

Abstract
Proteus mirabilis is a common urinary tract pathogen, and may induce various inflammation symptoms. Its notorious ability to resist multiple antibiotics and to form urinary tract stones makes its treatment a long and painful process, which is further challenged by the frequent horizontal gene transferring events in P. mirabilis genomes. Three strains of P. mirabilis C02011/C04010/C04013 were isolated from a local outbreak of a food poisoning event in Shenzhen, China. Our hypothesis is that new genes may have been acquired horizontally to exert the digestion tract infection and toxicity. The functional characterization of these three genomes shows that each of them independently acquired dozens of virulent genes horizontally from the other microbial genomes. The representative strain C02011 induces the symptoms of both vomit and diarrhea, and has recently acquired a complete type IV secretion system and digestion tract toxic genes from the other bacteria.
Introduction
The gram-negative anaerobic bacterium Proteus mirabilis is one of the major Proteus infectious factors, and may cause severe pain by forming stones in the urinary tract [1]. P. mirabilis may induce various pathogenic symptoms, including fever, chills and chest pain, etc [2]. Its motility and adhesion to solid surface makes P. mirabilis extremely easy to propagate through medical devices in hospitals [2]. P. mirabilis is also notorious for its ability to actively acquire antibiotic resistance and infectious toxin genes from other microbial genomes through horizontal gene transferring [3, 4].
Quite a number of molecular typing technologies have been developed for a prompt screening of Proteus mirabilis in clinical samples. The protein UreC is the alpha subunit of the urea degradation pathway encoded by P. mirabilis [5]. The second metabolite repressor gene rsmA is involved in the swarming motility and the expression regulation of virulence factors in P. mirabilis [6]. P. mirabilis also encodes a kidney damage inducer, the hemolysin HpmA, which shows significant cytotoxicity [7]. The virulence factor metalloprotease ZapA is demonstrated to regulate the IgA hydrolysis process by the inhibiting profiles [8]. A number of social network analyzing techniques were applied to characterize the disease-associated microRNAs [9, 10], and the improved detection performance suggests that techniques from different areas may be complementary to each other for the challenging detection problem for the disease-microRNA association [11, 12], which may benefit the detection problem of pathogenicity genes in P. mirabilis.
This study hypothesized that P. mirabilis exerts its infectious and toxic functions in the new location, digestion tract, through site specific genes. Three P. mirabilis strains isolated from a local outbreak of food poison, and two more non-toxic strains were isolated from the same physical location after the outbreak event. The genes and repetitive elements were annotated for all the P. mirabilis strains. A comprehensive screening of the digestion tract toxicity (DTT) specific genes is carried out for the P. mirabilis infectious strains. The comparative study supports our hypothesis that some strain specific genes may serve as candidates for rapid strain typing and treatment selection.
Material and Methods
Strain isolations
During an outbreak of a food-poisoning event in Shenzhen in June of 2002, three strains of P. mirabilis were collected from samples of different patients. The strain C02011 was isolated from the vomit sample of a patient, and it has the same Pulsed-Field Gel Electrophoresis (PFGE) band of the P. mirabilis strain isolated from the diarrhea stool. So the strain C02011 is kept as a representative strain for further analysis. The strain C04010 was isolated from the diarrhea stool sample of a patient in the same event. Another strain C04013 was isolated from the food taken by the patients immediately before this food-poisoning event.
These three strains of P. mirabilis were compared with two locally isolated strains and two reference strains. Another two strains of P. mirabilis C02034 and B02005 were isolated from the food and food worker’s stool during non-outbreak time at the same physical location. The two reference strains HI4320 [2] and BB2000 [13] were urinary tract infection strains, and were also chosen as the control samples for the comparative genomics investigation.
This study was approved by the Ethics Committee of Shenzhen Center for Disease Control and Prevention on June 9, 2002. The authors did not collect the samples directly from the patients, and received the samples from the local hospitals. This study investigated the genome sequencing data of the pathogens and did not access the patients’ personal data. So no informed consent forms were collected from the patients.
Genomic sequencing and gene annotation
The five newly isolated strains were sequenced and assembled by the BGI-Shenzhen. Two insert fragment lengths 500 and 2000 bps were chosen for the paired-end (PE) sequencing using the Illumina HiSeq-2000 machine. The read lengths were set as PE-90 and PE-49 for the 500-bp and 2000-bp libraries, respectively. The 500-bp paired-end reads were de novo assembled using the program SOAPdenovo version 1.06, and the contigs were further connected into scaffolds using both 500-bp and 2000-bp PE reads [14]. The genes were annotated using the program Glimmer version 3.02 [15]. The functions of each gene were annotated using the NCBI BLAST based on the databases NCBI NR [16], SwissProt/Trembl [17], COG [18] and KEGG [19], respectively. The data are summarized in the Results and discussion section. Transcriptome is not investigated in this study and the microRNA precursor genes will be characterized by both in silico techniques [20] and RNA-seq experiments [21].
