

Abstract
The activity of Rac in leukocytes is essential for immunity. However, its role in NK cell-mediated anti-microbial signaling remains unclear. In this study, we investigated the role of Rac in NK cell mediated anti-cryptococcal killing. We found that Cryptococcus neoformans independently activates both Rac and SFK pathways in NK cells, and unlike in tumor killing, Cryptococcus initiated a novel Rac → PI3K → Erk cytotoxicity cascade. Remarkably, Rac was not required for conjugate formation, despite its essential role in NK cytotoxicity against C. neoformans. Taken together, our data show that, unlike observations with tumor cells, NK cells use a novel Rac cytotoxicity pathway in conjunction with SFK, to kill C. neoformans.
Keywords: adhesion, fungi, natural killer cells (NK cells), phosphatidylinositide 3-kinase (PI 3-kinase), Rac (Rac GTPase), Src, Cellular signaling, Cryptococcus
Introduction
NK3 cells are a subset of lymphocytes that have well characterized anti-tumor and anti-viral activity. However, it is now apparent that NK cells also have the capacity to kill microbial targets, which contribute to host defense, and differ in important ways from tumor killing (1). Receptor-mediated NK anti-tumor signaling depends on sequential activation of PI3K → Rac → Erk (2); however, because the receptors required for cryptococcal killing differ from those required for tumor killing (3), we predicted that anti-microbial signaling would also differ. Our previous studies have shown that a PI3K → Erk signaling pathway was essential for cryptococcal killing (4). Because other laboratories showed that PI3K can activate Erk via either Rac or phospholipase Cγ (PLCγ), we sought to examine the pathway used in NK cell-mediated cryptococcal killing by investigating both PI3K → Rac → Erk and PI3K → PLCγ → Erk pathways (5–9).
Rac is a subfamily of Ras homology (Rho) GTPases consisting of Rac1, Rac2, Rac3, and RhoG. They are activated by guanine nucleotide exchange factors by exchanging GDP for GTP (reviewed in Ref. 10). Activation of Rac in NK cells is crucial for conjugate formation between NK and tumor targets (11, 12). During tumor killing, Rac-GTP regulates actin remodeling by activating downstream effectors such as WAVE (WASP family verprolin-homologous protein), SRA1 (specifically RAC1-associated protein 1), and p21-activated kinases (PAKs) (13–16). In addition to its important role in cytoskeletal modulation, Rac plays a role in the activation of Erk and NK cell-mediated tumor cytotoxicity (9). Against Raji tumor targets, the human NK cell line, NK92 and primary NK cells required Rac to initiate a PAK1 →Mek1/2 → Erk1/2 signaling cascade that allows for cytotoxic granule mobilization and release (2). Rac activation was found to be dependent on PI3K, because NK92 cells transfected with a constitutively active Rac were able to continue killing tumor targets in the presence of PI3K inhibitors, whereas control cells were not (2).
In addition to Rac, PLCγ can also become activated by PI3K and lead to Erk activation. PLCγ is best known for the production of diacyl glycerol and inositol 3-phosphate, which activates downstream kinases and regulates intracellular calcium (reviewed in Ref. 17). In certain cells, such as platelets, PLCγ activity is dependent on PI3K (8). PLCγ activity is also required for the activation of cytokine-mediated Erk signaling (5). Similar to Rac, PLCγ is also essential in NK anti-tumor cytotoxicity (7). However, it is not clear whether Rac and PLCγ participate in NK cell microbial killing and where they are positioned in the signaling cascade.
The microbial target, Cryptococcus neoformans is a globally endemic fungal pathogen that affects HIV-infected patients, causing ∼1 million meningitis cases annually; 600,000 of these cases result in death within 3 months of presentation to the health care system (18). Protection against cryptococcal infection is provided by a combination of leukocytes, including macrophages, dendritic cells, T and B cells, granulocytes, and NK cells (reviewed in Ref. 19). The importance of NK cell activity against Cryptococcus was highlighted in murine studies, where mice with defective or depleted NK cells were susceptible to cryptococcal infection (20–22). It is known that human or mouse NK cells or human NK cell lines killed Cryptococcus (1, 21, 23, 24). Inhibition of cytotoxic effector molecules, such as perforin, in NK cells led to a reduction in anti-cryptococcal activity (1). These studies outline the important role of NK cells in protecting against cryptococcal infection and highlight the need to study NK cell anti-cryptococcal signaling to lay the groundwork for therapies to restore defective NK function in HIV patients (24).
Tumor and viral ligands activated NK cell signaling pathways with multiple points of convergence and divergence (25, 26). For example, NKG2D (natural killer group 2 member D) and 2B4 are two NK cell receptors that initiated different signaling pathways: YINM or immunoreceptor tyrosine-based switch motif signaling, respectively (25). Although two different pathways were initiated, both pathways converged into a Vav1 → PLCγ pathway that led to degranulation (25). In cryptococcal killing, the SFK → PI3K → Erk cytotoxicity pathway has been identified (4, 27). We considered the possibility that multiple anti-cryptococcal signaling pathways converge on to this central pathway. Because Rac and PLCγ are activated by PI3K and led to Erk signaling, convergence of Rac and PLCγ could be required for NK cryptococcal killing (5, 9, 17). Additionally, Rac activated PI3K in epithelial cells (28). This raises the possibility that Rac and SFK signaling converge to activate PI3K. By studying Rac and PLCγ, this study aims to elucidate the interconnections between the pathways that are activated by Cryptococcus.
To examine signaling in NK cell-mediated cryptococcal killing, we used the human NK cell line, YT (29), and primary blood-derived NK cells isolated from healthy adults. Rac activation was assessed by precipitation with the p21 binding domain (PBD) of PAK. The requirement for Rac in the anticryptococcal activity of NK cells was assessed using two independent pharmacologic inhibitors and siRNA knockdown and examining cryptococcal cfu. A flow cytometric assay was used to assess conjugate formation between NK cells and Cryptococcus. To determine the proximal and distal signaling elements, the interplay between Rac, SFK, PI3K, and Erk1/2 was assessed using loss of function approaches and performing immunoblots for activation of each element. We found that NK cell mediated anti-cryptococcal cytotoxicity required both Rac- and SFK-mediated activation of PI3K → Erk signaling. Our results suggest a novel role for Rac as an upstream regulator of PI3K activity in NK cells.
