

Abstract
The physiological role of the TGR5 receptor in the pancreas is not fully understood. We previously showed that activation of TGR5 in pancreatic β cells by bile acids induces insulin secretion. Glucagon released from pancreatic α cells and glucagon-like peptide 1 (GLP-1) released from intestinal L cells regulate insulin secretion. Both glucagon and GLP-1 are derived from alternate splicing of a common precursor, proglucagon by PC2 and PC1, respectively. We investigated whether TGR5 activation in pancreatic α cells enhances hyperglycemia-induced PC1 expression thereby releasing GLP-1, which in turn increases β cell mass and function in a paracrine manner. TGR5 activation augmented a hyperglycemia-induced switch from glucagon to GLP-1 synthesis in human and mouse islet α cells by GS/cAMP/PKA/cAMP-response element-binding protein-dependent activation of PC1. Furthermore, TGR5-induced GLP-1 release from α cells was via an Epac-mediated PKA-independent mechanism. Administration of the TGR5 agonist, INT-777, to db/db mice attenuated the increase in body weight and improved glucose tolerance and insulin sensitivity. INT-777 augmented PC1 expression in α cells and stimulated GLP-1 release from islets of db/db mice compared with control. INT-777 also increased pancreatic β cell proliferation and insulin synthesis. The effect of TGR5-mediated GLP-1 from α cells on insulin release from islets could be blocked by GLP-1 receptor antagonist. These results suggest that TGR5 activation mediates cross-talk between α and β cells by switching from glucagon to GLP-1 to restore β cell mass and function under hyperglycemic conditions. Thus, INT-777-mediated TGR5 activation could be leveraged as a novel way to treat type 2 diabetes mellitus.
Keywords: bile acid, cell signaling, G protein-coupled receptor, metabolic syndrome, pancreatic islet, reprogramming
Introduction
Bile acids have emerged as important mediators of metabolic homeostasis. In addition to their lipid emulsification properties, bile acids engage a number of receptors such as the farnesoid X receptor and the Takeda G-protein-coupled receptor-5 (TGR5) to mediate their metabolic functions (1–7). TGR5 receptors are plasma membrane receptors and have been largely characterized in the intestine where they are present in L cells (8). Activation of TGR5 receptors on L cells promotes secretion of glucagon-like peptide-1 (GLP-1),4 a key insulin-sensitizing and trophic hormone (9, 10). Following meal-stimulated gallbladder contraction and bile release into the intestinal lumen, bile acid engagement of TGR5 receptors on L cells and the subsequent release of GLP-1 represent an important component of the intestinal endocrine response to meals (11, 12). Recently, TGR5 receptors have been identified on both pancreatic islet α and β cells (13, 14). We have previously demonstrated that TGR5 activation on β cells can increase insulin secretion (14). However, the function and physiological role of TGR5 in α cells remain obscure.
Glucagon and GLP-1 are produced from a common precursor proglucagon by alternate splicing mediated by proconvertase-2 (PC2) and PC1, respectively (15, 16). Whereas the L cells express PC1 only, the pancreatic islet α cells express both PC1 and PC2 (17). Under euglycemic conditions, PC2 activity predominates, and α cells secrete mainly glucagon (18). We hypothesized that TGR5 activation by bile acids under hyperglycemia would activate PC1 as seen in L cells and switch the α cell secretory phenotype from glucagon to GLP-1, which would have a trophic effect on adjacent β cells in a paracrine manner. A combination of decreased systemic glucagon secretion and increased local GLP-1 production would be expected to improve insulin resistance and maintain islet β cell mass. Indeed, several mouse models of diabetes such as streptozotocin-induced diabetes, prediabetic NOD mice, and both ob/ob and db/db mice are associated with increased α cell PC1 and GLP-1 expression, although the mechanisms are not fully understood (19, 20).
We tested this hypothesis by the following: 1) defining the expression of TGR5, PC1, and PC2 in pancreatic islet cells under low and high glucose conditions and the effects of TGR5 agonists on isolated human and mouse islets as well as the αTC1-6 cell line; 2) elucidation of the mechanisms by which TGR5 activation increases PC1 expression and GLP-1 release from α cells; 3) evaluation of the paracrine effect of TGR5-mediated α cell release of GLP-1 on β cells by pretreatment with the GLP-1 receptor antagonist exendin(9–39) (21) in vitro; and 4) performing a controlled mouse clinical trial of the TGR5-specific agonist INT-777 (22) versus vehicle alone in db/db mice that become obese, insulin-resistant, and represent a model of hyperglycemia reminiscent of that seen with type 2 diabetes mellitus (23). The effects of INT-777 on weight gain, insulin resistance, fasting hyperglycemia, and glucose tolerance were evaluated. Simultaneously, the pancreatic α cell PC1 and GLP-1 expression was measured along with α and β cell mass and β cell proliferation index.
Experimental Procedures
Materials
NF449 was obtained from Santa Cruz Biotechnology; antibodies to PC2, p-CREB, and CREB were from Cell Signaling Technology. Collagenase P was obtained from Roche Diagnostics; HEPES, Lipofectamine 2000, and RPMI 1640 medium were obtained from Invitrogen; U73122 and myristoylated PKI were obtained from Calbiochem; ESI-05 was from Biolog; Western blotting and chromatography materials were obtained from Bio-Rad. Dulbecco's modified Eagle's medium (DMEM), 2-mercaptoethanol, 8-pCPT-2′-O-Me-cAMP, 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid-acetoxymethyl ester, exendin(9–39), Histopaque (10771 and 11191), and all other reagents were obtained from Sigma.
Animals
7–8-Week-old male C57BL/6J wild type and male db/db mice (the Jackson Laboratory) were maintained on a 12-h light/dark cycle with ad libitum access to water and normal chow diet. The mice were treated with INT-777 (30 mg/kg/day) or carrier solution (DMSO) intraperitoneally for 7 weeks, and body weight was monitored. The animals were housed in the animal facility administered by the Division of Animal Resources, Virginia Commonwealth University. All procedures were approved by the Institutional Animal Care and Use Committee of Virginia Commonwealth University.
Cell Culture
For the pancreatic β cell line, MIN6 cells were cultured in DMEM containing l-glutamine, sodium carbonate, 2.5 mm 2-mercaptoethanol, and for the glucagon-secreting pancreatic α cell line, αTC1-6 cells (obtained from ATCC) were cultured in DMEM containing HEPES, non-essential amino acids, bovine serum albumin (BSA), sodium carbonate. All the media were supplemented with 10% fetal bovine serum and 100 units/ml penicillin/streptomycin, and the cells were incubated at 37 °C in 5% CO2.
Isolation and Maintenance of Mouse Islets
Pancreatic islets from mice were isolated by sequential enzyme digestion of pancreas, filtration, and centrifugation as described previously (24). The isolated islets were maintained in RPMI 1640 medium supplemented with 10% FBS and 100 units/ml penicillin/streptomycin and incubated at 37°C in 5% CO2. Human islets were obtained from National Disease Research Interchange (NDRI), Philadelphia.
RNA Isolation and Quantitative RT-PCR Analysis
Total RNA was isolated from cells (αTC1-6 and MIN6) and human and mouse islets using RNeasy Plus universal mini kit (Qiagen) following the manufacturer's instructions. The purified RNA was reverse-transcribed to single-stranded cDNA, and conventional PCR was carried out as described previously (25). The amplified PCR products were analyzed on 2% agarose gel containing ethidium bromide using Gel DocTMEZ imager. Real time PCR was carried out using StepOneTM real time PCR system (Applied Biosystems). Cycle threshold (Ct) values were obtained, and the relative fold change in gene expression was calculated as 2−ΔΔCt. The change in mRNA expression was calculated using differences of Ct values compared with housekeeping genes (β actin, Mm00607939_s1; Hs01060665_g1). The probes (TaqMan Gene Expression Assays, Applied Biosystems) used were as follows: TGR5 (Mm04212121_s1; Hs01937849_s1), PC1 (Mm00479023_m1; hs01026107_m1), and PC2 (Mm00500981_m1; Hs00159922_m1).
