

Abstract
Xanthophyllomyces dendrorhous β-fructofuranosidase (XdINV)is a highly glycosylated dimeric enzyme that hydrolyzes sucrose and releases fructose from various fructooligosaccharides (FOS) and fructans. It also catalyzes the synthesis of FOS, prebiotics that stimulate the growth of beneficial bacteria in human gut. In contrast to most fructosylating enzymes, XdINV produces neo-FOS, which makes it an interesting biotechnology target. We present here its three-dimensional structure, which shows the expected bimodular arrangement and also a long extension of its C terminus that together with an N-linked glycan mediate the formation of an unusual dimer. The two active sites of the dimer are connected by a long crevice, which might indicate its potential ability to accommodate branched fructans. This arrangement could be representative of a group of GH32 yeast enzymes having the traits observed in XdINV. The inactive D80A mutant was used to obtain complexes with relevant substrates and products, with their crystals structures showing at least four binding subsites at each active site. Moreover, two different positions are observed from subsite +2 depending on the substrate, and thus, a flexible loop (Glu-334–His-343) is essential in binding sucrose and β(2–1)-linked oligosaccharides. Conversely, β(2–6) and neo-type substrates are accommodated mainly by stacking to Trp-105, explaining the production of neokestose and the efficient fructosylating activity of XdINV on α-glucosides. The role of relevant residues has been investigated by mutagenesis and kinetics measurements, and a model for the transfructosylating reaction has been proposed. The plasticity of its active site makes XdINV a valuable and flexible biocatalyst to produce novel bioconjugates.
Keywords: enzyme mechanism, enzyme structure, glycoside hydrolase, glycosylation, kinetics, oligomerization, X-ray crystallography
Introduction
Invertases, or β-fructofuranosidases (EC 3.2.1.26), are biotechnologically important enzymes secreted by many fungi showing wide applications in the food and pharmaceutical industries. β-Fructofuranosidase (XdINV)5 from the basidiomycota yeast Xanthophyllomyces dendrorhous (also Phaffia rhodozyma) is a highly glycosylated protein with a content of 60–70% N-linked carbohydrates and a homodimeric active form of 320–380 kDa (1, 2). As other β-fructofuranosidases, XdINV catalyzes the release of β-fructose from the nonreducing termini of various β-d-fructofuranoside substrates such as sucrose, 1-kestose, or nystose (1), and it may also catalyze the synthesis of short-chain fructooligosaccharides (FOS), in which one or two fructosyl moieties are linked to the sucrose skeleton. Most fructosylating enzymes, such as levansucrases (3), inulosucrases (4), and the majority of fructofuranosidases (5), are able to form either β(2–1) (1F-FOS) or β(2–6) (6F-FOS) linkages between fructose units. However, the distinctive property of XdINV is its ability to transfer fructosyl moieties to the glucose unit of sucrose, thus forming neokestose and neonystose as the main transglycosylation products, both neo-FOS with a levan-type structure (6G-FOS series). FOS are considered as functional food, acting as prebiotics that selectively stimulate growth/activity of lactobacilli and bifidobacteria in the digestive tract and prevent growth of pathogenic microorganisms. They exert a beneficial effect on human health, contributing to the prevention of cardiovascular diseases, colon cancer, or osteoporosis (6). Neo-FOS possess enhanced properties and chemical stability compared with 1F-FOS series (e.g. 1-kestose or nystose) typically used as prebiotics (7–9). In addition, XdINV is also able to fructosylate a variety of carbohydrates containing glucose, in particular disaccharides (maltose, isomaltulose, isomaltose, and trehalose) and higher oligosaccharides (maltotriose, raffinose, and maltotetraose) but not monosaccharides (glucose, fructose, and galactose) (10). Thus, novel heterofructooligosaccharides with potential applications in the functional food and pharmaceutical industries can be obtained with this enzyme, which makes it an interesting biotechnological target. Further structural analysis of XdINV is necessary to fully understand its particular specificity and to improve its biosynthetic potential.
On the basis of its amino acid sequences, XdINV was classified into the glycoside hydrolase (GH) family GH32 (1), a family including other invertases, β-fructofuranosidases, inulinases, and fructosyltransferases (11), which along with proteins in family GH68 constitute clan GH-J. The enzymes within this clan share a five-blade β-propeller N-terminal domain, in which β-sheets are arranged around a central pocket that accommodates the active site (catalytic domain). Three key acidic residues located in the active site and surrounded by conserved sequences in the GH32 family, NDPNG (D acts as a nucleophile), RDP (D acts as a stabilizer of the transient state), and EC (E acts as an acid base catalyst), are implicated in substrate binding and hydrolysis (12). These enzymes can have hydrolytic or synthetic activity, depending on whether water or an oligosaccharide (the acceptor substrate) accepts the fructose released by fructan hydrolysis (the donor substrate). A distinctive trait of GH32 enzymes is the presence of an additional β-sandwich structure fused at the C terminus of the catalytic domain. In the last decade, GH32 enzymes from bacteria (13, 14), fungi (15–17), and plants (18–20) have been the subject of many structural studies that showed that all are monomeric enzymes. Surprisingly, studies on Schwanniomyces occidentalis fructofuranosidase (SoFfase) (21) and Saccharomyces cerevisiae invertase (ScINV) (22) showed that these enzymes from yeast are unique in that they form dimers mediated by their β-sandwich domain, which in the case of ScINV associate into higher oligomers. The structural studies have delineated the main structural determinants of activity, the loops and turns connecting the different elements of the β-propeller domain, in the surroundings of the active site, defining the specificity unique to each enzyme (21, 23). However, and remarkably, oligomerization of the yeast enzymes regulates their functionality. Thus, the analysis of the structure of a complex of SoFfase with the polymeric inulin showed the direct involvement of the β-sandwich domain of an adjacent subunit in substrate binding (24).
In this study, we report the three-dimensional structure of the yeast XdINV that discloses novel features and an unusual dimer. Complexes with relevant substrates and products have been obtained, which allowed mapping the catalytic pocket. The role of identified leading residues has been investigated by mutagenesis. Our results give the experimental evidence of the direct involvement of glycosylation in dimerization and activity of a GH32 enzyme and suggest the role of a flexible loop surrounding the catalytic pocket in discriminating substrate specificity.
Experimental Procedures
Organisms, Media, Plasmids, and Mutagenesis
X. dendrorhous ATCC MYA-131 was grown in MMM medium as referred previously (1). Pichia pastoris GS115 (his4−, Invitrogen) was used as expression host for the different enzyme variants. It was grown in YEPD medium (yeast extract 1%, peptone 2%, and glucose 2%) at 30 °C. MD media (YNB 1.34%, biotin 4 × 10−5 %, and glucose 2%) was used to select transformants. Expression and induction of proteins in P. pastoris were analyzed using BMG and BMM media (both are the same as MD but in potassium phosphate, pH 6.0, and glycerol 1% or methanol 0.5% as carbon sources, respectively). Yeast growth was followed spectrophotometrically at a 600-nm wavelength. Escherichia coli DH5α was used for DNA manipulation and amplification using the standard techniques.
The β-fructofuranosidase Xd-INV gene from X. dendrorhous (GenBankTM accession number FJ539193.2) comprises an open reading frame of 1995 bp corresponding to a 665-amino acid protein. The QDNS-pIB4 construction contained the 1902-bp fragment of Xd-INV responsible for the synthesis of the last 634 amino acids of this protein fused to the 267-bp fragment of the S. cerevisiae MFα secretion signal sequence (25). This construct was used as a template to obtain all the mutants generated in this work. Site-directed mutagenesis was carried out using specific primers, including substitutions responsible for mutations N58S, D80A, N107S, E303A, E334S/E334N/E334V/E334Q, Q341N, N342S, H343A/H343T, N471S, and Y659STOP, and following the method described previously (21). DNA sequencing was used to verify that only the desired mutations were present in all the obtained constructs.
Protein Expression and Purification
X. dendrorhous β-fructofuranosidase was purified using tangential concentration and DEAE-Sephacel chromatography, as described previously (26). Active fractions were concentrated using Microcon YM-10 (Amicon) filters and stored at −70 °C. β-Fructofuranosidase variants from X. dendrorhous (wild type and mutants) expressed in P. pastoris were purified as described elsewhere (25). ProtoBlue Safe Colloidal Coomassie-stained (National Scientific) denaturing gel electrophoresis (SDS-PAGE; 8% polyacrylamide) of the samples confirmed the purity of the protein fractions. Protein concentration was determined using the Bio-Rad microprotein determination assay and bovine serum albumin as standard.
After purification of XdINV, protein deglycosylation was performed using endoglycosidase H (Endo H; New England Biolabs) as described previously (26). Deglycosylated protein was subjected to a 1-min ultrafiltration using an Amicon Ultra-4 50K device (Millipore) to remove the Endo H present in the sample and was subsequently concentrated to about 8 mg ml−1 using a 10-kDa cutoff membrane.