Experimental Procedures
Chemicals
FITC was purchased from Sigma-Aldrich (catalog no. 3326-32-7), EHT 1864 was purchased from Tocris (Bristol, UK; catalog no. 3872). Rac inhibitor II was purchased from Millipore (Etobicoke, Canada; catalog no. 553511). Ly294002 was purchased from Calbiochem (Etobicoke, Canada; catalog no. 440202). Dasatinib was from New England BioLabs (Whitby, Canada; catalog no. 9052s). DMSO was obtained from Sigma-Aldrich (catalog no. 472301). PP2 was purchased from Enzo Life Sciences (Farmingdale, NY; catalog no. BML-El297-0001). PP3 was purchased from Calbiochem (catalog no. 529574). Methyl β-cyclodextran (MBCD) was obtained from Sigma-Aldrich (catalog no. C4555). Carboxyfluorescein succinimidyl ester was obtained from Millipore (catalog no. 4500-0270). Trypan blue stain 0.4% was purchased from Invitrogen (catalog no. 15250).
Antibodies
Protein bands in immunoblots were revealed with specific antibodies: rabbit polyclonal anti-phospho-AKT1/2/3 (serine 473) (Santa Cruz, Dallas, TX; catalog no. sc79885R), mouse anti-Akt2 (Santa Cruz; catalog no. sc-81436), mouse anti-phosphotyrosine (Millipore; catalog no. 05-321), rabbit anti-phospho-Src family kinase (Y416) (Cell Signaling, Whitby, Canada; catalog no. 2101S), rabbit anti-phospho-Erk1/2 (T202/Y204) (Cell Signaling; catalog no. 9101S), mouse anti-Erk1/2 (Cell Signaling; catalog no. 9107S), mouse anti-Fyn (BD Transduction Laboratories, San Jose, CA; catalog no. 610163), mouse anti-Rac1 (Thermo Scientific, Waltham, MA; catalog no. 1862341), goat anti-rabbit IgG infrared dye 700DX (Rockland, Limerick, PA; catalog no. 611-130-002), and goat anti-mouse IgG infrared dye 800 (Licor, Lincoln, NE; catalog no. 923-32210). Cells for flow cytometry were labeled with specific antibody for mouse anti-CD11a PE-Cy5 (BD Biosciences; catalog no. 551131).
Cells and Cryptococcus
The human NK-like leukemia cell line YT (gift from C. Clayberger, Emeritus Faculty, Stanford University, Stanford, CA) was used to model human NK cells. YT cells were validated by its expression of NKp30 and NKp44 and lack of CD3. YT cells were maintained in complete medium containing RPMI 1640 supplemented with 10% fetal calf serum (Invitrogen), 1% nonessential amino acids (Invitrogen; catalog no. 11140), 1% sodium pyruvate (Invitrogen; catalog no. 11360), and 1% penicillin-streptomycin, in a 37 °C 5% CO2 incubator. Primary NK cells were isolated from healthy donors using a NK isolation kit (Miltenyi Biotec, San Diego, CA; catalog no. 130–092-657) as per the manufacturer's instructions. Isolated cells were routinely >92% CD56+, <0.5% CD3+. C. neoformans strain B3501 (ATCC, Manassas, VA; catalog no. 34873) and strain 145 (ATCC; catalog no. 62070) were grown to log phase in Sabouraud dextrose broth (Becton Dickinson; catalog no. 238230) on a 32 °C shaker overnight.
Immunoblotting
YT cells (3 × 105 to 3 × 106) were preincubated with varying inhibitors for 1 h in 37 °C CO2 incubator. YT cells were then co-incubated with C. neoformans strain B3501 at and an effector to target (E:T) ratio of 100:1 for varying time points in a 37 °C water bath. The cells were lysed in Nonidet P-40 lysis buffer containing 50 mm Tris, pH 7.4, 250 mm NaCl, 5 mm EDTA, 50 mm NaF, 1 mm Na3VO4, 1% Nonidet P-40, and 0.02% NaN3. Lysis buffer was supplemented with phosphatase (Roche) and protease inhibitors (Roche). Lysates were separated on a 4–12% Bis-Tris NuPAGE gels (Invitrogen; catalog no. NP0335BOX). After separation, the samples were transferred to a nitrocellulose membrane and revealed with indicated antibodies. Bands were recorded using ODYSSEY infrared imaging system (Licor). Densitometry was calculated by measuring the area under the intensity plot, using ImageJ (National Institute of Health; version 1.48). Fold increase in signaling compared with unstimulated was calculated as (intensity of stimulated condition normalized to loading control)/(intensity of unstimulated condition normalized to loading control) − 1.
NK Anti-cryptococcal Killing Assay
C. neoformans strain B3501 and strain 145 were grown to log phase overnight in Sabouraud dextrose broth on an orbital shaker at 32 °C. YT cells were co-cultured with the indicated strain of C. neoformans at an E:T ratio of 150:1 in round bottom 96-well plates (Thermo Scientific; catalog no. 163320). cfu were determined at 24 and 48 h postinoculation. The anti-cryptococcal activity of primary NK cells were determined by co-culture with C. neoformans at an E:T ratio of 1000:1 in round bottom 96-well plates. cfu were determined 24 h postinoculation. In experiments where EHT 1864, Rac inhibitor II, or MBCD were used, the inhibitors were added to the YT or primary NK cells at the same time that Cryptococcus was added. In addition, an equivalent volume of sterile H2O was added to control wells to control for the highest levels of EHT1864 used, an equivalent concentration of DMSO was added to control for the highest levels of Rac inhibitor II used, and PBS was added to control for MBCD. YT cells were preincubated with varying concentrations of U73122 for 1 h, which has been shown to block lytic granule convergence in a similar NK cell line (YTS) (30). YT cells were then washed with complete medium and incubated with Cryptococcus as described above. Primary NK cell and YT cell viability was determined by trypan blue staining. The percentage of viability was calculated as (number of trypan blue positive cells)/(total number of cells) × 100%. The concentrations of inhibitors used did not affect viability of YT and primary NK cells.
