locus pair. Significant pairs are highlighted in red; (c) Significant locus pairs identified across a region of chromosome 6; (d) 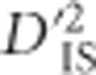 for every locus pair across the same region of chromosome 6.
for every locus pair across the same region of chromosome 6.
In other instances, the pattern of significant 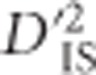 values does not correspond as clearly to the expectation under a large sweep, and it is plausible that epistatic selection has taken place. An example of this is shown in Figures 4c and d, which depict a region on chromosome 6 where significant values of
values does not correspond as clearly to the expectation under a large sweep, and it is plausible that epistatic selection has taken place. An example of this is shown in Figures 4c and d, which depict a region on chromosome 6 where significant values of 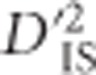 were identified. Importantly, for this region there does not appear to be an overall elevation of
were identified. Importantly, for this region there does not appear to be an overall elevation of 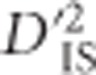 values. However, the evidence that this is a case of epistatic selection is not overwhelming: it may simply reflect a weak selective sweep and therefore less of an abundance of significant signals.
values. However, the evidence that this is a case of epistatic selection is not overwhelming: it may simply reflect a weak selective sweep and therefore less of an abundance of significant signals.
Linked epistatic selection suggested by pathway information
To further evaluate the possibility that some of the locus pairs identified as putatively under selection correspond to cases of linked epistatic selection, pathway information was investigated. Our hypothesis was that genes within the same pathway are more likely to be epistatic to each other than are genes in different pathways, and therefore we may find an excess of significant 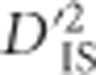 values in regions of the genome with multiple genes belonging to the same pathway. Since pathway information is incomplete, and because there is no guarantee that neighboring genes in the same pathway are epistatic, this analysis should be considered an approximation. We identified 803 groups of genes in chickens that are within 500 kb of other genes in the same pathway. Of these groups, 68 encompass at least 1 locus that is part of a pair with a significant value of
values in regions of the genome with multiple genes belonging to the same pathway. Since pathway information is incomplete, and because there is no guarantee that neighboring genes in the same pathway are epistatic, this analysis should be considered an approximation. We identified 803 groups of genes in chickens that are within 500 kb of other genes in the same pathway. Of these groups, 68 encompass at least 1 locus that is part of a pair with a significant value of 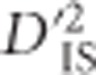 from our scan. Supplementary Table S2 contains information describing each of these 68 groups of genes. Since the groups identified pertain to loci with significant
from our scan. Supplementary Table S2 contains information describing each of these 68 groups of genes. Since the groups identified pertain to loci with significant 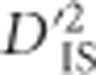 values, this is evidence that they may have been subjected to some sort of dispersive selection. Moreover, because the highlighted groups contain genes previously predicted to correspond to the same pathway, these represent candidates for gene clusters that are epistatic.
values, this is evidence that they may have been subjected to some sort of dispersive selection. Moreover, because the highlighted groups contain genes previously predicted to correspond to the same pathway, these represent candidates for gene clusters that are epistatic.
Similarly, we computed 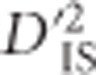 values to test for epistatic selection between pairs of genic SNPs for all sets of genes that are on the same chromosome and members of the same pathway. This analysis was not limited by the 200-marker distance limit imposed for the genome scan, yet due to computational limitations it was restricted to include only markers located within the start and end positions of known gene models. Four locus pairs that exceeded the thresholds derived from the null distribution of
values to test for epistatic selection between pairs of genic SNPs for all sets of genes that are on the same chromosome and members of the same pathway. This analysis was not limited by the 200-marker distance limit imposed for the genome scan, yet due to computational limitations it was restricted to include only markers located within the start and end positions of known gene models. Four locus pairs that exceeded the thresholds derived from the null distribution of 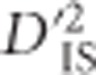 were identified (Table 1). These four pairs of loci represent the most likely among those we tested to have been subjected to epistatic selection in chickens. Interestingly, one of the four pairs consisted of two SNPs ~8-Mb apart, a distance unlikely to be the result of a single-locus sweep. The corresponding genes were PIK3CB and IL1RAP, both in the apoptosis pathway (gga04210) (Kanehisa et al., 2014). Previous studies have shown an indirect interaction between PIK3CB and IL1RAP via both genes interacting with a common gene (PIK3R1) (Sizemore et al., 1999; Havugimana et al., 2012). PIK3CB also appeared as significant in a pair with CAB39 in the mTOR signaling pathway (gga04150) (Kanehisa et al., 2014).