Western Blot Analysis
The cells were solubilized in RIPA buffer containing protease inhibitor mixture (104 mm 4-(2-aminoethyl)benzenesulfonyl fluoride, 80 μm aprotinin, 4 mm bestatin, 1.4 mm E-64, 2 mm leupeptin, 1.5 mm pepstatin A). The supernatant was collected after centrifuging the lysate at 10,000 × g for 15 min at 4 °C, and the protein concentration was determined by DC protein assay kit from Bio-Rad. Equivalent amounts of protein were resolved by SDS-PAGE and transferred onto a nitrocellulose membrane. Blots were blocked in 5% nonfat dry milk for 1 h followed by immunoblotting with specific antibodies and visualized on film using horseradish peroxidase-conjugated secondary antibodies and advanced ECL Western blotting detection reagents. Western blot images were scanned and analyzed with ImageJ software for densitometric measurements. The average intensity obtained for each band was normalized to its respective band of β-actin. The band intensity was then presented as relative fold changes compared with the corresponding control.
Phosphoinositide (PI) Hydrolysis Assay
αTC1-6 cells were labeled with myo-[3H]inositol (0.5 μCi/ml) in DMEM for 24 h. After 24 h, cells were washed with PBS and treated with INT-777 or a selective Epac ligand, 8-pCPT-2′-O-Me-cAMP in the presence or absence of NF449 (a selective Gαs inhibitor), U73122 (PI hydrolysis inhibitor) or ESI-05 (Epac2 inhibitor) for 1 min. The reaction was terminated using 940 μl of chloroform/methanol/HCl (50:100:1 v/v) as described previously (26). The upper aqueous phase was applied to the column prepared with Dowex AG-50W-X8 resin and water (1:1), and the [3H]inositol triphosphate was eluted with 0.8 m ammonium formate plus 0.1 m formic acid. Radioactivity was determined by liquid scintillation counting, and the results were expressed as percent increase above basal.
Transfection and Promoter Activity Assay
αTC1-6 cells were plated at a density of 5 × 105 cells in 96-well plates in DMEM and cultured until confluent. One day before the transfection, the media were changed to antibiotic-free medium. The cells were then transiently transfected with 0.2 μg of PC1 plasmid (human PC1 luciferase promoter construct, −971 to −1 bp relative to the translation initiation codon inserted in the pGL2-basic vector) and Renilla luciferase expression plasmid for transfection control using Lipofectamine 2000. Control cells were transfected with vector (pGL2-basic vector) alone. All transfections were done in quadruplet. After 24 h of transfection, cells were treated with 5 or 25 mm glucose with or without INT-777 in the presence of specific inhibitors for 24 h. Luciferase activity was measured using dual-luciferase reporter assay kit (Promega), and firefly luciferase activity was normalized by Renilla luciferase activity.
Measurement of GLP-1 Secretion
αTC1-6 cells were plated at a density of 2 × 105 cells in 24-well plates with DMEM containing 5 or 25 mm glucose and cultured for 6–7 days. The cells were then washed and incubated in DMEM containing 5 or 25 mm glucose with or without OA, INT-777, or LCA for 24 h. The supernatants were collected and assayed for GLP-1 using an ELISA kit (Millipore). For GLP-1 secretion from control mouse islets, the islets were cultured in RPMI 1640 medium containing 5 or 25 mm glucose for 6–7 days and then treated with or without OA, INT-777, or LCA for 24 h. The islets isolated from db/db mice were cultured in RPMI 1640 medium with or without OA, INT-777, or LCA for 24 h. The supernatants were collected and assayed for GLP-1 secretion. 25–30 mouse islets were used in each experimental condition. DPP-4 inhibitor (EMD Millipore, DPP4–010) was added to prevent GLP-1 degradation at a concentration of 10 μl/ml medium.
Measurement of Insulin Secretion
Human islets were cultured under low (5 mm) or high (25 mm) glucose conditions for 7 days. The islets were washed and incubated in Hanks' balanced salt solution containing 3 mm glucose for 2 h at 37 °C. Human islets (30–35 islets/condition) were then treated for 1 h in Hanks' balanced salt solution containing 3 mm (basal) or 25 mm glucose (stimulated) with INT-777 or exendin(9–39) or both. The supernatants were collected and assayed for insulin using ELISA kit (Mercodia).
Glucose and Insulin Tolerance Tests
Mice were fasted overnight, and baseline blood glucose levels were measured in tail vein blood using an Accu-Chek Compact Plus glucometer (Roche Diagnostics). Glucose (1 mg of dextrose/g of body weight) in sterile phosphate-buffered saline was injected intraperitoneally, and blood glucose levels were measured prior to and 15, 30, 60, 90, and 120 min after glucose injection as described previously (27). For the insulin tolerance test, mice were injected with insulin (0.75 units/kg body weight) intraperitoneally, and blood glucose levels were measured prior to and 15, 30, 45, 60, 75, and 90 min after injection.
Immunofluorescence
The pancreatic tissues were fixed with 10% neutral buffered formalin, and the paraffin-embedded tissues were sectioned using a microtome to obtain the maximum footprint of the pancreas. The tissue sections were deparaffinized and rehydrated through a series of ethanol concentrations. After antigen retrieval with citrate buffer (pH 6.0) at 94 °C for 15 min, the sections were blocked using Dako protein block and incubated with primary antibody as appropriate. Mouse anti-glucagon (Abcam, ab133195, 1:100), rabbit anti-insulin (Santa Cruz Biotechnology, sc-9168, 1:400), mouse anti-insulin (Sigma, I2018, 1:400), rabbit anti-Ki67 (Abcam, ab15580, 1:100), or rabbit anti PC1/3 (Millipore, ab10553, 1:50) were used overnight at 4 °C. The sections were then incubated with Alexa Fluor secondary antibodies (Life Technologies, Inc.) as appropriate for 2 h at room temperature. In addition, the cell nuclei were stained with DAPI and mounted, and the slides were visualized using an epifluorescent microscope (Axiophot, Zeiss, Jena, Germany).
Pancreatic Islet Morphometry
Morphometric analysis of pancreatic sections was performed as described previously (28, 29). In brief, a composite picture covering an entire section of pancreas was captured using the mosaic function of AxioVision analysis software (version 4.7.10, Zeiss). Then the border of a pancreatic section was defined as an area above background for each composite image. Areas positive for insulin or glucagon were defined by setting a threshold signal for each antigen. The size of the positive area was quantified automatically based upon these thresholds using AxioVision analysis software. Nuclei positive for Ki67 were counted in >1500 β cells per mouse. Colocalization was analyzed using Image-Pro Plus 6 software and expressed as Pearson's colocalization coefficient. To quantitate the insulin content, images (islets of similar sizes) were analyzed using Zeiss software by determining the average pixel value of staining per islet. Background staining was subtracted from each value.
MTT Cell Proliferation Assay
MIN6 cells were treated with different concentrations of INT-777 (10, 25, or 50 μm) for 24, 48, or 72 h. 10 μl of 5 mg/ml MTT solution was added to each well of a 96-well plate and incubated for 4 h at 37 °C. The formazan granules formed were then dissolved in dimethyl sulfoxide (DMSO). The absorbance values were measured with a microplate spectrophotometer at a wavelength of 600 nm.
Statistical Analysis
Results were calculated as means ± S.E., and the experiments were performed at least three times. The experiments were performed on islets isolated from different animals. Statistical significance was analyzed using Student's t test or analysis of variance with post hoc Bonferroni correction for multiple comparisons. GraphPad Prism software (version 6) was used for all statistical analyses, and p values < 0.05 were considered significant.
Results
TGR5 Activation Increases Hyperglycemia-induced PC1 Expression in Pancreatic α Cells
PC1 mRNA was initially measured in pancreatic α cells (αTC1-6) following 7 days of culture under low (5 mm) and high (25 mm) glucose conditions. The expression of PC1 was higher in αTC1-6 cells cultured under high glucose conditions as compared with cells cultured under low glucose conditions (Fig. 1A). Similar results were obtained in isolated mouse and human islets exposed to high glucose as compared with low glucose conditions (Fig. 1, B and C).
FIGURE 1.

INT-777 up-regulates PC1 expression in human and mouse islets and αTC1-6 cells under hyperglycemic conditions. Expression of PC1 mRNA after 48 h of incubation with or without INT-777 (25 μm) in αTC1-6 cells (A), control mouse islets (B), and human islets (C) cultured under low (5 mm) or high (25 mm) glucose (G) conditions for 7 days. D, PC1 mRNA expression in db/db mouse islets treated with or without INT-777 (25 μm) for 48 h. Whole cell lysates were prepared, and immunoblot analysis was performed for PC1 in αTC1-6 cells (E) and db/db mouse islets (F) treated with or without INT-777. Bar graphs show the densitometric values calculated after normalization to loading control (β-actin). Data are expressed as the mean ± S.E. of three or four experiments. ##, p < 0.001, compared with 5 mm glucose basal or control basal; *, p < 0.05, or **, p < 0.001, compared with 25 mm glucose basal or db/db basal.