Conjugate Assay
C. neoformans strain B3501 was labeled following the procedure for Cryptococcus gattii as described (31). Briefly, C. neoformans was cultured overnight to the exponential phase of proliferation and labeled with 2.5 μg/ml of FITC per 108 cells at 22 °C for 10 min. C. neoformans was then washed three times with PBS. YT cells or primary NK cells were co-incubated with 5 μl of anti-CD11a PE-Cy5 antibody and 100 μm EHT1864 or vehicle control for 30 min in a 37 °C CO2 incubator. YT cells or primary NK cells and different amounts of Cryptococcus were incubated together for 10 min at 37 °C in 200 μl of complete medium. YT cells or primary NK cells were then agitated by pipetting. Conjugates were detected by Guava EasyCyte flow cytometer (Cytosoft version 5.3, Guava Technologies, Millipore, Danvers, MA), and the data were analyzed by FlowJo software (Tree Star, Ashland, OR). The percentage of NK cells in conjugates with C. neoformans were determined as follows: (number of green and red event)/(total number of red events) × 100%. For conjugate assays testing SFK, YT cells were preincubated with dasatinib for 60 min or PP2 or PP3, which served as the control for PP2 for 120 min in a 37 °C CO2 incubator. Other steps were performed the same as below.
Peripheral Blood Mononuclear Cell (PBMC) Proliferation
PBMC were isolated from healthy donors by Ficol-Paque gradient (GE Healthcare Life Sciences; catalog no. 17-1440-02). PBMC (2 × 106/ml) was labeled with carboxyfluorescein succinimidyl ester as per the manufacturer's instructions and stimulated with 5 μg/ml of phytohemeagglutinin (Sigma-Aldrich) in the presence of 1 μm, 2.5 μm U73122, or DMSO control equivalent to the highest amount of U73122 used. PBMC were allowed to proliferate for 5 days in 24-well plates in 37 °C CO2 incubator. The levels of carboxyfluorescein succinimidyl ester labeling were determined by Guava EasyCyte flow cytometer.
Rac-GTP Precipitation
YT cells were precultured with 10 μm EHT 1864 or 100 μm Rac inhibitor II or 50 μm Ly294002 or DMSO vehicle control for 1 h at 37 °C. YT cells were unstimulated or stimulated with C. neoformans for 4 min in a 37 °C water bath. YT cells centrifuged at 6000 × g for 30 s, and the supernatant was decanted. YT cells were then lysed and Cryptococcus was added to the unstimulated conditions to control for the additional volume of Cryptococcus in the stimulated conditions. Rac-GTP was then extracted according to the manufacturer's instructions with two modifications (Millipore; catalog no. 17-10394). Protease inhibitor (Roche) was substituted for leupeptin, and 5 μl of the PBD-conjugated magnetic beads of PAK was used. Aliquots of whole cell lysate were saved before the addition of PBD-coated beads, so that the total levels of Rac1 could be determined.
Rac1 siRNA Knockdown
Two different sequences of siRNA specific against Rac1 (UAAGGAGAUUGGUGCUGUA and AUGAAAGUGUCACGGGUAA), one of which bears no resemblance to Rac2, and one nontargeting control sequence were purchased from Dharmacon RNAi Technologies (Lafayette, CO). Transfection of each of the siRNA sequences was performed by resuspending 3 million YT cells in 100 μl of Nucleofector solution (kit V; Amaxa, Walkersville, MD) and adding 2 μg of the corresponding siRNA. The solution was then transferred to cuvettes, and nucleofection was performed using the O-017 program. A total of 500 μl of prewarmed complete medium was added, and the cells were transferred to 1.5-ml Eppendorf tubes and placed in a 37 °C incubator for 10 min. The cells were then transferred to a 6-well plate containing 5 ml of prewarmed complete medium.
Statistics
GraphPad Prism was used to evaluate statistics. Error bars represent the S.E. The data were analyzed by one-way analysis of variance with Bonferroni correction. p < 0.05 is considered to be a statistically significant different between conditions. The percentage of reduction in conjugates compared with H2O or DMSO was analyzed by the column statistics program in GraphPad Prism.
Ethics
Experimental protocols were approved and performed following the guidelines from the Conjoint Health Research Ethics Board of the University of Calgary (protocol number REB15-0600).
Results
Rac but Not PLCγ Is Required for NK Cell-mediated Cryptococcal Killing
NK cell tumor and cryptococcal killing both depend on a PI3K → Erk signaling pathway (4, 9). This suggests that the cytotoxicity pathway may be similar between tumor and cryptococcal killing. During tumor killing, PI3K was found to activate Rac, which then triggers Erk activity (9). Alternatively, PI3K has also been shown to activate PLCγ leading to Erk signaling (32, 33). To determine whether cryptococcal killing depends on Rac or PLCγ, we first investigated Rac activation in YT cells. We stimulated YT cells with C. neoformans at varying time points and found that cryptococcal stimulation activated Rac1 (Rac1-GTP) as assessed by binding to the PBD of PAK conjugated to magnetic beads (Fig. 1, A and B). Rac activation occurred by 5 min and returned to base line after 15 min. Thus, YT cells responding to cryptococcal stimulation activated Rac, suggesting that Rac may play a role in anti-cryptococcal signaling. We also investigated PLCγ by pretreating YT cells with the PLCγ inhibitor, U-73122, at concentrations found to inhibit tumor killing (34). The anti-cryptococcal activity was examined, and we found that the addition of YT cells significantly reduced the number of cryptococcal cfu, which is consistent with our previous observations of killing (1, 27, 35). Despite using concentrations of U-73122 in excess to inhibit PLCγ, there was no impact on YT anti-cryptococcal killing, suggesting that PLCγ was not involved in NK cell-mediated cryptococcal killing (Fig. 1C). This lack of inhibition was not due to inactivity of U-73122, because a lower concentration of the inhibitor was capable of blocking proliferation of PBMC (Fig. 1D).
FIGURE 1.