were identified (Table 1). These four pairs of loci represent the most likely among those we tested to have been subjected to epistatic selection in chickens. Interestingly, one of the four pairs consisted of two SNPs ~8-Mb apart, a distance unlikely to be the result of a single-locus sweep. The corresponding genes were PIK3CB and IL1RAP, both in the apoptosis pathway (gga04210) (Kanehisa et al., 2014). Previous studies have shown an indirect interaction between PIK3CB and IL1RAP via both genes interacting with a common gene (PIK3R1) (Sizemore et al., 1999; Havugimana et al., 2012). PIK3CB also appeared as significant in a pair with CAB39 in the mTOR signaling pathway (gga04150) (Kanehisa et al., 2014).
Table 1. Four locus pairs displaying putative evidence of epistatic selection.
| Chr. | Marker_1 | Marker_2 | Marker_1 position | Marker_2 position | Number pops | D′2IS | Pathway | Gene_1 | Gene_2 |
|---|---|---|---|---|---|---|---|---|---|
| 7 | AX-77016330 | AX-77017217 | 26503508 | 26843408 | 41 | 1.00306 | gga04270 | ADCY5 | MYLK |
| 9 | AX-77173228 | AX-77173761 | 4913635 | 5110804 | 18 | 1.22067 | gga04150 | CAB39 | PIK3CB |
| 9 | AX-77173228 | AX-77173770 | 4913635 | 5113445 | 17 | 1.26847 | gga04150 | CAB39 | PIK3CB |
| 9 | AX-77173770 | AX-77134125 | 5113445 | 13345894 | 27 | 1.12192 | gga04210 | PIK3CB | IL1RAP |
Abbreviation: SNP, single-nucleotide polymorphism.
SNP pairs shown here correspond to pairs of distinct genes predicted to be in the same pathway (Kanehisa et al., 2014). The corresponding 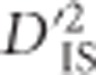 values exceed the critical thresholds computed from the null distribution.
values exceed the critical thresholds computed from the null distribution.
Discussion
Haplotype-based scan
Methods commonly used to identify selection from dense marker or sequencing data at the whole-genome level can be broadly divided into two classes. First, there are approaches that seek to identify selection based on variability between populations, usually based on tests such as that proposed by Lewontin and Krakauer (1973). Second, there are methods tailored for studying variability within a single population, which tend to utilize patterns developed due to genetic hitchhiking (Maynard Smith and Haigh, 1974). Another subset of methods is based on a combination of ideas from each of these classes. These include the XP-CLR method, which is based on differences in multi-locus allele frequencies between populations (Chen et al., 2010), the XP-EHH method, involving a search for long-range haplotypes that have been generated from differential hard sweeps between populations (Sabeti et al., 2007), and more recently the hapFLK approach, which uses haplotype information and seeking to explicitly incorporate population stratification (Fariello et al., 2013). The use of 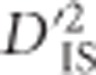 to detect selection is similar to this latter group of methods given that evaluating gametic frequencies of locus pairs is a form of haplotype analysis, with haplotypes defined based on two loci. However, the roots of
to detect selection is similar to this latter group of methods given that evaluating gametic frequencies of locus pairs is a form of haplotype analysis, with haplotypes defined based on two loci. However, the roots of 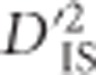 are grounded in its analogy with FST (Ohta, 1982a, b). Therefore, it is comparable to an FST-based scan as well. The scan may be used to identify pairs of loci depicting correlated patterns of selection while retaining much of the straight-forward interpretability of an FST approach. Similar to other haplotype-based approaches, however,
are grounded in its analogy with FST (Ohta, 1982a, b). Therefore, it is comparable to an FST-based scan as well. The scan may be used to identify pairs of loci depicting correlated patterns of selection while retaining much of the straight-forward interpretability of an FST approach. Similar to other haplotype-based approaches, however, 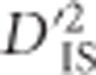 as we implement it has no power to detect selection after alleles have become fixed in all subpopulations.
as we implement it has no power to detect selection after alleles have become fixed in all subpopulations.