β-Fructofuranosidase Activity and Kinetics Assays
β-Fructofuranosidase hydrolytic activity was determined by the dinitrosalicylic acid method adapted to a 96-well microplate scale as described previously (25). Standard enzymatic assays were performed using 2% sucrose in 100 mm sodium phosphate buffer, pH 5.5, at 60 °C. Inulin (Orafti GR, BENEO Ibérica, S.L. Barcelona, Spain) and fructans from Agave tequilana (kindly supplied by Dr. Georgina Sandoval, CIATEJ, Guadalajara, Mexico) at 1% were also used in this assay. One unit of activity was defined as that catalyzing the formation of 1 μmol of reducing sugar/min. Hydrolase activity analysis at 55-80 °C was carried out under the aforementioned conditions. The thermostability refers to the temperature required for 50% enzyme inactivation (enzyme half-life) after heating about 0.5 units of the pure enzyme at different temperatures (60-85 °C) during 10-120 min and was determined by removing samples at regular intervals and estimating the residual β-fructofuranosidase activity.
For kinetic analysis, the initial velocity was measured in triplicate with 45–30 μg ml−1 of enzyme and using 0–80 mm sucrose, 0–60 mm raffinose, or 0–60 mm nystose. The reaction time was 20 min. The plotting and analysis of the curves were carried out using the kinetic module of SigmaPlot software (version 12). Kinetic parameters were calculated by fitting the initial rate values to the Michaelis-Menten equation.
The activity of several XdINV mutants was analyzed by zymogram analysis using non-denaturing gels (6% polyacrylamide) stained with 2,3,5-triphenyltetrazolium-chloride, as described previously (1). Bovine serum albumin was used as reference and negative control.
Crystallization and Data Collection
Crystals of XdINV were grown as described before (26). For data collection, native crystals were transferred to cryoprotectant solutions consisting of mother liquor plus 10% (v/v) glycerol before being cooled in liquid nitrogen. Diffraction data were collected using synchrotron radiation at the European Synchrotron Radiation Facility (ESRF, Grenoble) on ID23-1 beamline. Diffraction images were processed with iMOSFLM (27) and merged using the CCP4 package (28). Crystals of XdINV were indexed in the P21212 space group with two molecules in the asymmetric unit and 70% solvent content within the unit cell (Table 1).
TABLE 1.
Crystallographic statistics (Values in parentheses are for the high resolution shell)

†Rmerge=Σhkl Σi|Ii(hkl) − [I(hkl)]|/Σhkl ΣiIi(hkl), where Ii(hkl) is the ith measurement of reflection hkl and [I(hkl)] is the weighted mean of all measurements.
†† Rpim = Σhkl (1/(N − 1)) 1/2 Σi | Ii(hkl) − [I(hkl)]|/Σhkl Σi Ii(hkl), where N is the redundancy for the hkl reflection.
††† Rwork/Rfree = Σhkl | Fo − Fc |/Σhkl|Fo|, where Fc is the calculated and Fo is the observed structure factor amplitude of reflection hkl for the working/free (5%) set, respectively.
The inactive D80A mutant was deglycosylated as reported for native enzyme and tested for crystallization. The best diamond-shaped crystals were grown by mixing equal amounts of 8 mg ml−1 protein (in 20 mm Tris-HCl, pH 7) and a solution consisting of 1.3 m sodium citrate and 0.1 m Hepes or 0.1 m BisTris propane, pH 7.5. D80A complexes with 1-kestose or nystose (both from TCI-Europe), raffinose (Sigma), neokestose or neoerlose, both purified from reactions including XdINV as described in references (1, 10), were obtained by soaking into solutions containing 5–30 mm ligand for 5–30 min, whereas the complex with sucrose was obtained by soaking into a 25% (w/v) sucrose solution. Finally, the ternary complexes of D80A-Fru with the corresponding buffer, Hepes or Bistris propane, were obtained in crystals pre-incubated with 30 mm fructose for 5–10 min and then adding different acceptors as maltotriose, isomaltose, or isomaltulose (all from Sigma) that were not found in the crystal. For data collection, all D80A crystals (except the D80A-sucrose complex) were transferred to cryoprotectant solutions consisting of mother liquor plus 10% (v/v) ethylene glycol. Diffraction data were collected using synchrotron radiation on different sources, and images were processed with XDS (29) and merged using AIMLESS from the CCP4 package (28). The data collection statistics are given in Table 1.
Structure Solution and Refinement
The structure of XdINV was solved by molecular replacement using the MOLREP program (30). The best model was the invertase from Aspergillus japonicus (PDB code 3LF7), having 34% identity (48% similarity), from which a template was prepared using the program Chainsaw (31) and a protein sequence alignment of XdINV onto the template. A solution containing two molecules in the asymmetric unit was found using reflections up to 2.5 Å resolution range and a Patterson radius of 40 Å, which after rigid body fitting led to an R-factor of 49%. Crystallographic refinement was performed using the program REFMAC (32) within the CCP4 suite with flat bulk-solvent correction, maximum likelihood target features, and local non-crystallographic symmetry. Free R-factor was calculated using a subset of 5% randomly selected structure-factor amplitudes that were excluded from automated refinement. Extensive model building using the program COOT (33) was combined with several rounds of refinement leading to a model showing a continuous density for the whole polypeptide chain. At the later stages, carbohydrate and water molecules were included in the model, combined with more rounds of restrained refinement that led to a final R-factor of 16.9 (Rfree = 18.8) for all data set up to 2.14 Å resolution. Refinement parameters are reported in Table 1.
The structure of D80A mutant was solved by molecular replacement using MOLREP and the coordinates of native XdINV as the search model. A first cycle of refinement was performed using REFMAC, combined with model building with COOT. The structures of the D80A complexes were solved by difference Fourier synthesis using these refined coordinates. The ligands were manually built into the electron density maps and were refined similarly, to reach the R-factors listed in Table 1. For neokestose and neoerlose, not present in the Protein Data Bank, a model was built by the on-line biomolecule building program GLYCAM (34). Then PRODRG (35) was used to automatically generate molecular topologies suitable for REFMAC refinement.
Stereochemistry of the models was checked with PROCHECK (36). The figures were generated with PyMOL (37). Root mean square deviation analysis was made using the program SUPERPOSE within the CCP4 package (28).
Polysaccharide Chain Model Building
A model of a β(2–6)-linked fructan chain was constructed using the program GLYCAM (34) and exported in its lowest energy state. This chain was manually docked into the molecular surface of the XdINV dimer. Models containing β(2–6) fructose units, connected with both gt or gg conformations, were found to fit properly into the cleft connecting the two binding sites.
Results
XdINV Folding Displays Novel Structural Features
We have purified and crystallized the deglycosylated form of the wild-type β-fructofuranosidase XdINV, as reported previously (26). Treatment of this highly glycosylated protein (monomer of 160–200 kDa) with Endo H led to a molecular mass of 66 kDa and reduced drastically the heterogeneity of the sample, allowing us to obtain high quality crystals. We present here its three-dimensional structure solved by molecular replacement at 2.14 Å resolution. Experimental and structural determination details are given under “Experimental Procedures” and in Table 1. It should be noted that the deglycosylated enzyme retains activity, and heterogeneity was reduced without affecting protein quaternary structure, as shown below. The crystals belong to the space group P21212, with two molecules per asymmetric unit that are related by a non-crystallographic 2-fold symmetry axis. No significant structural differences were observed between the two chains (Ser-38–Tyr-665) that present a root mean square deviation of 0.325 Å on 627 matched Cα atoms.
In common with the GH32 enzymes, XdINV is bimodular (Fig. 1) and folds into a catalytic β-propeller domain (residues 57–461) and a β-sandwich domain (residues 473–637), linked by a short α-helix (residues 462–472). The β-propeller has five blades, each consisting of four antiparallel β-strands (A, B, C, and D) with the classical “W” pattern around the central axis, enclosing the active site. The loops linking the different blades (L1–L5), and strand B to C (TI–TV) within each blade, are shaping a deep cavity and are the most variable regions within family GH32 conferring specificity to the different enzymes (Fig. 2). The C-terminal β-sandwich domain is organized in two β-sheets with five strands. However, a unique feature of XdINV is a sort of lace made by an extension of the N terminus (residues 38–56) and a long 30-residue extension at the C-terminal chain (residues 635–665), which increases the interaction between both domains (Fig. 1, A and B). Remarkably, the long C-terminal region makes an extended arm that folds onto blade 1 from the catalytic domain, contouring the active site pocket at its end. As will be explained below, this region is essential for activity and oligomerization.
FIGURE 1.