Rac is involved in NK anti-cry1ptococcal killing. A, YT cells were stimulated with Cryptococcus, and levels of Rac-GTP were measured by immunoblotting. B, densitometry of normalized Rac-GTP/total Rac of the mean of three experiments. C, YT cells were preincubated with U73122 or DMSO control and co-cultured with Cryptococcus overnight. The data are representative of three experiments. D, PBMC were labeled with carboxyfluorescein succinimidyl ester and stimulated with phytohemeagglutinin for 5 days in the presence of DMSO or U73122. A reduction in fluorescence intensity indicates proliferation. The data are representative of three experiments using PBMC from three donors. E, YT cells were preincubated with EHT 1864 or Rac inhibitor II and stimulated with C. neoformans. The levels of Rac-GTP were determined. The data are representative of two experiments. F and G, YT cells were co-incubated with Cryptococcus overnight in the presence or absence of EHT 1864 (F) or Rac (G) inhibitor II. F and G are representative of three and two independent experiments, respectively. H, primary NK cells were co-cultured with Cryptococcus in the presence of EHT 1864 or H2O control. I, levels of Rac1 in transfected YT cells were analyzed by immunoblotting. J, YT cells were transfected with nontargeting siRNA or two different sequences of Rac1 specific siRNA. Transfected cells were used in a killing assay with C. neoformans. The data were representative of three experiments. The data were all analyzed using one-way analysis of variance with Bonferroni correction. Cryptococcus strain B3501 was used in all experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001. Error bars represent S.E. C+YT, YT co-cultured with Cryptococcus; CA, Cryptococcus alone; YT, unstimulated YT cells.
Having determined that Rac is activated by Cryptococcus, we explored whether Rac is required for NK cytotoxicity by using two different inhibitors of Rac: EHT 1864 and Rac inhibitor II. EHT 1864 binds to Rac and facilitates the release of GTP, preventing the association of Rac with downstream effectors (36). By contrast, Rac inhibitor II prevents Rac activation by blocking the association of Rac with guanine nucleotide exchange factor (37). To confirm that the Rac inhibitors blocked Rac activation, YT cells were pretreated with both Rac inhibitors and stimulated with Cryptococcus. Immunoblotting confirmed that both inhibitors successfully reduced the levels of Rac-GTP (Fig. 1E). YT cells were co-incubated with two different strains of C. neoformans in the presence of varying concentrations of EHT1864, Rac inhibitor II, or DMSO vehicle control. We found that the addition of YT cells to C. neoformans cultures reduced the number of cfu for both strain B3501 (Fig. 1, F and G) and strain 145 (data not shown), demonstrating NK cell killing of Cryptococcus (1, 24, 27) and demonstrating that the vehicle control did not affect this activity. Treatment of YT cells with EHT1864 or Rac inhibitor II caused a dose-dependent increase in cfu compared with vehicle controls, indicating a loss of anti-cryptococcal activity (Fig. 1, F and G). Trypan blue viability staining showed that EHT 1864, Rac inhibitor II, and DMSO did not affect viability of YT cells. These results suggest that Rac was required for NK cell-mediated killing of Cryptococcus.
Studies were performed to determine whether Rac was required for primary NK cells to kill Cryptococcus. We isolated primary NK cells from healthy donors. Primary NK cells were rested overnight and co-cultured with C. neoformans strain B3501, in the presence of EHT1864 or control. We found that EHT 1864 inhibited the anti-cryptococcal killing of primary NK cells (Fig. 1H), suggesting that the importance of Rac extended to primary NK cells.
Despite using two different pharmacological inhibitors, we considered the possibility that a nonspecific pharmacologic effect accounted for the reduced anti-cryptococcal activity. To exclude this possibility, experiments were performed using Rac1 siRNA knockdown. Two different sequences of Rac1 siRNA were used to knockdown Rac1 expression in YT cells to control for off target effects. Immunoblotting revealed that both sequences of siRNA reduced Rac1 expression, whereas the nontargeting siRNA did not (Fig. 1I). We observed that YT cells, in which Rac1 was knocked down, lost cytotoxic potential against C. neoformans compared with mock or nontargeting siRNA transfected YT cells (Fig. 1J), confirming that NK anticryptococcal activity was Rac-dependent.
Conjugate Formation between Cryptococcus and NK Cells Is Rac-independent
Having demonstrated that Rac was required in cryptococcal killing, we hypothesized that Rac would affect conjugate formation, similar to its role against K562 targets (11). We tested conjugate formation between NK cells and Cryptococcus by labeling YT cells with anti-CD11a conjugated to PE-Cy5 (because CD11a has previously been shown to be uninvolved in conjugate formation or killing (3)) and Cryptococcus with FITC. This allowed conjugate formation to be analyzed by flow cytometry as double positive events, as previously described (24). We found that increasing the proportion of Cryptococcus increased the ratio of YT cells that formed conjugates (Fig. 2, A and B). Despite the observation that EHT 1864 abrogated anti-cryptococcal activity, when we inhibited Rac activity with EHT 1864, surprisingly, we found no difference in conjugate formation. As a positive control, we tested whether disruption of lipid rafts would prevent conjugate formation and found that inhibiting lipid raft formation with MBCD modestly but significantly reduced conjugate formation and inhibited NK cell killing of Cryptococcus (Fig. 2, C and D). These observations suggested that, unlike the requirement for lipid rafts, Rac was not involved in the formation of the conjugates between NK cells and Cryptococcus.
FIGURE 2.

Rac inhibition did not impact NK-cryptococcal conjugate formation. A and B, YT cells were labeled with anti-CD11a conjugated to PE-Cy5 in the presence of 10 μm EHT1864 or H2O control. C. neoformans was labeled with FITC. YT cells and Cryptococcus were then co-incubated together at varying effector to target (E:T) ratios. Conjugates were analyzed by flow cytometry. A, events that matched the profile of YT cells by forward and side scatter were analyzed for the presence of those that had bound FITC-labeled Cryptococcus. B, percentage of cells forming conjugates was calculated by (number of green and red events)/(total number of red events). % reduction in conjugate formation compared with H2O was calculated by (1 − (% conjugates in EHT 1864 treated)/(% conjugates in H2O)) * 100%. The data are representative of three experiments. n.s., no statistically significant difference compared with H2O. C, MBCD treatment of NK cells reduces conjugate formation at E:T ratio of 1:3. YT cells were labeled with anti-CD11a conjugated to PE-Cy5 and either EHT1864 or DMSO control. C. neoformans was labeled with FITC. YT cells and Cryptococcus were then co-incubated together at 1:3 E:T ratio. Conjugates were analyzed by flow cytometry. The bar graph depicts the mean of three separate experiments ± S.E. D, YT cells were co-incubated with Cryptococcus in the presence of MBCD or PBS. cfu was counted at 24 h. The data are representative of two experiments. *, p < 0.05; **, p < 0.01. C+YT, YT co-cultured with Cryptococcus.