Annotation of repetitive elements
Repetitive elements are not annotated by default for a genome sequencing service, and a comprehensive annotation is conducted for the five sequenced strains and the two reference strains of P. mirabilis investigated in this study.
Repetitive elements were annotated by the RepeatMasker version 4.05 [22]. The tandem repeats were screened by the de novo program TRF version 4.04 [23]. Prokaryotic genomes have transposable elements like Insertion Sequence (IS) elements [24, 25] and Miniature Inverted-repeat Transposable Elements (MITEs) [26–28]. Full copies of IS elements were detected and curated by multiple sequence alignments of IS encoded transposase genes, using the program MEGA version 6.06 [29]. Full copies of detected IS elements and all other full-length IS copies from the database ISfinder [30] were used as templates to screen for all the IS copies in the seven strains of P. mirabilis. No MITEs were detected by the program MUST version 1.0 [31].
CRISPRs are a series of consecutive short repeats with spacers in between, and the spacer sequences are usually acquired from the invasive foreign elements [32, 33]. The program CRISPRfinder [34] was used to detect CRISPRs in the seven investigated P. mirabilis genomes.
Generation of the phylogenetic tree
A phylogenetic tree based on the 16S ribosomal RNA (rRNA) genes is generated for the five P. mirabilis strains sequenced by this study and the three additional P. mirabilis reference strains published elsewhere. The 16S rRNA genes of the five newly sequenced P. mirabilis strains are annotated as described in the above sections. The 16S rRNA genes of the two P. mirabilis strains HI4320 and BB2000 are collected from the NCBI database. C05028 is another publicly available strain with DTT function [35], and its 16S rRNA gene is extracted using NCBI MegaBlast version 2.2.32+ [36] with the 16S rRNA of P. mirabilis HI4320 as template. The phylogenetic tree is rooted at Proteus penneri ATCC 35198, whose 16S rRNA gene is detected with the template gene from the strain HI4320 using the program NCBI BLASTN version 2.2.32+ [36]. The E-value 0.0 supports that the two microbial genomes are close relatives with enough statistical significance. The phylogenetic tree is generated using Phylogeny.fr [37]. All the programs are executed using the default parameters.
Screening for the strain specific genes
A highly sensitive screening of whether a gene has a homolog in a given genome is conducted using the NCBI MegaBLAST version 2.2.32+ [36] with the default parameters. Only those genes with no homologs are kept as strain specific candidates.
Results and Discussion
In vitro PFGE strain typing
Three isolated P. mirabilis strains demonstrate an abnormal infectious ability and toxicity to the digestion tract, but two other strains don’t have such ability. So the first question is whether the existing molecular typing technologies may differentiate these strains by their pathogenesis.
PFGE is a widely used rapid subtyping technique for the macrorestriction analysis of pathogens, as recommended by the PulseNet laboratories [38]. 50 U of the restriction enzyme SfiI in the 200 ml buffer was used to generate the genomic fragment profile, and a standard protocol was followed [39]. After the gel was scanned as the TIFF format image, a dendrogram tree was calculated using the software BioNumerics version 5.0 (Applied Maths BVBA, Belgium), as similar in [40].
The restriction enzyme SfiI cuts the genomic sequence into fragments of lengths between 20–700 kbps, and the PFGE profiles in Fig 1 shows significant differences even between closely related P. mirabilis strains. The inconsistency of PFGE profiles of these six P. mirabilis strains with their pathogenicity suggests that these genomes may under strong selection pressures for genomic changes, and may undergo both intra-genome structural variations and inter-genome/strain horizontal transferring.
Fig 1. The dendrogram tree of six P. mirabilis strains.

Strains C05028 and C02011 are digestion tract infectious. Strains HI4320 and ATCC29906 are urinary tract infectious. Strains B02005 and C02034 are non-infectious strains.
In vitro PCR strain typing
The polymerase chain reaction (PCR) is also a common technique to subtype pathogens based on the existences of known pathogenic factors [41]. The PCR-based screening of the genes ureC, rsmA, hpmA and zapA were conducted in the five newly isolated strains of P. mirabilis. As shown in Table 1, the primers for these four genes were designed in-house, and a standard PCR protocol was followed.