Structure of XdINV. A, ribbon diagram of XdINV monomer showing the bimodular molecular architecture, colored in rainbow sequence-code, which folds into a catalytic β-propeller domain and a β-sandwich domain. Two well-ordered glycan chains are observed at positions Asn-58 and Asn-107, whereas Asn-471 is the only position conserved within GH32 from fungi. B, molecular surface of XdINV with the same color code, and the N-glycan chains shown as sticks. C, XdINV is a highly glycosylated protein containing 18 potential glycosylation sites; apart from the two long N-glycans, GlcNAc units have been found in the crystal N-attached to other Asn residues, their location being indicated by small yellow spheres.
FIGURE 2.

Structural alignment of GH32 from fungi. The catalytic domain of XdINV is superimposed to the A. japonicus fructosyltransferase, AjFT (PDB code 3LEM), A. awamorii exo-inulinase, AaEI (PDB code 1Y9G), S. cerevisiae invertase, ScINV (PDB code 4EQV), and S. occidentalis β-fructofuranosidase, SoFfase (PDB code 3U75), as given by the DALI server (44). The red squares indicate amino acid similarity as calculated by ESPript (45). A–D indicate the β-strands, from the axis toward the outside of the propeller, within each blade. Loops L1–L4, connecting the different blades of the propeller, and turns TI–TV, linking strands B to C within each blade, are underlined with an orange bar. XdINV glycosylation sites are highlighted in yellow. Catalytic residues are marked with a black asterisk. Residues of XdINV involved in polar (angle) and hydrophobic (asterisk) interactions within the dimer interface are indicated in green. Residues of XdINV mutated in this work are marked with a cyan asterisk.
Endo H treatment is known to cleave the oligosaccharide moieties leaving single N-acetylglucosamine (GlcNAc) units, although poorly accessible glycosylation sites may remain partially glycosylated. XdINV is a highly glycosylated protein that contains 18 potential glycosylation sites in its analyzed sequence. From the electron density maps, GlcNAc units were modeled at 17 Asn positions (represented in Fig. 1C). However, a remarkable feature are the two well-ordered glycan chains that were observed at positions Asn-58, placed at the N terminus extension, and Asn-107, located at TI loop in blade 1. Interestingly, the long C-terminal arm is sandwiched between both glycan chains, shaping a platform that is crucial for oligomerization, as described below.
XdINV Is a Dimeric Enzyme
Analytical ultracentrifugation of native and deglycosylated XdINV samples showed an average molecular mass of 260 and 130 kDa, respectively (data not shown), which are consistent with the dimeric state observed by crystallography. The dimer is a flat oblate with dimensions 135 × 75 × 45 Å (Fig. 3), which is made up of two identical subunits related by a crystallographic 2-fold axis. Its molecular surface is 43733 Å2, with an interface of 1037 Å2. The interaction between subunits is made by blades 1 and 2 from each catalytic domain, through loops L1 (residues 135–141), TII (residues 178–182), and L2 (residues 200–210), and also by the C-terminal region (residues 650–662) (see Figs. 2 and 3). Although the buried surface area is moderate (20 residues are more than 50% buried), and only four direct hydrogen bonds are linking both subunits (Table 2), the interface is further reinforced by the glycan chains linked to each Asn-107, which make a net of intermolecular hydrogen bonds, mainly to loops L1 and L2 (Table 2) and also to each other (Fig. 3D). These glycans are filling a tunnel at the dimer interface (Fig. 3C), which results in an increment of the contact surface up to 4150 Å2 that should contribute to its stabilization. Moreover, the glycans attached to Asn-58 are packed over the C-terminal region extending further the contact interface to 6140 Å2 (Fig. 3B). The contribution of the glycans at the interface and the importance of the atomic interactions explain why treatment of XdINV with Endo H does not remove the glycan chains attached to Asn-58 and Asn-107.
FIGURE 3.

XdINV dimer. A, molecular surface view of the dimer that is made by association of two subunits through their catalytic domains (dark orange/green) and the C-terminal region of the corresponding β-sandwich domains (light orange/green). A glycerol molecule found in the native protein crystals is shown in sphere representation at each active site pocket. The glycan chain is shown as sticks. B, same view of the dimer in schematic representation, highlighting the regions involved in the interface. Inset, a zoom showing the detailed intermolecular atomic interaction found around the glycan chain attached to Asn-58. C and D the opposite view of A and B showing the dimer and the atomic interactions at the interface around the glycan chain attached to Asn-107. NAG, GlcNAc; MAN, α-mannose; BMA, β-mannose.
TABLE 2.
Hydrogen bonds at the dimer interface
| Molecule A | Molecule B | Distance |
|---|---|---|
| Å | ||
| Intermolecular | ||
| Tyr-200 (OH) | Thr-655 (OG1) | 2.66 |
| Ala-202 (N) | Trp-651 (O) | 3.04 |
| Trp-651 (O) | Ala-202 (N) | 2.92 |
| Thr-655 (OG1 ) | Tyr-200 (OH) | 2.68 |
| Trp-134 (NE1) | MAN111 (O3) | 2.77 |
| Gln-137 (OE1) | NAG107 (O3) | 2.60 |
| Gln-137 (NE2) | NAG107 (O4) | 3.00 |
| Gln-137 (NE2) | NAG108 (O7) | 3.01 |
| Asp-155 (OD2) | MAN115 (O4) | 2.70 |
| Lys-197 (N) | MAN111 (O6) | 3.05 |
| Intramolecular | ||
| Glu-128 (OE1) | NAG108 (N2) | 2.85 |
| Glu-128 (OE1) | NAG107 (O6) | 2.70 |
| Lys-131 (NZ) | NAG107 (O5) | 2.74 |
| Lys-131 (NZ) | NAG107 (O6) | 2.96 |
| Lys-405 (N) | NAG059 (O6) | 2.90 |
| NAG108 (O3) | BMA109 (O6) | 2.98 |
| NAG58 (O6) | NAG59 (O6) | 3.45 |
| NAG59 (O3) | BMA60 (O6) | 3.30 |
N-Linked Glycans Are Crucial for Oligomerization and Activity
To investigate the precise role that glycosylation plays in the activity of XdINV, positions Asn-58 and Asn-107 were mutated to serine, and properties of the modified protein were explored by zymogram analysis and thermal stability assays. Enzymatic activity of these mutants was also tested. Additionally, Asn-471 was included in this analysis taking into account that this position is conserved among structurally known GH32 enzymes from fungi. Fig. 4A and Table 3 show that N58S and N471S mutations result in active dimeric forms of XdINV. In contrast, mutation N107S causes a total loss of activity that is possibly due to a defective protein folding resulting in a distorted variant of the protein that cannot be detected even by native PAGE (Fig. 4A). Moreover, the lack of a band on SDS-PAGE (Fig. 4B) suggests that mutant N107S might have not been recognized by the secretory pathway. This fact points to the conclusion that glycosylation at Asn-107 is essential for dimer formation, and its removal could be deleterious for proper protein folding. Glycosylation and thermostability are closely related properties in a protein. Thus, the effect of the N58S and N471S substitutions on the thermostability of XdINV was evaluated. Accordingly, these protein variants were purified, and their sensitivities to different temperatures were studied as described under “Experimental Procedures.” The N471S variant behaved similarly to the wild-type protein, displaying maximum activity levels (75–100%) in the range of 60–75 °C, whereas the N58S variant activity decreased to about 40 and 20%, at 70 and 75 °C, respectively (Fig. 4C). In addition, when variants were incubated without substrate in the range of 60–85 °C for 10–120 min and their activity was assayed, wild-type, N471S, and N58S variants maintained 50% of their activity (half-life) at 71–76, 68–76, and 62–64 °C, respectively (results not shown). The N471S mutation apparently did not affect the thermal stability of the enzyme nor its apparent catalytic efficiency (kcat/Km) (Table 3), whereas the thermal stability of N58S reduced both thermostability (about 10 °C) and catalytic efficiency with sucrose (4-fold) due to an elevated Km value. These results indicate that glycosylation at Asn-58 contributes to stabilize the structure of XdINV, whereas the role of that attached to Asn-471 is unclear.
FIGURE 4.