Erk-mediated Anti-cryptococcal Activity Is Rac-dependent
Having demonstrated that Rac was required for NK cell killing but not for NK cell-cryptococcal conjugate formation, we examined its role in NK cell microbial cytotoxicity. Previous studies showed that Erk was essential in both NK cell-mediated tumor and microbial killing and that Rac was required to activate Erk in tumor killing (4, 9). Because Rac and Erk are important in cryptococcal killing, we examined whether Rac also activated Erk in response to Cryptococcus. We treated YT cells with EHT 1864 or Rac inhibitor II and then stimulated them with C. neoformans. Both Rac inhibitors reduced Cryptococcus-dependent Erk phosphorylation (Fig. 3, A and B). We also knocked down the expression of Rac1 in YT cells and found that the reduction of Rac1 expression reduced phosphorylation of Erk in response to cryptococcal stimulation (Fig. 3C). Both knockdown and inhibitor data suggest that Rac activity was necessary for Erk activation in response to Cryptococcus.
FIGURE 3.
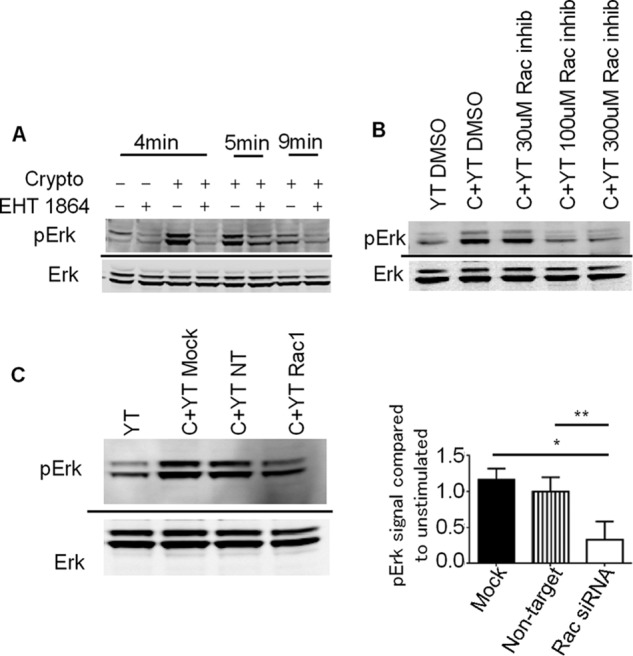
Rac is required for Erk activation. A, YT cells were preincubated with the Rac inhibitor EHT 1864 and then stimulated with C. neoformans for the indicated times. YT cells were then lysed and immunoblotted for pErk and Erk. The immunoblot is representative of two experiments. B, YT cells were preincubated with various concentrations of Rac inhibitor II. YT cells were then stimulated with Cryptococcus and lysed. The lysates were immunoblotted for pErk and Erk. The immunoblot is representative of two experiments. C, YT transfected with nontargeting or Rac1 specific siRNA. Transfected YT cells were stimulated with Cryptococcus, and levels of pErk and Erk were determined by immunoblots. The immunoblot is representative of three experiments. YT, unstimulated YT cells alone; C+YT, YT cells stimulated with Cryptococcus; NT, nontargeting
PI3K Anti-cryptococcal Activity Is Rac-dependent
Because Rac was required for activation of Erk in NK cells stimulated by C. neoformans and previous studies found that PI3K was also responsible for Erk activation (4), we sought to determine the relationship between Rac and PI3K. In NK cell-mediated tumor killing, PI3K activates Rac, which in turn activates Erk (9). Activation of Rac by PI3K is shared in numerous cell types including: endothelial cells, T cells, and neutrophils (reviewed in Ref. 38). However, other studies raised the possibility of a different pathway where Rac may be proximal to PI3K, thus regulating PI3K activity (28). To investigate the interaction between PI3K and Rac, we pretreated YT cells with EHT 1864 or Rac inhibitor II and assessed Rac-α serine/threonine-protein kinase (Akt) phosphorylation, which is a commonly used as a surrogate for PI3K activity (4, 39) in response to stimulation with C. neoformans. Rac inhibition reduced Akt phosphorylation in response to Cryptococcus (Fig. 4, A and B). Additionally, YT cells transfected with Rac1 siRNA had reduced activation of Akt when stimulated by Cryptococcus (Fig. 4C). Together, these data indicate that unlike the signaling pathway in tumor cytotoxicity, Rac was proximal and required for PI3K activation.
FIGURE 4.

Rac activity is required for PI3K activation. A, YT cells were preincubated with EHT 1864 or control. YT cells were stimulated with C. neoformans for the indicated times. The levels of pAkt and Akt2 were determined by immunoblots. The immunoblot is representative of two experiments. B, YT cells were preincubated with Rac inhibitor II. YT cells were then stimulated with Cryptococcus, and levels of pAkt and Akt2 were determined by immunoblots. Densitometry is the mean of four experiments ± S.E. C, YT cells were transfected with Rac siRNA. Transfected YT cells were stimulated with Cryptococcus, and the levels of pAkt were determined by immunoblots. The immunoblot is representative of three experiments. Densitometry is the mean of three experiments ± S.E. D and E, YT cells were preincubated with 50 μm Ly294002 or DMSO control. D, levels of active Rac bound to GTP were measured by Rac-GTP pulldown assay and Rac1 immunoblotting. Aliquots of whole cell lysate were immunoblotted to determine the total amount of Rac1. The immunoblot is representative of three experiments. E, levels of phosphorylated Akt and Akt 2 were determined by immunoblots. The immunoblot is representative of two experiments. YT, unstimulated YT cells alone; C+YT, YT cells stimulated with Cryptococcus; ns, no significant difference. *, p < 0.05; **, p < 0.01.
Although Rac was required for activation of PI3K, we considered the possibility that PI3K and Rac operated in a self-amplifying loop in which PI3K also activated Rac, as occurs in the leading edge of migrating neutrophils (41). To test this possibility, PI3K activity was inhibited by pretreating YT cells with Ly294002. YT cells were then stimulated with C. neoformans for 5 min, and the level of active Rac was determined by precipitation with PAK PBD. We found that inhibition of PI3K activity did not affect Cryptococcus-induced Rac activation (Fig. 4D). To ensure that PI3K was adequately inhibited, we performed immunoblots to examine the activation of Akt in the presence of Ly294002. We found that Ly294002 significantly inhibited phosphorylation of Akt, suggesting that PI3K activity is sensitive to Ly294002 (Fig. 4E). This datum agrees with previous publications where Ly294002 also inhibited PI3K activity in YT cells (4). Therefore, our data indicate that Rac was proximal to PI3K and not in a self-amplifying loop.
