

Abstract
The E3 transcription unit of human species C adenoviruses (Ads) encodes immunomodulatory proteins that mediate direct protection of infected cells. Recently, we described a novel immunomodulatory function for E3/49K, an E3 protein uniquely expressed by species D Ads. E3/49K of Ad19a/Ad64, a serotype that causes epidemic keratokonjunctivitis, is synthesized as a highly glycosylated type I transmembrane protein that is subsequently cleaved, resulting in secretion of its large ectodomain (sec49K). sec49K binds to CD45 on leukocytes, impairing activation and functions of natural killer cells and T cells. E3/49K is localized in the Golgi/trans-Golgi network (TGN), in the early endosomes, and on the plasma membrane, yet the cellular compartment where E3/49K is cleaved and the protease involved remained elusive. Here we show that TGN-localized E3/49K comprises both newly synthesized and recycled molecules. Full-length E3/49K was not detected in late endosomes/lysosomes, but the C-terminal fragment accumulated in this compartment at late times of infection. Inhibitor studies showed that cleavage occurs in a post-TGN compartment and that lysosomotropic agents enhance secretion. Interestingly, the cytoplasmic tail of E3/49K contains two potential sorting motifs, YXXΦ (where Φ represents a bulky hydrophobic amino acid) and LL, that are important for binding the clathrin adaptor proteins AP-1 and AP-2 in vitro. Surprisingly, mutating the LL motif, either alone or together with YXXΦ, did not prevent proteolytic processing but increased cell surface expression and secretion. Upon brefeldin A treatment, cell surface expression was rapidly lost, even for mutants lacking all known endocytosis motifs. Together with immunofluorescence data, we propose a model for intracellular E3/49K transport whereby cleavage takes place on the cell surface by matrix metalloproteases.
Keywords: adenovirus, intracellular trafficking, protein sorting, shedding, viral immunology, Adenovirus E3 proteins, CD45, E3/49K, Epidemic Keratoconjunctivitis, Immune evasion
Introduction
Human adenoviruses (Ads)2 comprise more than 60 different types that are classified into seven species, A–G (1, 2). In immunocompetent individuals, Ads typically induce self-limiting infections associated with acute disease, whereas in a fraction of cases, persistent infections may ensue (3, 4). In immunocompromised patients, diseases tend to be severe or even fatal, demonstrating the significant impact of the immune system on Ad disease. Although overlapping, the disease pattern differs between Ad species. Species A, F, and G are associated with gastroenteritis, whereas species B, C, and E are mainly associated with respiratory diseases, and species D is associated with eye diseases (3). However, only rarely has a clear link been demonstrated between a defined disease and infection with specific Ad types. One such case is epidemic keratoconjunctivitis (5), a highly contagious and severe eye disease that presents with typical subepithelial corneal infiltrates of dendritic cells, keratocytes, and inflammatory leukocytes that may persist for months or years (5–7). Only a few Ad types of species D (Ad8, Ad19a (recently renamed Ad64 (2), Ad37, Ad53, Ad54, and Ad56) cause this disease, yet the reasons for this association remain obscure (8–10). Because the cellular receptors of the epidemic keratoconjunctivitis-causing (sero)types are widely distributed or ubiquitous (11–13), differential receptor usage does not explain the specific pathogenesis in the eye. Hence, the disease may be due to selective events occurring post-attachment (e.g. differential triggering of innate and adaptive immune responses) (3, 14–16) or their differential manipulation by specific immunomodulatory functions encoded in the early transcription unit 3 (E3) of epidemic keratoconjunctivitis Ads (14, 17).
The E3 region is not required for Ad replication in vitro, yet it is preserved in all human Ads, indicating an important role for virus-host interaction in vivo (18). Supporting this view, previous studies of species C Ad E3 proteins unraveled multiple immune evasion mechanisms that seem to facilitate persistent infections (3, 14, 18, 19). E3 is one of the most divergent regions of the Ad genome (17, 20–22), differing considerably in size, gene composition, and sequence both between and within Ad species. Species D Ads have the largest E3 region, encoding eight open reading frames (ORFs). Of these, the E3/10.4K, 14.5K, and 14.7K ORFs are present in all species and down-regulate various apoptosis receptors from the cell surface or affect their signaling (3, 14, 23, 24), whereas E3/19K is only present in Ads of species B–E that do not cause gastroenteritis. E3/19K retains MHC class I molecules (MHC-I) and MHC-I-related chain A and B in the endoplasmic reticulum (ER), thereby suppressing recognition by cytotoxic T-lymphocytes (25–27) and natural killer (NK) cells (28, 29). A few E3 genes are unique to a particular species and hence may allow for species-specific immunomodulation and differential disease outcome (3, 17, 18, 30, 31). However, with the exception of E3/49K (32), no immune evasion function for species-specific E3 proteins has been identified to date.
The E3/49K ORF was initially identified in the E3 region of the epidemic keratoconjunctivitis-causing Ad19a/Ad64 (33). This gene is unique for species D Ads, and all species D Ads tested expressed the corresponding protein (34), implicating it in their pathogenesis. Interestingly, E3/49K (also called CR1-β) is the protein with the highest frequency of amino acid substitutions, presumably due to a recombination hot spot (22). E3/49K is a highly glycosylated type I transmembrane protein that migrates with an apparent molecular mass of 70–100 kDa and as such is by far the largest E3 protein. Ad19a E3/49K is abundantly synthesized in the early phase of infection but continues to be produced in the late phase, albeit only with immature carbohydrates. The sequence of the extracellular domain revealed three internal repeats designated conserved regions 1–3 that are predicted to form immunoglobulin-like domains. Interestingly, similar domains seem to be present in some other E3 proteins and members of the RL11 family in cytomegalovirus (33, 35, 36). E3/49K exhibits a novel processing pathway for E3 proteins. Approximately 1 h after synthesis, it is cleaved by an unknown cellular protease N-terminal to the transmembrane domain, generating a small membrane-integrated 12-14-kDa C-terminal fragment and a large ectodomain (sec49K) that is secreted or shed (32, 37). sec49K is the first secreted E3 protein and the first secreted adenovirus protein known to date. Unlike the other E3 proteins that act directly on infected cells, sec49K can affect host immune functions over a distance by targeting leukocytes via binding to the cell surface phosphatase CD45. This impairs activation of CD4 T cells and NK cells, inhibiting cytokine production and cytotoxicity, respectively, most likely by modulating signal transduction. Thus, for the first time, an immunomodulatory E3 function of a non-species C adenovirus was described. Because species D-based Ad vectors have considerable potential for applications in humans (38, 39), further characterization of E3/49K would be of great importance.
At steady state, the Ad19a E3/49K protein is predominantly localized in the Golgi/trans-Golgi-network (TGN) and in early endosomes (EEs) but is also present on the cell surface (32, 37). Its 19-residue cytoplasmic tail contains two conserved putative sorting motifs: a tyrosine-based YXXΦ motif (where Φ represents a bulky hydrophobic amino acid), and a dileucine-based (LL) motif (see Fig. 3) that may be involved in targeting E3/49K to these compartments (40–42). Such motifs are recognized by adaptor proteins (AP-1 to AP-5), thereby recruiting transmembrane cargo into transport vesicles at various locations within the cell (43). AP-1 to AP-3 can associate with the coat protein clathrin (e.g. in endosomes, at the plasma membrane, or at the TGN), determining trafficking pathways and ultimately the distribution of membrane proteins (41, 44, 45). However, it remains elusive what role these motifs may have in E3/49K trafficking, proteolytic processing, and secretion. It is also unclear which protease is involved and in which cellular compartment cleavage takes place.
FIGURE 3.
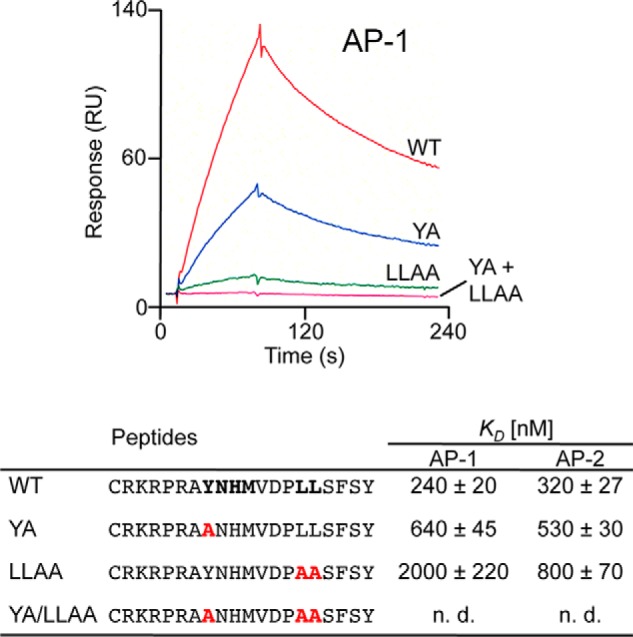
Efficient binding of clathrin adaptor proteins AP-1 and AP-2 to cytoplasmic tail peptides of E3/49K depends on the presence of the YXXΦ and LL motifs. The bottom panel shows the different cytoplasmic tail peptides used for the surface plasmon resonance spectroscopy studies with putative sorting signals in boldface type. YA is a mutant version with alanine substitution of the YXXΦ motif; LLAA is an altered version with alanine substitution of the LL motif; and YA/LLAA is a peptide with both motifs eliminated. Altered amino acids are shown in red. The equilibrium constants (KD) were determined in at least three independent experiments and are given in nm/liter ± S.D. The top panel shows the response versus time in seconds for the incubation of the different peptides with purified AP-1. n.d., not detectable. RU, resonance units.
Here we show that the cytoplasmic tail of E3/49K binds to AP-1 and AP-2 in vitro in a motif-dependent fashion. Mutation of the LL motif alone or in combination with YXXΦ profoundly increased E3/49K cell surface expression and sec49K secretion. Surprisingly, however, proteolytic cleavage was independent of their presence, suggesting that it requires only the default pathway and is executed rather inefficiently by a sheddase of the matrix metalloprotease (MMP) family at the cell surface. Although most E3/49K was recycled to the TGN, a minor fraction may be sorted to lysosomes where C-terminal fragments undergo a second cleavage. Based on these data, we propose a model for E3/49K trafficking that has implications for sec49K production and function.
Experimental Procedures
Chemicals
Chloroquine, leupeptin, brefeldin A (BFA), bafilomycin A1, tunicamycin, and monensin were purchased from Sigma, cycloheximide was from Calbiochem, and ammonium chloride was from Merck.
DNA Constructs
The expression vector pSG5-E3/49K encoding wild type E3/49K has been described previously (37). Fragments encoding mutations in the cytoplasmic tail of E3/49K were amplified by PCR using the following primers, all given in the 5′–3′ direction: universal sense primer, ggtacaatcatcaaggaacccagag; individual antisense primer (LL/AA), gtccagatctggatccgtcttagtaagagaagctggctgctgggtctaccata; ΔCT, gtccagatctggatccgtcttaacgcttgcggcagcagatgtagc; Y/A, gtccagatctggatccgtcttagtaagagaagctgagtagtgggtctaccatatgattggctgccctgggacgctt; YLL/AAA, gtccagatctggatccgtcttagtaagagaagctggctgctgggtctaccatatgattggctgccctgggacgctt. PCR fragments were cloned into the pSG5-E3/49K vector using BglII restriction sites (underlined) in the E3/49K coding sequence and the pSG5 multiple cloning site. Correct construction was verified by sequencing both strands of the E3/49K ORF. In the ΔCT construct, the stop-anchor sequence RKR was retained to ensure correct orientation and stable membrane integration.