Table 1. PCR primers for the four genes.
| Target | pName | Primer (5’-3’) | Product (bps) |
|---|---|---|---|
| ureC | ureC-F | GTATTCGTCGCGAAACCATT | 553 |
| ureC-R | ATATGACGACAGCCCTCCAC | ||
| rsmA | rsmA-F | TAGCGAGTGTTGACGAGTGG | 562 |
| rsmA-R | AGCGAGGTGAAGAACGAGAA | ||
| hpmA | hpmA-F | CCAGTGAATTAACGGCAGGT | 654 |
| hpmA-R | CGTGCCCAGTAATGGCTAAT | ||
| zapA | zapA-F | AACGCAGGTCAGAATGTTCC | 553 |
| zapA-R | TATCTTGTCCACGACCACCA |
Column pName lists the names of the primers.
Unfortunately, there is no difference between the digestion tract toxic strains and the other strains, since all the five strains encode all these four genes, as shown in Table 2 and S1 Fig. So the existing pathogen subtyping techniques are not precise enough to differentiate these five P. mirabilis strains.
Table 2. Currently adopted molecular typing technologies of biomarker genes in the infectious bacterial strains.
This study carried out a comprehensive screening of functional elements that are specific to these three digestion tract toxic strains, compared against the other two digestion tract infectious strains. These strain specific genes may serve as candidate subtyping targets.
Summary of the seven P. mirabilis strains
P. mirabilis strain C02011 induces the symptoms of both vomit and diarrhea, and is chosen as the reference genome for the screening of DTT genes. Among the seven P. mirabilis strains, C02034 and HI4320 have the genome sizes larger than 4 Mbps, and all the other strains have a genome size between 3.80 and 3.85 Mbps, as shown in Table 3. It’s interesting to observe that these two largest genomes also have the largest numbers of IS elements, with 28 and 24 in C02034 and HI4320, respectively. This supports the observation that IS number tends to be positively correlated with the genome size [25, 42]. All the seven strains have a similar GC content between 38.38–38.88%. The Pearson’s test generates the P-value = 7.341e-4 for the correlation between the Gsize and Gene# for the seven strains of P. mirabilis. So a large genome tends to encode more genes. Only the two strains C02011 and C02034 have 2 CRISPRs, respectively.
Table 3. Annotation summary of the seven P. mirabilis strains investigated in this study.
| Strain | Gsize (bps) | GC (%) | Gene# | IS | CRISPR | rDrug | Toxin | T4SS | DTT specific |
|---|---|---|---|---|---|---|---|---|---|
| C02011 | 3802255 | 38.45 | 3,463 | 20 | 2 | 45 | 17 | 14 | 62 |
| C04010 | 3803688 | 38.38 | 3,465 | 17 | 0 | 46 | 30 | 3 | 66 |
| C04013 | 3844051 | 38.46 | 3,511 | 11 | 0 | 46 | 10 | 3 | 59 |
| B02005 | 3830296 | 38.56 | 3,516 | 12 | 0 | 45 | 0 | 3 | - |
| C02034 | 4166171 | 38.62 | 3,761 | 28 | 2 | 51 | 0 | 3 | - |
| BB2000 | 3846754 | 38.60 | 3,455 | 15 | 0 | 42 | - | 3 | - |
| HI4320 | 4063606 | 38.88 | 3,607 | 24 | 0 | 41 | - | 3 | - |
Columns “Gsize (bps)” and “GC” give the estimated genome size and the G+C content of each strain. The numbers of annotated genes and IS elements are given in the columns “Gene#” and “IS”. The annotated CRISPR number is given in the column “CRISPR”. The numbers of drug resistant genes and toxin genes are based on the gene function annotation and listed in the columns “rDrug” and “Toxin”, respectively. The gene number in the T4SS is in the column “T4SS”, which is summarized from the gene annotations. The column “DTT specific” gives the number of genes with no homologs in the non-toxin P. mirabilis strains. “-”means that this item is not analyzed for this strain. The gene annotation of the strain C05028 is not publicly available, and is not included in this table.
All the seven P. mirabilis strains encode drug resistance genes, but only the three strains with DTT function encode toxin genes, as shown in Table 3. The type IV secretion system (T4SS) does not seem to be a major secretion system in P. mirabilis, with 3 T4SS genes in each strain except for C02011. And each of the three digestion tract toxic strains encodes ~60 genes, which are not detected in the other P. mirabilis strains.
This study will focus on the DTT specific genes that may facilitate the radical pathogenesis during the local food poison event.
Phylogenetic traces
P. mirabilis is notorious for its infection and stone forming in the urinary tract, and usually causes severe pain. The two publicly available strains BB2000 and HI4320 may induce various whole body symptoms when infecting the urinary tract [2, 13]. But the five strains isolated from Shenzhen and the strain C05028 seem to have gained the ability to infect the digestion tract, and four of them demonstrate DTT function during their infections.
