

Abstract
Organogels can be prepared by immobilizing an organic phase into a three-dimensional network coming from the self-assembly of a low molecular weight gelator molecule. In this work, an injectable subcutaneous organogel system based on safflower oil and a modified-tyrosine organogelator was evaluated in vivo for the delivery of rivastigmine, an acetylcholinesterase (AChE) inhibitor used in the treatment of Alzheimer’s disease. Different implant formulations were injected and the plasmatic drug concentration was assayed for up to 35 days. In parallel, the inhibition of AChE in different brain sections and the biocompatibility of the implants were monitored. The pharmacokinetic profiles were found to be influenced by the gel composition, injected dose and volume of the implant. The sustained delivery of rivastigmine was accompanied by a significant prolonged inhibition of AChE in the hippocampus, a brain structure involved in memory. The implant induced only a minimal to mild chronic inflammation and fibrosis, which was comparable to poly(D,L-lactide-co-glycolide) in situ-forming implants. These findings suggest that tyrosine-based organogels could represent an alternative approach to current formulations for the sustained delivery of cholinesterase inhibitors.
Keywords: Organogel, Rivastigmine, Alzheimer’s disease, Pharmacokinetic, Biocompatibility, AChE inhibition
1. Introduction
An organogel is a non-glassy thermoreversible semi-solid system composed of an organic liquid phase entrapped in a three-dimensionally cross-linked network. The liquid phase can be an organic solvent or an oil, whereas the structuring network is formed by self-assembled low molecular weight or polymeric organogelator molecules. Organogels have been investigated in fields as diverse as molecular photonics [1], art conservation [2] and food industry [3]. In the area of pharmaceutical sciences, organogels have only received a marked interest in recent years [4–6]. They have been tested with more or less success for the administration of inflammatory/analgesic drugs [7–9], cardiovascular drugs [4], antipsychotics [10] as well as nucleic acids [11] and peptides [12] by the transdermal, rectal, oral and buccal routes. In drug delivery, organogels are generally prepared using biocompatible and safe gelating molecules such as lecithin [8,13–15], sorbitan monostearate (SMS) [4,12,16] and amino acid derivatives [17]. A promising avenue for organogels lies in their use as depot formulations following parenteral extravascular injection. Compared to polymeric hydrogels, organogels have the ability to better retain low molecular weight polar compounds in their matrix [18]. Also, as opposed to implants based on lactic acid copolymers [19], their inner structure does not acidify upon degradation, which could be an advantage for the formulation of acid-sensitive pharmacological agents. Injectable organogels based on SMS or glyceryl esters of fatty acids have been successfully employed as vaccine adjuvants [16] and depot formulations for contraceptive steroids [20].
Over the past 6 years, we have been studying the ability of hydrophobized amino acids such as L-alanine [21] and L-tyrosine [22] to self-assemble in vegetable oils and form semi-solid/solid systems at body temperature. In order to partially inhibit gelation at room temperature and allow the injection of the formulation, a small amount of N-methyl pyrrolidone (NMP), a biocompatible and water-soluble hydrophilic organic solvent [23], is added to the oil/organogelators/drug mixture. Gel formation occurs after the subcutaneous (s.c.) injection of the formulation upon diffusion of the gelation inhibitor NMP into the surrounding tissues. The implants based on N-stearoyl L-alanine methyl ester (SAM) (in safflower oil) were found to be well tolerated in rats [24] and effective to deliver some drugs in a prolonged manner. The incorporation of leuprolide, an inhibitor of testosterone secretion, into these implants resulted in an efficient chemical castration of male rats for up to 50 days [25]. More recently, the same SAM organogels were assessed for the parenteral sustained delivery of rivastigmine, an AChE inhibitor prescribed to patients suffering from mild-to-moderate Alzheimer’s disease [26]. Rivastigmine is currently marketed as oral solution, capsules or transdermal patch requiring daily dosing [27]. It is therefore of potential interest to develop a sustained-release formulation that would only require a few injections per year. In vivo, the SAM gels were found to release the rivastigmine during only 10 days [25]. Moreover, the analytical assay involved the use of tritium-labelled rivastigmine and therefore the plasma concentration could have been partly biased by radiolabeled metabolites or degradation products.
Recently, we discovered that organogels prepared with L-tyrosine derivatives yielded implants with better mechanical properties, higher gel–sol transition temperatures and lower burst release in vitro [22]. In the present study, the sustained-release properties of such implants following s.c. injections were investigated for the first time in rats. The effects of gel composition (L-alanine vs. L-tyrosine), dose (15 vs. 25 mg kg−1) and implant volume (300 vs. 500 μL) on rivastigmine plasma concentrations were evaluated by LC/MS/MS. In addition, the AChE activity of the formulation in different sections of the brain as well as the biocompatibility of the rivastigmine-loaded implants were assessed in rats (Fig. 1).
Fig. 1.

Schematic representation of the experimental design: implant preparation, formation and biodegradation, and in vivo experiments.
2. Materials and methods
2.1. Materials
SAM and N-behenoyl L-tyrosine methyl ester (BTM) were synthesized as previously described [22]. NMP and 7-(β hydroxyethyl) theophyllin were purchased from Sigma–Aldrich Canada Ltd. (Oakville, ON, Canada) and used as supplied. Super-refined safflower oil (fatty acid composition of the triglycerides: 72.0% linoleic acid (C18:2), 16.6% oleic acid (C18:1), 7.4% palmitic acid (C16:0), 2.5% stearic acid (C18:0), and 1.5% v/v others) was kindly provided by Croda Inc. (Toronto, ON, Canada). Rivastigmine hydrogen tartrate was purchased from LGM Pharmaceuticals Inc. (Boca Raton, FL). Eligard® (22.5 mg, 3 months) was from Sanofi Aventis (Laval, QC, Canada).