Rac and SFK Activate Anti-cryptococcal Activity through Independent Signaling Pathways
Previous studies have established that SFK are required for the activation of the PI3K → Erk signaling cascades in NK cell-mediated cryptococcal killing (4, 27). Because inhibition of Rac prevented PI3K activity, we hypothesized that Rac would be distal to SFK but proximal to PI3K. Therefore, we examined the interactions between Rac and SFK. YT cells were pretreated with EHT1864 and then stimulated with C. neoformans. We performed immunoblots for phosphorylated SFK and found that Rac inhibition had no effect on SFK activation (Fig. 5A), but remarkably, SFK were not required to activate Rac in response to cryptococcal stimulation (Fig. 5B). These results suggest that SFK and Rac are independent pathways that are both necessary for PI3K-dependent NK cell cytotoxicity against Cryptococcus.
FIGURE 5.
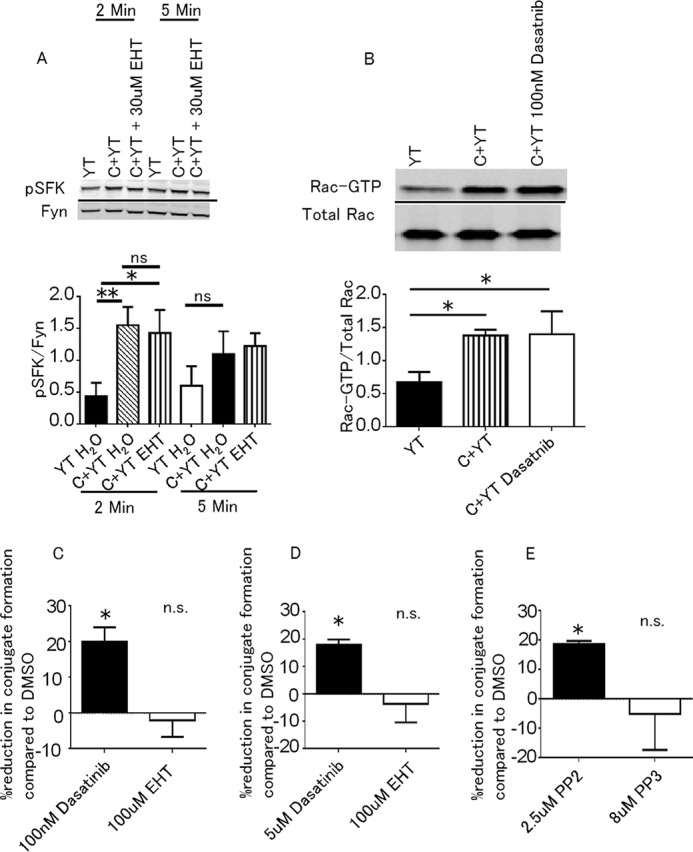
Rac and SFK activate PI3K by independent pathways. A, YT cells were preincubated with EHT1864 and then stimulated with Cryptococcus. The cells were lysed, and immunoblots for pSFK and Fyn were performed. The data are representative of three experiments. B, YT cells were preincubated with dasatnib or control. The levels of Rac-GTP were determined by Rac-GTP pulldown assay and Rac1 immunoblotting. The levels of total Rac were measured from whole cell lysate. The data are representative of three experiments. Intensity was measured using ImageJ. C+YT, YT cells stimulated with Cryptococcus. C and D, YT cells (C) or primary NK cells (D) were preincubated with dasatinib, EHT1864, or vehicle control. E, YT cells were preincubated with PP2, PP3, or DMSO. YT cells and primary NK cells were then labeled with anti-CD11a (red), and Cryptococcus was labeled with FITC. The percentage of cells forming conjugates was calculated by (number of green and red events)/(total number of green events). The percentage reduction in conjugate formation compared with DMSO was calculated by (1 − (% conjugates in treated)/(% conjugates in DMSO)) * 100%. The data are representative of three experiments (C and E) or two experiments (D). *, p < 0.05; **, p < 0.01. n.s., no statistically significant difference.
Conjugate Formation between NK and Cryptococcus Is SFK-dependent
We found that conjugate formation between NK cells and Cryptococcus was independent of Rac and that Cryptococcus stimulation independently activates SFK and Rac pathways. This raised the possibility that conjugate formation depended on SFK signaling rather than Rac. Previously, our lab had found that dasatinib caused a minor reduction in conjugate formation between fixed YT cells and Cryptococcus that did not achieve statistical significance (27). Using a flow cytometry based assay and live cells, we found that preincubating YT cells or primary NK cells with the SFK inhibitor, dasatinib, reduced the proportion of NK cells in conjugate with Cryptococcus compared with control, whereas EHT 1864 did not (Fig. 5, C and D). Another SFK inhibitor, PP2, also reduced conjugate formation between YT cells and Cryptococcus compared with control PP3 (Fig. 5E). These findings suggest that Cryptococcus initiates a SFK signaling pathway in NK cells that leads to enhanced conjugate formation.
Discussion
In this study, we made four important observations exploring the noncanonical role of Rac: (i) C. neoformans stimulates NK cells and activates Rac; (ii) in contrast to tumor cytotoxicity, Rac is upstream and required for the activation of the PI3K → Erk signaling pathway that leads to fungal cytotoxicity; (iii) Rac and SFK independently and nonredundantly activate PI3K; and (iv) NK cell cryptococcal conjugate formation is dependent on SFK and occurs independent of Rac.
Previous studies have shown that NK cell anti-cryptococcal killing and NK anti-tumor killing have many similarities. NK cells required PI3K → Erk signaling in both tumor and cryptococcal killing. NK microbial killing also depended on the NK activating receptor, NKp30, and the effector molecule, perforin (4, 24), which are both used in tumor killing. We hypothesized that Rac may be involved in cryptococcal killing, because Rac was shown to be involved in actin polymerization and granule polarization in tumor killing. However, there were also fundamental differences between NK cell microbial killing compared with tumor killing. The binding interface between NK cells and fungal targets was different. NK cells bound tumor targets with a tight interface in close apposition, whereas fungal targets were bound more distantly and appeared to use microvilli to penetrate the fungal target (42). Additionally, whereas LFA-1 played a critical role in tumor cell killing, it did not play a role in killing of Cryptococcus (3). In this paper, we showed that Rac was involved in cryptococcal killing; however, its role was distinct from tumor killing because it was proximal rather than distal to PI3K.