The five locally isolated strains of P. mirabilis together with the strain C05028 originate from the same common ancestor, whereas the other two urinary tract infectious strains BB2000 and HI4320 originate from another common ancestor, based on the phylogenetic tree in Fig 2. The two non-toxic strains C02034 and B02005 do not show a clear separation from the three digestion tract toxic (DTT) strains on the phylogenetic tree. This suggests that genes specific to DTT strains may play an essential role in infecting and inducing toxicity to the digestion tract.
Fig 2. The phylogenetic tree of the seven P. mirabilis strains based on the 16S rRNA genes.
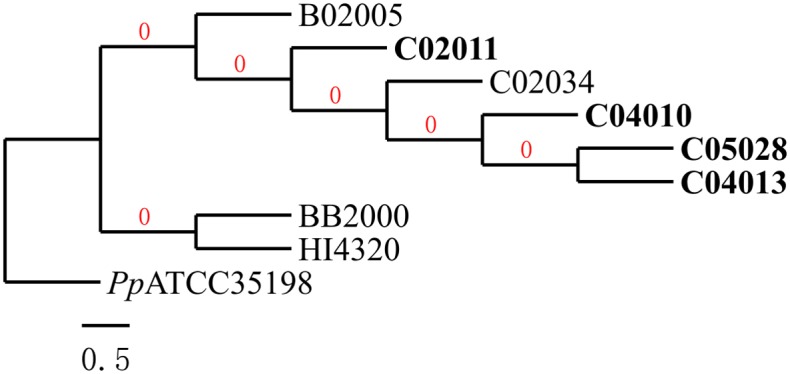
The Cladogram tree is rooted at Proteus penneri ATCC 35198, and generated by the Phylogeny.fr. The three P. mirabilis strains inducing DTT function are highlighted in the bold font.
DTT specific elements
Most of the DTT specific genes are also strain specific, as shown in Fig 3. The DTT specific genes have no homologs in the two non-toxic strains C02034 and B02005 and the two urinary tract infectious strains HI4320 and BB2000, and over 70% of them are only detected in one of the three DTT strains. And all 45 genes specific in the strain C02011 does not have detectable homologs in the publicly available strain P. mirabilis C05028. By considering the phylogenetic relationships among the three DTT strains (as in Fig 2), the current data strongly suggests that all the three P. mirabilis DTT strains are actively and independently acquiring foreign genes from the environmental neighboring microbes.
Fig 3. Venn plot of the strain specific genes in the three P. mirabilis DTT strains.

The numbers in the brackets are the numbers of DTT specific genes compared against the other P. mirabilis strains.
Core module for DTT function
The most infectious and toxic strain C02011 encodes 45 genes with no homologs in all the seven other strains. This suggests that these genes may have been acquired through the mechanism of horizontal gene transferring. The function annotation shows that these 45 genes do not introduce new antibiotic resisting abilities into the strain C02011, as shown in Table 4 and S1 Table.
Table 4. 45 genes specific in P. mirabilis C02011.
| Scaffold | Start | End | Strand | gene_id | Annotated protein |
|---|---|---|---|---|---|
| fa_scaffold3 | 465840 | 465962 | + | C02011GL001038 | |
| fa_scaffold3 | 465952 | 466815 | + | C02011GL001039 | |
| fa_scaffold3 | 466833 | 467849 | + | C02011GL001040 | Putative phage gene |
| fa_scaffold3 | 467846 | 468379 | + | C02011GL001041 | |
| fa_scaffold3 | 468467 | 468952 | + | C02011GL001042 | Putative phage gene |
| fa_scaffold3 | 468949 | 469179 | + | C02011GL001043 | |
| fa_scaffold3 | 469224 | 471878 | + | C02011GL001044 | Putative inner membrane protein |
| fa_scaffold3 | 472166 | 472393 | + | C02011GL001045 | Putative phage gene |
| fa_scaffold3 | 472383 | 472538 | + | C02011GL001046 | |
| fa_scaffold3 | 473119 | 473679 | + | C02011GL001047 | |
| fa_scaffold3 | 473682 | 475016 | + | C02011GL001048 | Retro-type reverse transcriptase |
| fa_scaffold3 | 1068445 | 1069563 | - | C02011GL001596 | |
| fa_scaffold3 | 1069568 | 1070161 | - | C02011GL001597 | |
| fa_scaffold3 | 1535544 | 1536122 | - | C02011GL002036 | |
| fa_scaffold3 | 1536545 | 1537150 | + | C02011GL002037 | Integrase |
| fa_scaffold3 | 1538064 | 1538282 | + | C02011GL002039 | |
| fa_scaffold3 | 1538847 | 1539260 | - | C02011GL002041 | |
| fa_scaffold3 | 1543281 | 1543748 | - | C02011GL002049 | |
| fa_scaffold3 | 1543975 | 1544586 | - | C02011GL002050 | |