An advantage of 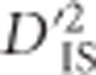 is that it allows the construction of a null distribution from the data. This null distribution captures most of the variability of
is that it allows the construction of a null distribution from the data. This null distribution captures most of the variability of 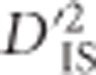 that may be explained by invoking only stochastic processes such as sampling error and genetic drift. It has proven useful for defining a lower boundary for significance. This lower boundary represents a substantial improvement over most single-locus tests for selection, especially those that employ FST, since these tests usually rely on arbitrary outlier thresholds, for example, the upper 99% quantile of the data distribution (Akey, 2009), which guarantees false positives if selection has not taken place. The null distribution described here does not eliminate the possibility of drift-generated values that appear to be significant, so it should be considered a hybrid approach between the commonly employed outlier thresholds and true statistical significance. This is because our null eliminates most values that are explainable without selection, yet values above the threshold can potentially be explained without invoking selection as well. Therefore, such locus pairs should be treated as outliers for which selection is likely, but not necessarily, the explanation. An important advantage of this approach, however, is that unlike other typical outlier approaches, there is no guarantee of false positives in the absence of selection—if
that may be explained by invoking only stochastic processes such as sampling error and genetic drift. It has proven useful for defining a lower boundary for significance. This lower boundary represents a substantial improvement over most single-locus tests for selection, especially those that employ FST, since these tests usually rely on arbitrary outlier thresholds, for example, the upper 99% quantile of the data distribution (Akey, 2009), which guarantees false positives if selection has not taken place. The null distribution described here does not eliminate the possibility of drift-generated values that appear to be significant, so it should be considered a hybrid approach between the commonly employed outlier thresholds and true statistical significance. This is because our null eliminates most values that are explainable without selection, yet values above the threshold can potentially be explained without invoking selection as well. Therefore, such locus pairs should be treated as outliers for which selection is likely, but not necessarily, the explanation. An important advantage of this approach, however, is that unlike other typical outlier approaches, there is no guarantee of false positives in the absence of selection—if 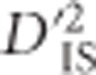 is highly variable due to only drift and sampling error, it is possible that few or no experimental locus pairs will exceed the null.
is highly variable due to only drift and sampling error, it is possible that few or no experimental locus pairs will exceed the null.
Epistasis
Although Ohta's introduction of the variance components of LD was for the study of epistatic selection (Ohta, 1982a, b), we have demonstrated that 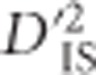 is not exclusively limited to that phenomenon. In particular, it is capable of identifying loci that are in LD due to epistasis, or it may identify pairs of loci that are members of a haplotype undergoing a hard sweep. Our results suggest that ascertaining the phenomenon taking place is not straight forward. Specifically, when it is known or hypothesized a priori that epistasis for a selected trait may exist for a set of loci, using
is not exclusively limited to that phenomenon. In particular, it is capable of identifying loci that are in LD due to epistasis, or it may identify pairs of loci that are members of a haplotype undergoing a hard sweep. Our results suggest that ascertaining the phenomenon taking place is not straight forward. Specifically, when it is known or hypothesized a priori that epistasis for a selected trait may exist for a set of loci, using 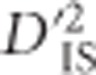 to test this hypothesis seems appropriate and, in such a situation, the null distribution developed here can be used to better establish significance than previous approaches. Using this idea, we isolated and tested pairwise combinations of SNPs within sets of genes on the same chromosome and in the same pathway. We identified four pairs of SNPs on which epistatic selection is suggested, although it cannot be ruled out that these genes simply fall within a selected haplotype.
to test this hypothesis seems appropriate and, in such a situation, the null distribution developed here can be used to better establish significance than previous approaches. Using this idea, we isolated and tested pairwise combinations of SNPs within sets of genes on the same chromosome and in the same pathway. We identified four pairs of SNPs on which epistatic selection is suggested, although it cannot be ruled out that these genes simply fall within a selected haplotype.