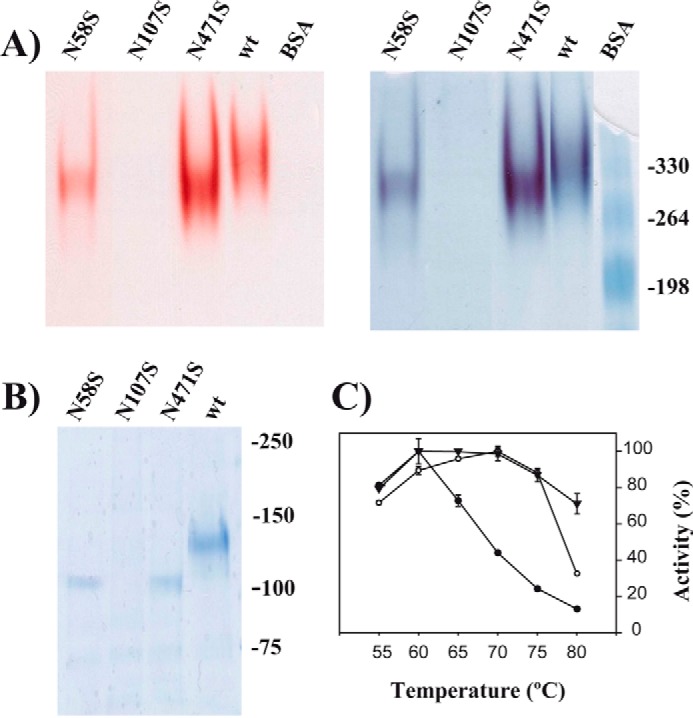
Role of glycosylation in XdINV. A, PAGE analysis of the XdINV variants. Culture filtrates (200 ml) of the P. pastoris transformants expressing the wild-type (wt) or the indicated XdINV mutants were concentrated through 50000 Mr cutoff (PES membrane) and applied to DEAE-Sephacel chromatography. Active fractions eluting at 0.1 m NaCl were concentrated to 0.2–0.5 mg/ml. The enzyme activities (10 μl; about 2–5 μg of proteins) were revealed in situ (left) and subsequently stained using colloidal Coomassie (right). The mutant N107S was processed in the same way, but 20 μl of the final concentrate was analyzed. B, referred proteins were analyzed by SDS-PAGE. Numbers at the right in A and B indicate the positions of the bovine serum albumin (BSA) molecular masses and the weight markers used as control (in kDa), respectively. C, temperature dependence profiles. Activity of the wild-type (filled triangles), N58S (filled circles), and N471S (open circles) protein variants were evaluated at the indicated temperatures. Each value represents the average for four independent measurements. Standard errors are indicated.
TABLE 3.
Kinetic analysis of XdINV mutants
The kcat values were calculated assuming a protein molecular mass of 66 kDa (unglycosylated monomer). The ± sign refers to standard error curve fit using the kinetic module of SigmaPlot version 12. The mutants D80A, N107S, E303A, E334S/E334V/E334N, Y659STOP gave no activity using sucrose. WT, wild type; ND, not detected; blank, not evaluated.
| XdINV variant |
Km (mm) |
kcat (s−1) |
kcat/Km (mm−1 s−1) |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Sucrose | Raffinose | Nystose | Sucrose | Raffinose | Nystose | Sucrose | Raffinose | Nystose | |
| WT | 1.9 ± 0.3 | 7.7 ± 1.6 | 21.7 ± 3.6 | 911 ± 47 | 291 ± 51 | 148 ± 25 | 479 ± 68 | 56 ± 10 | 72 ± 12 |
| N58S | 6.1 ± 1.7 | 711 ± 200 | 117 ± 33 | ||||||
| E334Q | 3.6 ± 0.6 | 15.5 ± 3.4 | 1.9 ± 0.4 | 91 ± 15 | 7 ± 1 | 46 ± 10 | 25 ± 4 | 0.4 ± 0.1 | 23 ± 5 |
| Q341N | 1.8 ± 0.4 | 8.4 ± 2.0 | 30.6 ± 6.9 | 377 ± 76 | 171 ± 40 | 71 ± 16 | 212 ± 43 | 20 ± 5 | 2 ± 1 |
| N342S | 7.3 ± 1.3 | 2.9 ± 0.7 | 12.7 ± 2.1 | 1070 ± 189 | 165 ± 41 | 65 ± 11 | 147 ± 26 | 58 ± 14 | 5 ± 1 |
| H343A | 8.3 ± 1.5 | 7.5 ± 1.5 | ND | 105 ± 19 | 32 ± 6 | ND | 13 ± 2 | 4 ± 1 | ND |
| H343T | 12.6 ± 3.3 | 12.9 ± 3.5 | ND | 855 ± 223 | 101 ± 27 | ND | 68 ± 18 | 8 ± 2 | ND |
| N471S | 3.4 ± 0.9 | 1151 ± 307 | 339 ± 91 | ||||||
XdINV Presents Unique Traits within Known GH32 Enzymes from Fungi
The three-dimensional structure of five other GH32 members from fungi has been reported as follows: the Aspergillus awamorii exo-inulinase (AaEI (15)), the A. japonicus fructosyltransferase (AjFT (16)), the Aspergillus ficuum endo-inulinase (17), the S. occidentalis β-fructofuranosidase (SoFfase (21)), and the S. cerevisiae invertase (ScINV (22)). XdINV shares the highest identity (37%) with AjFT, whereas 20–25% identity is found with respect to the other enzymes, a value equivalent to that observed when XdINV is compared with the GH32 enzymes from bacteria and plants. Structural alignment of the catalytic domain of XdINV onto the fungal exo-enzymes (all but A. ficuum endo-inulinase) is shown in Fig. 2. As observed, both XdINV and AjFT present long loops linking the different β-sheets, and remarkably, AjFT presents also the inserted region at the N terminus described in XdINV but not the C-terminal extension. In contrast, Asn-471, at the segment linking the β-structure to its catalytic domain, is the sole conserved Asn residue in all the sequences, although Asn-52 and Asn-58 are present in AjFT and Asn-107 is only conserved in AaEI. Consequently, the glycosylation pattern of XdINV and the concurrence of Asn-107 and the C-terminal extension, both shown to be key determinants in oligomerization, are unique to this protein. Moreover, as can be observed in Fig. 2, loops L1, TII, and L2 are highly variable in sequence, and the residues involved in polar links or interatomic interactions at the dimer interface are not conserved.
It is worth noting that from all GH32 enzymes, only the reported structures from yeasts, SoFfase and ScINV, revealed the existence of dimeric forms. However, the dimerization pattern of those enzymes was different from that observed in XdINV (Fig. 5), with blades 4 and 5 involved in the interface of the SoFfase and ScINV dimers through loops L3, L4, and the segment linking C5-D5 (Fig. 2). Nevertheless, the β-sandwich domain is involved in the assembly of the two types of dimers and also in shaping the active site cavity, although through an evident different pattern. Thus, this feature seems to be a common trend of the GH32 enzymes from yeast, and remarkably, it is the only case in which a role has been assigned to the β-sandwich domain within the GH32 family enzymes. A notable feature of XdINV is that, unlike SoFfase, the particular architecture of its dimer houses the active sites at the same face and separated 30 Å away. A close inspection of the molecular surface reveals a long crevice encompassing the two active sites and beyond. Therefore, it is appealing to suggest that this disposition might be conferring the XdINV dimer the additional ability to accommodate a putative polymeric substrate, as illustrated in Fig. 5. In this way, the XdINV cleft could be able to allocate, for instance, a linear β(2–6)-linked fructose chain with β(2–1) branches, characteristic of mixed fructans or graminans (38), its β(2–1) branches being hydrolyzed at each catalytic pocket. In agreement with this, XdINV showed a small but significant hydrolase activity on fructan polymers, showing a moderate activity on inulin (72 units/mg versus 960 units/mg on sucrose) as is observed in other GH32 enzymes. Furthermore, XdINV is also able to hydrolyze agave fructans (15 units/mg), which consist of a complex mixture of fructose-based polymers containing principally β(2–1) linkages but also β(2–6) (39). Although this is not evidence that branched substrates can bind to both sites, the channel-like topology of the dimer surface is, indeed, a remarkable trait of XdINV.
FIGURE 5.
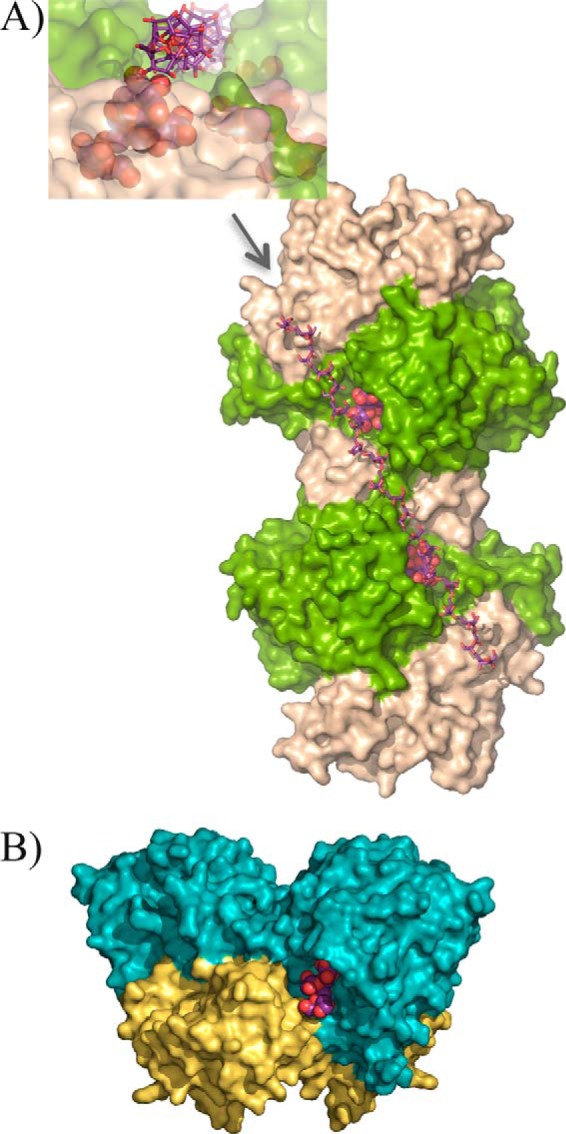
XdINV versus SoFfase dimers. A, molecular surface of XdINV dimer showing the two catalytic domains in light green, and the corresponding β-sandwich domains are colored in sand. Each active site cavity is filled with a nystose molecule (in spheres) as found in the complexes obtained in this work and described below. A modeled β(2–6)-linked fructose chain (in sticks) has been manually docked into the long crevice that encompasses the two active sites and beyond. Inset, a detail of this crevice as viewed from the position indicated by the arrow, i.e. along the modeled levan-type fructose chain; a nystose molecule (spheres) is visible at one of the active sites. B, molecular surface of the SoFfase dimer showing the catalytic domains in cyan and their β-sandwich domains in yellow. Each active site is located at one face of the dimer, and only a fructosyl-nystose molecule found in the crystal (24) is visible as spheres.