| fa_scaffold3 | 1544614 | 1545003 | - | C02011GL002051 | |
| fa_scaffold3 | 1809941 | 1810177 | - | C02011GL002280 | |
| fa_scaffold3 | 1810202 | 1810420 | - | C02011GL002281 | |
| fa_scaffold3 | 1810632 | 1811384 | - | C02011GL002282 | Putative phage regulatory protein |
| fa_scaffold3 | 1811504 | 1812076 | - | C02011GL002283 | BRO family protein |
| fa_scaffold3 | 1812403 | 1813122 | + | C02011GL002284 | |
| fa_scaffold3 | 1823562 | 1824128 | - | C02011GL002300 | Putative phage terminase |
| fa_scaffold3 | 1824196 | 1824423 | + | C02011GL002301 | |
| fa_scaffold3 | 1824634 | 1824879 | - | C02011GL002302 | |
| fa_scaffold3 | 1825050 | 1825649 | - | C02011GL002303 | KilA-N domain-containing protein |
| fa_scaffold6 | 547377 | 548129 | - | C02011GL003316 | MobC |
| fa_scaffold6 | 548138 | 549889 | - | C02011GL003317 | MobB |
| fa_scaffold6 | 549859 | 550503 | - | C02011GL003318 | |
| fa_scaffold6 | 550993 | 551298 | - | C02011GL003319 | ORF15 |
| fa_scaffold6 | 551334 | 551708 | - | C02011GL003320 | |
| fa_scaffold6 | 551729 | 552754 | - | C02011GL003321 | T4SS virB11 |
| fa_scaffold6 | 552744 | 554012 | - | C02011GL003322 | T4SS virB10 |
| fa_scaffold6 | 554056 | 554961 | - | C02011GL003323 | T4SS virB9 |
| fa_scaffold6 | 554961 | 555644 | - | C02011GL003324 | T4SS virB8 |
| fa_scaffold6 | 555866 | 556936 | - | C02011GL003325 | T4SS virB6 |
| fa_scaffold6 | 557184 | 557885 | - | C02011GL003326 | T4SS virB5 |
| fa_scaffold6 | 557903 | 560641 | - | C02011GL003327 | T4SS virB4 |
| fa_scaffold6 | 560654 | 560833 | - | C02011GL003328 | T4SS virB2 |
| fa_scaffold6 | 560947 | 561699 | - | C02011GL003329 | T4SS virB1 |
| fa_scaffold6 | 562549 | 563136 | - | C02011GL003330 | |
| fa_scaffold6 | 590486 | 591754 | - | C02011GL003343 | Integrase |
The three large genomic islands are highlighted by a bold font in the column “gene_id”. The gene annotations are summarized from the S1 Table.
Three large genomic islands with at least 5 consecutively distributed genes are detected in the strain C02011. The largest genomic island encodes 15 genes, including a complete T4SS. The transmembrane secretion assembly T4SS consists of multiple component proteins, and is utilized by various gram-negative bacteria to transport virulent factors between the pathogen and the host cells [43]. Among the sequenced P. mirabilis strains, T4SS is only observed in the urinary tract infecting strain HI4320 [2, 44, 45]. And the T4SS in the strain C02011 does not show any similarity on the primary sequence level to that in HI4320, which supports the observation of little sequence conservation of T4SS modules from different bacteria [43, 46–49]. This genomic island also encodes three genes with significant homologs in the plasmid pMET-1 from the Gammaproteobacterium Klebsiella pneumoniae[50]. Among the three genes, mobB encodes three transmembrane domains and ORF15 has a 5’ signal peptide.
Another two genomic islands encode genes with various phage functionalities, as shown in the S1 Table.
Besides these transmembrane transporting genes, the strain C02011 also encodes a gene C02011GL002300 homologous to the phage terminase from a food poison pathogen Yersinia enterocolitica 8081, as shown in the S1 Table. Y. enterocolitica 8081 belongs to the serotype 0:8 and biotype 1B of Y. enterocolitica, and may induce severe diarrhea [51]. The most recent common ancestor between P. mirabilis and Y. enterocolitica on the phylogenetic tree is Enterobacteriaceae on the family level. The data suggests that P. mirabilis C02011 may have horizontally acquired severe digestion tract toxic genes from another genus Yersinia.