Conversely, in extreme cases where epistatic selection between linked loci is ancient, a unique pattern of 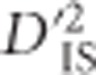 is expected to form which may indicate linked epistatic selection. However, excluding this pattern (rarely seen in this data set), it is probable that
is expected to form which may indicate linked epistatic selection. However, excluding this pattern (rarely seen in this data set), it is probable that 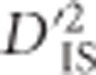 generally identifies major sweeps that vary between populations and arise from selection on a single, non-epistatic, locus. As such, an evaluation of the pairwise patterns of significant loci that were observed in this experiment (for example, Figure 4a and b),
generally identifies major sweeps that vary between populations and arise from selection on a single, non-epistatic, locus. As such, an evaluation of the pairwise patterns of significant loci that were observed in this experiment (for example, Figure 4a and b), 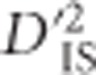 appears to define the boundaries of selected haplotypes well. In particular, overlap between pairs of loci that are deemed significant may suggest the extent of any sweep that has occurred (a lack of overlap may be indicative of separate sweeps).
appears to define the boundaries of selected haplotypes well. In particular, overlap between pairs of loci that are deemed significant may suggest the extent of any sweep that has occurred (a lack of overlap may be indicative of separate sweeps).
Future directions
While 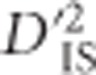 can be employed to identify pairs of loci that have undergone selection, there are components of this work that would benefit from further exploration. One aspect involves our use of the Burrows approximation (Cockerham and Weir, 1977) to estimate gametic frequencies. Without family data, utilizing known gametic frequencies was not possible, but if this information were available it would likely improve the accuracy of the test. In addition, computation is intensive, which poses a challenge. Ideally,
can be employed to identify pairs of loci that have undergone selection, there are components of this work that would benefit from further exploration. One aspect involves our use of the Burrows approximation (Cockerham and Weir, 1977) to estimate gametic frequencies. Without family data, utilizing known gametic frequencies was not possible, but if this information were available it would likely improve the accuracy of the test. In addition, computation is intensive, which poses a challenge. Ideally, 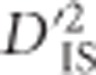 values would be computed for all pairs of loci that are potentially linked. Since this was not feasible without increased computational power, our solution was to compute values for sets of loci that are most likely to be linked. This was done by allowing the distance between them to span up to 200 markers, or ~341 kb. With advancements in computational methods and in high throughput computing, extending the distance between pairs of markers will be possible in the future. In addition, an analysis at an even larger scale may become more feasible if coded in a lower-level language than R, as was used here. However, as the number of locus pairs considered is expanded, the number of pairs used to develop the null distribution needs to increase as well, which adds to the computational cost.
values would be computed for all pairs of loci that are potentially linked. Since this was not feasible without increased computational power, our solution was to compute values for sets of loci that are most likely to be linked. This was done by allowing the distance between them to span up to 200 markers, or ~341 kb. With advancements in computational methods and in high throughput computing, extending the distance between pairs of markers will be possible in the future. In addition, an analysis at an even larger scale may become more feasible if coded in a lower-level language than R, as was used here. However, as the number of locus pairs considered is expanded, the number of pairs used to develop the null distribution needs to increase as well, which adds to the computational cost.
Finally, we have mentioned the difficulty of distinguishing whether a significant 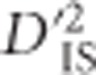 value for a pair of loci is the result of a single-locus sweep or epistatic selection between pairs of loci. We also discussed a unique pattern of
value for a pair of loci is the result of a single-locus sweep or epistatic selection between pairs of loci. We also discussed a unique pattern of 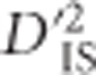 that may establish, more conclusively, if epistatic selection is at play. A more formal investigation of the differences between patterns generated by each type of selection may prove fruitful.
that may establish, more conclusively, if epistatic selection is at play. A more formal investigation of the differences between patterns generated by each type of selection may prove fruitful.
Data archiving
Data are available from figshare: http://dx.doi.org/10.6084/m9.figshare.1497961.