Monoclonal Antibodies (mAbs) and Antisera
The polyclonal rabbit antiserum R25050 (anti-49Kct) raised against the C-terminal 15 amino acids of E3/49K was described previously (34), as were the polyclonal rabbit antiserum R48 (anti-49K-N) and the rat mAb 4D1, both raised against amino acids 20–382 of Ad19a E3/49K (32). The sheep and rabbit antisera against TGN46 were published previously (77). The mouse mAb 2D5 against LAMP-2 (46) was a kind gift of A. Hasilik (University of Marburg, Germany).
Viruses, Infection, Transfection, and Cell Culture
For growing Ad19a stocks, for plaque assays and most infection experiments, the lung epithelial carcinoma cell line A549 (ATCC CCL-185) was used. For some infection experiments, primary human foreskin fibroblasts (SeBu) were employed (24). Both cell types were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 100 units/ml penicillin, 0.1 mg/ml streptomycin (Invitrogen), and 10% fetal calf serum (Roche Diagnostics). In general, infection was performed using 5 pfu of Ad19a/cell for 1 h. Then the virus was removed, and this time point was defined as the start of infection.
Generation of A549 cells stably expressing mutant E3/49K molecules was done as for the cells expressing wild-type E3/49K (37) using the calcium phosphate method essentially as described (47) except that the glycerol shock was omitted. Briefly, three 10-cm diameter dishes of A549 cells were co-transfected with 20 μg each of the relevant pSG5-E3/49K construct and 2 μg of the pSV2-neor plasmid conferring G418 resistance as described (48). The next day, the medium was changed, and 2 days later, cells were split 1:4 or 1:8 into medium containing 1 mg/ml G418. After 10–14 days, ∼30 G418-resistent clones were isolated using cloning cylinders and maintained in medium containing 1 mg/ml G418. For transient transfection of A549 cells, principally the same protocol was followed except that 6-cm diameter dishes of cells were incubated with 0.5 ml of calcium-DNA precipitate (5–8 μg of pSG5 construct).
Metabolic Labeling, Immunoprecipitation, and SDS-PAGE
Labeling of A549 cells with [35S]methionine alone or in combination with [35S]cysteine (Promix, GE Healthcare), immunoprecipitation, and SDS-PAGE were performed as described previously (25, 37) using 14C-methylated proteins as molecular weight markers (GE Healthcare). Radioactive bands were quantified using a Storm 860 molecular imager (Molecular Dynamics).
Immunofluorescence
Subconfluent layers of A549 or primary SeBu cells were grown on glass coverslips and processed for immunofluorescence exactly as described (37, 49). Secondary antibodies were purchased from Dianova (Germany) donkey anti-mouse IgG fluorescein isothiocyanate (FITC), donkey anti-rabbit IgG FITC, donkey anti-mouse IgG Rhodamine Red-X, donkey anti-rat IgG Texas Red, and donkey anti-sheep IgG Rhodamine Red-X or from Life Technologies (goat anti-rat IgG Alexa Fluor 488, goat anti-sheep IgG Alexa Fluor 594, and goat anti-rabbit IgG Alexa Fluor 633). Mounted cells were analyzed with a laser-scanning confocal microscope (Leitz DM IRB scanner, Leica TCS NT). Images shown are single sections in z.
Flow Cytometry
Flow cytometry (FACS) was carried out essentially as described (50), with some modifications (49). Because E3/49K is sensitive to trypsin-mediated proteolysis, cells were washed with PBS and then detached with 1 mm EDTA in PBS. 300,000–500,000 cells were incubated with 1 μg of purified or 100 μl of undiluted supernatant of mAb 4D1 directed to the ectodomain of Ad19a E3/49K. Following several washes in FACS buffer, cells were incubated with 50 μl of FITC-labeled (1:50; Sigma) or Alexa Fluor 488-labeled goat anti-rat IgG (1:100; Invitrogen). Subsequently, 5000 cells were analyzed in a FACScan or FACSCalibur (BD Biosciences).
Detection of Secreted E3/49K Using the Quantitative FACS-based Binding Assay
The amount of sec49K in the supernatant of cells was quantitatively determined by binding to CD45+ Jurkat T cells as a read-out (32). WT- or mutant E3/49K-expressing A549 cell clones were incubated for 15–18 h with the broad spectrum MMP inhibitors BB-94 (5 μm; Tocris Biosciences) and CT1746 (51) (10 μm), kindly provided by D. Edwards (University of East Anglia, Norwich, UK). The more selective inhibitor GI254023X (52), targeting primarily ADAM10 and less well ADAM17, was kindly provided by A. Ludwig (RWTH Aachen University) and was used at 5 μm. All MMP inhibitors were solubilized in DMSO; hence, data from inhibitor-treated cells were related to those of mock-treated cells using the same final concentration of DMSO (0.1%). For detection of sec49K in supernatants of mutant cell lines, cells were grown in 6-cm dishes or 6-well dishes for 4 days. Cell supernatants were harvested and centrifuged for 10 min at 1500 × g prior to incubation with Jurkat cells or storage at 4 °C. Subsequently, cells were treated with trypsin/EDTA to determine the number of cells in the culture. Data were collated from at least two independent supernatants and four independent FACS measurements. Production of sec49K was calculated as mean fluorescence intensity of sec49K binding/106 producer cells. The different expression level was taken into account by relating this value to the intracellular expression after 5 h of BFA treatment.
Surface Plasmon Resonance
In order to test for the capacity of the cytoplasmic tail of E3/49K to bind the clathrin adaptor complexes AP-1 and AP-2, peptides comprising the WT cytoplasmic tail and mutant forms of E3/49K with mutations of the potential sorting motifs, LL and YXXΦ, were coupled to a CM5 sensor chip. The in vitro interaction was analyzed using a BIAcore 2000 (BIAcore AB) as described (53). Briefly, AP-1 and AP-2 were purified from porcine brain and injected at 100 nm in buffer A (20 mm HEPES, pH 7.0, 150 mm NaCl, 10 mm KCl, 2 mm MgCl2, 0.2 mm dithiothreitol) at a flow rate of 20 μl/min. Association (2 min) was followed by dissociation (2 min), during which buffer A was perfused, and the equilibrium constant (KD) was determined (53).
Statistical Analysis
Statistical significance was assessed by using Student's t test. Differences in means were considered statistically significant when p was <0.05.
Results
Differential Localization of N-terminal and C-terminal Domains of E3/49K during Early and Late Times of Infection
Previous studies using an antibody against the E3/49K cytoplasmic tail (anti-49Kct) showed that E3/49K is localized in the ER, the Golgi/TGN, early endosomes, and the plasma membrane at steady state (32, 37). To address the fate of the large N-terminal ectodomain and the full-length protein, we raised the antiserum anti-49K-N and mAb 4D1 directed to this domain (32). In the early phase of infection, co-staining with antibodies to the N-terminal domain and the C terminus revealed a large overlap (Fig. 1A, 12 h p.i.). By contrast, in the late phase of infection (36 h p.i.), no co-localization was visible with the exception of a prominent perinuclear structure (Fig. 1B, merge, yellow) that also stained for the TGN marker TGN46 (see below; Fig. 2C), implying that E3/49K was still intact in the TGN. In the early phase, the E3/49K C terminus showed only limited overlap with LAMP-2, a marker for late endosomes (LE)/lysosomes (Fig. 1C), whereas in the late phase, it was present in many peripheral vesicles that were positive for this marker (Fig. 1D). By contrast, the N-terminal domain/full-length molecule was rarely detected in LE/lysosomes either early or late after infection (Fig. 1, E and F). Thus, E3/49K C-terminal fragments accumulate in late endosomes/lysosomes at late times of infection, whereas the N-terminal domain/full-length molecule is either largely excluded from these compartments or immediately degraded. Taken together, this indicates that the C-terminal fragments and the full-length E3/49K protein may follow distinct trafficking pathways.
FIGURE 1.
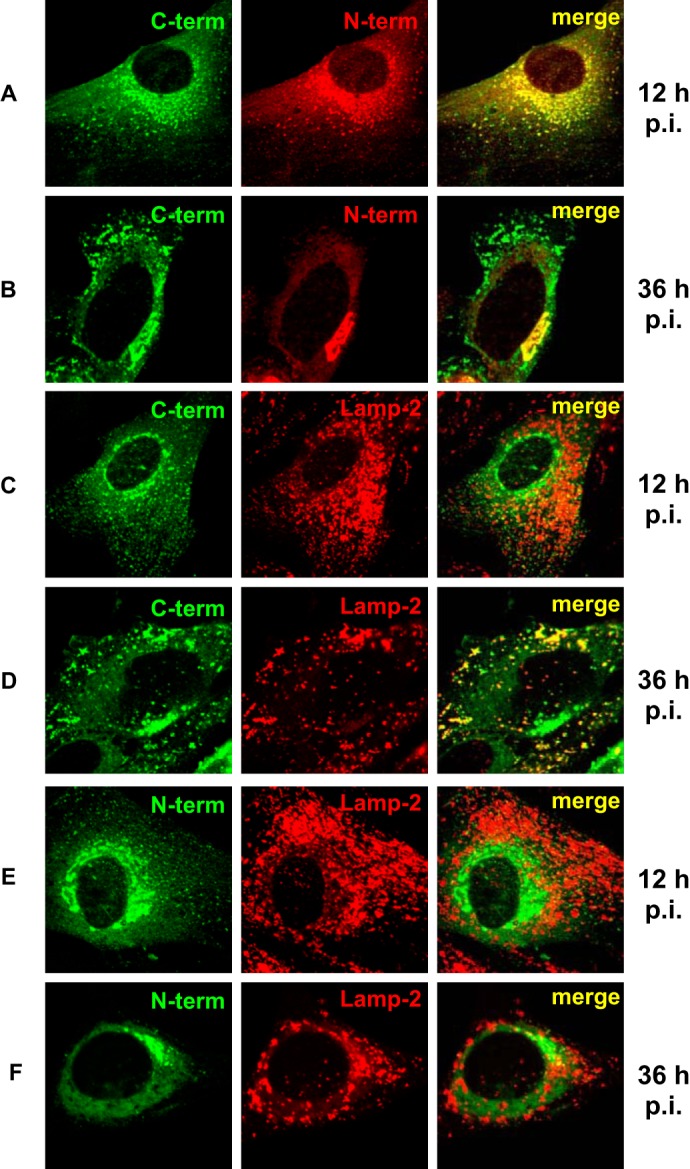
Differential localization of the ectodomain and the cytoplasmic tail of E3/49K in the Golgi/TGN and late endosomes/lysosomes during early and late phases of infection. Primary fibroblasts (SeBu) were infected with Ad19a and processed for confocal laser microscopy at 12 or 36 h postinfection (p.i.), corresponding to the early and late phase of the infection cycle in these primary fibroblasts, respectively. The subcellular localization of E3/49K was analyzed with rabbit antiserum R25050 directed against the C terminus (C-term; A–D) and antiserum R48-7B (anti-49K-N; E and F), followed by FITC-labeled goat anti-rabbit IgG. For detection of the N-terminal part of E3/49K (N-term) the rat mAb 4D1 was employed (A and B, middle). This was compared with the late endosomal/lysosomal marker LAMP-2, using mouse mAb 2D5 (C and F, middle). Subsequent staining was done with donkey anti-rat IgG Texas Red and donkey anti-mouse IgG Rhodamine Red-X, respectively. One typical experiment of at least three is shown.
FIGURE 2.