Having established that Rac was required for cryptococcal killing, we questioned how Rac affected NK cell function. Studies using other lymphocytes, as well as NK cells, showed that Rac was important for conjugate formation (11, 43, 44). Surprisingly, inhibiting Rac did not affect the ability of NK cells to form conjugates to Cryptococcus (Fig. 2). As mentioned above, LFA-1, which has also been implicated in conjugate formation of NK cells, was not involved in cryptococcal killing (45, 46). Although conjugate formation can be achieved by other signaling pathways, the lack of Rac and LFA-1 involvement may suggest an unidentified actin remodeling process (45).
In addition to conjugate formation, we investigated the role of Rac in Erk activation, which is required for cryptococcal killing (4). We found that inhibition of Rac prevented Cryptococcus from stimulating Erk phosphorylation, suggesting that Rac was required to activate Erk, as it is in tumor killing (Fig. 3). Surprisingly, we also found that Rac was required for PI3K activity, but PI3K was not required for Rac activation (Fig. 4), which is distinct from tumor killing. Although we cannot absolutely exclude a requirement for another pathway in Erk activation, these data are most consistent with our model that Rac is upstream of PI3K in anti-cryptococcal signaling. These findings demonstrated a noncanonical NK cell Rac signaling pathway. PI3K activation of Rac is well documented; however, Rac activation of PI3K is less understood. Previous studies suggest models of Rac-mediated activation of PI3K. Rac activation of PI3K has found that in mammalian epithelial cells (T47D), Rac activated PI3K, leading to increased mobility and invasive properties (28). In the chicken B cell line (DT40), B cell receptor stimulation initiated a Vav3 → Rac1 → PI3K pathway (47). Also, active Rac-GTP bound to PI3K directly in neutrophils and fibroblasts (48). Rac and PI3K could also activate each other in a positive feedback loop, which allowed neutrophils to establish a leading edge (41). Lastly, Rho family GTPases downstream of PI3K could regulate PI3K activation through a positive feedback loop (49). Our data added to this body of literature by showing that Rac activation was proximal, required, but not sufficient for PI3K activation in cryptococcal killing. Additionally, we showed that in the Rac → PI3K pathway, knockdown of the Rac1 isoform prevented cryptococcal killing by NK cells. Although NK cells express both Rac1 and Rac2 isoforms (50), which share redundancy in T cells (51), loss of Rac1 was sufficient to prevent cytotoxicity. This suggested that Rac2 was not able to compensate for Rac1 in NK-mediated cytotoxicity.
SFK have been shown to play a vital role in NK cell-mediated cryptococcal killing, by activating the PI3K → Erk signaling pathway (27). Because both SFK and Rac were required for PI3K activation and SFK and Rac did not activate each other (Fig. 5), these data indicated that SFK and Rac activated PI3K via independent signaling pathways. Interestingly, NKp30 relied on SFK-dependent CD3ζ signaling in tumor killing (52). Although there are no examples of natural cytotoxicity receptors, such as NKp30, activating both SFK and Rac-dependent pathways, we acknowledge that following TCR ligation and CD3ζ activation, ZAP70 activated Vav1 via linker of activated T cells (LAT), leading to activation of Rac1 (53). Nevertheless, we believe our results are more consistent with a model whereby NKp30 activates SFK, whereas Rac is activated by an unidentified receptor (Fig. 6).
FIGURE 6.
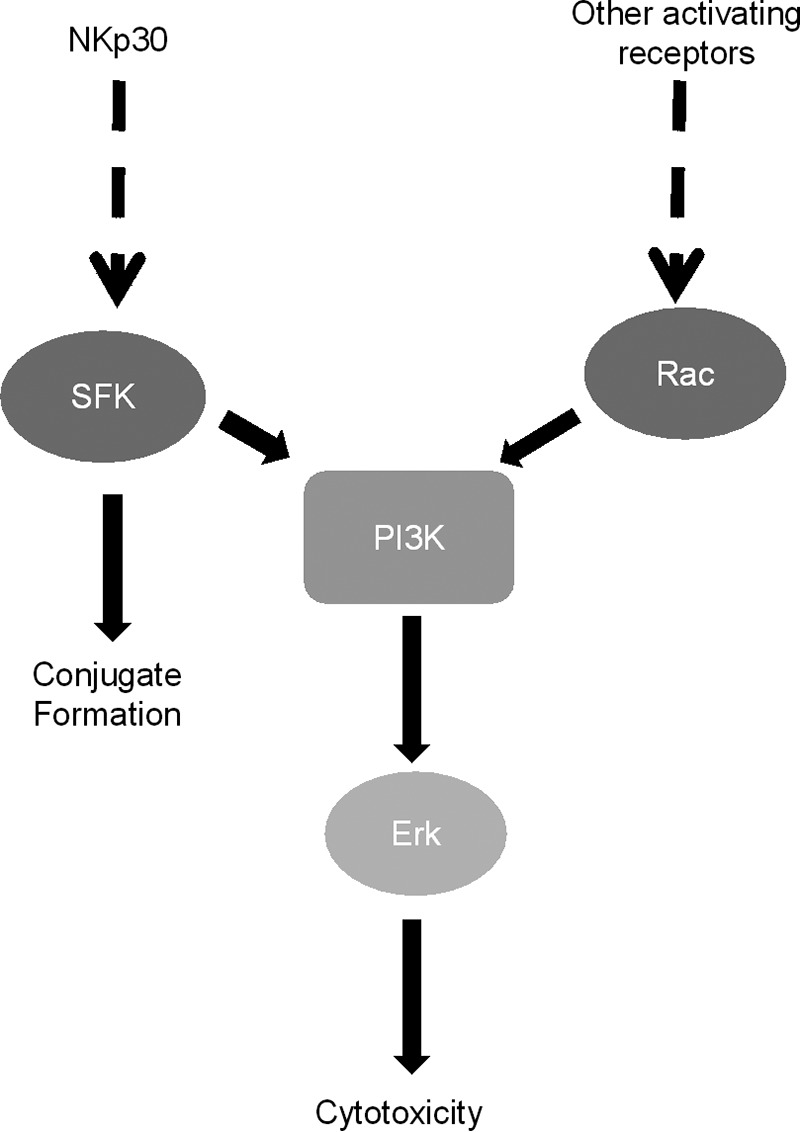
Model of NK anti-cryptococcal signaling. Cryptococcal capsule and cell wall components are recognized by activating NK receptors such as NKp30. Receptor binding initiates numerous independent signaling cascades, including SFK and Rac. Independent signaling cascades converge on PI3K. PI3K results in the activation of Erk, which leads to NK cytotoxicity. In addition, SFK is also required for conjugate formation.