2.2. Formulation preparation
BTM, SAM and rivastigmine were sterilized by γ-irradiation at 25 kGy using a 60Co source (Nordion Inc., Laval, QC, Canada). Stability after sterilization of the organogelators and drug was confirmed by 1H NMR. The chemical shifts (ppm) of the organogelator in CDCl3 obtained with a Bruker ARX-400 spectrometer (400 MHz, Bruker, Milton, ON, Canada) before and after γ-irradiation did not change: BTM: 0.9 (triplet t, 3H), 1.2 (multiplet m, 16H), 1.6 (quintuplet q, 2H), 2.2 (t, 2H), 3.0 (m, 2H), 3.7 (singlet s, 3H), 4.9 (doublet of triplets dt, 1H), 5.5 (s, 1H), 5.8 (doublet d, 1H), 6.7–7.0 (m, 4H); SAM: 0.9 (t, 3H), 1.2 (m, 32H), 1.4 (d, 3H), 1.6 (q, 2H), 2.2 (t, 2H), 3.7 (s, 3H), 4.6 (q, 1H), 6.0 (d, 1H). Safflower oil and NMP were sterilized on 0.2-μm polytetrafluoroethylene filters. All formulations were prepared under aseptic conditions. Organogelator (5% w/w) and safflower oil were weighed into a polyethylene flask, mixed and heated to 90 °C. Once the organogelator was dissolved, the temperature was decreased to 80 °C and rivastigmine powder was physically dispersed in the oily solution by magnetic stirring. NMP (3% w/w of oil) was then added and syringes (20G1-gauge needle) were immediately filled with the hot dispersion (400 or 600 μL) and placed on ice for 30 min. Control formulations included an oil/NMP/rivastigmine solution devoid of organogelator (pharmacokinetic study, PK), a saline solution of rivastigmine (0.045% w/v) (enzymatic titration study, ET) and gel formulation devoid of rivastigmine (biocompatibility study, BC). Table 1 lists the composition of each formulation and controls tested in vivo.
Table 1.
The formulations for the pharmacokinetic (PK), enzymatic titration (ET) and biocompatibility (BC) studies.
| Study | Formulation | Organogelator | Corga (% w/w) | Implant volume (μL) | CRivb (% w/w) | Rivastigmine dose (mg kg−1) |
|---|---|---|---|---|---|---|
| PK | PK-Oil-300-5 | – | 0 | 300 | 0.6 ± 0.1 | 5.9 ± 1.0 |
| PK-BTM-300-15 | BTM | 5 | 300 | 1.6 ± 0.4 | 14.9 ± 0.7 | |
| PK-BTM-300-25 | BTM | 5 | 300 | 2.8 ± 0.3 | 25.9 ± 2.9 | |
| PK-BTM-500-25 | BTM | 5 | 500 | 1.4 ± 0.2 | 21.8 ± 3.2 | |
| PK-SAM-300-15 | SAM | 5 | 300 | 1.4 ± 0.2 | 13.2 ± 2.3 | |
| ET | ET-saline-1.5 | – | 0 | 1000 | 0.045c | 15d |
| ET-BTM-300-15 | BTM | 5 | 300 | 1.8 ± 0.1 | 14.9 ± 1.8 | |
| ET-BTM-500-25 | BTM | 5 | 500 | 2.2 ± 0.9 | 26.8 ± 3.6 | |
| BC | BC-BTM-500-0 | BTM | 5 | 500 | 0 | 0 |
| BC-BTM-500-25 | BTM | 5 | 500 | 1.7 ± 0.1 | 22.9 ± 3.0 |
Organogelator concentration in safflower oil (% w/w).
Rivastigmine concentration in safflower oil (% w/w).
0.045% (w/v) rivastigmine dissolved in 1 mL saline.
Final dose given to rats after 10 days.
For drug content analysis a 100-μL sample from each syringe was dissolved in 1 mL of NMP, mixed with 9 mL of phosphate-buffered saline (PBS) (104 mM NaH2PO4, 36 mM NaCl, 0.1% w/v NaN3, pH 7.4), and assayed by HPLC. A Gilson Model 302 HPLC system (Gilson, Middletown, WI) equipped with a Gilson 234 autoinjector, a Gilson 106 pump and a Gilson 151 dual-wavelength UV-detector was employed. An Altima guard column (C18, 4.6 × 7.5 mm, 5 μm; Mandel Scientific Company Inc., Guelph, ON, Canada) was placed upstream of the XTerra (RP-18, 4.6 × 250 mm, 5 μm) analytical column (Waters, Mississauga, ON, Canada). The mobile phase consisted of water/acetonitrile (78/22 v/v) with 0.1% (v/v) trifluoroacetic acid. The flow rate was set at 1 mL min−1. Injection volume and detection wavelength were 20 μL and 210 nm, respectively. Each formulation was analyzed in triplicate. Table 1 displays measured rivastigmine concentrations and injected doses for individual formulations.
2.3. Pharmacokinetic study
All experimental procedures involving animals were conducted following a protocol approved by the Animal Care Committee of the University of Montreal and complied with The Guide for the Care and Use of Laboratory Animals (NIH Publication no. 85-23, revised 1996). Male Sprague Dawley rats (300–325 g; Charles Rivers Inc., St-Constant, QC, Canada) were housed for 1 week under controlled conditions (12-h light/dark schedule, 24 °C) prior to experiments. Tap water was provided ad libitum while chow was controlled at 5 g/day/100 g rat body weight. Rats were randomly divided into 5 groups (n = 8/group) and received a single s.c. injection of the appropriate formulation (Table 1, PK study) in the higher dorsal area under isoflurane anaesthesia. After the injection, the needle was held in place for 10 s to prevent the semi-solid implant from leaking out from the injection site. The exact amount of injected formulation was obtained by weighing the syringes before and after injection. Blood samples (400–450 μL) were periodically collected from the subclavian vein under isoflurane anaesthesia into EDTA-K2 vials. Blood samples were centrifuged (15 min, 1600g, 4 °C) to collect plasma. The latter (200 μL) was mixed with 20 μL of internal standard (IS) solution (7-(β hydroxyethyl) theophyllin, 4 μg mL−1 in water) and 400 μL of methanol. The mixture was vortexed and centrifuged (20 min, 8000g, 25 °C). Three hundred μL of supernatant was added to 500 μL of water before quantification of the rivastigmine concentration by LC/MS/MS (see below). The final composition in water/methanol was 75/25 (v/v) and the IS concentration was 48.4 ng mL−1. For the calibration curve, the rivastigmine concentrations spanned from 0.1 to 500 ng mL−1.
2.4. LC/MS/MS method
Separations were performed on a Surveyor LC system coupled to a Quantum Ultra AM triple quadrupole mass spectrometer (Thermo Fisher, San Jose, CA). The chromatographic column was a Luna C18(2), 50 × 3.0 mm, 2.5 μm particle size (Phenomenex, Torrance, CA) operating at 0.2 mL min−1. A water/acetonitrile linear gradient, containing 0.1% (v/v) formic acid, was used for elution. Separation was performed by increasing the acetonitrile concentration from 10 to 50% over 3 min. From 3 to 4 min, 90% acetonitrile was delivered through the column followed by an equilibration step with the starting mobile phase for a total run time of 8 min. Positive ions were generated using a heated electrospray ionization source and mass spectra were acquired over two different scan events. In the first scan event, set up in selected reaction monitoring mode, the transition 251.3 → 206.3 was acquired for rivastigmine and 225.3 → 181.0 was acquired for the IS. The collision energies used were 14 and 18 V and the Tune Lens were set to 90 and 75 for rivastigmine and the IS respectively. In the second scan event, set up in scan mode, mass spectra were acquired from m/z 100 to 1000. The electrospray voltage was set at 3300 V and the capillary temperature at 350 °C. For the first 1.6 min, the chromatographic flow was diverted to waste in order to prevent residual salts or un-retained small molecules from plasma from entering the mass spectrometer. Useful LC–MS data was acquired from 1.6 to 4.5 min after which the flow was directed once again to waste in order to prevent residual plasma proteins from contaminating the mass spectrometer. The dynamic range of rivastigmine quantitation was evaluated using twenty calibration solutions with concentrations ranging from 0.1 to 500 ng mL−1. A linear response with a correlation coefficient r2 of 0.9964 was obtained for concentrations up to 350 ng mL−1. The lower limit of quantitation of this method was 0.1 ng mL−1.