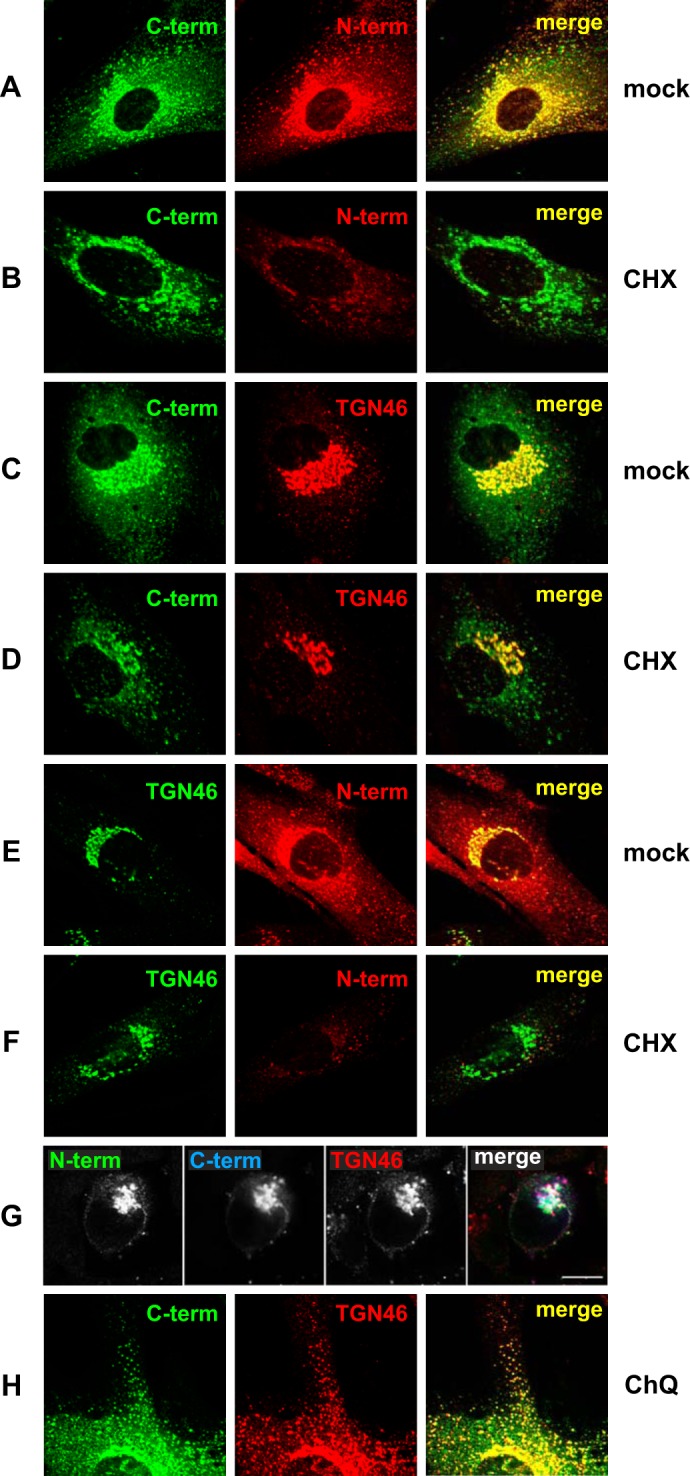
The steady state localization of E3/49K in the Golgi/TGN comprises newly synthesized uncleaved E3/49K as well as recycled C-terminal fragments that co-localize extensively with TGN46. Primary fibroblasts (SeBu) infected with Ad19a for 12 h were treated for 6 h with 50 μg/ml CHX. Subsequently, cells were processed for confocal laser microscopy by staining E3/49K with anti-49Kct (A–D) and rat mAb 4D1 against the ectodomain (N-term; A, B, E, and F). Golgi/TGN localization was determined by co-staining for the TGN marker TGN46 using either sheep polyclonal Abs (C, D, G, and H) or rabbit polyclonal Abs (E and F), followed by FITC-labeled donkey anti-rabbit IgG or rhodamine-labeled donkey anti-sheep IgG, anti-rat IgG, and anti-mouse IgG. For the triple labeling experiment (G), A549 cells infected with Ad19a for 9 h were fixed and stained with antibodies against TGN46 and the N and C termini of E3/49K. Secondary Abs were Alexa Fluor 488-labeled goat anti-rat IgG, Alexa Fluor 633-labeled goat anti-rabbit IgG, and Alexa Fluor 594-labeled donkey anti-sheep IgG. Images are single optical sections. Each channel was imaged separately under single laser illumination to prevent cross-over and overlaid using Leica confocal software. Scale bar, 10 μm. H, primary fibroblasts infected with Ad19a for 12 h were treated for 3 h with 100 μm chloroquine (CHQ) prior to staining with antibodies against TGN46 and anti-49Kct. The figure shows one typical experiment of three (A–F) and two (G and H), respectively.
Evidence for E3/49K Recycling to the TGN
The prominent perinuclear structure positive for both the N- and C-terminal domains of E3/49K was identified as the TGN by colocalization with TGN46, a marker for the TGN (Fig. 2, C and E). We next investigated whether or not the strong TGN staining of E3/49K at steady state represented newly synthesized E3/49K molecules en route to the cell surface or E3/49K recycled from the cell surface via early endosomes, a pathway demonstrated for TGN46. To this end, Ad19a-infected SeBu cells were treated from 12 to 18 h postinfection with cycloheximide (CHX) to block further protein synthesis. Subsequently, the localization of E3/49K was compared with that of TGN46 (Fig. 2). After a 6-h treatment with CHX, the perinuclear staining of the N-terminal E3/49K domain was drastically diminished (Fig. 2, N-term; compare B with A and F with E). This contrasts with the C-terminal staining, which was only slightly reduced (Fig. 2, C-term; compare B with A and D with C), similar to the limited changes noted for TGN46 under these two conditions (Fig. 2, compare D with C and F with E). The rapid CHX-mediated depletion of E3/49K molecules containing the N-terminal ectodomain indicated that these represented newly synthesized full-length E3/49K molecules, whereas the (largely) CHX-resistant C terminus-positive species represented predominantly C-terminal fragments of E3/49K that had been recycled to the TGN presumably from the plasma membrane. To confirm the presence of full-length E3/49K in the TGN, we pursued triple labeling experiments staining for both N- and C termini of E3/49K and the TGN, followed by secondary Abs linked to three distinct dyes. In A549 cells infected with Ad19a, both the N terminus and C terminus of E3/49K were present in the TGN (Fig. 2G), with a slight trend of the C termini being in more peripheral areas of the TGN. When Ad19a-infected SeBu cells were treated with the lysosomotropic agent chloroquine for 3 h to inhibit acidification, a considerable fraction of E3/49K C termini were found together with TGN46 in peripheral vesicles (Fig. 2H, CHQ). Considering that TGN46 is known to recycle from the cell surface to the TGN via EEs (54, 55), these data suggest that chloroquine inhibits recycling to the TGN (56, 57), trapping both molecules in EEs. Thus, the two proteins appear to utilize at least in part overlapping trafficking pathways. Taken together, we conclude that the prominent steady-state localization of E3/49K in the Golgi/TGN is due primarily to newly synthesized full-length proteins en route to the cell surface and to C terminus-positive E3/49K species that might include both full-length and C-terminal fragments recycled from the cell surface via endosomes.
The Cytoplasmic Tail of E3/49K Binds to Clathrin Adaptor Complexes in Vitro
To determine whether the dileucine motif LL and the tyrosine-based motif, YNHM, in the cytoplasmic tail are involved in sorting and intracellular trafficking of E3/49K, we initially used surface plasmon resonance spectroscopy to examine whether immobilized peptides representing the WT cytoplasmic tail or mutated versions of it (Fig. 3, mutations highlighted in red) would bind to purified clathrin adaptor complexes AP-1 and AP-2 in vitro. Indeed, a significant interaction with both AP-1 and AP-2 was detected (Fig. 3, WT). Replacing the tyrosine with alanine (YA) inhibited binding to AP1 and AP2 by 2.7- and 1.6-fold, respectively. Mutation of the dileucine motif (LLAA) reduced AP-1 binding by 8.3-fold and AP-2 binding by 2.5-fold. Interaction was completely abolished when both motifs were mutated (YA/LLAA). Thus, both motifs can be recognized by AP-1 and AP-2 in vitro, although the interaction with the dileucine motif seems to be more critical than that of the YXXΦ motif.
Increased Cell Surface Expression of E3/49K Proteins with Mutated Cytoplasmic Tails
To test the function of these putative sorting motifs in vivo, stable A549 cell lines were established that constitutively expressed E3/49K proteins with either or both of the motifs mutated by alanine substitution (YA and LLAA). Additionally, the influence on trafficking of the entire cytoplasmic tail was examined by creating the E3/49K mutant ΔCT that lacked all cytoplasmic residues except the stop-anchor sequence RKR. Relative cell surface expression of each molecule was determined by flow cytometry comparing the mean fluorescence intensities (MFIs) of multiple clones in the absence (cell surface) and presence of the detergent saponin (representing mostly internal E3/49K). To allow a comparison of clones with different E3/49K protein expression levels, the MFI ratios (surface/intracellular) were analyzed rather than the absolute surface MFIs. For WT E3/49K clones, this MFI ratio ranged from 0.24 to 0.29 (Fig. 4), suggesting that the majority of E3/49K is localized inside of the cell, consistent with the immunofluorescence analysis (Fig. 1). This is in contrast to a typical cell surface molecule, such as HLA, where the majority is found on the cell surface. E3/49K mutants YA/LLAA and ΔCT, lacking both motifs, showed 2.9- and 2.3-fold higher ratios, respectively. Thus, a larger proportion of E3/49K is displayed on the cell surface in the absence of these C-terminal tail motifs. This is statistically significant as compared with WT, whereas the difference between the two mutants is not. For the single mutant LLAA, the ratio was also increased (1.7-fold), whereas the YA mutants were comparable with WT (0.9-fold). Thus, a higher proportion of YA/LLAA and ΔCT and, to a lesser extent, LLAA were expressed on the cell surface as compared with WT and the YA mutant of E3/49K. These data are consistent with the notion that the LL motif is important for E3/49K internalization.
FIGURE 4.

Elimination of the LL motif in the cytoplasmic tail significantly enhances relative cell surface expression of E3/49K. A549 cell lines stably expressing WT E3/49K or mutants in which the LL and YXXΦ motif, respectively, in the cytoplasmic tail were mutated to AA (LLAA) and AXXΦ (YA). In YA/LLAA, both putative sorting motifs were mutated, whereas ΔCT lacked the entire cytoplasmic tail except a basic RKR stop-transfer sequence. Expression on the cell surface and internally was quantitatively analyzed by flow cytometry in the absence and presence of the detergent saponin, respectively. By determining the ratio of cell surface versus intracellular MFIs rather than the absolute cell surface values, E3/49K cell surface expression becomes independent of its general synthesis level in each transfectant clone. MFIs for cell surface and intracellular staining were determined in 4–6 independent experiments for 2–5 isolated clones, of which 2–3 are shown. Error bars, S.D. A significant difference between cell lines expressing wild type and mutated E3/49K is indicated by an asterisk (p < 0.05). The difference in the ratios between double mutant YA/LLAA and ΔCT is not statistically significant.