To determine whether TGR5 activation could regulate PC1 expression, PC1 mRNA and protein were measured in α cells after exposure to INT-777, a specific TGR5 agonist for 48 h under both low and high glucose conditions. INT-777 significantly (p = 0.0012) increased PC1 mRNA (Fig. 1A) and protein (Fig. 1E) in αTC1-6 cells as well as in mouse and human islets under high glucose conditions (Fig. 1, B and C). In contrast, INT-777 had no effect on PC1 mRNA in cells exposed to low glucose conditions (Fig. 1). Consistent with these observations, the expression of PC1 was significantly higher in islets from db/db mice compared with islets from wild type (C57BL/6J) control mice (Fig. 1D). Treatment of islets from db/db mice with INT-777 further augmented the PC1 mRNA (Fig. 1D) and protein (Fig. 1F) expression. However, INT-777 did not affect PC2 mRNA expression under either low or high glucose conditions in αTC1-6 cells and control mouse and human islets as well as db/db mouse islets (Fig. 2).
FIGURE 2.

Effect of INT-777 on PC2 expression. PC2 mRNA expression in αTC1-6 cells (A), control mouse islets (B), and human islets (C) cultured for 7 days under low (5 mm) or high (25 mm) glucose (G) conditions in the presence or absence of INT-777 (25 μm) for 48 h. D, PC2 mRNA expression in db/db mouse islets treated with or without INT-777 (25 μm) for 48 h. Data are expressed as the mean ± S.E. of three experiments.
To investigate whether the glucose-mediated effects on PC1 were a function of TGR5 expression, TGR5 mRNA was also measured at low (5 mm) and high (25 mm) glucose conditions. There was no difference in TGR5 mRNA and protein levels under low versus high glucose conditions in either αTC1-6 cells or mouse islets (Fig. 3, A and B). Similar results were obtained in db/db mouse islets as compared with wild type (C57BL/6J) control mouse islets (Fig. 3D). Thus, hyperglycemia had no effect on the expression of α cell TGR5 receptors. In addition, INT-777 also had no effect on TGR5 mRNA and protein expression (Fig. 3). Furthermore, TGR5 expression on β cells (MIN6) was not altered by ambient glucose levels either (Fig. 3C). Taken together, these results suggest that TGR5 activation induces PC1 expression in pancreatic α cells under hyperglycemic conditions.
FIGURE 3.

Effect of hyperglycemia and INT-777 on TGR5 expression. TGR5 protein (top panel) and mRNA expression in αTC1-6 cells (A), control mouse islets (B), and MIN6 cells (C) cultured for 7 days under low (5 mm) or high (25 mm) glucose (G) conditions in the presence or absence of INT-777 (25 μm) for 48 h. D, TGR5 protein (top panel) and mRNA expression in db/db mouse islets treated with or without INT-777 (25 μm) for 48 h. Data are expressed as the mean ± S.E. of three experiments.
Signaling Mechanisms Involved in TGR5-mediated PC1 Expression in α Cells
To examine the mechanisms underlying the TGR5-mediated increase in PC1 expression under hyperglycemic conditions, αTC1-6 cells were transfected with the human PC1 luciferase promoter construct. Subsequent treatment of αTC1-6 cells with 25 mm glucose caused a significant increase in the promoter activity that was further augmented by INT-777 (25 μm) (Fig. 4A). In contrast, neither glucose nor INT-777 treatment had an effect on the promoter activity of control vector. The INT-777-induced increase in PC1 promoter activity at high (25 mm) glucose conditions was significantly inhibited by both NF449 (Gαs inhibitor, 10 μm, 75.6 ± 3.87% inhibition) and myristoylated PKI (PKA inhibitor, 1 μm, 100% inhibition) (Fig. 4A). Furthermore, treatment of αTC1-6 cells with INT-777 caused activation of CREB (Fig. 4B). Similarly, treatment of islets from db/db mice with INT-777 caused activation of CREB as compared with treatment of islets from control mice (Fig. 4C), thus suggesting that INT-777-induced PC1 expression in α cells is via cAMP/PKA-dependent pathway leading to phosphorylation of CREB.
FIGURE 4.
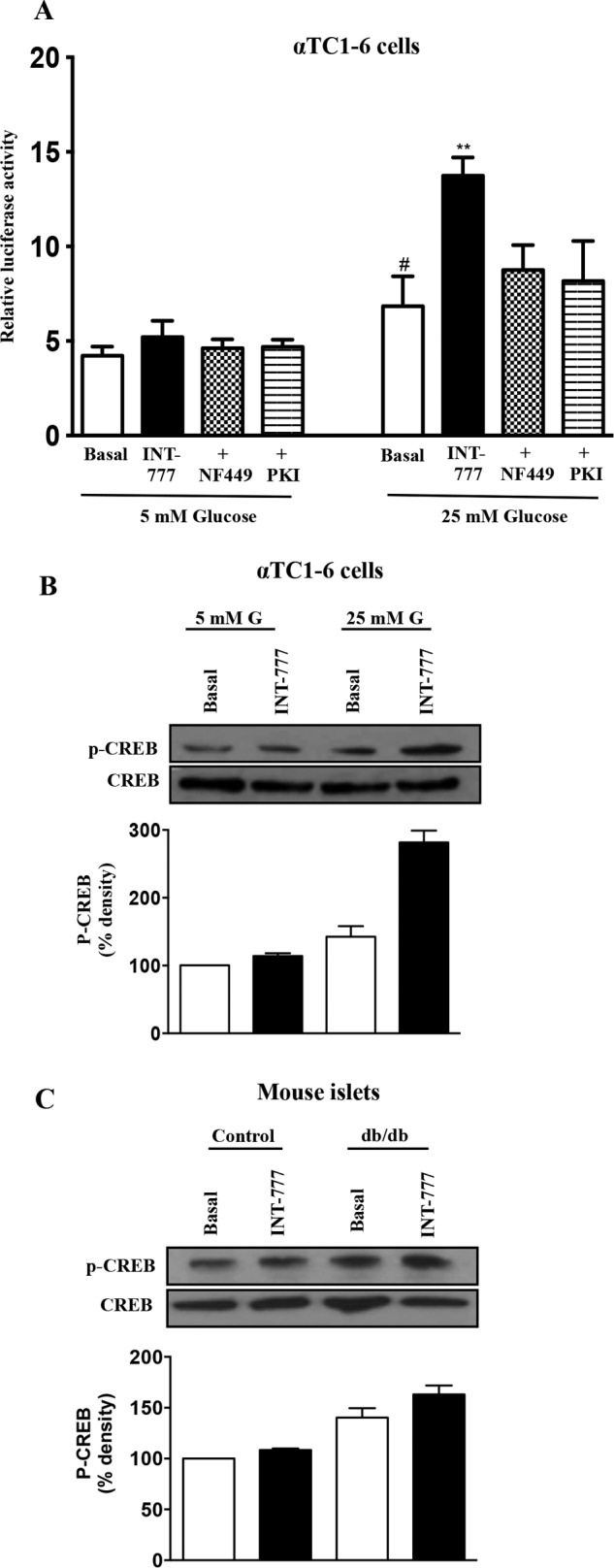
Signaling mechanism involved in INT-777-induced increased PC1 expression in α cells. A, PC1 promoter activity in αTC1-6 cells treated with or without INT-777 in the presence (+) of Gαs inhibitor (NF449, 10 μm) or PKA inhibitor (Myr-PKI, 1 μm). Immunoblot analysis was performed for phosphorylated and total CREB (pCREB and CREB) in αTC1-6 cells (B) and db/db mouse islets (C) treated with or without INT-777. Bar graphs show the densitometric values calculated after normalization to CREB. G, glucose. Data are expressed as the mean ± S.E. of three or four experiments. *, p < 0.05, compared with 5 mm glucose basal; **, p < 0.001, compared with 25 mm glucose basal.