2.5. In vivo titration of AChE activity
Rats were randomly divided into 3 groups (Table 1, ET study), which were sacrificed at different time points (n = 8 per time point per group) and were given the rivastigmine formulation (Table 1) by s.c. injection. Rats receiving the gel formulation were injected only once with 15 or 25 mg/kg rivastigmine whereas the control treated animals were given a daily dose of 1.5 mg/kg for 10 days (total injected dose: 15 mg/kg). Three non-treated control groups were used for baseline value assessment (Day 0). About 30 min prior to decapitation, rats received an intra-peritoneal injection of acepromazine (0.05 mg kg−1). After decapitation, the brain was dissected and the following brain structures were extracted on ice: frontal, temporal and parietal cortices, striatum, and hippocampus. They were frozen immediately in 2-methylbutane on dry ice and stored at −80 °C. For analysis, homogenates of the different brain structures were prepared on ice in 25 mM Na/K/PO4 buffer (pH 7.4) using potter-elvehjem tissue grinders (Wheaton Science Products, Millville, NJ) and teflon pestles (Kontes Glass Co., Vineland, NJ). The protein concentration of each homogenate was assayed with a colorimetric test using the bicinchoninic acid (BCA) protein assay kit (Pierce, Rockford, IL) according to the supplier’s instructions.
The AChE activity was evaluated with a photometric test based on Ellman et al.’s method [28]. This method relies on the production of thiocholine after acetylthiocholine hydrolysis. Thiol groups of thiocholine then react with 5,5′-dithiobis[2-nitrobenzoate] (DTNB) to form 5-thio(2-nitrobenzoic acid) (yellow color). Homogenate samples, 25 mM Na/K/PO4 (pH 7.4) buffer and 5 mM acetylthiocholine were added in a 96-well plate. The plate was incubated at room temperature and under stirring during 30 min, followed by the addition of 500 μL DTNB solution composed of 125 mM of DTNB in ethanol/H2O/100 mM Na/K/PO4 buffer (pH 7.4) (48/32/20 v/v/v). The optical density (OD) was immediately measured at 411 nm. The AChE activity ( ) was calculated as:
with εl = 13.6 mmol−1, mprotein being the protein amount measured by the BCA method (mg) and t = 30 min. Results were reported in %AChE activity, relative to the baseline activity.
2.6. Biocompatibility study
Rats were randomly divided into 2 groups (gels with or without rivastigmine) (Table 1, BC study), which were sacrificed at different time points (n = 3 per time point per group). The data were compared to 2 control groups: one group of rats did not receive any formulation (negative control) while the other group was given the Eligard® formulation (in situ-forming implant consisting of leuprolide, poly(D,L-lactide-co-glycolide) (PLGA) and NMP). Rats were sacrificed by carbon dioxide asphyxia. Tissues adjacent to the injection site and the remaining implants were collected and fixed with 10% neutral-buffered formalin. All samples were processed and embedded in paraffin. Glass slides with 4-μm tissue sections were prepared using a Leica 2155 microtome (Leica Microsystems Inc., Richmond Hill, ON, Canada). Slides of each implant were stained according to a hematoxylin–phloxin–safran (HPS) standard procedure, using hematoxylin (nucleus coloration) and phloxin (cytoplasmic coloration) for cellularity and cell number, and safran for collagen deposition and fibrous tissue formation [29]. The local inflammatory response was assessed by histological evaluation of the surrounding tissue for signs of acute inflammation (neutrophils and eosinophils) and chronic inflammation (histocytes, plasmocytes, lymphocytes, multinucleated giant cells, fibroblasts, neoangiogenesis, and collagen deposition) [30]. The intensity of the response was graded on a scale from absent, minimal, mild, moderate, and significant to severe, depending on the number of cells and general appearance of the tissue.
2.7. Statistical analysis
For AChE titration, normal distribution was verified for each data set. Significant differences between means of %AChE activity were analyzed by one-way analysis of variance (ANOVA), followed by Tukey–Kramer’s post hoc test for pairwise comparisons. Differences were considered statistically significant for p < 0.05.
3. Results
3.1. Pharmacokinetic study
In the present study, a new in situ-forming implant composed of BTM and safflower oil was evaluated for the sustained delivery of rivastigmine after s.c. injection. Table 1 lists all formulations which were administered for the pharmacokinetic study (PK group) and the exact doses that were actually given (target doses were 15 or 25 mg kg−1 for the implants). In order to assess the extent of drug release, a threshold concentration of 1 ng mL−1 was chosen as it corresponds to a level still providing therapeutic benefits in humans [27]. Fig. 2 shows the effect of the formulation composition (SAM vs. BTM gels) at 15 mg kg−1 rivastigmine on the plasma levels as measured by LC/MS/MS. The control group received an oily dispersion of the drug at 5 mg kg−1. A higher dose was not injected due to strong side-effects such as high shaking, lifeless state and harderian porphyrin excretion, probably related to the high Cmax, the maximal value of the rivastigmine plasmatic concentration. In this experiment, the volume of formulation administered was 300 μL. As shown in Fig. 2 and Table 2, the addition of SAM or BTM to safflower oil decreased substantially the burst release and provided sustained drug levels (>1 ng mL−1) for ca. 10 days (compared to 2 days for the control PK-Oil-300-5). The Cmax was reduced by 5 and 10 fold, respectively for a total drug dose which was 3 times higher than the control. BTM was twice more effective at reducing Cmax than SAM. However, the BTM formulation was not better at prolonging drug release.
Fig. 2.