The role of the E3/49K cytoplasmic tail motifs in its localization was investigated further by confocal microscopy using transient transfection of E3/49K expression plasmids. The YA/LLAA mutant exhibited much stronger cell surface staining with anti-C-terminal antibody than either the WT or other mutants (Fig. 5A). Generally, all mutants lacking the LL motif seemed to be present in substantially smaller vesicles, whereas the phenotype of the YA mutant was basically identical to WT. In contrast, cell surface staining for the ectodomain was considerably weaker (Fig. 5A, YA/LLAA) and only modestly increased from WT, as was also the case for the ΔCT and LLAA mutants. Within the cell, the number of vesicles containing the N-terminal domain was considerably reduced in all LL mutant molecules compared with staining with anti-49Kct, indicating that the ectodomain may have been removed at the cell surface or during endocytosis. These data support the idea that the dileucine motif may be involved in endocytosis of E3/49K.
FIGURE 5.

Removal of both putative sorting motifs enhances cell surface display of full-length E3/49K and C-terminal fragments and reduces their accumulation in swollen vesicles during chloroquine treatment. A549 cells were transiently transfected with pSG5 plasmids encoding WT and mutant E3/49K proteins as described under “Experimental Procedures.” Cells were treated with 100 μm chloroquine for 12 h (B) or left untreated (A). After 36 h, cells were processed for confocal laser microscopy. Staining was performed with rat mAb 4D1 directed against the N-terminal ectodomain (αN) and rabbit serum R25050 against the C terminus of E3/49K (αC-term), followed by donkey anti-rabbit IgG FITC and donkey anti-mouse IgG Rhodamine Red-X. The figure shows a representative experiment of three.
To trap E3/49K molecules in early endosomes after internalization from the cell surface and prevent their potential degradation, we also assayed their localization in chloroquine-treated cells. For wild type and YA and LLAA mutant E3/49K, most vesicles stained with both N-terminal and C-terminal antibodies and appeared swollen under these conditions (Fig. 5B, Chloroquine), hence representing predominantly full-length E3/49K species in endosomes. In contrast, for both the YA/LLAA and ΔCT mutants, N-terminal and C-terminal 49K antibodies gave distinct staining patterns, apart from a few swollen vesicular structures in the center of the cell that stained with both antibodies and are likely to represent the TGN (Fig. 5B). For YA/LLAA, the C-terminal antibody labeled the plasma membrane and many, mostly smaller, intracellular vesicles. By contrast, staining with the antibody against the ectodomain detected hardly any intracellular vesicles apart from the centrally located presumed TGN structure, and surface staining was less prominent. For the ΔCT mutant, N-terminal staining was similar to the double mutant, but the surface display was further enhanced; C-terminal antibody cannot recognize the ΔCT mutant. Taken together, these data indicate that internalization of the YA/LLAA and the ΔCT mutants of E3/49K was indeed strongly impaired and that the ectodomain may be cleaved at the cell surface. In the absence of functional trafficking motifs, C terminus-specific antibodies label apart from the cell surface intracellular vesicles that are largely not swollen and thus may represent mostly full-length E3/49K species en route to the cell surface. Conversely, this suggests that full-length E3/49K lacking transport motifs is not efficiently internalized and is not present in endo/lysosomes at steady state.
Impact on E3/49K Processing of Pharmacological Agents Affecting Acidification, Lysosomal Proteases, Glycosylation, and Intracellular Trafficking
To investigate whether proteolytic processing of E3/49K could be manipulated and to gain first insight into the compartments where cleavage of E3/49K was occurring, Ad19a-infected A549 cells were pulse-labeled with [35S]methionine (0 h) and chased for 2 and 6 h in the presence of various pharmacological agents. Detergent extracts (lysate) were used to monitor intracellular processing and generation of the C-terminal fragment by immunoprecipitation with antibodies against the cytoplasmic tail, whereas concomitantly, the release of the ectodomain into the supernatant was followed with N-terminal specific antibodies (32). In mock-treated cells, the secreted ectodomain was detected in the supernatant after 2 h of chase, whereas three low molecular weight C-terminal species (h1–3) were visualized in the lysates, with h3 being typically a minor species. After 6 h of chase, h1 was hardly visible anymore, whereas the smallest fragment, h3, had considerably increased in intensity (Fig. 6A, lanes 2–4). This indicates that following the initial cleavage(s) releasing the N-terminal ectodomain, the C-terminal fragment(s) is further trimmed. This second cleavage seems to occur in a different compartment from that of the initial cleavage, because h3 appeared later (at >2–3 h of chase) than fragments h1 and h2, which became visible at ∼1 h of chase.
FIGURE 6.

Proteolytic processing and secretion of E3/49K in the presence of agents affecting glycosylation, endosomal/lysosomal proteases, the pH of intracellular compartments, or trafficking. Ad19a-infected A549 cells were labeled with [35S]methionine for 30 min (0 h) and chased with medium containing non-radioactive methionine for 2 and 6 h. Subsequently, detergent extracts were prepared (lysate), and E3/49K was immunoprecipitated with anti-49Kct, whereas the supernatant was processed and immunoprecipitated with anti-49K-N as described (32). The different agents indicated at the top were included both in the starvation medium and the following incubations. A, mock-treated, with the same medium changes but without inhibitors (lanes 2–7) and leupeptin (200 μm; lanes 8–13); B, chloroquine (100 μm; lanes 1–6) and ammonium chloride (10 mm; lanes 7–12); C, bafilomycin A1 (100 nm; lanes 1–6) and monensin (10 μm; lanes 7–12); D, tunicamycin (10 μg/ml; lanes 1–6) and brefeldin A (5 μg/ml; lanes 7–12). The high molecular weight E3/49K species (49K) and the C-terminal fragments h1–h3 are denoted on the right. E3/49K-CHO and E3/19K-CHO, the unglycosylated species of E3/49K and E3/19K. E, deglycosylated E3/49K colocalizes with the ER marker calreticulin upon tunicamycin treatment (3 h) in permanently transfected A549 cells expressing codon-optimized E3/49K (A549E3/49K-co15) and in infected cells (data not shown). Calreticulin was detected with a rabbit antiserum (Stressgen). F, A549 cells were infected with Ad19a for 6 h and treated for 3 h with tunicamycin (bottom panels) or were mock-treated (DMSO; top panels). Fixed cells were stained for E3/49K N terminus and the ER-resident Ad19a protein E3/19K using rabbit antiserum R22612 and imaged as in Fig. 2G. Scale bar, 20 μm. All experiments were carried out at least twice.
When cells were treated with leupeptin, an inhibitor of lysosomal cysteine and serine proteases (Fig. 6A, lanes 8–13), fragment h3 was not detected (compare lanes 10 and 4), suggesting that h3 is generated by a lysosomal cysteine or serine protease. Consistent with this notion, very little if any h3 was detected after treatment with agents that elevate the pH of endosomal/lysosomal compartments, such as chloroquine (Fig. 6B, lanes 1–6) or bafilomycin A1, an inhibitor of the vacuolar type H+-ATPase (Fig. 6C, lanes 1–6). Treatment with 10 mm ammonium chloride also reduced the amount of h3 but was less effective (Fig. 6B, lanes 7–12). Upon treatment with the Na+ ionophore monensin (58), which affects primarily the TGN cisternae and late processing events, such as terminal glycosylation and proteolytic cleavages, but may also impact other acidic compartments, such as lysosomes, h3 was also not produced (Fig. 6C, lanes 7–12). Collectively, these results substantiate the hypothesis that the second cleavage that gives rise to the h3 fragment is pH-dependent and takes place in LE/lysosomes.
Surprisingly, none of these treatments used to perturb intracellular processing prevented initial cleavage and secretion of E3/49K (Fig. 6, A–C). Only some minor effects on carbohydrate processing were noted. Chloroquine and to a lesser extent ammonium chloride reduced the kinetics of carbohydrate processing, whereas monensin impaired glycan modification, as indicated by the minimally increased molecular weight of the chased E3/49K. Such an effect of monensin has been previously noted for a number of glycoproteins and is thought to be due to either direct inhibition of the modifying enzymes or inhibition of further transport through the medial/trans-Golgi apparatus (58). The latter can be excluded in this case because E3/49K is still secreted in the presence of monensin (Fig. 6C, lanes 10–12). The impact of these agents on the efficiency of sec49K secretion was quantitatively assessed by determining the amount of radioactive sec49K released after 6 h of chase using phosphorimaging analysis and relating this to the amount of intracellular E3/49K synthesized after the pulse (radioactivity in the high molecular weight forms at 0 h chase). Taking into account the relative frequency of methionine in sec49K versus full-length E3/49K (4 versus 5), 80% of the radioactivity was expected to be in the N-terminal ectodomain and 20% in the cytoplasmic tail fragments. Potential differences in the affinities of the anti-49Kct and anti-N antibodies were ignored. Unexpectedly, chloroquine and bafilomycin A1 treatment increased the fraction secreted considerably, from 25–30% in untreated cells to ∼53 and 54%, respectively, whereas leupeptin promoted secretion only slightly to ∼36%, and ammonium chloride had no significant effect. In line with previous reports (58), monensin reduced secretion slightly by ∼25% (data not shown).
Strikingly, treatment with tunicamycin, an inhibitor of N-glycosylation, and BFA, which causes the redistribution/absorption of Golgi membranes and proteins into the ER (59) blocking transport to the cell surface, abolished proteolytic processing of E3/49K altogether. In the presence of these drugs, neither low molecular weight C-terminal fragments were detected in the lysates nor was the ectodomain sec49K found in the supernatant (Fig. 6D). As expected, tunicamycin reduced the relative molecular mass of E3/49K from 80–100 to 49 kDa (49K-CHO). Under these conditions, a prominent band of a size consistent with non-glycosylated Ad19a E3/19K (an ER-resident protein; 19K-CHO) was co-precipitated with E3/49K lacking N-linked sugars, suggesting that E3/49K was retained in the ER, probably due to misfolding. This was confirmed by immunofluorescence studies showing that in the presence of tunicamycin, E3/49K colocalizes with the ER marker calreticulin in transfected cells (Fig. 6E) and with E3/19K in infected cells (Fig. 6F). Thus, consistent with the timing of the appearance of the C-terminal fragments, cleavage occurs in a post-ER compartment. This does not seem to be the Golgi/TGN because E3/49K was still cleaved and secreted in the presence of monensin. However, the redistribution of Golgi membranes into the ER and the block of transport by BFA prevented cleavage and sec49K secretion although limited glycan processing was evident, as indicated by the marginal molecular weight shift of E3/49K during the chase period (Fig. 6D, lanes 7–9). Taken together, we conclude that proteolytic processing of E3/49K requires transport through the medial/trans-Golgi apparatus and occurs in a post-Golgi/TGN compartment.
E3/49K Cleavage Does Not Require Trafficking Motifs and Involves Matrix Metalloproteases at the Cell Surface
We next investigated whether the above post-Golgi compartment where ectodomain cleavage putatively occurs might be the plasma membrane. To this end, two cell clones of WT and each E3/49K mutant were treated with BFA for 4 h, and subsequently the level of E3/49K surface staining was quantified by flow cytometry using antibodies against the ectodomain. Strikingly, cell surface staining was rapidly lost when transport of newly synthesized protein to the cell surface was blocked by BFA (Fig. 7A). Only between ∼5 and 15% of the E3/49K levels remained at the cell surface after 4 h. By contrast, the impact of BFA on typical cellular plasma membrane protein levels was very modest, reducing their expression in comparison with mock-treated cells only to ∼75% for HLA, ∼86% for Fas, and 80% for the EGF receptor (Fig. 7A). Thus, the rapid loss of E3/49K and its mutants from the cell surface despite a partial or complete removal of putative endocytosis motifs supports the notion that rapid loss reflects proteolytic cleavage on the cell surface.