Acknowledgments
We are indebted to all the breeders who supported sampling from their animals. This research was funded by the German Federal Ministry of Education and Research (BMBF) within the AgroClustEr ‘Synbreed—Synergistic plant and animal breeding' (FKZ 0315528). In addition, we used the computational resources and assistance of the University of Wisconsin—Madison Center For High Throughput Computing (CHTC) in the Department of Computer Sciences. The CHTC is supported by UW-Madison and the Wisconsin Alumni Research Foundation, and is an active member of the Open Science Grid, which is supported by the National Science Foundation and the US Department of Energy's Office of Science. TMB is supported by the University of Wisconsin Graduate School and by funding to the University of Wisconsin–Madison Plant Breeding and Plant Genetics program from Monsanto.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on Heredity website (http://www.nature.com/hdy)
Supplementary Material
References
- Akey JM. (2009). Constructing genomic maps of positive selection in humans: where do we go from here? Genome Res 19: 711–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akey JM, Zhang G, Zhang K, Jin L, Shriver MD. (2002). Interrogating a high-density SNP map for signatures of natural selection. Genome Res 12: 1805–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beissinger TM, Hirsch CN, Vaillancourt B, Deshpande S, Barry K, Buell CR et al. (2013). A genome-wide scan for evidence of selection in a maize population under long-term artificial selection for ear number. Genetics 196: 829–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black WC IV, Krafsur ES. (1985). A FORTRAN program for the calculation and analysis of two-locus linkage disequilibrium coefficients. Theor Appl Genet 70: 491–496. [DOI] [PubMed] [Google Scholar]
- Chen H, Patterson N, Reich D. (2010). Population differentiation as a test for selective sweeps. Genome Res 20: 393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockerham CC, Weir BS. (1977). Digenic descent measures for finite populations. Genet Res 30: 121–147. [Google Scholar]
- Eriksson J, Larson G, Gunnarsson U, Bed'hom B, Tixier-Boichard M, Stromstedt L et al. (2008). Identification of the yellow skin gene reveals a hybrid origin of the domestic chicken. PLoS Genet 4: e1000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fariello MI, Boitard S, Naya H, SanCristobal M, Servin B. (2013). detecting signatures of selection through haplotype differentiation among hierarchically structured populations. Genetics 193: 929–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnier-Gere P, Dillmann C. (1992). A computer program for testing pairwise linkage disequilibria in subdivided populations. J Hered 83: 239–239. [DOI] [PubMed] [Google Scholar]
- Havugimana PC, Hart GT, Nepusz T, Yang H, Turinsky AL, Li Z et al. (2012). A census of human soluble protein complexes. Cell 150: 1068–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill WG, Weir BS. (1988). Variances and covariances of squared linkage disequilibria in finite populations. Theor Popul Biol 33: 54–78. [DOI] [PubMed] [Google Scholar]
- Hirsch CN, Flint-Garcia SA, Beissinger TM, Eichten SR, Deshpande S, Barry K et al. (2014). Insights into the effects of long-term artificial selection on seed size in maize. Genetics 198: 409–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imsland F, Feng C, Boije H, Bed'hom B, Fillon V, Dorshorst B et al. (2012). The rose-comb mutation in chickens constitutes a structural rearrangement causing both altered comb morphology and defective sperm motility. PLoS Genet 8: e1002775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. (2014). Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res 42: D199–D205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly JK. (1997). A test of neutrality based on interlocus associations. Genetics 146: 1197–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. (1965). Attainment of quasi linkage equilibrium when gene frequencies are changing by natural selection. Genetics 52: 875–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranis A, Gheyas AA, Boschiero C, Turner F, Yu L, Smith S et al. (2013). Development of a high density 600K SNP genotyping array for chicken. BMC Genomics 14: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewontin RC, Krakauer J. (1973). Distribution of gene frequency as a test of the theory of the selective neutrality of polymorphisms. Genetics 74: 175–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo GP, Isken A, Hoff S, Babino D, von Lintig J. (2012). BCDO2 acts as a carotenoid scavenger and gatekeeper for the mitochondrial apoptotic pathway. Dev Camb Engl 139: 2966–2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X-F, Hall D, Onge KRS, Jansson S, Ingvarsson PK. (2010). Genetic differentiation, clinal variation and phenotypic associations with growth cessation across the populus tremula photoperiodic pathway. Genetics 186: 1033–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard Smith J, Haigh J. (1974). The hitch-hiking effect of a favourable gene. Genet Res 23: 23–35. [PubMed] [Google Scholar]
- Miyashita NT, Aguadé M, Langley CH. (1993). Linkage disequilibrium in the white locus region of Drosophila melanogaster. Genet Res 62: 101–109. [DOI] [PubMed] [Google Scholar]
- Nagylaki T. (1993). The evolution of multilocus systems under weak selection. Genetics 134: 627–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta T. (1982. a). Linkage disequilibrium due to random genetic drift in finite subdivided populations. Proc Natl Acad Sci USA 79: 1940–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta T. (1982. b). Linkage disequilibrium with the island model. Genetics 101: 139–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parts L, Cubillos FA, Warringer J, Jain K, Salinas F, Bumpstead SJ et al. (2011). Revealing the genetic structure of a trait by sequencing a population under selection. Genome Res 21: 1131–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R. Core TeamR: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria.