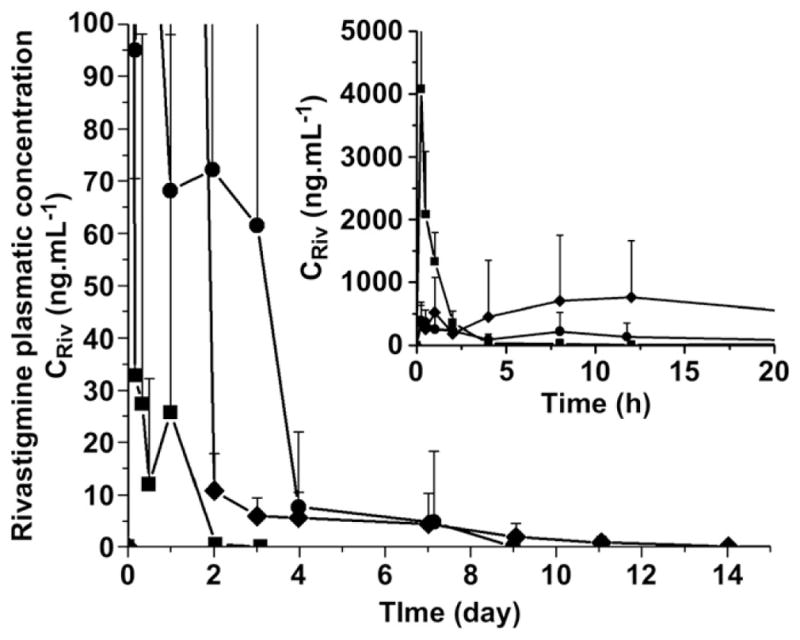
Pharmacokinetic study (plasmatic rivastigmine concentration CRiv vs. time) of the following formulations: PK-Oil-300-5 (■), PK-BTM-300-15 (●) and PK-SAM-300-15 (◆) (Mean ± SEM, n = 8). Inset represents the early time points.
Table 2.
Pharmacokinetic parameters of the different formulations injected to rats (n = 8). Cmax is the maximal value of the rivastigmine plasmatic concentration and tmax the corresponding time. t1ng corresponds to the time when the rivastigmine concentration was less than 1 ng mL−1. AUC corresponds to the area under the plasma concentration vs. time curve between days 0 and the last day of the study.
| Formulation | Cmax (ng mL−1) | tmax (h) | t1ng (day) | AUC ± SD (h ng mL−1) |
|---|---|---|---|---|
| PK-Oil-300-5 | 4077 | 0.25 | 2 | 4100 ± 1900 |
| PK-BTM-300-15 | 397 | 0.25 | 9 | 8100 ± 6600 |
| PK-BTM-300-25 | 482 | 8 | 14 | 11,600 ± 10,100 |
| PK-BTM-500-25 | 334 | 8 | >35 | 14,700 ± 7000 |
| PK-SAM-300-15 | 758 | 12 | 11 | 20,300 ± 17,600 |
In the second experiment, the effects of the volume of implant and drug loading on the rivastigmine plasma levels were investigated using the BTM gel. The rats received 25 mg kg−1 rivastigmine by either raising the volume of the injected formulation from 300 to 500 μL or by increasing the drug loading in the gel from ca.1.5–2.5% while maintaining the injected volume to 300 μL. As illustrated in Fig. 3, the 300-μL gel with the higher drug content provided sustained drug levels for 14 days and exhibited a 20% increase in Cmax and 40% increase in area under the plasma concentration vs. time curve (AUC) compared to the 15 mg kg−1 dose (Table 2). There was also a shift in tmax (time corresponding to Cmax) from 15 min to 8 h. As shown in Fig. 3, the volume of the implant had a more pronounced impact on the pharmacokinetic profile than the drug content. Indeed, by raising the injected volume from 300 to 500 μL (PK-BTM-300-25 vs. PK-BTM-500-25), sustained plasma levels were achieved for more than 35 days. Interestingly, despite the fact that the total dose was increased from 15 to 25 mg kg−1, the Cmax value did not change (397 vs. 334 ng mL−1) (Table 2). In this case also, the tmax shifted from 15 min to 8 h.
Fig. 3.
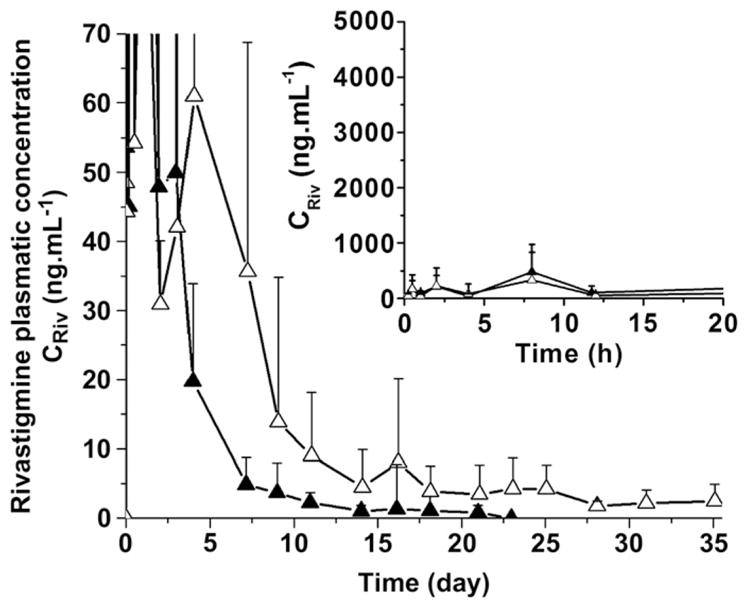
Pharmacokinetic study (plasmatic rivastigmine concentration CRiv vs. time) of the following formulations: PK-BTM-300-25 (▲) and PK-BTM-500-25 (△) (Mean ± SEM, n = 8). Inset represents the early time points.