Structure of the XdINV Complexes Discloses the Binding Subsites
Inactivated XdINV D80A mutant was crystallized and used for soaking experiments with different donor substrates, including sucrose, 1-kestose, neokestose, and nystose; from these, 1-kestose and neokestose are also transfructosylation products. As has been reported recently (10), XdINV is able to fructosylate glucose-containing oligosaccharides as maltose, which produces neoerlose. To depict this activity, this trisaccharide has been also included in the soaking experiments. Furthermore, in an attempt to capture a putative intermediate complex, mutant crystals were pre-incubated with fructose before being transferred to solutions containing tested acceptors as maltotriose, isomaltulose, isomaltose, and raffinose. However, only raffinose was found at the active site, but it was occupying the catalytic pocket in a donor substrate mode. Interestingly, fructose and a molecule of Hepes or Bistris propane, both from the buffer, were found at the active site in some of the crystals mimicking the location of an acceptor for the fructose in a putative intermediate complex, as will be commented on below.
The catalytic site is buried in a deep pocket 20 Å deep and about 10 × 10 Å width. All the obtained complexes show clear electron density at the active site, which allowed unambiguous ligand modeling that situated the terminal fructose at the bottom and in contact with the catalytic residues (Fig. 6). This fructose at subsite −1 keeps the same atomic interactions and features previously described for other complexes from GH32 enzymes (16, 19, 24). Remarkably, a net of well ordered water molecules is found in all complexes, contributing to substrate recognition. The only significant differences found in the protein upon substrate binding are located at loop TIV (residues Glu-334–His-343), which is rather disordered in the free enzyme and is stabilized in the complexes making crucial interactions, as explained below.
FIGURE 6.

Structure of the XdINV complexes. Close-up view of the active site showing the relevant residues making interactions with each ligand. Main polar interactions are represented as dashed lines. The interactions at subsite −1 are conserved and only shown in the XdINV-sucrose complex. The catalytic residues are highlighted in purple and the referred ligands in yellow. Chemical structure of the sugars is given in the scheme, the 1F-FOS is 1-kestose or nystose, when n = 1 or 2; sucrose, n = 0.
The glucose bound at subsite +1 in the complex with sucrose, neokestose, and raffinose is tightly fixed by direct hydrogen bonds to the acid/base catalytic Glu-303 (through its O2), and to Glu-334 (O2 and O3) and His-343 (O2), both from loop TIV. The glucose is further stabilized by interaction of its O4 to Leu-170 and Ala-172 from loop TII, through several water molecules. In the case of 1-kestose and nystose, having fructose at this subsite, and neoerlose, with a β(2–6) linked glucose, a water molecule is occupying the position of the glucose O2 observed in the sucrose complex, therefore mediating the same interaction pattern to Glu-334 and His-343.
Subsite +2 is made by stacking interaction to Trp-105 from loop TI, which allocates the corresponding sugar unit in a variety of modes revealing a noticeable plasticity. Apart from this hydrophobic interaction, there are many hydrogen bonds to different residues, always through numerous water molecules and, also, many intramolecular hydrogen bonds fixing each substrate in a precise conformation. First of all, the C-terminal extension is making a polar interaction through Leu-661 main chain, conserved in all the complexes. But additional interactions depend on the type of substrate and, thus, neokestose, raffinose, and neoerlose are accommodated between loop TII and the C-terminal segment by multiple polar interactions to Leu-170, Ala-172, Gly-660, and Leu-661. By contrast, 1-kestose and nystose are oriented toward loop TIV and interacting with Gln-341. Finally, subsite +3, occupied only in the nystose complex, is mainly shaped by loop TIV and shows direct polar interactions of the terminal glucose with Gln-335, Asn-342, and His-343.
In summary, subsite +1 is common to all complexes, being shaped by TII through Leu-170 and Ala-172 main chain and by TIV through Glu-334 and His-343. Subsite +2 is created by stacking to Trp-105 and polar interaction to the main chain of the C-terminal region (Gly-660 and Leu-661), but two different sites are observed depending on the substrate, and thus, inulin-type ligands are accommodated by loop TIV through Gln-341, whereas levan or neo-type ligands are allocated by loop TII (Leu-170 and Ala-172) (Fig. 7). An additional subsite +3 for inulin-type substrate is formed essentially by loop TIV (Gln-335, Gln-341, Asn-342, and His-343). In summary, the structure of the XdINV complexes presented here suggests a role of loop TIV in regulating specificity of this protein, especially through residues Glu-334, Gln-341, and His-343.
FIGURE 7.
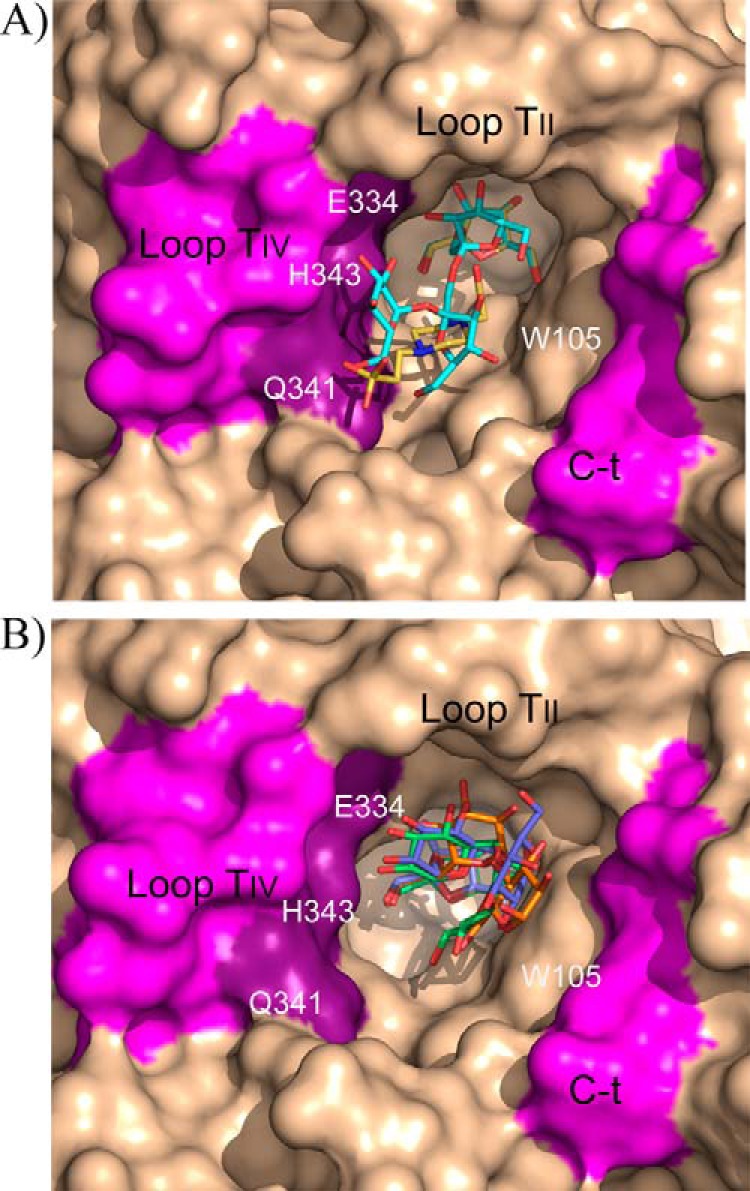
Two binding modes of XdINV. Detail of the XdINV active site representing main structural features as follows: subsite +1 is common to all complexes, being shaped by TII and by TIV (light violet, key residues highlighted in dark violet); subsite +2 is created by stacking to Trp-105 and polar interaction to the main chain of the C-terminal region (light violet), but two different sites are observed depending on the substrate. A, inulin-type ligands (represented by nystose in cyan and Hepes in yellow) are accommodated by loop TIV. B, levan or neo-type ligands are allocated by loop TII (represented by neokestose in green, raffinose in slate, and neoerlose in orange). Additional subsite +3 for inulin-type substrates is formed essentially by loop TIV (A).