Mechanisms Involved in TGR5-mediated Increase in GLP-1 Release by Pancreatic α Cells
We next examined the effect of TGR5 agonists on GLP-1 secretion in αTC1-6 cells as well as in human and mouse islets cultured under low (5 mm) or high (25 mm) glucose conditions. Consistent with increased PC1 expression, TGR5 agonists (INT-777, a specific TGR5 agonist, or LCA, a natural bile acid) augmented GLP-1 release in αTC1-6 cells, control mouse islets, and human islets cultured only at high glucose conditions (Fig. 5, A–C). Similarly, INT-777 significantly stimulated GLP-1 release from diabetic (db/db) islets but not in control mouse islets (Fig. 5D).
FIGURE 5.

INT-777 increases GLP-1 release in human and mouse islets and αTC1-6 cells under hyperglycemic conditions via Epac in a PKA-independent mechanism. GLP-1 release after 24 h of incubation with or without INT-777 (25 μm) or LCA (25 μm) in αTC1-6 cells (A), control mouse islets (B), and human islets (C) cultured under low (5 mm) or high (25 mm) glucose (G) condition for 7 days. D, GLP-1 release from db/db mouse islets treated with or without INT-777 (25 μm) for 24 h. Data are expressed as the mean ± S.E. of three experiments. ##, p < 0.001 compared with 5 mm glucose basal or control basal; *, p < 0.05, or **, p < 0.001, compared with 25 mm glucose basal or db/db basal. E, PI hydrolysis measured by ion exchange chromatography in αTC1-6 cells labeled with myo-[3H]inositol for 24 h and treated with INT-777(25 μm) or Epac ligand (10 μm) with or without inhibitors of Gαs (NF449,10 μm), PI hydrolysis (U73122, 10 μm), or Epac2 (ESI-05, 10 μm). Data are expressed as mean ± S.E. of three experiments. *, p < 0.05, or **, p < 0.001, significant inhibition in PI hydrolysis compared with INT-777 or Epac ligand. F, signaling pathway involved in TGR5-mediated up-regulation of PC1 expression and GLP-1 release from αTC1-6 cells under hyperglycemia.
We have previously shown that bile acids via activation of TGR5 stimulates insulin secretion from pancreatic β cells via a Gs/Epac/PLC-ϵ/Ca2+ pathway (14). To test whether the same pathway is involved in GLP-1 secretion, the effect of INT-777 and Epac-selective ligand 8-pCPT-2′-O-Me-cAMP on phosphoinositol hydrolysis in αTC1-6 cells was examined. Both INT-777 and Epac ligand caused a significant increase in PI hydrolysis (Fig. 5E). The INT-777-induced increase in PI hydrolysis (1201 ± 38 cpm/mg of protein; 185 ± 3% increase above basal levels) was blocked by selective inhibitors of Gαs protein (NF449, 76 ± 5% inhibition), PI hydrolysis (U73122, 77 ± 6% inhibition), or Epac2 (ESI-05, 80 ± 4% inhibition). These data suggest that stimulation of PI hydrolysis by INT-777 was mediated via activation of Gαs proteins and cAMP-dependent Epac/PLC-ϵ activity. Similarly, the selective Epac ligand also stimulated PI hydrolysis (1154 ± 20 cpm/mg of protein; 173 ± 4% increase above basal levels) that was inhibited by U73122 (66.6 ± 3.5% inhibition) or ESI-05 (65.0 ± 2.35% inhibition) but not by NF449 (1033 ± 10 cpm/mg of protein; 143 ± 35% increase above basal levels) (Fig. 5E).
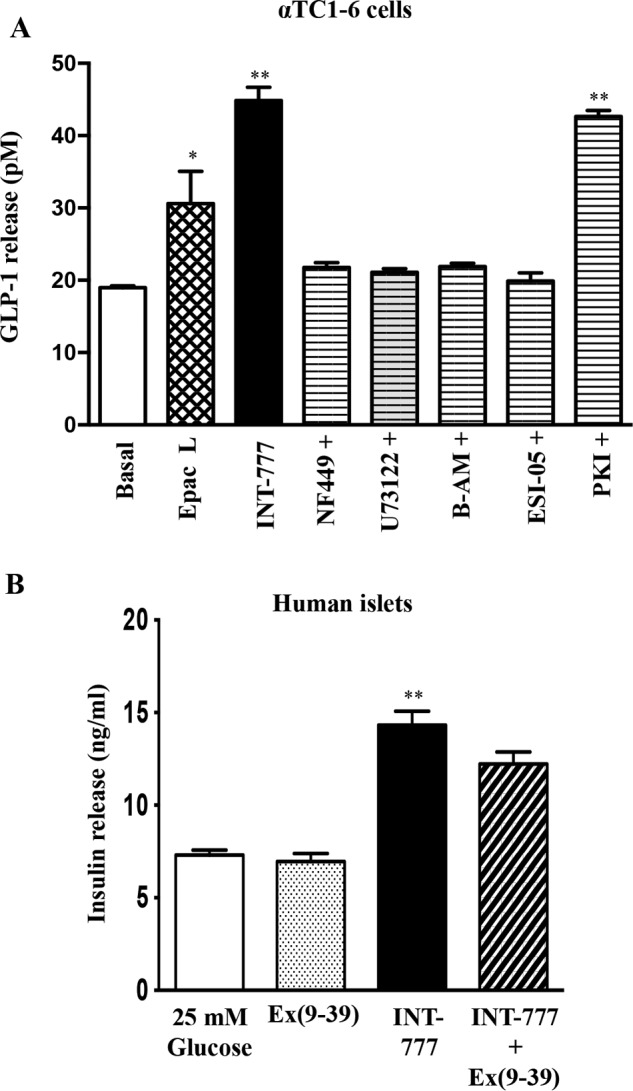
To further confirm the signaling pathway involved in TGR5-mediated GLP-1 secretion in pancreatic α cells, we measured the effect of INT-777 on GLP-1 release in the presence or absence of different inhibitors (Fig. 6A). INT-777-induced GLP-1 secretion was abolished by incubation of cells with NF449 (Gαs inhibitor), U73122 (PI hydrolysis inhibitor), BAPTA-AM (calcium chelator), or ESI-05 (Epac 2 inhibitor) but not by PKI (PKA inhibitor). Consistent with these results, Epac2 and PLC-ϵ was found to be expressed in αTC1-6 cells. Thus, INT-777-augmented hyperglycemia-induced GLP-1 secretion involves sequential activation of Gs, cAMP-dependent Epac, and Epac-mediated PLC-ϵ activity and Ca2+ release (Fig. 5F).
FIGURE 6.
A, INT-777 induces GLP-1 release in α cells via Epac. GLP-1 release from αTC1-6 cells cultured under low (5 mm) or high (25 mm) glucose conditions for 7 days treated with INT-777 (25 μm) or Epac ligand (10 μm) with or without inhibitors of Gαs (NF449, 10 μm), PI hydrolysis (U73122, 10 μm), or Epac2 (ESI-05, 10 μm). Data are expressed as mean ± S.E. of four experiments. *, p < 0.05, or **, p < 0.001, compared with 25 mm glucose basal. B, effect of exendin(9–39) (Ex(9–39)) on insulin secretion in human islets. Human islets were cultured in high (25 mm) glucose for 7 days. Glucose-stimulated insulin secretion after 1 h of incubation with or without INT-777 in the presence or absence of GLP-1 receptor antagonist, exendin(9–39) (0.25 μm). Data are expressed as mean ± S.E. of three experiments. **, p < 0.001 compared with 25 mm glucose.
Effect of TGR5-mediated Increase in α Cell GLP-1 Secretion on Pancreatic β Cell Function
To determine whether the observed increase in PC1 activity and GLP-1 release by TGR5 activation under hyperglycemic conditions affected pancreatic islet β cell function, isolated human islets were cultured under hyperglycemic (25 mm) conditions. Glucose-stimulated insulin secretion was measured with and without INT-777 (25 μm). INT-777 increased insulin secretion, and this effect was blocked by pretreatment with the GLP-1 receptor antagonist, exendin(9–39), indicating that GLP-1 released by α cells stimulates insulin secretion by adjacent β cells under hyperglycemic conditions (Fig. 6B).