FIGURE 7.

E3/49K cleavage occurs independent of the putative transport motifs at the cell surface and is mediated by MMPs. A, the mean fluorescence intensity of E3/49K WT or mutant clones (indicated at the bottom) on the cell surface was determined by flow cytometry using mAb 4D1 after mock (black bars) and 4-h BFA treatment (5 μg/ml; white bars). Mean values and the S.D. were calculated from 4–6 independent experiments except for Fas and EGF receptor expression that was measured only once. No BFA-induced cytotoxicity or increased background staining was detectable within this time frame. B, cells expressing WT E3/49K (K27S) or the double mutant YA/LLAA (clone 32) lacking all motifs were incubated with the broad spectrum MMP inhibitors BB-94 and CT1746 and the more selective ADAM10 inhibitor GI254023X in 0.1% DMSO (final concentration) for 15–18 h. Cells were also incubated with medium alone (mock) and 0.1% DMSO as solvent control (DMSO). Supernatants were harvested and centrifuged at 1500 × g. Jurkat T cells (6 × 106) were incubated with 200 μl of supernatant, followed by detection of sec49K binding using FACS analysis. The data show the mean plus S.D. (error bars) of five independent staining experiments and three independently produced supernatants. C, mutation of the transport motifs does not prevent initial cleavage but rather secondary cleavage. Transfected clones indicated at the top expressing E3/49K WT and mutant proteins were metabolically labeled for 2 h. E3/49K was precipitated with anti-49Kct antiserum (lanes 1–11) and anti-49K-N (lanes 12 and 13). The figure shows one of two experiments. Only the relevant sections of the SDS-PAGE with the 80–100- and 10–14-kDa E3/49K species are shown. The arrow marks the C-terminal fragment h3.
A major class of proteases involved in cleavage of ectodomains on the cell surface, a process named shedding, comprises MMPs. To investigate their role in sec49K release, wild-type E3/49K-expressing cells (K27S) and cells expressing E3/49K devoid of all sorting motifs (YA/LLAA32) were treated with various MMP inhibitors, and release of sec49K into the medium was monitored by the sensitive T lymphocyte binding assay (32). Secretion of sec49K in WT cells was reduced to only ∼30% using the broadly acting MMP inhibitors BB-94 and CT1746 (Fig. 7B). Strikingly, a similar reduction was observed for GI254023X, an inhibitor of ADAM10 (a distintegrin and metalloproteinase 10) and ADAM17. These inhibitory effects were further increased for cells expressing the E3/49K mutant YA/LLAA lacking putative endocytosis motifs, consistent with the notion that such a molecule would exhibit a prolonged residence time on the cell surface, and hence the impact of an inhibitor acting on a cell surface protease is expected to be more severe. Concomitantly, inhibitor treatment resulted in a substantial increase in E3/49K cell surface expression, which was further enhanced for mutant YA/LLAA lacking the motifs (data not shown).
If cleavage occurs at the cell surface, it is predicted that it is independent of the presence of sorting motifs and should occur by default. Indeed, the small C-terminal E3/49K fragments visualized upon metabolic labeling were detected in all cell lines except for those that express the mutant that lacks the cytoplasmic tail and hence has lost the antibody epitope (Fig. 7C). Thus, the primary cleavage resulting in ectodomain secretion is independent of the sorting motifs. Moreover, whereas the migration of the low molecular weight fragments in each mutant differs slightly (Fig. 7C, lanes 2–9), it is obvious that only WT and YA proteins produced significant amounts of the smallest h3 fragment (arrow). Thus, the generation of this fragment that occurs subsequent to the primary h1/2 products requires the LL but not the YXXΦ motif, consistent with the importance of the endo/lysosomal compartment for this secondary cleavage (Fig. 6).
Increased Secretion/Ectodomain Shedding of sec49K in E3/49K Proteins Lacking the LL Motif
The impact of the E3/49K sorting motifs on glycan maturation kinetics and sec49K secretion was further investigated by pulse-chase analysis of two individual transfectant clones with similar expression levels. No significant differences in the processing kinetics between WT and mutant E3/49K proteins were observed (data not shown). As examples, immunoprecipitations of the WT and mutants YA/LLAA and ΔCT that lack both known transport motifs are shown (Fig. 8A). The carbohydrate processing of WT (lanes 3–5), YA/LLAA (lanes 9–11), and ΔCT mutants (lanes 15–17) was very similar. By 45 min of chase, all E3/49K molecules had been converted to the higher molecular weight form containing mature carbohydrates, although detection of the double mutant by anti-49Kct was inefficient (lanes 9–11) due to reduced recognition of the mutated cytoplasmic tail. Furthermore, the kinetics of sec49K secretion, first visible in the supernatant at 45 min of chase, was also not altered significantly between WT and mutant E3/49K proteins (compare lanes 6–8 with lanes 12–14 and 18–20). However, the amount of sec49K appeared to be increased in the YA/LLAA and ΔCT mutants (compare lanes 14 and 20 with lane 8). Therefore, we quantitatively assessed the extent of sec49K secretion for all mutants (two clones each) using pulse/chase analysis combined with phosphorimaging analysis. For WT E3/49K, an average of 21% was secreted. Setting this secretion of WT as 100%, sec49K secretion from the YA/LLAA and the ΔCT mutants was increased to ∼350%, whereas that of LLAA mutants was 3-fold enhanced (Fig. 8B). In contrast, secretion of the YA mutant was similar to WT. Secretion of WT protein was also drastically increased upon treatment of infected cells with chloroquine and bafilomycin A1 (4- and 3.5-fold, respectively; Fig. 8B).
FIGURE 8.

Elimination of the LL motif alone or in combination with the YXXΦ motif enhances secretion. A, the processing kinetics of WT and mutant E3/49K proteins was determined by a 30-min pulse with [35S]methionine followed by a 45- and 180-min chase. E3/49K was precipitated from untransfected (mock), WT, and YA/LLAA mutant cell lysates with anti-49Kct (lanes 1–5 and 9–11) and anti-49K-N from ΔCT mutant cell lysates (lanes 15–17) and all supernatants (lanes 6–8, 12–14, and 18–20). B, the proportion of E3/49K secreted was quantitatively determined in a separate pulse (0.5 h)-chase (4 h) experiment followed by immunoprecipitation with anti-49K-N and SDS-PAGE. Bands corresponding to sec49K were quantified by phosphorimaging analysis and related to the radioactivity in E3/49K high molecular weight species after the pulse that was defined as 100%. The maximum amount is secreted when 80% of the total E3/49K labeling is released because the cytoplasmic tail contains 20% of the total [35S]methionine labeling. Mean values were calculated from two independent experiments except for the values obtained upon chloroquine and bafilomycin A1 treatment, which were derived from single experiments. The mean value of WT clone K9S was set as 100%. Error bars, S.D. A significant difference between mutants and WT is indicated by asterisks (p < 0.05). C, enhanced lymphocyte binding activity (sec49K) is observed in supernatants of mutants lacking the LL motif. One clone from WT and each mutant was assessed for the efficiency of secretion as measured using the Jurkat T lymphocyte binding assay. Cells were cultured for 4 days, and the supernatant was harvested and centrifuged. Three independently collected supernatants were analyzed for the presence of sec49K in at least four different experiments as described under “Experimental Procedures.” Error bars, S.D. A significant difference between mutants and WT is indicated by asterisks (p < 0.05).
To obtain independent evidence for the amount of sec49K secreted in the different mutant cell lines, we also used the Jurkat binding assay to quantify sec49K in the supernatant of mutant cells. One clone from WT and each mutant was selected. The amount of sec49K detected was normalized to the number of cells in the cell culture plate and the differential synthesis level that was assessed by intracellular staining of E3/49K in the presence of BFA (Fig. 8C). As can be seen, whereas the YA-expressing cell clone secreted similar amounts of sec49K as WT over a 4-day period, the amount of sec49K was ∼2-fold higher in cell clones that lack the LL motif alone or in combination with the tyrosine-based motif. Thus, the LL motif has a significant impact on shedding of the E3/49K ectodomain.
Interestingly, the increased secretion of mutant E3/49K proteins correlated well with the increased cell surface ratio of these mutants (Fig. 4), consistent with the hypothesis that proteolytic cleavage occurs at the cell surface. To further test this hypothesis, we examined the fate of surface-exposed E3/49K in cell lines expressing either WT or mutant E3/49K by incubating live cells with the E3/49K N-terminal antibody at 0 °C and then warmed to 37 °C for different periods of time prior to fixation and immunofluorescence detection (Fig. 9). At time 0 (no incubation at 37 °C), WT, YA/LLAA, and ΔCT cells each showed exclusively surface labeling, as expected. Within 10 min at 37 °C, much of the fluorescent WT E3/49K had been internalized into smaller vesicles in the periphery, some of which co-localized with TGN46. At 45 min, the vesicles appeared slightly larger and in a more central location, partially overlapping with the TGN (Fig. 9) (data not shown). By contrast, neither of the mutant proteins showed any internalization, even after 45 min, yet the level of surface staining still diminished such that most cells became essentially unlabeled by 45 min. Thus, this loss of E3/49K cell surface staining can only be explained by shedding (i.e. direct cleavage at the cell surface). Taken together, the removal of the dileucine motif alone or in concert with the YXXΦ motif enhances secretion, presumably by impairing rapid endocytosis and subsequent transport to the TGN and LE/lysosomes. Consequently, this would lead to a prolonged exposure to the sheddase on the cell surface and hence a more efficient secretion.
FIGURE 9.

Surface fluorescence of mutated E3/49K is lost in the absence of any internalization, whereas WT E3/49K is internalized. A549 cells stably expressing either WT, YA/LLAA mutant, or ΔCT truncated E3/49K were surface-labeled on ice with E3/49K N-terminal mAb 4D1. Excess antibody was removed, and then cells were warmed to 37 °C and incubated for the times indicated. Upon fixation, the location of prebound antibody was then revealed by staining with secondary antibody and imaged as in Fig. 2G using the same settings throughout to ensure comparability of image intensity. Scale bar, 25 μm. One representative experiment of at least two is shown.
Discussion
The Ad19a E3/49K protein has been shown to exhibit a novel immunomodulatory mechanism for Ad E3 proteins. Unlike other E3 proteins, it can modulate activities of leukocytes through secretion and binding to CD45 (32). Interestingly, this is not achieved by the synthesis of a genuine secreted protein but by synthesizing first a transmembrane glycoprotein that is subsequently proteolytically cleaved into a small membrane-integrated C-terminal fragment and a large N-terminal ectodomain, sec49K, that is shed (32, 37). This processing pathway is associated with a complex subcellular distribution of E3/49K at steady state in the ER, Golgi/TGN, and early endosomes and on the cell surface (37).