Supporting Information
(a) ureC, (b) rsmA, (c) hpmA, and (d) zapA. The lanes M and C are the 100-bp ladder and the control ddH2O lanes, respectively. The lanes 1 through 5 are the P. mirabilis strains C02011, C04010, C04013, C02034 and B02005, respectively.
(DOCX)
The three candidate genomic islands are highlighted by bold font in the column “gene_id”. The annotation is provided by the BGI, using the reference databases NCBI NR, SwissProt, Trembl, COG and KEGG.
(DOCX)
Acknowledgments
This work was supported by grants from National Natural Science Foundation of China (Grant 81302434), Science and Technology Planning Project of Shenzhen, Guangdong, China (Grant 201302239, JCYJ20130329103949651) and Shenzhen Public Service Platform of Pathogenic Microorganisms Repository. The authors do not have any conflicts of interest. The reviewers’ positive and constructive comments were appreciated.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by grants from National Natural Science Foundation of China (Grant 81302434), Science and Technology Planning Project of Shenzhen, Guangdong, China (Grant 201302239, JCYJ20130329103949651) and Shenzhen Public Service Platform of Pathogenic Microorganisms Repository.
References
- 1.Li X, Zhao H, Lockatell CV, Drachenberg CB, Johnson DE, Mobley HL. Visualization of Proteus mirabilis within the matrix of urease-induced bladder stones during experimental urinary tract infection. Infection and immunity. 2002;70(1):389–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pearson MM, Sebaihia M, Churcher C, Quail MA, Seshasayee AS, Luscombe NM, et al. Complete genome sequence of uropathogenic Proteus mirabilis, a master of both adherence and motility. Journal of bacteriology. 2008;190(11):4027–37. 10.1128/JB.01981-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leverstein-van Hall MA, MB HE, TD AR, Paauw A, Fluit AC, Verhoef J. Multidrug resistance among Enterobacteriaceae is strongly associated with the presence of integrons and is independent of species or isolate origin. The Journal of infectious diseases. 2003;187(2):251–9. 10.1086/345880 . [DOI] [PubMed] [Google Scholar]
- 4.Pagani L, Migliavacca R, Pallecchi L, Matti C, Giacobone E, Amicosante G, et al. Emerging extended-spectrum beta-lactamases in Proteus mirabilis. Journal of clinical microbiology. 2002;40(4):1549–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sriwanthana B, Mobley HL. Proteus mirabilis urease: histidine 320 of UreC is essential for urea hydrolysis and nickel ion binding within the native enzyme. Infection and immunity. 1993;61(6):2570–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liaw SJ, Lai HC, Ho SW, Luh KT, Wang WB. Role of RsmA in the regulation of swarming motility and virulence factor expression in Proteus mirabilis. Journal of medical microbiology. 2003;52(Pt 1):19–28. 10.1099/jmm.0.05024-0 . [DOI] [PubMed] [Google Scholar]
- 7.Cestari SE, Ludovico MS, Martins FH, da Rocha SP, Elias WP, Pelayo JS. Molecular detection of HpmA and HlyA hemolysin of uropathogenic Proteus mirabilis. Current microbiology. 2013;67(6):703–7. 10.1007/s00284-013-0423-5 . [DOI] [PubMed] [Google Scholar]
- 8.Carson L, Cathcart GR, Scott CJ, Hollenberg MD, Walker B, Ceri H, et al. Comprehensive inhibitor profiling of the Proteus mirabilis metalloprotease virulence factor ZapA (mirabilysin). Biochimie. 2011;93(10):1824–7. 10.1016/j.biochi.2011.06.030 . [DOI] [PubMed] [Google Scholar]
- 9.Zou Q, Li J, Wang C, Zeng X. Approaches for recognizing disease genes based on network. BioMed research international. 2014;2014:416323 10.1155/2014/416323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zou Q, Li J, Hong Q, Lin Z, Wu Y, Shi H, et al. Prediction of MicroRNA-Disease Associations Based on Social Network Analysis Methods. BioMed research international. 2015;2015:810514 10.1155/2015/810514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zeng X, Zhang X, Zou Q. Integrative approaches for predicting microRNA function and prioritizing disease-related microRNA using biological interaction networks. Briefings in bioinformatics. 2015. 10.1093/bib/bbv033 . [DOI] [PubMed] [Google Scholar]
- 12.Zou Q, Li J, Song L, Zeng X, Wang G. Similarity computation strategies in the microRNA-disease network: a survey. Briefings in functional genomics. 2016;15(1):55–64. 10.1093/bfgp/elv024 . [DOI] [PubMed] [Google Scholar]