Mutational Analysis Unveils Key Residues Modulating Activity
The role of the leading residues identified in the complex analysis was investigated by kinetic analysis of mutants that is given in Table 3. First, the expected lack of activity observed in mutants D80A and E303A is consistent with its catalytic role as nucleophile and electron/proton donors, respectively. But, remarkably, the presence of glutamate at position 334 seems to be also required for sucrose hydrolysis, as its replacement by serine, valine, or glutamine is deleterious for activity and leads to fully inactivated enzymes, revealing the major role of Glu-334 in binding sucrose. Only the mild E334Q replacement gives detectable activity on sucrose, and interestingly, this mutant keeps a significant activity on nystose. As described above, Glu-334 is making a direct hydrogen bond to the sucrose O2 and O3 atoms, from the glucose located at site +1, and therefore, some effect upon its replacement was expected in the hydrolysis of this substrate. The presence of an acidic residue at this position seems necessary for sucrose binding and hydrolysis in many clan GH-J enzymes, and as such, the equivalent D239A mutant of Arabidopsis thaliana invertase exhibited a 6-fold increase in Km against sucrose (40). Moreover, although this Asp/Ala replacement compromised its ability to degrade sucrose, the enzyme retained fructan hexohydrolase activity (41). Thus, from sequence analysis of plant GH32 enzymes, it was proposed that members having an Asp-239 homolog have sucrose as the preferential donor, whereas enzymes lacking this residue use fructans as their donor substrate (41). In agreement with this role in regulating specificity, kinetic analysis of SoFfase mutants at Gln-228 position, structurally related to the A. thaliana Asp-239, revealed that Gln-228 is essentially responsible for conferring affinity for acceptor sucrose in the transfructosylation reaction (24). However, in that case, even the SoFfase Q228V and Q228T mutants retained moderate invertase activity. Consequently, the XdINV Glu-334 plays a more essential role in sucrose binding than that reported for the yeast SoFfase, resembling the behavior described for plant GH32 enzymes.
Mutants H343A/H343T also showed drastically reduced activity on the assayed substrates. As observed in the complexes, His-343 establishes a direct hydrogen bond to the glucose O2 from sucrose, and in agreement, there is a concomitant decreased binding affinity as revealed by its Km value. This position is equivalent to Lys-242 from the A. thaliana invertase that, together with Asp-239, is proposed to be a hot spot mediating sucrose binding. Furthermore, this position is structurally equivalent to the SoFfase Asn-254, which was reported to be involved in altering the hydrolase/transferase ratio but mostly in modulating the transfructosylating product specificity (24). According to this observation, preliminary analysis shows that the H343T mutant shows an altered profile of FOS production (data not shown). In contrast, the altered catalytic efficiency observed in the Q341N and N342S mutants might be explained in terms of a different mobility of the Glu-334–His-343 loop, as none of these two residues interact with sucrose. As commented previously, mobility must be a feature of this loop, as it appears highly disordered in the free enzyme being clearly defined upon ligand binding. Consequently, changes at this loop may modify the substrate access to the active site thus altering catalysis. However, the marked impaired activity of the Q341N and N342S mutants on nystose, which is not apparent on raffinose, is consistent with that observed in the complexes and clearly supports the role of these residues and loop TIV in shaping subsites +2 and +3 only for inulin-type substrates.
Finally, a truncated enzyme lacking the last residues comprising the 660GLFAGY sequence yields a fully inactive enzyme (Table 3). This segment, which is an extended arm following the small helix at the C terminus, surrounds the active site cavity and harbors Gly-660 and Leu-661 shaping subsite +2. Removal of this region probably produces a distortion in the active site architecture of XdINV that is detrimental for activity or even for proper folding of the dimer, as this truncated enzyme is not detected by PAGE (data not shown).
Structural Basis of the Neo/Hetero-FOS Production of XdINV
As commented on previously, 1-kestose and neokestose are the main transglycosylation products obtained with XdINV from sucrose. The experimental complexes here presented show the binding mode of product 1-kestose in the binding site, from which we can infer how the acceptor sucrose would be positioned in subsites +1 and +2 of an enzyme-fructose intermediate complex for the subsequent transfer of this sugar to form a β(2–1)-linkage. Thus, the terminal glucose of acceptor sucrose would be mostly oriented through polar interactions to loop TIV, as shown in the schematic representation of the transfructosylation mechanism given in Fig. 8. In contrast, the complex obtained by soaking into neokestose shows this sugar in a donor substrate mode, i.e. its Fru-β(2–1)-Glu moiety is occupying the −1/+1 subsites. Instead, the XdINV-neoerlose complex represents the binding mode of a true neo-type product, with its Fru-β(2–6)-Glu moiety spanning the −1/+1 subsites, and therefore the binding of acceptor sucrose to produce neokestose can be extrapolated from this complex (Fig. 8A). Thus, although there are also polar interactions stabilizing the hydroxyls, subsite +2 would be mostly determined by stacking of the sugar ring to Trp-105. Interestingly, this trait explains why XdINV is able to fructosylate α-linked glucosides, including maltose, isomaltulose, isomaltose, trehalose, and higher oligosaccharides as maltotriose or maltotetraose, as reported previously (10). In fact, many of these α-glucosides are better acceptors than sucrose, which might be explicated in terms of a more efficient hydrophobic interaction to Trp-105 at subsite +2. In turn, this feature explains why oligosaccharides such as lactose and lactulose did not act as fructose acceptors due to their β(1–4) linkage, which is unable to accommodate properly through staking to Trp-105.
FIGURE 8.

Proposed mechanism of XdINV transfructosylating activity. A, view of the XdINV-neoerlose complex, showing the ligand in cyan and the main interactions of its Glu-α(1–4)Glu moiety located at subsites +1 and +2. The position of this portion represents a potential binding mode of acceptor maltose for the formation of a β(2–6) linkage of glucose to fructose. The position of a putative sucrose acceptor molecule (white) has been inferred by superposition of its glucose to the glucose found at subsite +1 of neoerlose and adjusting manually the fructose unit by small torsion of its glycosidic bond, to best packing to Trp-105 side chain. B, scheme of the proposed mechanism. The donor-substrate sucrose is hydrolyzed by nucleophilic attack forming the fructosyl-XdINV intermediate; a subsequent sucrose molecule (the acceptor substrate) can enter the active site pocket with binding mode 1 generating 1-kestose or, alternatively, with binding mode 2 and generating neokestose.
The two binding sites described here for acceptor sucrose to form inulin or neo-type products (Fig. 8B) are supported by the observed XdINV-Fru-Hepes and XdINV-Fru-Bistris propane ternary complexes, as shown in Fig. 6. Thus, the Hepes molecule is located similarly to the nystose and mimics a putative donor substrate for a β(2–1) transfer to fructose. In contrast, the large and flexible Bistris propane molecule seems apparently able to span both subsites, but in any case, both buffer molecules have a hydroxyl oxygen pointing to the fructose O6 atom, located 3.6 Å away, and thus apparently available to create a linkage. This is also consistent with the altered FOS profile observed in versions of XdINV mutated at loop TIV as commented on previously. Further studies are underway to fully characterize the broad synthetic specificity of this enzyme and to investigate new potential acceptors yielding novel bioconjugates of biomedical interest.
Discussion
Fructans, the fructose-rich polymers biosynthetically derivedfrom sucrose, are important non-structural storage compounds in many bacteria, fungi, and numerous plant species. They are associated with essential biological processes as sucrose metabolism and vegetative development. Also, fructans are biotechnologically relevant compounds due to its prebiotic effect. Consequently, the enzymes involved in fructans and sucrose processing are subject to extensive research with the aim to uncover their biological role and also to improve their enzymatic efficiency.
In this study, we described the structure of β-fructofuranosidase from X. dendrorhous that exhibits novel features regulating activity. The crystal structure shows a bimodular arrangement of its polypeptide chain similar to that described previously for other GH32 enzymes. However, a long 30-residue extension at its C terminus, together with a glycan chain attached to a non-conserved Asn, mediates the formation of an unusual dimer. This dimer shows a peculiar topology that locates both active sites in the same face of the protein and apparently connected by a long crevice. A blast search shows about 60 entries showing 30–60% similarity with XdINV sequence. These sequences include hydrolases or transferases mostly from Basidiomycota but also from Ascomycota (data not shown). Alignment of the corresponding sequences reveals that they all display both determinants that are shown to be key determinants for the oligomerization state in XdINV, i.e. the C-terminal extension and the N-glycosylation sequon at a position equivalent to Asn-107, and consequently, the dimer described here would be a model for this group of GH32 enzymes.