3.2. AChE activity titration
In a subsequent study, the pharmacological activity of the two BTM formulations presenting the lowest burst release (Table 1, ET-BTM-300-15 and ET-BTM-500-25 for a total drug dose of 15 and 25 mg kg−1, respectively) was assessed in vivo and compared to that of a daily s.c. injection of a 1.5 mg kg−1 rivastigmine saline solution (×10 days for a total administered dose of 15 mg kg−1). The animals were sacrificed at different time points and the AChE activity was assayed in the frontal, temporal and parietal cortices and the striatum (Supplementary Figures S1–S3) and hippocampus (Fig. 4). Except for temporal and parietal cortices at day 7, the daily injection of rivastigmine solution was not able to significantly inhibit the activity of AChE in the analyzed sections of the brain (Supplementary Figure S1 and Fig. 4). Moreover, rats receiving this control solution seemed to experience an initial increase of AChE activity both in the hippocampus (Fig. 4) and in other brain structures studied here (Supplementary Figure S1) at day 3. Such an initial amplification could be explained by an autologous counter-regulation of AChE levels. The injection of the rivastigmine solution and subsequent sudden exposure to high drug levels might have triggered AChE release from cerebral neurons stored to counterbalance the rivastigmine effect. Shapira et al. [31] and Kaufer et al. [32] already showed the possibility of an AChE accumulation in brain in response to anti-AChE intoxication and acute stress respectively. Such a trend was not observed in the implant-treated groups possibly because of the slower drug release. The ET-BTM-300-15 formulation decreased significantly the AChE activity in the frontal, temporal and parietal cortices and the striatum for the first 3 days (Supplementary Figure S2). After 7 days of rivastigmine release, AChE activity returned to the baseline level. In the striatum, some activity seemed to persist after 10 days, but due to the relatively important variability, statistical significance could not be established. For ET-BTM-500-25, the same trend was observed over a longer period of time (35 days for the striatum) but statistical significance could not be demonstrated after 10 days (Supplementary Figure S3). The most noticeable AChE inhibition was observed in the hippocampus. While the saline solution was ineffective, BTM implants were able to inhibit the AChE activity in a prolonged fashion (Fig. 4). The ET-BTM-300-15 formulation produced a 20% inhibition of enzymatic activity 3 days after administration (p < 0.05). Return to baseline levels occurred between days 3 and 7. The ET-BTM-500-25 system displayed more sustained enzymatic inhibition. The effect lasted up to 14 days with a significant difference, after which the AChE activity progressively returned to its initial level (Fig. 4).
Fig. 4.

%AChE activity (AChE activity after x days/AChE activity for healthy rat at day 0) (Mean ± SD, n = 8) in hippocampus at day 0 (▭) and after 3 (
 ), 7 (
), 7 (
 ), 10 (▬), 14 (
), 10 (▬), 14 (
 ), 21 (
), 21 (
 ), 28 (
), 28 (
 ) and 35 days (
) and 35 days (
 ). Rats received daily subcutaneous injections of ET-saline-1.5, and two organogel formulation injections (at day 0): ET-BTM-300-15 and ET-BTM-500-25. (*) p < 0.05.
). Rats received daily subcutaneous injections of ET-saline-1.5, and two organogel formulation injections (at day 0): ET-BTM-300-15 and ET-BTM-500-25. (*) p < 0.05.
3.3. Biocompatibility study
In the last part of the work, the biocompatibility of the BTM implants after s.c. injection was investigated. The impact of rivastigmine on the inflammatory reaction was measured (Table 1, BC study) and the data were compared to a negative control and to the inflammatory reaction produced by a commercial formulation of an in situ-forming implant composed of PLGA and NMP (Eligard®) (Figs. 5 and 6). At the macroscopic level, the injection of the drug-free and rivastigmine-loaded BTM organogels did not cause any redness of swelling (data not shown). Likewise, during the whole study period animals did not show external signs of side-effects such as weight loss or loss of appetite. The histological analysis of the injection site revealed that subcutaneous tissues presented a granulomatous chronic inflammatory reaction as for foreign body, accompanied by cicatricial-aspect fibrosis near the organogel implant (Fig. 5B). The inflammatory reaction to the formulation was rivastigmine-independent. In addition to fibrosis (Fig. 5D), the lesions included inflammatory infiltrates with epitheloid cells, macrophages, multi-nucleus giant cells, and lymphocytes around the cholesteronilic cleft (Fig. 5C). No difference in fibrosis and chronic inflammatory infiltration was observed between BC-BTM-500-0 and BC-BTM-500-25. Compared to negative controls (no treatment for the animals), fibrosis increased slightly in intensity from minimal to minimal-mild at some time points but did not exceed this level (Fig. 6A). For both formulations, inflammatory chronic infiltrate rapidly increased until a mild-moderate level was reached at day 7 (Fig. 6B). Then, the inflammation decreased and the characteristic infiltrates returned to a minimal-mild level grade at day 14 and remained constant thereafter. Interestingly, organogels produced an inflammatory reaction that was comparable to that of Eligard®. The optical histological micrograph did not reveal any difference between both formulations (compare Fig. 5E and F). The hypoderm reaction displayed the same evolution for both formulations after 30 and 70 days: a minimal-mild level of the inflammatory chronic infiltrate and fibrosis responses (Fig. 6).
Fig. 5.

Microscopic section of (A) normal skin and (B) subcutaneous tissue 7 days after injection of BC-BTM-500-25 with chronic inflammation (white arrow) and fibrosis (red arrow) in the vicinity of the implant (black arrow). (C) Higher magnification of the inflammatory infiltrate displayed in (B). The inflammatory cells consist of lymphocytes, macrophages (black rectangle) and multinucleated giant cells (yellow arrow). Cholesterol clefts are also observed focally (grey arrow). (D) Fibrosis typically surrounds the implant site and the inflammatory area for subcutaneous tissue 7 days after injection of BC-BTM-500-25. (E) Subcutaneous tissue 70 days after injection of Eligard® (22.5 mg, 3 months) formulation. (F) Subcutaneous tissue 70 days after injection of BC-BTM-500-25 formulation.
Fig. 6.

Hypoderm lesions evaluation: (A) fibrosis and (B) chronic inflammatory infiltrate, graded from absent to severe, after subcutaneous injection of BC-BTM-500-0, BC-BTM-500-25 and Eligard® (22.5 mg, 3 months) formulations after 7 (
), 14 (
), 30 (
) and 70 days (
 ). The negative control (▭) corresponds to non-treated rats. (Mean ± SD, n = 3).
). The negative control (▭) corresponds to non-treated rats. (Mean ± SD, n = 3).
4. Discussion
Alzheimer’s disease is characterized, among other neuropathological features, by the degeneration of cholinergic neurons in the cortex and hippocampus, resulting in lower levels of acetylcholine and altered cholinergic transmission [33]. Barnes et al. showed that the overall hippocampal atrophy rate is 1.4% in healthy patients from 69 to 83 years; this rate increased to 4.6% for subjects with Alzheimer’s disease [34]. Rivastigmine is a selective cholinesterase inhibitor. It links to the esteratic and ionic site of AChE, preventing the enzyme from metabolizing acetylcholine. This inhibition while generalized to all brain structures expressing the cholinergic phenotype is particularly relevant in regions associated with Alzheimer’s disease neuropathology such as the fronto-parietal cortex and the hippocampus. This is the basis for the use of AChE inhibitors in this disease [35], the role of the hippocampus in cognitive processes being well established [36,37].
Until 2007, AChE inhibitors were only available as oral formulations taken once or twice daily in order to achieve adequate and consistent inhibition of cholinesterase [33]. Recently, a once-a-day rivastigmine patch received approval in the US. The patch exhibited an efficacy similar to that of the oral dosage form with significantly less gastro-intestinal side-effects [38] and was found more convenient for the caregiver [39]. Persistence to oral AChE inhibitors is reported to be poor [40] and the adherence to the treatment, although apparently good, requires a high level of intervention and including the use of pill boxes and assistance in taking prescribed drugs [41].