- Sabeti PC, Reich DE, Higgins JM, Levine HZP, Richter DJ, Schaffner SF et al. (2002). Detecting recent positive selection in the human genome from haplotype structure. Nature 419: 832–837. [DOI] [PubMed] [Google Scholar]
- Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E, Cotsapas C et al. (2007). Genome-wide detection and characterization of positive selection in human populations. Nature 449: 913–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer SW, Goetting-Minesky MP, Kovacevic M, Peoples JR, Graybill JL, Miller JM et al. (2003). Evolutionary genomics of inversions in Drosophila pseudoobscura: Evidence for epistasis. Proc Natl Acad Sci USA 100: 8319–8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaid DJ. (2004). Linkage disequilibrium testing when linkage phase is unknown. Genetics 166: 505–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sizemore N, Leung S, Stark GR. (1999). Activation of phosphatidylinositol 3-kinase in response to interleukin-1 leads to phosphorylation and activation of the NF-kappaB p65/RelA subunit. Mol Cell Biol 19: 4798–4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song B-H, Windsor AJ, Schmid KJ, Ramos-Onsins S, Schranz ME, Heidel AJ et al. (2009). Multilocus patterns of nucleotide diversity, population structure and linkage disequilibrium in Boechera stricta, a wild relative of Arabidopsis. Genetics 181: 1021–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz JF, Kelly JK. (2008). Effects of spatially varying selection on nucleotide diversity and linkage disequilibrium: insights from deer mouse globin genes. Genetics 180: 367–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang K, Thornton KR, Stoneking M. (2007). A new approach for using genome scans to detect recent positive selection in the human genome. PLoS Biol 5: e171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner TL, Stewart AD, Fields AT, Rice WR, Tarone AM. (2011). Population-based resequencing of experimentally evolved populations reveals the genetic basis of body size variation in Drosophila melanogaster. PLoS Genet 7: e1001336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitalis R, Riba M, Colas B, Grillas P, Olivieri I. (2002). Multilocus genetic structure at contrasted spatial scales of the endangered water fern Marsilea strigosa Willd. (Marsileaceae, Pteridophyta). Am J Bot 89: 1142–1155. [DOI] [PubMed] [Google Scholar]
- Voight BF, Kudaravalli S, Wen X, Pritchard JK. (2006). A map of recent positive selection in the human genome. PLoS Biol 4: e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigend S, Janssen-Tapken U, Erbe M, Ober U, Weigend A, Preisinger R et al. (2014). Biodiversität beim huhn–potenziale für die praxis. Züchtungskunde 86: 25–41. [Google Scholar]
- Wisser RJ, Murray SC, Kolkman JM, Ceballos H, Nelson RJ. (2008). Selection mapping of loci for quantitative disease resistance in a diverse maize population. Genetics 180: 583–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S. (1949). The genetical structure of populations. Ann Eugen 15: 323–354. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.