TGR5 Agonists Improve Insulin Sensitivity and Pancreatic β Cell Mass and Function under Hyperglycemic Conditions in Vivo
To further evaluate the physiological relevance of TGR5-mediated reprogramming of pancreatic islets under hyperglycemic conditions, the TGR5 agonist INT-777 (30 mg/kg/day) or vehicle was administered intraperitoneally to hyperglycemic db/db or euglycemic wild type (C57BL/6J) mice for 7 weeks.
A: INT-777 Improves Insulin Sensitivity and Glucose Tolerance
As expected, over the time course of the study, db/db mice gained weight and developed insulin resistance and hyperglycemia, although wild type mice did not demonstrate these features (Fig. 7A). INT-777 reduced fasting plasma glucose in hyperglycemic db/db mice but not in wild type mice (Fig. 7B). This was accompanied by a decrease in fasting insulin levels in the db/db mice alone (Fig. 7C) and was reflected in a significant decrease in insulin resistance as measured by the Homeostatic Model Assessment for Insulin Resistance (HOMA-IR) model (Fig. 7D) (30). Formal assessment of glucose tolerance by an intraperitoneal glucose tolerance test demonstrated that INT-777 reduced peak glucose levels as compared with vehicle in db/db mice (Fig. 7E). In wild type mice, INT-777 had no effect on the glucose tolerance test results (Fig. 7E). On a separate day, an insulin tolerance test (ITT) was performed in these mice and confirmed that INT-777 improved insulin sensitivity in hyperglycemic db/db mice compared with vehicle alone but had no effect in wild type mice (Fig. 7F). However glucagon levels were not altered in db/db mice treated with INT-777 (Fig. 8).
FIGURE 7.

INT-777 improves insulin sensitivity and glucose tolerance in db/db mice. Wild type and db/db mice were treated with vehicle (Veh) (DMSO) or INT-777 (30 mg/kg/day) intraperitoneally for 7 weeks (n = 10 per group). Body weights (A), fasting blood glucose concentration (B), fasting plasma insulin (C), and HOMA-IR values (D) were calculated based on the following formula: (fasting insulin (milliunits/liter) × fasting glucose (mmol/liter))/22.5. E, blood glucose concentration (mg/dl) after intraperitoneal injection of 1 g/kg glucose. F, blood glucose concentration (mg/dl) after administration of 0.75 units/kg insulin. Inset depicts the area under curve (AUC) with or without treatment of INT-777 in db/db mice. Data are expressed as the mean ± S.E. of three experiments. #, p < 0.05, or ##, p < 0.001, compared with wild type mice; *, p < 0.05, or **, p < 0.001, compared with db/db mice. HOMA-IR, Homeostatic Model Assessment for Insulin Resistance. AU, arbitrary units.
FIGURE 8.
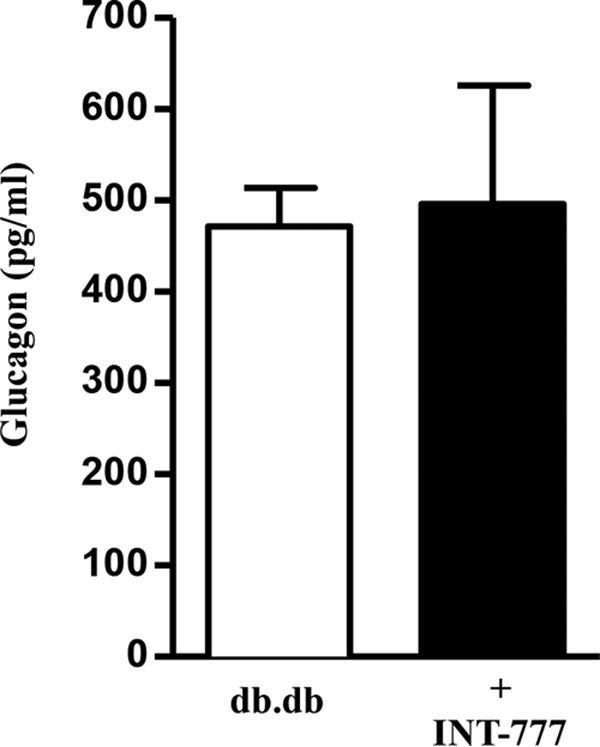
Effect of INT-777 on plasma glucagon in db/db mice. Fasting plasma glucagon in db/db mice treated with vehicle (DMSO) or INT-777 intraperitoneally for 7 weeks. Data are expressed as the mean ± S.E. (n = 5 mice per group).
B: INT-777 Increases PC1 Expression and GLP-1 Secretion in α Cells
At the end of the study period, the mice were euthanized, and the pancreatic islets were harvested in half the mice, and the entire pancreas removed in the others was fixed in formalin. Immunofluorescent staining of pancreatic tissue from db/db mice treated with INT-777 showed an increase in PC1 and glucagon colocalization when compared with wild type mice (Fig. 9, A and B). The co-localization with glucagon indicated that the observed PC1 expression was in pancreatic α cells. Pancreatic islets isolated from db/db mice treated with INT-777 confirmed an increase in PC1 mRNA expression and GLP-1 release as compared with vehicle alone (Fig. 9, C and D). Such effects were not noted in wild type mice. Also, of note, there were no significant differences in TGR5 mRNA expression in isolated islets between wild type or db/db mice, and INT-777 did not alter TGR5 mRNA in islets from either type of mice (Fig. 9E).
FIGURE 9.

INT-777 up-regulates PC1 expression and GLP-1 production in db/db mice. Wild type (Wt) and db/db mice were treated with vehicle (Veh) (DMSO) or INT-777 (30 mg/kg/day) intraperitoneally for 7 weeks (n = 10 per group). A, representative immunohistochemistry images (n = 5) of pancreatic tissue sections using antibodies against glucagon (red) and PC1 (green). Arrows indicate glucagon/PC1 colocalization. Scale bars, 100 μm. B, PC1/glucagon colocalization expressed as Pearson's coefficient of colocalization. Expression of PC1 mRNA (C), GLP-1 release over a 24-h incubation (D), and TGR5 protein (top panel) and mRNA expression (E) in pancreatic islets from wild type and db/db mice after treatment with or without INT-777 for 7 weeks. Data are expressed as the mean ± S.E. of three experiments. ##, p < 0.001, compared with wild type mice; *, p < 0.05, or **, p < 0.001, compared with db/db mice.
C: INT-777 Increases Pancreatic β Cell Area and Proliferation in db/db Mice
Islet morphometry indicated increased islet size in db/db mice as compared with wild type mice. Furthermore, INT-777 increased both the total islet size and the β cell area within the islets in hyperglycemic db/db mice but not in wild type mice (Fig. 10, A–E). β cell proliferation was also increased significantly by INT-777 in db/db mice but not in wild type control mice (Fig. 11, A and B). Furthermore, β cells in islets from db/db mice stained more intensely for insulin in mice receiving INT-777 (Fig. 11C).
FIGURE 10.

INT-777 increases pancreatic β cell area in db/db mice. A, representative immunohistochemistry images (n = 5) of pancreatic tissue sections using antibodies against insulin (green) and glucagon (red). Effect of INT-777 on β cell area, expressed as % of pancreas area (B), pancreas area, expressed as μm2 (C), α cell area, expressed as % of pancreas (D), and β cell area per islet, expressed as μm2 (E) in wild type (Wt) and db/db mice with or without INT-777 treatment for 7 weeks. Data are expressed as the mean ± S.E. for 5 mice per group; *, p < 0.05, or **, p < 0.001, compared with db/db mice.
FIGURE 11.

INT-777 increases pancreatic β cell proliferation in db/db mice. A, immunohistochemistry of pancreatic tissue sections using antibodies against Ki67 (green) and insulin (red). Representative image of n = 5; arrows indicate Ki67-positive cells. Scale bars, 100 μm. B, effect of INT-777 on β cell proliferation, expressed as % of β cells analyzed. C, INT-777 increases insulin staining intensity of β cells in db/db mice. Representative immunohistochemistry images of pancreatic tissue sections using antibody against insulin (green) in db/db mice treated with vehicle (DMSO) or INT-777 (30 mg/kg/d) intraperitoneally for 7 weeks and quantitation of the intensity of insulin staining. Data are expressed as mean fluorescence intensity (n = 6). *, p < 0.05, compared with db/db mice. Scale bars, 100 μm.