Compared to oral formulations, injectable controlled-release dosage forms generally provide greater dosing precision, and lower inter-subject variability due to the absence of erratic absorption. Parenteral depot also reduce the large differences in peak and trough plasma levels, which ultimately prevents the side-effects associated with repeated rapid increases in concentrations [42]. The literature on parenteral sustained-release dosage forms for AChE inhibitors is scarce. Yang et al. [43] demonstrated that PLGA microspheres could sustain tacrine delivery in vitro for several weeks. Recently, several studies reported the use of biodegradable microspheres to sustain the delivery of huperzine A, a natural AChE inhibitor isolated from the Chinese herb Huperzia serrata and approved for human use in China [44,45]. In rat [44,46] and dog models [47], it was found that the s.c. injection of huperzine A-loaded microspheres provided therapeutic plasma drug levels for ca. 15–60 days, and inhibited the rat cerebral cortex AChE by 10–20%. More recently, PLGA microspheres were investigated for the s.c. delivery of donepezil, another marketed AChE inhibitor [48]. When injected at a single drug dose of 90 mg/kg, microspheres were found as efficient as an oral daily dose of 3 mg/kg/day during one month.
In the present study, it was shown that optimized BTM gels could deliver rivastigmine for a period exceeding 4 weeks after s.c. administration. In order to facilitate the injection, the gels contained NMP at a NMP/BTM ratio of 3. This ratio was previously shown to fluidize the network while allowing the formation of reproducible gels after administration and diffusion of NMP in the surrounding environment [22]. Doses of 15 and 25 mg kg−1 could be injected without inducing immediate visible side-effects. Conversely, injecting more than 5 mg kg−1 of the control rivastigmine solution was associated with strong side-effects (data not shown). In terms of burst release in vivo, this system was also proven superior to the SAM implant which was initially investigated [26] (Fig. 2). Indeed, compared to L-alanine gels, the L-tyrosine derivatives possess higher gel–sol phase transition temperatures and elastic moduli [22]. The superior gelling ability of tyrosine derivatives could be explained by their well-structured 2-dimensional packing in the network. Such a packing was found to possibly involve van der Waals interactions and H-bonds in one direction and H-bonds between the phenol heads in the other direction [22]. In vitro release kinetics showed that the burst release from L-alanine based gels was indeed higher than that of the L-tyrosine counterpart [22].
Increasing the volume of injected implants was efficient to prolong the in vivo release of rivastigmine plasma levels. Indeed, at 25 mg kg−1, the 300-μL implant provided rivastigmine concentrations above 1 ng mL−1 for 14 days whereas this threshold was reached after 35 days for the 500-μL system (Table 2 and Fig. 3). Following the burst phase, it is thought that drug release is mostly driven by erosion and/or implant degradation by subcutaneous esterases and lipases [49,50]. The observed sustained release was accompanied by a measurable pharmacological effect as demonstrated by the significant inhibition of AChE in the hippocampus (i.e. brain region involved in episodic memory [36,37]) for at least 14 days (Fig. 4).
The present study also demonstrated that the BTM implants either empty or loaded with rivastigmine were biocompatible after s.c. administration. Indeed, the inflammatory reaction observed for up to 70 days was minimal to mild and in general comparable to that triggered by the marketed 3-month release in situ-forming PLGA implant, Eligard®. These data are in agreement with a previous study, which showed that SAM organogels elicited an inflammatory response similar to PLGA microspheres [24,51]. In the case of SAM (5% w/w in safflower oil), the implant completely degraded in vivo after about 30 days, whereas the BTM implants at the same organogelator concentration partly persisted for up to 70 days (data not shown). This greater in vivo persistence could be related to differences in the physical properties of these gels [22] but also in the degradation rates of organogelators. This point will have to be investigated in future studies.
5. Conclusion
This article reports the in vivo evaluation of an injectable sustained-release formulation of rivastigmine, an AChE inhibitor. This implant was composed of BTM, safflower oil and the gelation inhibitor, NMP. It formed in situ after the injection and was able to deliver rivastigmine for several weeks. The sustained rivastigmine concentrations in plasma were associated with the inhibition of AChE in various parts of the brain and in particular in the hippocampus, a structure well known to be affected early in the pathology of Alzheimer’s disease. The implant was well tolerated and therefore this approach could represent an alternative treatment for mild-to-moderate Alzheimer’s disease in patients who are not compliant to a daily therapy.
Supplementary Material
Acknowledgments
The Canadian Institutes of Health Research (JCL, RQ), the Natural Sciences and Engineering Council of Canada (Steacie Fellowship to JCL), the Canada Research Chair program and Ethypharm SA are acknowledged for their financial support. The authors thank Dr. Louis Gaboury and all the members of the Histology Core Facility of the Institute for Research in Immunology and Cancer of the University of Montreal for their help on the biocompatibility study. The authors also thank Karine Venne and Marie-Christine Tang for their help with LC/MS/MS analysis.
Appendix. Supplementary data
Supplementary data associated with this article can be found, in the on-line version, at doi:10.1016/j.biomaterials.2010.04.009.
Appendix
Figures with essential color discrimination. Figs. 1 and 5 of this article may be difficult to interpret in black and white. The full color images can be found in the on-line version, at doi:10.1016/j.biomaterials.2010.04.009.