- 13.Sullivan NL, Septer AN, Fields AT, Wenren LM, Gibbs KA. The Complete Genome Sequence of Proteus mirabilis Strain BB2000 Reveals Differences from the P. mirabilis Reference Strain. Genome announcements. 2013;1(5). 10.1128/genomeA.00024-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience. 2012;1(1):18 10.1186/2047-217X-1-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics. 2007;23(6):673–9. 10.1093/bioinformatics/btm009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O'Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic acids research. 2016;44(D1):D733–45. 10.1093/nar/gkv1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McMillan LE, Martin AC. Automatically extracting functionally equivalent proteins from SwissProt. BMC bioinformatics. 2008;9:418 10.1186/1471-2105-9-418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic acids research. 2000;28(1):33–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic acids research. 2016;44(D1):D457–62. 10.1093/nar/gkv1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wei L, Liao M, Gao Y, Ji R, He Z, Zou Q. Improved and Promising Identification of Human MicroRNAs by Incorporating a High-Quality Negative Set. IEEE/ACM transactions on computational biology and bioinformatics / IEEE, ACM. 2014;11(1):192–201. 10.1109/TCBB.2013.146 . [DOI] [PubMed] [Google Scholar]
- 21.McGettigan PA. Transcriptomics in the RNA-seq era. Current opinion in chemical biology. 2013;17(1):4–11. 10.1016/j.cbpa.2012.12.008 . [DOI] [PubMed] [Google Scholar]
- 22.Tempel S. Using and understanding RepeatMasker. Methods in molecular biology. 2012;859:29–51. 10.1007/978-1-61779-603-6_2 . [DOI] [PubMed] [Google Scholar]
- 23.Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic acids research. 1999;27(2):573–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siguier P, Gourbeyre E, Chandler M. Bacterial insertion sequences: their genomic impact and diversity. FEMS microbiology reviews. 2014;38(5):865–91. 10.1111/1574-6976.12067 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou F, Olman V, Xu Y. Insertion Sequences show diverse recent activities in Cyanobacteria and Archaea. BMC genomics. 2008;9:36 10.1186/1471-2164-9-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Y, Zhou F, Li G, Xu Y. A recently active miniature inverted-repeat transposable element, Chunjie, inserted into an operon without disturbing the operon structure in Geobacter uraniireducens Rf4. Genetics. 2008;179(4):2291–7. 10.1534/genetics.108.089995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou F, Tran T, Xu Y. Nezha, a novel active miniature inverted-repeat transposable element in cyanobacteria. Biochemical and biophysical research communications. 2008;365(4):790–4. 10.1016/j.bbrc.2007.11.038 . [DOI] [PubMed] [Google Scholar]
- 28.Brugger K, Redder P, She Q, Confalonieri F, Zivanovic Y, Garrett RA. Mobile elements in archaeal genomes. FEMS microbiology letters. 2002;206(2):131–41. . [DOI] [PubMed] [Google Scholar]
- 29.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Molecular biology and evolution. 2013;30(12):2725–9. 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M. ISfinder: the reference centre for bacterial insertion sequences. Nucleic acids research. 2006;34(Database issue):D32–6. 10.1093/nar/gkj014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen Y, Zhou F, Li G, Xu Y. MUST: a system for identification of miniature inverted-repeat transposable elements and applications to Anabaena variabilis and Haloquadratum walsbyi. Gene. 2009;436(1–2):1–7. 10.1016/j.gene.2009.01.019 . [DOI] [PubMed] [Google Scholar]
- 32.Sorek R, Kunin V, Hugenholtz P. CRISPR—a widespread system that provides acquired resistance against phages in bacteria and archaea. Nature reviews Microbiology. 2008;6(3):181–6. 10.1038/nrmicro1793 . [DOI] [PubMed] [Google Scholar]
- 33.Deveau H, Garneau JE, Moineau S. CRISPR/Cas system and its role in phage-bacteria interactions. Annual review of microbiology. 2010;64:475–93. 10.1146/annurev.micro.112408.134123 . [DOI] [PubMed] [Google Scholar]
- 34.Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic acids research. 2007;35(Web Server issue):W52–7. 10.1093/nar/gkm360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi X, Zhu Y, Li Y, Jiang M, Lin Y, Qiu Y, et al. Genome Sequence of Proteus mirabilis Clinical Isolate C05028. Genome announcements. 2014;2(2). 10.1128/genomeA.00167-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Z, Schwartz S, Wagner L, Miller W. A greedy algorithm for aligning DNA sequences. Journal of computational biology: a journal of computational molecular cell biology. 2000;7(1–2):203–14. 10.1089/10665270050081478 . [DOI] [PubMed] [Google Scholar]