Because TGR5 receptors are also present on β cells (14), we tested the possibility whether the increased β cell proliferation in vivo after INT-777 was a direct effect of the drug on β cells by evaluating the effects of INT-777 on β cell proliferation and viability in MIN6 cells. However, INT-777 had no effect on β cell proliferation in vitro (Fig. 12A).
FIGURE 12.
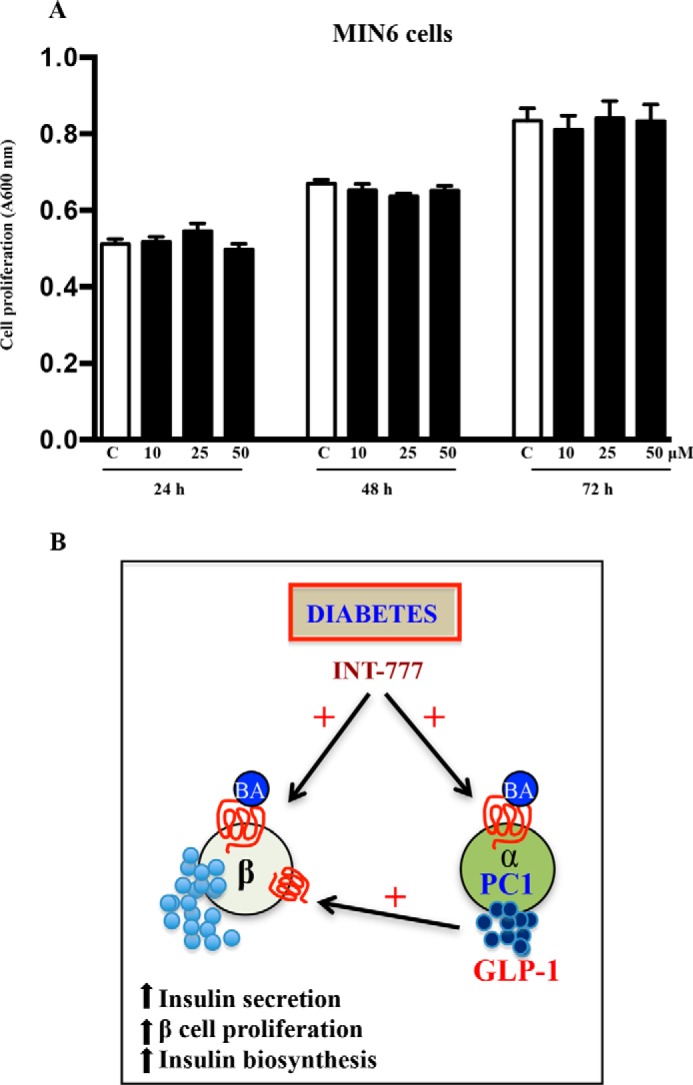
A, INT-777 does not affect MIN6 cell proliferation. MTT assay for cell proliferation of MIN6 cells with or without INT-777 (10, 25, and 50 μm) for 24, 48, or 72 h. Data are expressed as the mean ± S.E. of four experiments. C, untreated. B, mechanism of TGR5-mediated regulation of pancreatic α cells to promote glucose homeostasis. Under hyperglycemia or diabetes, activation of TGR5 receptors on α cells augments glucose-induced PC1 expression leading to GLP-1 secretion that mediates trophic effects on β cells. This results in increased insulin secretion, β cell proliferation, and increased insulin biosynthesis providing the evidence of improvement in glucose homeostasis in db/db mice treated with INT-777, a TGR5-specific agonist.
Discussion
A variety of factors can influence pancreatic islet β cell function and response to systemic metabolic stress associated with insulin-resistant states. These include hyperglycemia itself, circulating free fatty acids, and inflammatory cytokines such as IL-6 and hormones such as GLP-1 among others (31–33). Recently, although pancreatic α cells have been shown to modulate insulin release from β cells by a paracrine mechanism involving acetylcholine release, their role in the β cell response to insulin-resistant states is unclear (34). Here, we demonstrate that under hyperglycemic conditions, activation of TGR5 receptors reprogram α cells to make and secrete GLP-1 locally to promote β cell proliferation and glucose-stimulated insulin secretion.
A principal implication of these findings is that bile acids, the endogenous agonists for TGR5 receptors, may mediate an adaptive response to hyperglycemia by TGR5-mediated metabolic reprogramming of α cells to switch from glucagon to GLP-1 as its principal secretory product with GLP-1-mediated trophic effects on β cells and β cell function. It is indeed known that PC1 expression and GLP-1 release from α cells is increased in a variety of mouse models of hyperglycemia (19, 35–37). Also, circulating bile acid concentrations are elevated in type 2 diabetes (38–41) and could potentially mediate such effects. The failure to prevent diabetes by these endogenous mechanisms however indicates that this adaptive response is not sufficient to normalize the aberrant pathophysiology associated with insulin resistance and type 2 diabetes. In contrast, our data with INT-777 administration in db/db mice suggest the possibility of pharmacologically leveraging this mechanism to improve prediabetes or type 2 diabetes mellitus.
A central player in mediating the effects of α cell TGR5 activation on β cell mass and function is GLP-1. GLP-1 is a well known insulinotropic hormone and produces a dose-dependent increase in glucose-stimulated release of insulin and β cell proliferation (42–44). It also mediates differentiation of progenitor cells into β cells (45). Many elegant studies have demonstrated an important role for bile acid-mediated intestinal GLP-1 release in mediating the metabolic benefits of gastric bypass surgery (46–48). This study adds to the GLP-1 literature by providing a novel TGR5-mediated mechanism to increase GLP-1 in the islets locally and thus modulate β cell mass and function. This is supported by the observation that pretreatment with a GLP-1 receptor antagonist blocked TGR5 effects in isolated human islets. We also demonstrated that INT-777 had no direct effect on MIN6 pancreatic β cell proliferation. However, the potential caveat is that the response of MIN6 cells may differ from that of β cells of primary islets.
From a mechanistic point of view, our data indicate that neither hyperglycemia nor TGR5 agonists influence TGR5 expression in α or β cells. Therefore, the effects of TGR5 agonists are not simply a function of greater receptor expression. Rather, TGR5 agonists mediate their effects through enhancement of PC1. This is supported by augmentation of PC1 promoter activity, mRNA and protein expression by TGR5 agonists in vitro, and PC1 expression in isolated pancreatic islets from db/db mice and in db/db mice receiving INT-777 in vivo without affecting PC2 expression. This study further clarifies the mechanism of TGR5-mediated activation of PC1 in pancreatic α cells by demonstrating that it is Gαs-coupled and involves a Gαs/PKA/CREB pathway supporting the concept that binding of activated CREB to the cAMP-response element region on the PC1 gene promoter is responsible for the increase in the expression of PC1 in pancreatic α cells. Furthermore, it recapitulates our previous demonstration of the signaling pathway for stimulating insulin secretion in β cells in mediating GLP-1 secretion in pancreatic α cells.
It is interesting to note that these effects require a background of hyperglycemia. Studies in isolated islets and cell lines have shown increased PC1 expression with high glucose involving the cAMP pathway (49, 50). In this study, the increase in PC1 expression upon TGR5 activation by INT-777 may further augment cAMP levels and cAMP-mediated CREB activation in cells exposed to high glucose. These data are thus in line with the exciting data demonstrating amelioration of hyperglycemia and improved β cell function with cell therapy involving PC1 overexpressing α cells in db/db mice (51, 52).
The administration of TGR5 agonist, INT-777, to db/db mice exhibited a reduction in body weight and improved insulin sensitivity. This would reduce β cell work load and also account for the recovery of β cell function in the treated db/db mice. In this study, the potential contribution of systemic GLP-1 effects versus local GLP-1 effects could not be separated. INT-777 was administered intraperitoneally to minimize intestinal exposure to the drug with intestinal release of GLP-1, but it is recognized that intestinal exposure could not be avoided due to enterohepatic circulation of the drug. It is therefore likely that the effects in vivo involved both intestinal and pancreatic TGR5 activation. Although TGR5-mediated weight loss has shown to be due to increased brown adipose tissue energy expenditure in high fat diet-fed mice (53), it remains to be determined if this mechanism is also true in leptin receptor-deficient db/db mice.