By mutating putative sorting motifs in the cytoplasmic tail of E3/49K and the combined use of antibodies against the ectodomain and the C terminus, we investigated the role of these motifs for trafficking, proteolytic processing, and secretion. The results are summarized in the model shown in Fig. 10. In the early phase of infection, staining with antibodies against the cytoplasmic tail and the ectodomain overlaps greatly for the ER, Golgi/TGN, plasma membrane, and some internal vesicles that most likely represent EEs (37). The ER and the Golgi/TGN contain full-length E3/49K that represents primarily newly synthesized protein en route to the cell surface. The apparent concentration of E3/49K in the TGN at steady state may simply reflect the smaller membrane surface area of this compartment relative to others in the pathway; alternatively, the requirement for extensive modifications to be applied here may delay its exit. Collectively, the data suggest that E3/49K reaches the cell surface in an intact form, where it becomes cleaved by a sheddase (Fig. 10, 1). This cleavage does not appear to be very efficient because the molecule is clearly detectable on the cell surface by FACS. Upon internalization initiated by recognition of the LL motif by AP-2, a large proportion of the C-terminal fragment but also residual non-cleaved E3/49K is recycled from the cell surface via endosomes to the TGN from which they seem to be returned to the cell surface and re-exposed to the sheddase. This conclusion is based on the continuous staining of the TGN and vesicles with anti-49Kct antibody after inhibition of protein synthesis and the concomitant loss of ectodomain staining. It remains an open question whether internalized E3/49K species are also directly recycled to the cell surface from EEs or via recycling endosomes (RE; thin arrows in Fig. 10). During intracellular trafficking, a smaller fraction of the C-terminal fragments may be continuously diverted to LE/lysosomes, where these are cleaved a second time into the slightly smaller fragment h3 by an acidophil leupeptin-sensitive protease (Fig. 10, 2). The accumulation of C-terminal fragments in the LE/lysosomes at late times after infection might reflect selective trafficking relative to the uncleaved form, perhaps based on differential accessibility to adaptors due to conformation changes or differential acylation because E3/49K has a perfect acylation motif at the cytoplasmic tail/TMD border, but the observations may equally be explained simply by the continual loss of sec49K at the plasma membrane through successive trafficking cycles. In any case, trafficking of E3/49K to LE/lysosomes may only be a minor pathway because the great majority of full-length E3/49K and its C-terminal part are clearly entering a recycling pathway to the TGN (Fig. 10, thick arrow) as demonstrated by the partial overlap of cell surface-labeled E3/49K with the Golgi/TGN and the persistent staining with anti-49Kct after blocking protein synthesis.
FIGURE 10.

Model for E3/49K trafficking and proteolytic processing. At steady state, E3/49K is found in the Golgi/TGN, at the plasma membrane, and in EEs. At the TGN, E3/49K seems to be packaged by default into secretory vesicles (SV); however, a diversion to an EE prior to appearing on the cell surface cannot formally be ruled out. Proteolytic cleavage (dotted arrows) releasing the N-terminal ectodomain most likely occurs at the cell surface through “shedding” by MMPs (1). The C-terminal fragment and a considerable fraction of uncleaved E3/49K appear to be endocytosed through interaction of the LL motif in the C-tail with AP-2 (thick arrows). Full-length E3/49K may be sorted in the EEs into a recycling pathway to the plasma membrane, either directly from EEs or via the TGN or the recycling endosome (RE) for another round of cell surface transport possibly involving AP-1. A minor pathway may direct E3/49K or predominantly the cytoplasmic tail fragments to LE and lysosomes (Lys), where further proteolytic processing of h1/2 to h3 takes place (3). For further details and references, see “Results.” The extent of the proposed transport pathway is indicated by the thickness of the arrows.
This complex trafficking pathway of E3/49K seems to be governed by sorting motifs in the cytoplasmic tail because cytoplasmic tail peptides were capable of binding to the clathrin adaptor complexes AP-1 and AP-2 in vitro in a motif-dependent fashion (Fig. 3). Although the LL motif does not fit the consensus sequence of acidic (D/E)XXXL(L/I) dileucine signals recognized by AP-1 and AP-2 or DXXLL motifs that are bound by Golgi-localized, γ-ear-containing, ARF-binding proteins because the acidic residue is in position −2 in E3/49K (17), its mutation altered the subcellular distribution. Relative to their synthesis, mutant molecules lacking LL were expressed at higher levels on the cell surface compared with WT, consistent with a deficiency in endocytosis (Figs. 4 and 5). A role for the dileucine motif in endocytosis was further supported by the drastically reduced presence of full-length YA/LLAA and ΔCT in swollen endosomal vesicles after chloroquine treatment (Fig. 5), the complete lack of endocytosis of surface-labeled LL-mutated E3/49K, and a marked increase in sec49K secretion for all LL mutants. In contrast, mutation of the tyrosine motif had little effect in any of these assays (Figs. 4, 5, 7, and 8), suggesting that LL is the dominant internalization motif of E3/49K. Interestingly, the in vitro binding to AP-1 was more affected by mutation of the motifs than binding to AP-2. Therefore, an alternative explanation might be that the dileucine controls a critical sorting step subsequent to endocytosis (e.g. in endosomes, as proposed for the mannose-6 phosphate receptor 46 where LL was implicated in endocytosis and recycling to the TGN) (60, 61). Alternatively, LL may be responsible for diverting a fraction of E3/49K to LE/lysosomes, thereby reducing the amount of E3/49K available for secretion. The mechanism by which the C-terminal fragments are diverted to LE/lysosomes and accumulate there remains an open question, but we speculate that targeting depends on prior ectodomain cleavage. Considering the localization of AP-1 at the TGN and EEs and AP-2 at the plasma membrane (43), we propose that AP-2 mediates internalization primarily through binding to the LL motif, whereas AP-1 is involved in the recycling of E3/49K species from EEs to the TGN (Fig. 10).
The precise role of the YXXΦ motif for trafficking remains elusive. Whereas in vitro binding of E3/49K cytoplasmic tail peptides to AP-1 and AP-2 was in a similar range as for other cargo protein peptides (41, 44, 53) and was affected by the YA substitution (Fig. 3), no phenotype was noted upon expression of the corresponding E3/49K mutant in cells. It is conceivable that the close proximity (6 amino acids) of the YXXΦ motif to the transmembrane domain may prevent recognition by the adaptor proteins in vivo because there is a minimum separation of 7 residues among other proteins in which this motif is active, and in one of these, LAMP-1, reduction to less than 6 residues led to altered sorting (78). Also, the sequence context of the motif is not particularly favorable for a transport motif, and the amino acid methionine is rarely found in the bulky hydrophobic position in such motifs (17). On the other hand, the tyrosine mutation in addition to the LL mutation consistently gave a more robust phenotype, resulting in a higher surface/intracellular ratio, a stronger cell surface staining, and an enhanced secretion as compared with the LL mutation alone. This suggests that the tyrosine-based motif and the LL motif act in concert. The YXXΦ motif might have a role in recycling to the TGN, as shown for one of the tyrosine motifs in TGN46 (56). We also cannot rule out a role of the YXXΦ motif in basolateral sorting in polarized epithelial cells, as shown for other proteins (62, 63), or in sorting in specialized cell types, such as dendritic cells. These antigen-presenting cells are efficiently infected by Ad19a (38). Alternatively, YXXM may serve as a docking site for class I phosphatidylinositol 3-kinases (PI3Ks), thereby impacting endocytosis or signaling (or both) along the E3/49K trafficking pathway relevant for cell metabolism, cell survival/apoptosis, or functions of immune cells (64–66).
Our study provides strong evidence that ectodomain processing of E3/49K occurs at the plasma membrane. First, drugs that prevent ER export of E3/49K or promote redistribution of membrane proteins from the Golgi into the ER blocked proteolytic processing and secretion of E3/49K, whereas monensin, which acts primarily on late TGN compartments, had no impact on either process, strongly suggesting that ectodomain cleavage occurs in a post-Golgi compartment. Because full-length E3/49K reaches the cell surface in significant amounts (32), proteolytic processing in secretory vesicles (SV; Fig. 10) appears unlikely. Moreover, none of the lysosomotropic agents prevented cleavage, ruling out the involvement of any endosomal/lysosomal compartment. In support of this conclusion, mutation of either or both transport motifs did not prevent cleavage, indicating that no sorting step is required for cleavage. Because ΔCT lacks all known sorting motifs, it is expected to reach the cell surface by the default pathway, allowing the ectodomain to be cleaved and secreted. Supporting this suggestion, BFA treatment caused a rapid loss of WT and mutant E3/49K cell surface expression. Moreover, in the mutant cell lines, surface-labeled fluorescent E3/49K is lost within 10–45 min in the absence of any notable internalization (Fig. 9), strongly suggesting that ectodomain cleavage occurs at the cell surface.
We conclude that E3/49K cleavage is mediated by cellular sheddases of the MMP family (Fig. 10). Inhibitors of MMPs acting primarily on the cell surface, including one targeting ADAM10 and ADAM17, reduced secretion of sec49K dramatically (Fig. 7B) while enhancing cell surface expression of E3/49K (data not shown). In line with this conclusion, increased cell surface expression of LL-mutated molecules correlated with increased secretion (Figs. 4 and 8). This can be explained by the increased residence time on the cell surface upon elimination of the putative endocytosis motif, allowing for a more efficient cleavage of E3/49K by such proteases acting at the cell surface. However, increased ectodomain shedding of these molecules may also be due in part to a lack of intracellular degradation normally mediated by these motif(s). In support of this notion, chloroquine and bafilomycin enhanced secretion (Fig. 8), although no substantial staining of the ectodomain with late endosomes/lysosomes was noted even in the presence of chloroquine.
Many secreted cellular proteins, including cytokines, cytokine receptors, and growth factors, are initially synthesized as transmembrane proteins and are subsequently cleaved by sheddases (67), such as ADAMs (e.g. ADAM10 and ADAM17) (67–69), so this pathway clearly has advantages under some circumstances. Frequently, the initial cleavage of the ectodomain is followed by a second, regulated intramembrane cleavage of the C-terminal fragment that may have additional functions. For example, the large ectodomain of the amyloid precursor protein (70, 71) is shed by α- and β-secretases, whereby α-secretase activity includes ADAMs (72), whereas intramembrane cleavage is carried out by γ-secretase, leading to amyloid-β production (67, 72). The second cleavage of Notch, another ADAM target, results in a fragment that modulates transcription. Interestingly, the smallest C-terminal E3/49K fragment h3 is clearly the result of a second cleavage of h1/h2 in LE/lysosomes (Fig. 10, 2). The role of these C-terminal fragments, if any, remains elusive. Although our data indicate that the E3/49K ectodomain is shed by MMPs, including ADAM10 or ADAM17, more detailed studies using knockdown of individual MMPs are necessary to identify conclusively the relevant protease. Together with a precise analysis of the cleavage products, this knowledge will be very useful for putative exploitation of sec49K as a recombinant virus-derived immunomodulator for therapeutic applications (73).
It is puzzling as to why E3/49K is initially synthesized as a membrane protein only to give rise to a secreted ectodomain with binding activity to CD45 (32). Ectodomain shedding as opposed to direct secretion might be easier to regulate than conventional uncontrolled secretion. This could be relevant to limit the immune response to E3/49K. Alternatively, this points to an additional hitherto unknown function for the membrane-bound form or the C-terminal E3/49K stub on the cell surface or intracellularly. Cell surface E3/49K did not inhibit NK lysis (32); however, its activity on other leukocytes was not tested. The cycling of E3/49K between plasma membrane, endosomes, and TGN would be consistent with it rerouting and degrading cellular target proteins as shown for CMV M6 (74) or the Ad E3/10.4–14.5K complex (24, 49, 75). The latter down-regulates various apoptosis receptors and the EGF receptor, whereby the two E3 proteins involved each contain a sorting motif in its cytoplasmic tail. However, to date, we have not detected any significant interaction/co-precipitation of membrane-bound E3/49K with cellular proteins. An additional role of the C-terminal portion would be consistent with the relatively long half-life and the high conservation of its sequence. The last 50 amino acids exhibit 84–100% amino acid identity for different Ad types of species D, whereas that of the secreted N-terminal domain (∼383 amino acids) is considerably lower, ranging from ∼36 to 55% (34, 76). This N-terminal sequence diversity is consistent with the proposed recombination hot spot within E3/49K/CR1β, which seems to drive rapid evolution and diversification of sec49K (22). New evidence suggests that apart from the common interaction of species D E3/49Ks with CD45, this diversity results in differential, Ad type-specific interactions with various inhibitory receptors of the immune system, which could be relevant for the disease-causing potential.3 Further research into the specific interactions of E3/49K molecules of different Ads will be crucial to understand their individual functions and potential relationship to disease.