- 37.Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, et al. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic acids research. 2008;36(Web Server issue):W465–9. 10.1093/nar/gkn180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pichel M, Brengi SP, Cooper KL, Ribot EM, Al-Busaidy S, Araya P, et al. Standardization and international multicenter validation of a PulseNet pulsed-field gel electrophoresis protocol for subtyping Shigella flexneri isolates. Foodborne pathogens and disease. 2012;9(5):418–24. 10.1089/fpd.2011.1067 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Furuhata K, Ishizaki N, Umekawa N, Nishizima M, Fukuyama M. Pulsed-Field Gel Electrophoresis (PFGE) pattern analysis and chlorine-resistance of Legionella pneumophila isolated from hot spring water samples. Biocontrol science. 2014;19(1):33–8. . [DOI] [PubMed] [Google Scholar]
- 40.Yu J, Sun Z, Liu W, Xi X, Song Y, Xu H, et al. Multilocus sequence typing of Streptococcus thermophilus from naturally fermented dairy foods in China and Mongolia. BMC microbiology. 2015;15(1):236 10.1186/s12866-015-0551-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bartlett JM, Stirling D. A short history of the polymerase chain reaction PCR protocols: Springer; 2003. p. 3–6. [DOI] [PubMed] [Google Scholar]
- 42.Wang G, Zhou F, Olman V, Li F, Xu Y. Prediction of pathogenicity islands in enterohemorrhagic Escherichia coli O157:H7 using genomic barcodes. FEBS letters. 2010;584(1):194–8. 10.1016/j.febslet.2009.11.067 . [DOI] [PubMed] [Google Scholar]
- 43.Christie PJ. Agrobacterium tumefaciens T-complex transport apparatus: a paradigm for a new family of multifunctional transporters in eubacteria. Journal of bacteriology. 1997;179(10):3085–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Flannery EL, Antczak SM, Mobley HL. Self-transmissibility of the integrative and conjugative element ICEPm1 between clinical isolates requires a functional integrase, relaxase, and type IV secretion system. Journal of bacteriology. 2011;193(16):4104–12. 10.1128/JB.05119-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou F, Olman V, Xu Y. Barcodes for genomes and applications. BMC bioinformatics. 2008;9:546 Epub 2008/12/19. 10.1186/1471-2105-9-546 ; PubMed Central PMCID: PMCPmc2621371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Christie PJ. Type IV secretion: the Agrobacterium VirB/D4 and related conjugation systems. Biochimica et biophysica acta. 2004;1694(1–3):219–34. 10.1016/j.bbamcr.2004.02.013 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burns DL. Type IV transporters of pathogenic bacteria. Current opinion in microbiology. 2003;6(1):29–34. . [DOI] [PubMed] [Google Scholar]
- 48.Xie J, Zhou F, Xu G, Mai G, Hu J, Wang G, et al. Genome-wide screening of pathogenicity islands in Mycobacterium tuberculosis based on the genomic barcode visualization. Molecular biology reports. 2014;41(9):5883–9. Epub 2014/08/12. 10.1007/s11033-014-3463-4 . [DOI] [PubMed] [Google Scholar]
- 49.Zhou F, Xu Y. cBar: a computer program to distinguish plasmid-derived from chromosome-derived sequence fragments in metagenomics data. Bioinformatics. 2010;26(16):2051–2. Epub 2010/06/12. 10.1093/bioinformatics/btq299 ; PubMed Central PMCID: PMCPmc2916713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Soler Bistue AJ, Birshan D, Tomaras AP, Dandekar M, Tran T, Newmark J, et al. Klebsiella pneumoniae multiresistance plasmid pMET1: similarity with the Yersinia pestis plasmid pCRY and integrative conjugative elements. PloS one. 2008;3(3):e1800 10.1371/journal.pone.0001800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thomson NR, Howard S, Wren BW, Holden MT, Crossman L, Challis GL, et al. The complete genome sequence and comparative genome analysis of the high pathogenicity Yersinia enterocolitica strain 8081. PLoS genetics. 2006;2(12):e206 10.1371/journal.pgen.0020206 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(a) ureC, (b) rsmA, (c) hpmA, and (d) zapA. The lanes M and C are the 100-bp ladder and the control ddH2O lanes, respectively. The lanes 1 through 5 are the P. mirabilis strains C02011, C04010, C04013, C02034 and B02005, respectively.
(DOCX)
The three candidate genomic islands are highlighted by bold font in the column “gene_id”. The annotation is provided by the BGI, using the reference databases NCBI NR, SwissProt, Trembl, COG and KEGG.
(DOCX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.