The unusual topology of the XdINV dimer suggests the possibly that it might be able to accommodate a putative polymeric substrate as it could be a mixed fructan with branched β(2–1)/β(2–6) linkages (42, 43) that, indeed, is degraded by XdINV. Fructans present in plants are a mixture of heterogeneous oligomers and polymers with a composition depending on the environment and the development state of the plant (38). Key enzymes for plant fructan metabolism include a complex set of biosynthetic and hydrolytic enzymes, including different types of fructan exohydrolases and fructosyltransferases. Consequently, fungi isolated from plant environments may have evolved to interact or degrade these molecules, by expressing their own enzymes. Nevertheless, the possibility of the yeast producing these compounds cannot be discarded, as the biological function of microbial fructans has also been related to the creation of a physical barrier, by enhancing resistance to environmental stresses and assisting in nutrient assimilation (Ref. 43 and references therein). In fact, fructans have proved to act as virulence factors in colonization and to play a critical factor in the pathogenesis of disease-related microorganisms. Many studies have shown that bacteria that synthesize fructans interact with eukaryotic hosts and can be found in symbiotic association with plants. Although the reported studies have been performed in bacteria, it is conceivable that fungi develop a similar mechanism to adapt to the environment. In this sense, it should be noted that the yeast X. dendrorhous has been isolated from soil samples collected at different cold ecosystems. The exact physiological role of XdINV and the target substrate at its natural habitat are difficult to envisage, and therefore, it is hard to derive precise structure-function relationships, but clearly, the peculiar topology of its molecular surface is a remarkable and distinctive trait within GH32 enzymes.
The crystallographic analysis of the XdINV complexes described here gives a detailed picture of the main features of its active site. Previous studies showed that most GH32 enzymes present a shallow pocket with residues shaping a conserved fructose-binding pocket, at subsite −1, and several hydrophobic and polar interactions defining subsite +1. In contrast, the substrate is loosely bound at subsite +2 or dangling into the solvent. In agreement with this observation, short substrates such as 1-kestose or raffinose are accommodated in essentially the same conformation in all the formerly described complexes (13, 16, 18, 19). In contrast, XdINV presents a deep cavity showing multiple binding sites, a feature that has been previously described for two other fungal enzymes, the AjFT and the SoFfase. The reported complexes of these enzymes with long substrates reveal that the active sites diverge from subsite +2, and thus, nystose bound at the AjFT catalytic pocket locates its terminal glucose at a position that is occupied by Trp-105 in XdINV (see Fig. 7A) (16). A very short TI loop and the absence of the C-terminal segment make a very different active site pocket in AjFT, which is remarkable taking into account that this enzyme is phylogenetically close to XdINV. Likewise, Fru-nystose bound at the SoFfase accommodates in a precise conformation that mimics the polymeric substrate inulin (24). As commented previously, both yeast enzymes are distinctive within GH32 in being dimers, and both active sites must be considered in terms of their oligomeric state. In the case of SoFfase, the topology of the active site seemed designed to optimally fit the conformation of the polymer inulin, which is consistent with its high inulinase activity. Thus, the topology of sites +2 and further from the cleaved linkage must reflect the profile of its natural substrate and consequently the physiological role of each enzyme in its environment. Remarkably, XdINV presents both inulin- and levan-type binding sites and an open pocket filled with many water molecules that confer plasticity to the enzyme. Therefore, this plasticity might be consistent with a more complex substrate as are the mixed fructans commented on previously, in contrast to the rigid active site of SoFfase adapted to accommodate the more regular inulin.
Carbohydrate-processing enzymes have developed intricate molecular mechanisms to cope with their highly heterogeneous substrates. Understanding these mechanisms will be of general interest to uncover essential biological processes at the molecular level. In the case of XdINV, an insertion at the C-terminal polypeptide and glycosylation have been shown to be essential in oligomerization and thus in shaping a peculiar active site. In turn, the plasticity of its active site makes the enzyme a valuable and flexible biocatalyst to produce novel compounds with biotechnological relevance. The potential of XdINV to produce novel bioconjugates is underway.
Author Contributions
J. S. A. and M. F. L. conceived and coordinated the study. D. L. performed the production of the native protein. M. G. P., Z. M., and M. F. L. conducted molecular biology assays and kinetics measurements and analyzed the results. M. R. E., B. G., and J. S. A. designed and performed the crystallographic work and interpreted the results. J. S. A. wrote the paper, and all authors read and commented on the manuscript.
Acknowledgments
We thank the staff of the European Synchrotron Radiation Facility (ESRF), Grenoble, France, for providing access and for technical assistance at beamline ID23.1. We also thank the German Electron Synchrotron (Desy) at Hamburg, Germany, for assistance at Petra III beamline, and the Synchrotron Radiation Source at Alba (Barcelona, Spain) for assistance at BL13-XALOC beamline.
This work was supported in part by Spanish Ministry of Economy and Competitiveness Grants BIO2010-20508-C04-03/-04 and BIO2013-48779-C4-3/-4, BioStruct-X Program from the European Community's Seventh Framework Programme Grant FP7/2007-2013, and a Fundación Ramón Areces institutional grant (to the Centro de Biología Molecular “Severo Ochoa”). The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (codes 5ANN, 5FIX, 5FKB, 5FMD, 5FK7, 5FK8, 5FKC, 5FMB, and 5FMC) have been deposited in the Protein Data Bank (http://wwpdb.org/).
The nucleotide sequence(s) reported in this paper has been submitted to the GenBankTM/EBI Data Bank with accession number(s) FJ539193.2
- XdINV
- X. dendrorhous invertase
- Bistris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane
- FOS
- fructooligosaccharides
- GlcNAc
- N-acetylglucosamine
- AaEI
- A. awamorii exo-inulinase
- AjFT
- A. japonicus fructosyltransferase
- GH
- glycosyl hydrolase
- ScINV
- S. cerevisiae invertase
- SoFfase
- S. occidentalis β-fructofuranosidase
- Endo H
- endoglycosidase H.
References
- 1. Linde D., Macias I., Fernández-Arrojo L., Plou F. J., Jiménez A., and Fernández-Lobato M. (2009) Molecular and biochemical characterization of a β-fructofuranosidase from Xanthophyllomyces dendrorhous. Appl. Environ. Microbiol. 75, 1065–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Linde D., Rodríguez-Colinas B., Estévez M., Poveda A., Plou F. J., and Fernández Lobato M. (2012) Analysis of neofructooligosaccharide production mediated by the extracellular β-fructofuranosidase from Xanthophyllomyces dendrorhous. Bioresour. Technol. 109, 123–130 [DOI] [PubMed] [Google Scholar]
- 3. Tian F., and Karboune S. (2012) Enzymatic synthesis of fructooligosaccharides by levansucrase from Bacillus amyloliquefaciens: specificity, kinetics, and product characterization. J. Mol. Catal. B-Enzym. 82, 71–79 [Google Scholar]
- 4. Rodriguez-Alegria M. E., Enciso-Rodriguez A., Ortiz-Soto M. E., Cassani J., Olvera C., and Munguia A. L. (2010) Fructooligosaccharide production by a truncated Leuconostoc citreum inulosucrase mutant. Biocatal. Biotransfor. 28, 51–59 [Google Scholar]
- 5. Alvaro-Benito M., de Abreu M., Fernández-Arrojo L., Plou F. J., Jiménez-Barbero J., Ballesteros A., Polaina J., and Fernández-Lobato M. (2007) Characterization of a β-fructofuranosidase from Schwanniomyces occidentalis with transfructosylating activity yielding the prebiotic 6-kestose. J. Biotechnol. 132, 75–81 [DOI] [PubMed] [Google Scholar]
- 6. Franco-Robles E., and Lopez M. G. (2015) Implication of fructans in health: immunomodulatory and antioxidant mechanisms. Scientific World J. 10.1155/2015/289267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Marx S. P., Winkler S., and Hartmeier W. (2000) Metabolization of β-(2,6)-linked fructose-oligosaccharides by different bifidobacteria. FEMS Microbiol. Lett. 182, 163–169 [DOI] [PubMed] [Google Scholar]