Author Contributions
H.-G. B. conceived and coordinated the study and wrote the paper together with M. W. H.-G. B. also performed and analyzed the experiments shown in Figs. 2G, 6 (E and F), 7B, 8C, and 9. He designed with M. W. all other experiments. M. W. contributed to the design of the experiments in Figs. 1, 2, 4, 5, 6D, 7 (A and C), and 8 (A and B) and performed and analyzed the experiments. He also wrote the paper together with H.-G. B. S. H. performed and analyzed the experiments shown in Fig. 3 and contributed to the writing of the manuscript. S. P. provided anti-TGN antibodies, and L. B. provided the cell clones expressing codon-optimized E3/49K. K. N. L. contributed to the experiments shown in Figs. 2G, 6 (E and F), and 9 and to the revision of the paper. C. S. performed and analyzed part of the experiments provided in Fig. 8C. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank A. Osterlehner for excellent technical assistance. We are grateful to A. Hasilik for kindly providing antibodies and D. Edwards and A. Ludwig for providing MMP inhibitors. We also thank M. Messerle for sharing data prior to publication. For critical reading of the manuscript and helpful suggestions, we thank M. Sester, R. Spooner, and C. Smith.
This work was supported by Deutsche Forschungsgemeinschaft Grants BU 642/1 and SFB455 (to H.-G. B.). The authors declare that they have no conflicts of interest with the contents of this article.
N. Martinez-Martin and L. Gonzalez, personal communication.
- Ad
- adenovirus
- ER
- endoplasmic reticulum
- NK
- natural killer
- TGN
- trans-Golgi network
- EE
- early endosome
- BFA
- brefeldin A
- MMP
- matrix metalloprotease
- LE
- late endosomes
- CHX
- cycloheximide
- MFI
- mean fluorescence intensity.
References
- 1. King A. M. Q., Adams M. J., Carstens E. B., and Lefkowitz E. J. (eds) (2012) Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses, pp. 213–228, Elsevier Academic Press, Amsterdam [Google Scholar]
- 2. Zhou X., Robinson C. M., Rajaiya J., Dehghan S., Seto D., Jones M. S., Dyer D. W., and Chodosh J. (2012) Analysis of human adenovirus type 19 associated with epidemic keratoconjunctivitis and its reclassification as adenovirus type 64. Invest. Ophthalmol. Vis. Sci. 53, 2804–2811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wold W. S. M., and Horwitz M. S. (2007) in Fields Virology (Knipe D. M., Howley P. M., Griffin D. E., and Lamb R. A., eds) pp. 2395–2436, 5th Ed., Lippincott Williams & Wilkins, Philadelphia [Google Scholar]
- 4. Fox J. P., Hall C. E., and Cooney M. K. (1977) The Seattle virus watch. VII. Observations of adenovirus infections. Am. J. Epidemiol. 105, 362–386 [DOI] [PubMed] [Google Scholar]
- 5. Chodosh J., Miller D., Stroop W. G., and Pflugfelder S. C. (1995) Adenovirus epithelial keratitis. Cornea 14, 167–174 [PubMed] [Google Scholar]
- 6. Meyer-Rüsenberg B., Loderstädt U., Richard G., Kaulfers P.-M., and Gesser C. (2011) Epidemic keratoconjunctivitis: the current situation and recommendations for prevention and treatment. Dtsch Arztebl. Int. 108, 475–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dosso A. A., and Rungger-Brändle E. (2008) Clinical course of epidemic keratoconjunctivitis: evaluation by in vivo confocal microscopy. Cornea 27, 263–268 [DOI] [PubMed] [Google Scholar]
- 8. Kaneko H., Suzutani T., Aoki K., Kitaichi N., Ishida S., Ishiko H., Ohashi T., Okamoto S., Nakagawa H., Hinokuma R., Asato Y., Oniki S., Hashimoto T., Iida T., and Ohno S. (2011) Epidemiological and virological features of epidemic keratoconjunctivitis due to new human adenovirus type 54 in Japan. Br. J. Ophthalmol. 95, 32–36 [DOI] [PubMed] [Google Scholar]
- 9. Walsh M. P., Chintakuntlawar A., Robinson C. M., Madisch I., Harrach B., Hudson N. R., Schnurr D., Heim A., Chodosh J., Seto D., and Jones M. S. (2009) Evidence of molecular evolution driven by recombination events influencing tropism in a novel human adenovirus that causes epidemic keratoconjunctivitis. PLoS One 4, e5635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kemp M. C., Hierholzer J. C., Cabradilla C. P., and Obijeski J. F. (1983) The changing etiology of epidemic keratoconjunctivitis: antigenic and restriction enzyme analyses of adenovirus types 19 and 37 isolated over a 10-year period. J. Infect. Dis. 148, 24–33 [DOI] [PubMed] [Google Scholar]
- 11. Arnberg N., Edlund K., Kidd A. H., and Wadell G. (2000) Adenovirus type 37 uses sialic acid as a cellular receptor. J. Virol. 74, 42–48 [PMC free article] [PubMed] [Google Scholar]
- 12. Nilsson E. C., Storm R. J., Bauer J., Johansson S. M. C., Lookene A., Ångstrom J., Hedenström M., Eriksson T. L., Frängsmyr L., Rinaldi S., Willison H. J., Domellöf F. P., Stehle T., and Arnberg N. (2011) The GD1a glycan is a cellular receptor for adenoviruses causing epidemic keratoconjunctivitis. Nat. Med. 17, 105–109 [DOI] [PubMed] [Google Scholar]
- 13. Arnberg N. (2012) Adenovirus receptors: implications for targeting of viral vectors. Trends Pharmacol. Sci. 33, 442–448 [DOI] [PubMed] [Google Scholar]
- 14. Burgert H. G., Ruzsics Z., Obermeier S., Hilgendorf A., Windheim M., and Elsing A. (2002) Subversion of host defense mechanisms by adenoviruses. Curr. Top. Microbiol. Immunol. 269, 273–318 [DOI] [PubMed] [Google Scholar]
- 15. Gregory S. M., Nazir S. A., and Metcalf J. P. (2011) Implications of the innate immune response to adenovirus and adenoviral vectors. Future Virol. 6, 357–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hendrickx R., Stichling N., Koelen J., Kuryk L., Lipiec A., and Greber U. F. (2014) Innate immunity to adenovirus. Hum. Gene Ther. 25, 265–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Windheim M., Hilgendorf A., and Burgert H. G. (2004) Immune evasion by adenovirus E3 proteins: exploitation of intracellular trafficking pathways. Curr. Top. Microbiol. Immunol. 273, 29–85 [DOI] [PubMed] [Google Scholar]
- 18. Burgert H.-G., and Blusch J. H. (2000) Immunomodulatory functions encoded by the E3 transcription unit of adenoviruses. Virus Genes 21, 13–25 [PubMed] [Google Scholar]
- 19. Ginsberg H. S., Lundholm-Beauchamp U., Horswood R. L., Pernis B., Wold W. S., Chanock R. M., and Prince G. A. (1989) Role of early region 3 (E3) in pathogenesis of adenovirus disease. Proc. Natl. Acad. Sci. U.S.A. 86, 3823–3827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Robinson C. M., Seto D., Jones M. S., Dyer D. W., and Chodosh J. (2011) Molecular evolution of human species D adenoviruses. Infect. Genet. Evol. 11, 1208–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Davison A. J., Benko M., and Harrach B. (2003) Genetic content and evolution of adenoviruses. J. Gen. Virol. 84, 2895–2908 [DOI] [PubMed] [Google Scholar]
- 22. Robinson C. M., Singh G., Lee J. Y., Dehghan S., Rajaiya J., Liu E. B., Yousuf M. A., Betensky R. A., Jones M. S., Dyer D. W., Seto D., and Chodosh J. (2013) Molecular evolution of human adenoviruses. Sci. Rep. 3, 1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McNees A. L., and Gooding L. R. (2002) Adenoviral inhibitors of apoptotic cell death. Virus Res. 88, 87–101 [DOI] [PubMed] [Google Scholar]
- 24. Elsing A., and Burgert H.-G. (1998) The adenovirus E3/10.4K-14.5K proteins down-modulate the apoptosis receptor Fas/Apo-1 by inducing its internalization. Proc. Natl. Acad. Sci. U.S.A. 95, 10072–10077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Burgert H.-G., and Kvist S. (1985) An adenovirus type 2 glycoprotein blocks cell surface expression of human histocompatibility class I antigens. Cell 41, 987–997 [DOI] [PubMed] [Google Scholar]
- 26. Burgert H. G., Maryanski J. L., and Kvist S. (1987) “E3/19K” protein of adenovirus type 2 inhibits lysis of cytolytic T lymphocytes by blocking cell-surface expression of histocompatibility class I antigens. Proc. Natl. Acad. Sci. U.S.A. 84, 1356–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sester M., Ruszics Z., Mackley E., and Burgert H. G. (2013) The transmembrane domain of the adenovirus E3/19K protein acts as an endoplasmic reticulum retention signal and contributes to intracellular sequestration of major histocompatibility complex class I molecules. J. Virol. 87, 6104–6117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McSharry B. P., Burgert H. G., Owen D. P., Stanton R. J., Prod'homme V., Sester M., Koebernick K., Groh V., Spies T., Cox S., Little A. M., Wang E. C., Tomasec P., and Wilkinson G. W. (2008) Adenovirus E3/19K promotes evasion of NK cell recognition by intracellular sequestration of the NKG2D ligands major histocompatibility complex class I chain-related proteins A and B. J. Virol. 82, 4585–4594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sester M., Koebernick K., Owen D., Ao M., Bromberg Y., May E., Stock E., Andrews L., Groh V., Spies T., Steinle A., Menz B., and Burgert H. G. (2010) Conserved amino acids within the adenovirus 2 E3/19K protein differentially affect downregulation of MHC class I and MICA/B proteins. J. Immunol. 184, 255–267 [DOI] [PubMed] [Google Scholar]
- 30. Tollefson A. E., Scaria A., Hermiston T. W., Ryerse J. S., Wold L. J., and Wold W. S. (1996) The adenovirus death protein (E3–11.6K) is required at very late stages of infection for efficient cell lysis and release of adenovirus from infected cells. J. Virol. 70, 2296–2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Frietze K. M., Campos S. K., and Kajon A. E. (2010) Open reading frame E3–10.9K of subspecies B1 human adenoviruses encodes a family of late orthologous proteins that vary in their predicted structural features and subcellular localization. J. Virol. 84, 11310–11322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Windheim M., Southcombe J. H., Kremmer E., Chaplin L., Urlaub D., Falk C. S., Claus M., Mihm J., Braithwaite M., Dennehy K., Renz H., Sester M., Watzl C., and Burgert H. G. (2013) A unique secreted adenovirus E3 protein binds to the leukocyte common antigen CD45 and modulates leukocyte functions. Proc. Natl. Acad. Sci. U.S.A. 110, E4884–E4893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Deryckere F., and Burgert H.-G. (1996) Early region 3 of adenovirus type 19 (subgroup D) encodes an HLA-binding protein distinct from that of subgroups B and C. J. Virol. 70, 2832–2841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Blusch J. H., Deryckere F., Windheim M., Ruzsics Z., Arnberg N., Adrian T., and Burgert H. G. (2002) The novel early region 3 protein E3/49K is specifically expressed by adenoviruses of subgenus D: implications for epidemic keratoconjunctivitis and adenovirus evolution. Virology 296, 94–106 [DOI] [PubMed] [Google Scholar]