In summary, we demonstrate that activation of the TGR5 signaling pathway reprograms pancreatic α cells to produce GLP-1 under hyperglycemic conditions with a GLP-1-mediated increase in β cell proliferation and mass (Fig. 12B). Physiologically, this results in improved insulin sensitivity and glucose tolerance along with weight loss. This provides a novel potential mechanism to improve glycemic control and obesity in subjects with type 2 diabetes. Further studies are now indicated to test this possibility in more detail.
Author Contributions
D. P. K., A. A., S. H. P., S. L., and Y. I. contributed to the experimental work. F. M. and Y. I. contributed to the design of some of the experiments and data analysis. J. L. N. and J. R. G. provided scientific insights to the study. D. P. K., K. S. M., and A. J. S. designed the experiments, critically evaluated the results, and wrote the manuscript. A. J. S. and K. S. M. are the principal investigators, obtained funding, and designed the overall study. All the authors reviewed the final version of the manuscript submitted.
Acknowledgments
We thank Intercept Pharmaceuticals, New York, for the generous gift of INT-777. We also thank Dr. Lakshmi Devi (Icahn School of Medicine) and Dr. Theodore Friedman (Charles R. Drew University of Medicine and Science) for the PC1 antibody and PC1 plasmid, respectively.
This work was supported by National Institutes of Health Grants RO1 DK081450 (to A. J. S.) and RO1 DK28300 (to K. S. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- GLP-1
- glucagon like peptide-1
- INT-777 (S-EMCA)
- 6α-ethyl-23(S)-methylcholic acid
- LCA
- lithocholic acid
- PC1
- prohormone convertase-1
- CREB
- cAMP-response element-binding protein
- PI
- phosphoinositide
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- 8-pCPT-2′-O-Me-cAMP
- 8-(4-chlorophenylthio)-2′-O-methyladenosine 3′,5′-cyclic monophosphate monosodium
- OA
- oleanolic acid.
References
- 1. Makishima M., Okamoto A. Y., Repa J. J., Tu H., Learned R. M., Luk A., Hull M. V., Lustig K. D., Mangelsdorf D. J., and Shan B. (1999) Identification of a nuclear receptor for bile acids. Science 284, 1362–1365 [DOI] [PubMed] [Google Scholar]
- 2. Parks D. J., Blanchard S. G., Bledsoe R. K., Chandra G., Consler T. G., Kliewer S. A., Stimmel J. B., Willson T. M., Zavacki A. M., Moore D. D., and Lehmann J. M. (1999) Bile acids: natural ligands for an orphan nuclear receptor. Science 284, 1365–1368 [DOI] [PubMed] [Google Scholar]
- 3. Maruyama T., Miyamoto Y., Nakamura T., Tamai Y., Okada H., Sugiyama E., Nakamura T., Itadani H., and Tanaka K. (2002) Identification of membrane-type receptor for bile acids (M-BAR). Biochem. Biophys. Res. Commun. 298, 714–719 [DOI] [PubMed] [Google Scholar]
- 4. Kawamata Y., Fujii R., Hosoya M., Harada M., Yoshida H., Miwa M., Fukusumi S., Habata Y., Itoh T., Shintani Y., Hinuma S., Fujisawa Y., and Fujino M. (2003) A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 278, 9435–9440 [DOI] [PubMed] [Google Scholar]
- 5. Lefebvre P., Cariou B., Lien F., Kuipers F., and Staels B. (2009) Role of bile acids and bile acid receptors in metabolic regulation. Physiol. Rev. 89, 147–191 [DOI] [PubMed] [Google Scholar]
- 6. Li T., and Chiang J. Y. (2014) Bile acid signaling in metabolic disease and drug therapy. Pharmacol. Rev. 66, 948–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hylemon P. B., Zhou H., Pandak W. M., Ren S., Gil G., and Dent P. (2009) Bile acids as regulatory molecules. J. Lipid Res. 50, 1509–1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lieu T., Jayaweera G., and Bunnett N. W. (2014) GPBA: a GPCR for bile acids and an emerging therapeutic target for disorders of digestion and sensation. Br. J. Pharmacol. 171, 1156–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Katsuma S., Hirasawa A., and Tsujimoto G. (2005) Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem. Biophys. Res. Commun. 329, 386–390 [DOI] [PubMed] [Google Scholar]
- 10. MacDonald P. E., El-Kholy W., Riedel M. J., Salapatek A. M., Light P. E., and Wheeler M. B. (2002) The multiple actions of GLP-1 on the process of glucose-stimulated insulin secretion. Diabetes 51, S434–S442 [DOI] [PubMed] [Google Scholar]
- 11. Houten S. M., Watanabe M., and Auwerx J. (2006) Endocrine functions of bile acids. EMBO J. 25, 1419–1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meier J. J., and Nauck M. A. (2005) Glucagon-like peptide 1(GLP-1) in biology and pathology. Diabetes Metab. Res. Rev. 21, 91–117 [DOI] [PubMed] [Google Scholar]
- 13. Whalley N. M., Pritchard L. E., Smith D. M., and White A. (2011) Processing of proglucagon to GLP-1 in pancreatic α-cells: is this a paracrine mechanism enabling GLP-1 to act on β-cells? J. Endocrinol. 211, 99–106 [DOI] [PubMed] [Google Scholar]
- 14. Kumar D. P., Rajagopal S., Mahavadi S., Mirshahi F., Grider J. R., Murthy K. S., and Sanyal A. J. (2012) Activation of transmembrane bile acid receptor TGR5 stimulates insulin secretion in pancreatic β cells. Biochem. Biophys. Res. Commun. 427, 600–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mojsov S., Heinrich G., Wilson I. B., Ravazzola M., Orci L., and Habener J. F. (1986) Preproglucagon gene expression in pancreas and intestine diversifies at the level of post-translational processing. J. Biol. Chem. 261, 11880–11889 [PubMed] [Google Scholar]
- 16. Tucker J. D., Dhanvantari S., and Brubaker P. L. (1996) Proglucagon processing in islet and intestinal cell lines. Regulatory Peptides 62, 29–35 [DOI] [PubMed] [Google Scholar]
- 17. Rouillé Y., Martin S., and Steiner D. (1995) Differential processing of proglucagon by the subtilisin-like prohormone convertases PC2 and PC3 to generate either glucagon or glucagon-like peptide. J. Biol. Chem. 270, 26488–26496 [DOI] [PubMed] [Google Scholar]
- 18. Rouillé Y., Westermark G., Martin S. K., and Steiner D. F. (1994) Proglucagon is processed to glucagon by prohormone convertase PC2 in aTC1-6 cells. Proc. Natl. Acad. Sci. U.S.A. 91, 3242–3246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kilimnik G., Kim A., Steiner D. F., Friedman T. C., and Hara M. (2010) Intraislet production of GLP-1 by activation of prohormone convertase 1/3 in pancreatic α-cells in mouse models of β-cell regeneration. Islets 2, 149–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thyssen S., Arany E., and Hill D. (2006) Ontogeny of regeneration of beta-cells in the neonatal rat after treatment with streptozotocin. Endocrinology 147, 2346–2356 [DOI] [PubMed] [Google Scholar]
- 21. Göke R., Fehmann H. C., Linn T., Schmidt H., Krause M., Eng J., and Göke B. (1993) Exendin-4 is a high potency agonist and truncated exendin-(9–39)-amide an antagonist at the glucagon-like peptide 1-(7–36)-amide receptor of insulin-secreting beta-cells. J. Biol. Chem. 268, 19650–19655 [PubMed] [Google Scholar]
- 22. Srinivasan K., and Ramarao P. (2007) Animal models in type 2 diabetes research: an overview. Indian J. Med. Res. 125, 451–472 [PubMed] [Google Scholar]
- 23. Pellicciari R., Gioiello A., Macchiarulo A., Thomas C., Rosatelli E., Natalini B., Sardella R., Pruzanski M., Roda A., Pastorini E., Schoonjans K., and Auwerx J. (2009) Discovery of 6α-ethyl-23(S)-methylcholic acid (S-EMCA, INT-777) as a potent and selective agonist for the TGR5 receptor, a novel target for diabesity. J. Med. Chem. 52, 7958–7961 [DOI] [PubMed] [Google Scholar]