- 8. Kilian S., Kritzinger S., Rycroft C., Gibson G., and du Preez J. (2002) The effects of the novel bifidogenic trisaccharide, neokestose, on the human colonic microbiota. World J. Microb. Biotechnol. 18, 637–644 [Google Scholar]
- 9. Lim J. S., Lee J. H., Kang S. W., Park S. W., and Kim S. W. (2007) Studies on production and physical properties of neo-FOS produced by co-immobilized Penicillium citrinum and neo-fructosyltransferase. Eur. Food Res. Technol. 225, 457–462 [Google Scholar]
- 10. Gimeno-Perez M., Santos-Moriano P., Fernandez-Arrojo L., Poveda A., Jimenez-Barbero J., Ballesteros A. O., Fernandez-Lobato M., and Plou F. J. (2014) Regioselective synthesis of neo-erlose by the β-fructofuranosidase from Xanthophyllomyces dendrorhous. Process Biochemistry 49, 423–429 [Google Scholar]
- 11. Henrissat B., Sulzenbacher G., and Bourne Y. (2008) Glycosyltransferases, glycoside hydrolases: surprise, surprise! Curr. Opin. Struct. Biol. 18, 527–533 [DOI] [PubMed] [Google Scholar]
- 12. Pons T., Naumoff D. G., Martínez-Fleites C., and Hernández L. (2004) Three acidic residues are at the active site of a β-propeller architecture in glycoside hydrolase families 32, 43, 62, and 68. Proteins 54, 424–432 [DOI] [PubMed] [Google Scholar]
- 13. Alberto F., Jordi E., Henrissat B., and Czjzek M. (2006) Crystal structure of inactivated Thermotoga maritima invertase in complex with the trisaccharide substrate raffinose. Biochem. J. 395, 457–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bujacz A., Jedrzejczak-Krzepkowska M., Bielecki S., Redzynia I., and Bujacz G. (2011) Crystal structures of the apo-form of β-fructofuranosidase from Bifidobacterium longum and its complex with fructose. FEBS J. 278, 1728–1744 [DOI] [PubMed] [Google Scholar]
- 15. Nagem R. A., Rojas A. L., Golubev A. M., Korneeva O. S., Eneyskaya E. V., Kulminskaya A. A., Neustroev K. N., and Polikarpov I. (2004) Crystal structure of exo-inulinase from Aspergillus awamori: the enzyme fold and structural determinants of substrate recognition. J. Mol. Biol. 344, 471–480 [DOI] [PubMed] [Google Scholar]
- 16. Chuankhayan P., Hsieh C. Y., Huang Y. C., Hsieh Y. Y., Guan H. H., Hsieh Y. C., Tien Y. C., Chen C. D., Chiang C. M., and Chen C. J. (2010) Crystal structures of Aspergillus japonicus fructosyltransferase complex with donor/acceptor substrates reveal complete subsites in the active site for catalysis. J. Biol. Chem. 285, 23251–23264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pouyez J., Mayard A., Vandamme A. M., Roussel G., Perpète E. A., Wouters J., Housen I., and Michaux C. (2012) First crystal structure of an endo-inulinase, INU2, from Aspergillus ficuum: discovery of an extra-pocket in the catalytic domain responsible for its endo-activity. Biochimie 94, 2423–2430 [DOI] [PubMed] [Google Scholar]
- 18. Verhaest M., Lammens W., Le Roy K., De Ranter C. J., Van Laere A., Rabijns A., and Van den Ende W. (2007) Insights into the fine architecture of the active site of chicory fructan 1-exohydrolase: 1-kestose as substrate vs sucrose as inhibitor. New Phytol. 174, 90–100 [DOI] [PubMed] [Google Scholar]
- 19. Lammens W., Le Roy K., Yuan S., Vergauwen R., Rabijns A., Van Laere A., Strelkov S. V., and Van den Ende W. (2012) Crystal structure of 6-SST/6-SFT from Pachysandra terminalis, a plant fructan biosynthesizing enzyme in complex with its acceptor substrate 6-kestose. Plant J. 70, 205–219 [DOI] [PubMed] [Google Scholar]
- 20. Verhaest M., Lammens W., Le Roy K., De Coninck B., De Ranter C. J., Van Laere A., Van den Ende W., and Rabijns A. (2006) X-ray diffraction structure of a cell-wall invertase from Arabidopsis thaliana. Acta Crystallogr. D Biol. Crystallogr. 62, 1555–1563 [DOI] [PubMed] [Google Scholar]
- 21. Alvaro-Benito M., Polo A., González B., Fernández-Lobato M., and Sanz-Aparicio J. (2010) Structural and kinetic analysis of Schwanniomyces occidentalis invertase reveals a new oligomerization pattern and the role of its supplementary domain in substrate binding. J. Biol. Chem. 285, 13930–13941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sainz-Polo M. A., Ramírez-Escudero M., Lafraya A., González B., Marín-Navarro J., Polaina J., and Sanz-Aparicio J. (2013) Three-dimensional structure of Saccharomyces invertase: role of a non-catalytic domain in oligomerization and substrate specificity. J. Biol. Chem. 288, 9755–9766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Trollope K. M., van Wyk N., Kotjomela M. A., and Volschenk H. (2015) Sequence and structure-based prediction of fructosyltransferase activity for functional subclassification of fungal GH32 enzymes. FEBS J. 282, 4782–4796 [DOI] [PubMed] [Google Scholar]
- 24. Álvaro-Benito M., Sainz-Polo M. A., González-Pérez D., González B., Plou F. J., Fernández-Lobato M., and Sanz-Aparicio J. (2012) Structural and kinetic insights reveal that the amino acid pair Gln-228/Asn-254 modulates the transfructosylating specificity of Schwanniomyces occidentalis β-fructofuranosidase, an enzyme that produces prebiotics. J. Biol. Chem. 287, 19674–19686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gimeno-Pérez M., Linde D., Fernández-Arrojo L., Plou F. J., and Fernández-Lobato M. (2015) Heterologous overproduction of β-fructofuranosidase from yeast Xanthophyllomyces dendrorhous, an enzyme producing prebiotic sugars. Appl. Microbiol. Biotechnol. 99, 3459–3467 [DOI] [PubMed] [Google Scholar]
- 26. Polo A., Linde D., Estévez M., Fernández-Lobato M., and Sanz-Aparicio J. (2010) Crystallization and preliminary x-ray diffraction analysis of the fructofuranosidase from Xanthophyllomyces dendrorhous. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 66, 1441–1444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Battye T. G., Kontogiannis L., Johnson O., Powell H. R., and Leslie A. G. (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67, 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vagin A., and Teplyakov A. (1997) MOLREP: an automated program for molecular replacement. J. Appl. Cryst. 30, 1022–1025 [Google Scholar]
- 31. Stein N. (2008) CHAINSAW: a program for mutating pdb files used as templates in molecular replacement. J. Appl. Crystallogr. 41, 641–643 [Google Scholar]
- 32. Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 33. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 34. Kirschner K. N., Yongye A. B., Tschampel S. M., González-Outeiriño J., Daniels C. R., Foley B. L., and Woods R. J. (2008) GLYCAM06: a generalizable biomolecular force field. Carbohydrates. J. Comput. Chem. 29, 622–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schüttelkopf A. W., and van Aalten D. M. (2004) PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 60, 1355–1363 [DOI] [PubMed] [Google Scholar]
- 36. Laskowski R. A., MacArthur M. W., Moss D. S., and Thornton J. M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26, 283–291 [Google Scholar]
- 37. DeLano W. L. (2002) The PyMOL Molecular Graphics System, version 1.6.0.0, DeLano Scientific, San Carlos, CA [Google Scholar]
- 38. Ritsema T., and Smeekens S. (2003) Fructans: beneficial for plants and humans. Curr. Opin. Plant Biol. 6, 223–230 [DOI] [PubMed] [Google Scholar]
- 39. Lopez M. G., Mancilla-Margalli N. A., and Mendoza-Diaz G. (2003) Molecular structures of fructans from Agave tequilana Weber var. azul. J Agric. Food Chem. 51, 7835–7840 [DOI] [PubMed] [Google Scholar]
- 40. Lammens W., Le Roy K., Van Laere A., Rabijns A., and Van den Ende W. (2008) Crystal structures of Arabidopsis thaliana cell-wall invertase mutants in complex with sucrose. J. Mol. Biol. 377, 378–385 [DOI] [PubMed] [Google Scholar]
- 41. Le Roy K., Lammens W., Verhaest M., De Coninck B., Rabijns A., Van Laere A., and Van den Ende W. (2007) Unraveling the difference between invertases and fructan exohydrolases: a single amino acid (Asp-239) substitution transforms Arabidopsis cell wall invertase1 into a fructan 1-exohydrolase. Plant Physiol. 145, 616–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cimini S., Locato V., Vergauwen R., Paradiso A., Cecchini C., Vandenpoel L., Verspreet J., Courtin C. M., D'Egidio M. G., Van den Ende W., and De Gara L. (2015) Fructan biosynthesis and degradation as part of plant metabolism controlling sugar fluxes during durum wheat kernel maturation. Front. Plant Sci. 6, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Velázquez-Hernández M. L., Baizabal-Aguirre V. M., Bravo-Patiño A., Cajero-Juárez M., Chávez-Moctezuma M. P., and Valdez-Alarcón J. J. (2009) Microbial fructosyltransferases and the role of fructans. J. Appl. Microbiol. 106, 1763–1778 [DOI] [PubMed] [Google Scholar]
- 44. Holm L., and Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Robert X., and Gouet P. (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–324 [DOI] [PMC free article] [PubMed] [Google Scholar]