References
- 1.Ajayaghosh A, Praveen VK, Vijayakumar C. Organogels as scaffolds for excitation energy transfer and light harvesting. Chem Soc Rev. 2008;37:109–22. doi: 10.1039/b704456a. [DOI] [PubMed] [Google Scholar]
- 2.Carretti E, Dei L, Weiss RG. Soft matter and art conservation Rheoreversible gels and beyond. Soft Matter. 2005;1:17–22. [Google Scholar]
- 3.Pernetti M, van Malssen KF, Flöter E, Bot A. Structuring of edible oil by alternatives to crystalline fat. Curr Opin Colloid Interf Sci. 2007;12:221–31. [Google Scholar]
- 4.Pisal S, Shelke V, Mahadik K, Kadam S. Effect of organogel components on in vitro nasal delivery of propranolol hydrochloride. AAPS Pharm Sci Tech. 2004;5:e63. doi: 10.1208/pt050463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Upadhyay KK, Tiwari C, Khopade AJ, Bohidar HB, Jain SK. Sorbitan ester organogels for transdermal delivery of sumatriptan. Drug Dev Ind Pharm. 2007;33:617–25. doi: 10.1080/03639040701199266. [DOI] [PubMed] [Google Scholar]
- 6.Jones DS, Muldoon BC, Woolfson AD, Andrews GP, Sanderson FD. Physicochemical characterization of bioactive polyacrylic acid organogels as potential antimicrobial implants for the buccal cavity. Biomacromolecules. 2008;9:624–33. doi: 10.1021/bm700597e. [DOI] [PubMed] [Google Scholar]
- 7.Agrawal GP, Juneja M, Agrawal S, Jain SK, Pancholi SS. Preparation and characterization of reverse micelle based organogels of piroxicam. Pharmazie. 2004;59:191–3. [PubMed] [Google Scholar]
- 8.Paice JA, Von Roenn JH, Hudgins JC, Luong L, Krejcie TC, Avram MJ. Morphine bioavailability from a topical gel formulation in volunteers. J Pain Symptom Manag. 2008;35:314–20. doi: 10.1016/j.jpainsymman.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 9.Shaikh IM, Jadhav KR, Gide PS, Kadam VJ, Pisal SS. Topical delivery of aceclofenac from lecithin organogels: preformulation study. Curr Drug Deliv. 2006;3:417–27. doi: 10.2174/156720106778559010. [DOI] [PubMed] [Google Scholar]
- 10.Lim PFC, Liu XY, Kang L, Ho PCL, Chan YW, Chan SY. Limonene GP1/PG organogel as a vehicle in transdermal delivery of haloperidol. Int J Pharm. 2006;311:157–64. doi: 10.1016/j.ijpharm.2005.12.042. [DOI] [PubMed] [Google Scholar]
- 11.Cui Z, Mumper RJ. Bilayer films for mucosal (genetic) immunization via the buccal route in rabbits. Pharm Res. 2002;19:947–53. doi: 10.1023/a:1016454003450. [DOI] [PubMed] [Google Scholar]
- 12.Murdan S, Andrysek T, Son D. Novel gels and their dispersions-oral drug delivery systems for ciclosporin. Int J Pharm. 2005;300:113–24. doi: 10.1016/j.ijpharm.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 13.Mahler P, Mahler F, Duruz H, Ramazzina M, Liguori V, Mautone G. Double-blind, randomized, controlled study on the efficacy and safety of a novel diclofenac epolamine gel formulated with lecithin for the treatment of sprains, strains and contusions. Drugs Exp Clin Res. 2003;29:45–52. [PubMed] [Google Scholar]
- 14.Spacca G, Cacchio A, Forgács A, Monteforte P, Rovetta G. Analgesic efficacy of a lecithin-vehiculated diclofenac epolamine gel in shoulder periarthritis and lateral epicondylitis: a placebo-controlled, multicenter, randomized, double-blind clinical trial. Drugs Exp Clin Res. 2005;31:147–54. [PubMed] [Google Scholar]
- 15.Aboofazeli R, Zia H, Needham TE. Transdermal delivery of nicardipine: an approach to in vitro permeation enhancement. Drug Deliv. 2002;9:239–47. doi: 10.1080/10717540260397855. [DOI] [PubMed] [Google Scholar]
- 16.Murdan S, Gregoriadis G, Florence AT. Sorbitan monostearate/polysorbate 20 organogels containing niosomes: a delivery vehicle for antigens? Eur J Pharm Sci. 1999;8:177–85. doi: 10.1016/s0928-0987(99)00014-7. [DOI] [PubMed] [Google Scholar]
- 17.Vintiloiu A, Leroux JC. Organogels and their use in drug delivery – a review. J Control Release. 2008;125:179–92. doi: 10.1016/j.jconrel.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 18.Ruel-Gariépy E, Shive M, Bichara A, Berrada M, Le Garrec D, Chenite A, et al. A thermosensitive chitosan-based hydrogel for the local delivery of paclitaxel. Eur J Pharm Biopharm. 2004;57:53–63. doi: 10.1016/s0939-6411(03)00095-x. [DOI] [PubMed] [Google Scholar]
- 19.Mäder K, Bittner B, Li Y, Wohlauf W, Kissel T. Monitoring microviscosity and microacidity of the albumin microenvironment inside degrading microparticles from poly(lactide-co-glycolide) (PLG) or ABA-triblock polymers containing hydrophobic poly(lactide-co-glycolide) A blocks and hydrophilic poly (ethyleneoxide) B blocks. Pharm Res. 1998;15:787–93. doi: 10.1023/a:1011939607573. [DOI] [PubMed] [Google Scholar]
- 20.Gao ZH, Crowley WR, Shukla AJ, Johnson JR, Reger JF. Controlled release of contraceptive steroids from biodegradable and injectable gel formulations: in vivo evaluation. Pharm Res. 1995;12:864–8. doi: 10.1023/a:1016261004230. [DOI] [PubMed] [Google Scholar]
- 21.Couffin-Hoarau AC, Motulsky A, Delmas P, Leroux JC. In situ-forming pharmaceutical organogels based on the self-assembly of L-alanine derivatives. Pharm Res. 2004;21:454–7. doi: 10.1023/B:PHAM.0000019299.01265.05. [DOI] [PubMed] [Google Scholar]
- 22.Bastiat G, Leroux JC. Pharmaceutical organogels prepared from aromatic amino acid derivatives. J Mater Chem. 2009;19:3867–77. doi: 10.1039/B822657A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Becci PJ, Gephart LA, Koschier FJ, Johnson WD, Burnette LW. Subchronic feeding study in beagle dogs of N-methylpyrrolidone. J Appl Toxicol. 1983;3:83–6. doi: 10.1002/jat.2550030206. [DOI] [PubMed] [Google Scholar]
- 24.Motulsky A, Lafleur M, Couffin-Hoarau AC, Hoarau D, Boury F, Benoit JP, et al. Characterization and biocompatibility of organogels based on L-alanine for parenteral drug delivery implants. Biomaterials. 2005;26:6242–53. doi: 10.1016/j.biomaterials.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 25.Plourde F, Motulsky A, Couffin-Hoarau AC, Hoarau D, Ong H, Leroux JC. First report on the efficacy of L-alanine-based in situ-forming implants for the long-term parenteral delivery of drugs. J Control Release. 2005;108:433–41. doi: 10.1016/j.jconrel.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 26.Vintiloiu A, Lafleur M, Bastiat G, Leroux JC. In situ-forming oleogel implant for rivastigmine delivery. Pharm Res. 2008;25:845–52. doi: 10.1007/s11095-007-9384-3. [DOI] [PubMed] [Google Scholar]