- 35. Davison A. J., Akter P., Cunningham C., Dolan A., Addison C., Dargan D. J., Hassan-Walker A. F., Emery V. C., Griffiths P. D., and Wilkinson G. W. G. (2003) Homology between the human cytomegalovirus RL11 gene family and human adenovirus E3 genes. J. Gen. Virol. 84, 657–663 [DOI] [PubMed] [Google Scholar]
- 36. Gabaev I., Steinbrück L., Pokoyski C., Pich A., Stanton R. J., Schwinzer R., Schulz T. F., Jacobs R., Messerle M., and Kay-Fedorov P. C. (2011) The human cytomegalovirus UL11 protein interacts with the receptor tyrosine phosphatase CD45, resulting in functional paralysis of T Cells. PLoS Pathog. 7, e1002432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Windheim M., and Burgert H. G. (2002) Characterization of E3/49K, a novel, highly glycosylated E3 protein of the epidemic keratoconjunctivitis-causing adenovirus type 19a. J. Virol. 76, 755–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ruzsics Z., Wagner M., Osterlehner A., Cook J., Koszinowski U., and Burgert H. G. (2006) Transposon-assisted cloning and traceless mutagenesis of adenoviruses: development of a novel vector based on species D. J. Virol. 80, 8100–8113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Uchino J., Curiel D. T., and Ugai H. (2014) Species D human adenovirus type 9 exhibits better virus-spread ability for antitumor efficacy among alternative serotypes. PLoS One 9, e87342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kirchhausen T. (1999) Adaptors for clathrin-mediated traffic. Annu. Rev. Cell Dev. Biol. 15, 705–732 [DOI] [PubMed] [Google Scholar]
- 41. Traub L. M. (2009) Tickets to ride: selecting cargo for clathrin-regulated internalization. Nat. Rev. Mol. Cell Biol. 10, 583–596 [DOI] [PubMed] [Google Scholar]
- 42. Heilker R., Spiess M., and Crottet P. (1999) Recognition of sorting signals by clathrin adaptors. Bioessays 21, 558–567 [DOI] [PubMed] [Google Scholar]
- 43. Robinson M. S. (2004) Adaptable adaptors for coated vesicles. Trends Cell Biol. 14, 167–174 [DOI] [PubMed] [Google Scholar]
- 44. Bonifacino J. S., and Traub L. M. (2003) Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 72, 395–447 [DOI] [PubMed] [Google Scholar]
- 45. Donaldson J. G., and Lippincott-Schwartz J. (2000) Sorting and signaling at the Golgi complex. Cell 101, 693–696 [DOI] [PubMed] [Google Scholar]
- 46. Diettrich O., Gallert F., and Hasilik A. (1996) Purification of lysosomal membrane proteins from human placenta. Eur. J. Cell Biol. 69, 99–106 [PubMed] [Google Scholar]
- 47. Arnold B., Burgert H. G., Hamann U., Hämmerling G., Kees U., and Kvist S. (1984) Cytolytic T cells recognize the two amino-terminal domains of H-2 K antigens in tandem in influenza A infected cells. Cell 38, 79–87 [DOI] [PubMed] [Google Scholar]
- 48. Körner H., Fritzsche U., and Burgert H.-G. (1992) Tumor necrosis factor α stimulates expression of adenovirus early region 3 proteins: implications for viral persistence. Proc. Natl. Acad. Sci. U.S.A. 89, 11857–11861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hilgendorf A., Lindberg J., Ruzsics Z., Höning S., Elsing A., Löfqvist M., Engelmann H., and Burgert H. G. (2003) Two distinct transport motifs in the adenovirus E3/10.4–14.5 proteins act in concert to down-modulate apoptosis receptors and the epidermal growth factor receptor. J. Biol. Chem. 278, 51872–51884 [DOI] [PubMed] [Google Scholar]
- 50. Sester M., and Burgert H.-G. (1994) Conserved cysteine residues within the E3/19K protein of adenovirus type 2 are essential for binding to major histocompatibility complex antigens. J. Virol. 68, 5423–5432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tape C. J., Willems S. H., Dombernowsky S. L., Stanley P. L., Fogarasi M., Ouwehand W., McCafferty J., and Murphy G. (2011) Cross-domain inhibition of TACE ectodomain. Proc. Natl. Acad. Sci. U.S.A. 108, 5578–5583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ludwig A., Hundhausen C., Lambert M. H., Broadway N., Andrews R. C., Bickett D. M., Leesnitzer M. A., and Becherer J. D. (2005) Metalloproteinase inhibitors for the disintegrin-like metalloproteinases ADAM10 and ADAM17 that differentially block constitutive and phorbol ester-inducible shedding of cell surface molecules. Comb. Chem. High Throughput Screen. 8, 161–171 [DOI] [PubMed] [Google Scholar]
- 53. Höning S., Sosa M., Hille-Rehfeld A., and von Figura K. (1997) The 46-kDa mannose 6-phosphate receptor contains multiple binding sites for clathrin adaptors. J. Biol. Chem. 272, 19884–19890 [DOI] [PubMed] [Google Scholar]
- 54. Ghosh R. N., Mallet W. G., Soe T. T., McGraw T. E., and Maxfield F. R. (1998) An endocytosed TGN38 chimeric protein is delivered to the TGN after trafficking through the endocytic recycling compartment in CHO cells. J. Cell Biol. 142, 923–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mallet W. G., and Maxfield F. R. (1999) Chimeric forms of furin and TGN38 are transported with the plasma membrane in the trans-Golgi network via distinct endosomal pathways. J. Cell Biol. 146, 345–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Banting G., and Ponnambalam S. (1997) TGN38 and its orthologues: roles in post-TGN vesicle formation and maintenance of TGN morphology. Biochim. Biophys. Acta 1355, 209–217 [DOI] [PubMed] [Google Scholar]
- 57. Ponnambalam S., Girotti M., Yaspo M. L., Owen C. E., Perry A. C., Suganuma T., Nilsson T., Fried M., Banting G., and Warren G. (1996) Primate homologues of rat TGN38: primary structure, expression and functional implications. J. Cell Sci. 109, 675–685 [DOI] [PubMed] [Google Scholar]
- 58. Mollenhauer H. H., Morré D. J., and Rowe L. D. (1990) Alteration of intracellular traffic by monensin: mechanism, specificity and relationship to toxicity. Biochim. Biophys. Acta 1031, 225–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Klausner R. D., Donaldson J. G., and Lippincott-Schwartz J. (1992) Brefeldin A: insights into the control of membrane traffic and organelle structure. J. Cell Biol. 116, 1071–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tikkanen R., Obermüller S., Denzer K., Pungitore R., Geuze H. J., von Figura K., and Höning S. (2000) The dileucine motif within the tail of MPR46 is required for sorting of the receptor in endosomes. Traffic 1, 631–640 [DOI] [PubMed] [Google Scholar]
- 61. Tortorella L. L., Schapiro F. B., and Maxfield F. R. (2007) Role of an acidic cluster/dileucine motif in cation-independent mannose 6-phosphate receptor traffic. Traffic 8, 402–413 [DOI] [PubMed] [Google Scholar]
- 62. Simonsen A., Bremnes B., Nordeng T. W., and Bakke O. (1998) The leucine-based motif DDQxxLI is recognized both for internalization and basolateral sorting of invariant chain in MDCK cells. Eur. J. Cell Biol. 76, 25–32 [DOI] [PubMed] [Google Scholar]
- 63. Hunziker W., and Fumey C. (1994) A di-leucine motif mediates endocytosis and basolateral sorting of macrophage IgG Fc receptors in MDCK cells. EMBO J. 13, 2963–2969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Okkenhaug K. (2013) Signaling by the phosphoinositide 3-kinase family in immune cells. Annu. Rev. Immunol. 31, 675–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Engelman J. A., Luo J., and Cantley L. C. (2006) The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 7, 606–619 [DOI] [PubMed] [Google Scholar]
- 66. Wu H., Windmiller D. A., Wang L., and Backer J. M. (2003) YXXM motifs in the PDGF-β receptor serve dual roles as phosphoinositide 3-kinase binding motifs and tyrosine-based endocytic sorting signals. J. Biol. Chem. 278, 40425–40428 [DOI] [PubMed] [Google Scholar]
- 67. Edwards D. R., Handsley M. M., and Pennington C. J. (2008) The ADAM metalloproteinases. Mol. Aspects Med. 29, 258–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Saftig P., and Reiss K. (2011) The “A Disintegrin And Metalloproteases” ADAM10 and ADAM17: novel drug targets with therapeutic potential? Eur. J. Cell Biol. 90, 527–535 [DOI] [PubMed] [Google Scholar]
- 69. Murphy G. (2008) The ADAMs: signalling scissors in the tumour microenvironment. Nat. Rev. Cancer 8, 929–941 [DOI] [PubMed] [Google Scholar]
- 70. Perez R. G., Soriano S., Hayes J. D., Ostaszewski B., Xia W., Selkoe D. J., Chen X., Stokin G. B., and Koo E. H. (1999) Mutagenesis identifies new signals for β-amyloid precursor protein endocytosis, turnover, and the generation of secreted fragments, including Aβ42. J. Biol. Chem. 274, 18851–18856 [DOI] [PubMed] [Google Scholar]
- 71. Caporaso G. L., Gandy S. E., Buxbaum J. D., and Greengard P. (1992) Chloroquine inhibits intracellular degradation but not secretion of Alzheimer β/A4 amyloid precursor protein. Proc. Natl. Acad. Sci. U.S.A. 89, 2252–2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. De Strooper B., Vassar R., and Golde T. (2010) The secretases: enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol. 6, 99–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Fallon P. G., and Alcami A. (2006) Pathogen-derived immunomodulatory molecules: future immunotherapeutics? Trends Immunol. 27, 470–476 [DOI] [PubMed] [Google Scholar]
- 74. Reusch U., Muranyi W., Lucin P., Burgert H.-G., Hengel H., and Koszinowski U. H. (1999) A cytomegalovirus glycoprotein re-routes MHC class I complexes to lysosomes for degradation. EMBO J. 18, 1081–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tollefson A. E., Hermiston T. W., Lichtenstein D. L., Colle C. F., Tripp R. A., Dimitrov T., Toth K., Wells C. E., Doherty P. C., and Wold W. S. (1998) Forced degradation of Fas inhibits apoptosis in adenovirus-infected cells. Nature 392, 726–730 [DOI] [PubMed] [Google Scholar]
- 76. Robinson C. M., Rajaiya J., Zhou X., Singh G., Dyer D. W., and Chodosh J. (2011) The E3 CR1-γ gene in human adenoviruses associated with epidemic keratoconjunctivitis. Virus Res. 160, 120–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Towler M. C., Prescott A. R., James J., Lucocq J. M., and Ponnambalam S. (2000) The manganese cation disrupts membrane dynamics along the secretory pathway. Exp. Cell Res. 259, 167–179 [DOI] [PubMed] [Google Scholar]
- 78. Rohrer J., Schweizer A., Russell D., and Kornfeld S. (1996) The targeting of Lamp1 to lysosomes is dependent on the spacing of its cytoplasmic tail tyrosine sorting motif relative to the membrane. J. Cell Biol. 132, 565–576 [DOI] [PMC free article] [PubMed] [Google Scholar]