- 24. Carter J. D., Dula S. B., Corbin K. L., Wu R., and Nunemaker C. S. (2009) A Practical Guide to Rodent Islet Isolation and Assessment. Biol. Proced. Online 11, 3–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hu W., Li F., Mahavadi S., and Murthy K. S. (2009) Upregulation of RGS4 expression by IL-1β in colonic smooth muscle is enhanced by ERK1/2 and p38 MAPK and inhibited by the PI3K/Akt/GSK3β pathway. Am. J. Physiol. Cell Physiol. 296, C1310–C1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Murthy K. S., and Makhlouf G. M. (1991) Phosphoinositide metabolism in intestinal smooth muscle: preferential production of Ins(1,4,5)P3 in circular muscle cells. Am. J. Physiol. 261, G945–G951 [DOI] [PubMed] [Google Scholar]
- 27. Ayala J. E., Samuel V. T., Morton G. J., Obici S., Croniger C. M., Shulman G. I., Wasserman D. H., McGuinness O. P., and NIH Mouse Metabolic Phenotyping Center Consortium. (2010) Standard operating procedures for describing and performing metabolic tests of glucose homeostasis in mice. Dis. Model. Mech. 3, 525–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Grzesik W. J., Nadler J. L., Machida Y., Nadler J. L., Imai Y., and Morris M. A. (2015) Expression pattern of 12-lipoxygenase in human islets with type 1 diabetes and type 2 diabetes. J. Clin. Endocrinol. Metab. 100, E387–E395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Imai Y., Dobrian A. D., Weaver J. R., Butcher M. J., Cole B. K., Galkina E. V., Morris M. A., Taylor-Fishwick D. A., and Nadler J. L. (2013) Interaction between cytokines and inflammatory cells in islet dysfunction, insulin resistance and vascular disease. Diabetes Obes. Metab. 15, 117–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mather K. (2009) Surrogate measures of insulin resistance: of rats, mice, and men. Am. J. Physiol. Endocrinol. Metab. 296, E398–E399 [DOI] [PubMed] [Google Scholar]
- 31. Boden G. (1999) Free fatty acids, insulin resistance, and type 2 diabetes mellitus. Proc. Assoc. Am. Physicians 3, 241–248 [DOI] [PubMed] [Google Scholar]
- 32. Ellingsgaard H., Hauselmann I., Schuler B., Habib A. M., Baggio L. L., Meier D. T., Eppler E., Bouzakri K., Wueest S., Muller Y. D., Hansen A. M., Reinecke M., Konrad D., Gassmann M., Reimann F., et al. (2011) Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat. Med. 17, 1481–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Donath M. Y., and Burcelin R. (2013) GLP-1 Effects on islets: hormonal, neuronal, or paracrine? Diabetes Care 36, S145–S148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rodriguez-Diaz R., Dando R., Jacques-Silva M. C., Fachado A., Molina J., Abdulreda M. H., Ricordi C., Roper S. D., Berggren P.-O., and Caicedo A. (2011) α cells secrete acetylcholine as a non-neuronal paracrine signal priming β cell function in humans. Nat. Med. 17, 888–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hansen A. M., Bödvarsdottir T. B., Nordestgaard D. N., Heller R. S., Gotfredsen C. F., Maedler K., Fels J. J., Holst J. J., and Karlsen A. E. (2011) Upregulation of α cell glucagon-like peptide 1 (GLP-1) in Psammomys obesus–an adaptive response to hyperglycaemia? Diabetologia 54, 1379–1387 [DOI] [PubMed] [Google Scholar]
- 36. Moffett R. C., Vasu S., Thorens B., Drucker D. J., and Flatt P. R. (2014) Incretin receptor null mice reveal key role of GLP-1 but not GIP in pancreatic β cell adaptation to pregnancy. PLoS ONE 9, e96863 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nie Y., Nakashima M., Brubaker P. L., Li Q.-L., Perfetti R., Jansen E., Zambre Y., Pipeleers D., and Friedman T. C. (2000) Regulation of pancreatic PC1 and PC2 associated with increased glucagon-like peptide 1 in diabetic rats. J. Clin. Invest. 105, 955–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vincent R. P., Omar S., Ghozlan S., Taylor D. R., Cross G., Sherwood R. A., Fandriks L., Olbers T., Werling M., Alaghband-Zadeh J., and le Roux C. W. (2013) Higher circulating bile acid concentrations in obese patients with type 2 diabetes. Ann. Clin. Biochem. 50, 360–364 [DOI] [PubMed] [Google Scholar]
- 39. Wewalka M., Patti M.-E., Barbato C., Houten S. M., and Goldfine A. B. (2014) Fasting serum taurine-conjugated bile acids are elevated in type 2 diabetes and do not change with intensification of insulin. J. Clin. Endocrinol. Metab. 99, 1442–1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Haeusler R. A., Astiarraga B., Camastra S., Accili D., and Ferrannini E. (2013) Human insulin resistance is associated with increased plasma levels of 12α-hydroxylated bile acids. Diabetes 62, 4184–4191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Prawitt J., Caron S., and Staels B. (2011) Bile acid metabolism and the pathogenesis of type 2 diabetes. Curr. Diab. Rep. 11, 160–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Drucker D. (2006) The biology of incretin hormones. Cell Metab. 3, 153–165 [DOI] [PubMed] [Google Scholar]
- 43. Ahrén B. (1998) Glucagon-like peptide-1 (GLP-1): A gut hormone of potential interest in the treatment of diabetes. Bioessays 20, 642–651 [DOI] [PubMed] [Google Scholar]
- 44. Yabe D., and Seino Y. (2011) Two incretin hormones GLP-1 and GIP: comparison of their actions in insulin secretion and β cell preservation. Prog. Biophys. Mol. Biol. 107, 248–256 [DOI] [PubMed] [Google Scholar]
- 45. Doyle M. E., and Egan J. M. (2007) Mechanisms of action of GLP-1 in the pancreas. Pharmacol. Ther. 113, 546–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nakatani H., Kasama K., Oshiro T., Watanabe M., Hirose H., and Itoh H. (2009) Serum bile acid along with plasma incretins and serum high-molecular weight adiponectin levels are increased after bariatric surgery. Metab. Clin. Exp. 58, 1400–1407 [DOI] [PubMed] [Google Scholar]
- 47. Patti M.-E., Houten S. M., Bianco A. C., Bernier R., Larsen P. R., Holst J. J., Badman M. K., Maratos-Flier E., Mun E. C., Pihlajamaki J., Auwerx J., and Goldfine A. B. (2009) Serum bile acids are higher in humans with prior gastric bypass: potential contribution to improved glucose and lipid metabolism. Obesity 17, 1671–1677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pournaras D. J., Glicksman C., Vincent R. P., Kuganolipava S., Alaghband-Zadeh J., Mahon D., Bekker J. H., Ghatei M. A., Bloom S. R., Walters J. R., Welbourn R., and le Roux C. W. (2012) The role of bile after Roux-en-Y gastric bypass in promoting weight loss and improving glycaemic control. Endocrinology 153, 3613–3619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jansen E., Ayoubi T. A., Meulemans S. M., and Van de Ven W. J. (1995) Neuroendocrine-specific expression of the human prohormone convertase 1 gene. Hormonal regulation of transcription through distinct cAMP response elements. J. Biol. Chem. 270, 15391–15397 [DOI] [PubMed] [Google Scholar]
- 50. McGirr R., Ejbick C. E., Carter D. E., Andrews J. D., Nie Y., Friedman T. C., and Dhanvantari S. (2005) Glucose dependence of the regulated secretory pathway in αTC1-6 cells. Endocrinology 146, 4514–4523 [DOI] [PubMed] [Google Scholar]
- 51. Wideman R. D., Covey S. D., Webb G. C., Drucker D. J., and Kieffer T. J. (2007) A switch from prohormone convertase (PC)-2 to PC1/3 expression in transplanted α-cells is accompanied by differential processing of proglucagon and improved glucose homeostasis in mice. Diabetes 56, 2744–2752 [DOI] [PubMed] [Google Scholar]
- 52. Wideman R. D., Gray S. L., Covey S. D., Webb G. C., and Kieffer T. J. (2009) Transplantation of PC1/3-expressing α-cells improves glucose handling and cold tolerance in leptin-resistant mice. Mol. Ther. 17, 191–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Watanabe M., Houten S. M., Mataki C., Christoffolete M. A., Kim B. W., Sato H., Messaddeq N., Harney J. W., Ezaki O., Kodama T., Schoonjans K., Bianco A. C., and Auwerx J. (2006) Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 439, 484–489 [DOI] [PubMed] [Google Scholar]