- 27.Kurz A, Farlow M, Lefevre G. Pharmacokinetics of a novel transdermal rivastigmine patch for the treatment of Alzheimer’s disease: a review. Int J Clin Pract. 2009;63:799–805. doi: 10.1111/j.1742-1241.2009.02052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ellman GL, Courtney KD, Andres V, Jr, Featherstone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 29.Luna LG. In: Manual of histologic staining methods of the armed forces institute of pathology. Luna LG, editor. New York: McGraw-Hill; 1960. [Google Scholar]
- 30.Anderson JM. Biological responses to materials. Ann Rev Mater Res. 2001;31:81–110. [Google Scholar]
- 31.Shapira M, Grant A, Korner M, Soreq H. Genomic and transcriptional characterization of the human AChE locus: complex involvement with acquired and inherited diseases. Isr Med Assoc J. 2000;2:470–3. [PubMed] [Google Scholar]
- 32.Kaufer D, Friedman A, Seidman S, Soreq H. Acute stress facilitates long-lasting changes in cholinergic gene expression. Nature. 1998;393:373–7. doi: 10.1038/30741. [DOI] [PubMed] [Google Scholar]
- 33.Onor ML, Trevisiol M, Aguglia E. Rivastigmine in the treatment of Alzheimer’s disease: an update. Clin Interv Aging. 2007;2:17–32. doi: 10.2147/ciia.2007.2.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barnes J, Barlett JW, van de Pol LA, Loy CT, Scahill RI, Frost C, et al. A meta-analysis of hippocampal atrophy rates in Alzheimer’s disease. Neurobiol Aging. 2009;30:1711–23. doi: 10.1016/j.neurobiolaging.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Williams BR, Nazarians A, Gill MA. A review of rivastigmine: a reversible cholinesterase inhibitor. Clin Ther. 2003;25:1634–53. doi: 10.1016/s0149-2918(03)80160-1. [DOI] [PubMed] [Google Scholar]
- 36.Cohen NJ, Ryan J, Hunt C, Romine L, Wszalek T, Nash C. Hippocampal system and declarative (relational) memory: summarizing the data from functional neuroimaging studies. Hippocampus. 1999;9:83–98. doi: 10.1002/(SICI)1098-1063(1999)9:1<83::AID-HIPO9>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 37.Moses SN, Ryan J. A comparison and evaluation of the predictions of relational and conjunctive accounts of hippocampal function. Hippocampus. 2006;16:43–65. doi: 10.1002/hipo.20131. [DOI] [PubMed] [Google Scholar]
- 38.Winblad B, Cummings J, Andreasen N, Grossberg G, Onofrj M, Sadowsky C, et al. A six-month double-bind, randomized, placebo-controlled study of a transdermal patch in Alzheimer’s disease – rivastigmine patch versus capsule. Int J Geriat Psychiatry. 2007;22:456–67. doi: 10.1002/gps.1788. [DOI] [PubMed] [Google Scholar]
- 39.Winblad B, Kawata AK, Beusterien KM, Thomas SK, Wimo A, Lane R, et al. Caregiver preference for rivastigmine patch relative to capsules for treatment of probable Alzheimer’s disease. Int J Geriat Psychiatry. 2007;22:485–91. doi: 10.1002/gps.1806. [DOI] [PubMed] [Google Scholar]
- 40.Mauskopf JA, Paramore C, Lee WC, Snyder EH. Drug persistency patterns for patients treated with rivastigmine or donepezil in usual care settings. J Managed Care Pharm. 2005;11:231–9. doi: 10.18553/jmcp.2005.11.3.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blais L, Kettani FZ, Perreault S, Leroux JC, Forget A, Kergoat MJ. Adherence to cholinesterase inhibitors in patients with Alzheimer’s disease. J Am Geriat Soc. 2009;57:366–7. doi: 10.1111/j.1532-5415.2009.02075.x. [DOI] [PubMed] [Google Scholar]
- 42.Cummings J, Lefèvre G, Small G, Appel-Dingemanse S. Pharmacokinetic rationale for the rivastigmine patch. Neurology. 2007;69:S10–3. doi: 10.1212/01.wnl.0000281846.40390.50. [DOI] [PubMed] [Google Scholar]
- 43.Yang Q, Williams D, Owusu-Ababio G, Ebube NK, Habib MJ. Controlled release tacrine delivery system for the treatment of Alzheimer’s disease. Drug Deliv. 2001;8:93–8. doi: 10.1080/107175401750177061. [DOI] [PubMed] [Google Scholar]
- 44.Gao P, Ding P, Xu H, Yuan Z, Chen D, Wei J, et al. In vitro and in vivo characterization of huperzine A loaded microspheres made from end-group uncapped poly(d, l-lactide acid) and poly(d, l-lactide-co-glycolide acid) Chem Pharm Bull. 2006;54:89–93. doi: 10.1248/cpb.54.89. [DOI] [PubMed] [Google Scholar]
- 45.Fu X, Ping Q, Gao Y. Effects of formulation factors on encapsulation efficiency and release behaviour in vitro of huperzine A-PLGA microspheres. J Microencapsulation. 2005;22:705–14. doi: 10.1080/02652040500162196. [DOI] [PubMed] [Google Scholar]
- 46.Gao P, Xu H, Ding P, Gao Q, Sun J, Chen D. Controlled release of huperzine A from biodegradable microspheres: in vivo and in vitro studies. Int J Pharm. 2007;330:1–5. doi: 10.1016/j.ijpharm.2006.08.030. [DOI] [PubMed] [Google Scholar]
- 47.Chu DF, Fu XQ, Liu WH, Liu K, Li YX. Pharmacokinetics and in vitro and in vivo correlation of huperzine A loaded poly(lactic-co-glycolic acid) microspheres in dogs. Int J Pharm. 2006;325:116–23. doi: 10.1016/j.ijpharm.2006.06.032. [DOI] [PubMed] [Google Scholar]
- 48.Zhang P, Chen L, Gu W, Xu Z, Gao Y, Li Y. In vitro and in vivo evaluation of donepezil-sustained release microparticles for the treatment of Alzheimer’s disease. Biomaterials. 2007;28:1882–8. doi: 10.1016/j.biomaterials.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 49.Coppack SW, Yost TJ, Fisher RM, Eckel RH, Miles JM. Periprandial systemic and regional lipase activity in normal humans. Am J Physiol Endocrinol Metab. 1996;270:E718–22. doi: 10.1152/ajpendo.1996.270.4.E718. [DOI] [PubMed] [Google Scholar]
- 50.Jeong B, Choi YK, Bae YH, Zentner G, Kim SW. New biodegradable polymers for injectable drug delivery systems. J Control Release. 1999;62:109–14. doi: 10.1016/s0168-3659(99)00061-9. [DOI] [PubMed] [Google Scholar]
- 51.Yamaguchi K, Anderson JM. In vivo biocompatibility studies of medisorb 65/35 D, L-lactide/glycolide copolymer microspheres. J Control Release. 1993;24:81–93. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.