The finding that SFK and Rac were activated independently also raised questions about the function of these pathways in NK cryptococcal killing. Although both proteins have been found to activate cytotoxicity through Erk, we found that only SFK was responsible for conjugate formation between NK cells and Cryptococcus (Fig. 5, C–E). This suggested that in addition to the Erk cytotoxicity pathway, SFK were required for the activation of a separate pathway that was responsible for conjugate formation. SFK have also been shown to be required for conjugate formation between NK cells and tumor targets (54). Additionally, in T cells, SFK have been shown to remodel the actin cytoskeleton and enhance conjugate formation between T cells and antigen-presenting cells via a PI3K → Erk independent pathway (55). Together, our data and previous findings pointed to the possibility that SFK-regulated conjugate formation between NK cells and Cryptococcus, in a PI3K → Erk independent manner.
Because the cryptococcal stimulus induced a Rac and SFK signaling pathway, which are independent of each other, our data uncovered a model of NK microbial cytotoxicity that was triggered by the simultaneous activation of multiple stimulatory pathways that worked together to activate PI3K. Although we did not determine the mechanism by which Rac and SFK activated PI3K, the p85 subunit of PI3K is essential in NK cryptococcal killing (4), and Rac and SFK have been shown to directly bind to this subunit and activate PI3K (48, 56). Alternatively, Rac has also been shown to directly bind and activate the p110β subunit of PI3K (57). Therefore, full activation of PI3K could require SFK to bind the p85 subunit and Rac to bind the p110β subunit. This model is similar to G-protein activation of the p110β subunit of PI3K, where full activation required both Gβγ and tyrosine kinase activity (58). Rac and SFK may also activate PI3K in different cellular compartments. Because Rac can activate p110β subunit of PI3K directly, it might be able to activate PI3K with different regulatory subunits (p85, p55, or p50). On the other hand, SFK requires the p85 subunit of PI3K. The smaller (p55 and p50) regulatory subunits of PI3K localize to different compartments of the cell. For example, p55 binds to tubulin and localizes to the microtubule network (59). The requirement for both SFK and Rac could be to allow the cell to regulate where the activation of PI3K occurs.
The requirement for multiple stimulatory pathways is supported by previous studies on NK tumor killing. P815 redirected activation assays demonstrated that the NK receptors NKp46, 2B4, NKG2D, DNAM, and CD2 act synergistically to induce calcium flux and degranulation (60). Individually, each of these pathways was not enough to trigger calcium flux or degranulation, but co-cross-linking of multiple receptors activated a calcium influx pathway that resulted in degranulation. Each of these receptors utilized its own independent signaling pathways, which converged to promote NK cell function. This convergence of independent signaling pathways is reminiscent of our results. Individually, SFK and Rac were not capable of initiating the PI3K cytotoxicity pathway. However, the requirement for the activation of multiple signaling pathways could hinder NK cell-mediated microbial cytotoxicity, because a microbe that is able to evade even a single pathway could escape NK cell cytotoxicity. This raises the question of why NK cells would require such stringent criteria to recognize and kill microbes. One possible explanation is that NK cells require multiple activation pathways to protect against targeting host cells if only one NK receptor was engaged nonspecifically (61, 62).
The involvement of Rac in NK cell-mediated cryptococcal killing may provide clinical insights into the susceptibility to cryptococcosis in patients taking azathioprine (63, 64). In addition to being a purine analogue, azathioprine functions by blocking GTP binding to Rac and prevents Rac activation (65, 66). Patients treated with azathioprine have been reported to develop both pulmonary cryptococcosis and cryptococcal meningitis (63, 64). Our data suggest that the increased susceptibility to Cryptococcus could be caused in part by defective NK cell function, because of azathioprine-induced blockage of the Rac → PI3K → Erk cytotoxicity pathway.
Originally, we hypothesized that Rac was the intermediate molecule that allowed PI3K to activate Erk. Because we have shown that Rac is not activated by PI3K, it raised questions about how PI3K activates Erk in cryptococcal killing. In addition, although PI3K is activated by Rac, this does not preclude other essential signaling pathways that might also be activated by Rac, especially if these alternative pathways synergize with cytotoxic functions that are controlled by Erk activity. Although we have shown that Rac was not involved in conjugate formation between Cryptococcus and YT cells, Rac has been shown to be involved in actin and microtubule remodeling, which could make it important for granule trafficking that occurs in later stages of cytotoxicity. One possibility is that Rac GTPases are involved in the phosphorylation of stathmin, which is required for microtubule remodeling (67). Additionally, stathmin also causes microtubule-organizing center polarization in activated T cells (40), so there is the possibility that cryptococcal mediated microtubule-organizing center polarization in NK cells is regulated by a Rac → stathmin pathway.
In summary, we have demonstrated a novel mechanism by which Rac activated PI3K in NK cells and demonstrated that this pathway was required for direct microbial cytotoxicity. This furthers our understanding of Rac as an upstream activator of PI3K, and defines a novel role for Rac in NK microbial cytotoxicity.
Author Contributions
R. F. X. developed the concepts, designed and performed the experiments, analyzed the data, and wrote the manuscript. D. S. isolated and provided the primary NK cells. S. M. H., S. S. L., H. O., and S. K. K. provided technical assistance and discussion. C. H. M. developed concepts, supervised the study, reviewed the data, and edited the manuscript. All authors reviewed drafts of this manuscript.
This work was supported by Canadian Institute for Health Research Grant 247301 (to C. H. M.). The authors declare that they have no conflicts of interest with the contents of this article.
- NK
- natural killer
- Rac
- Ras-related C3 botulinum toxin substrate
- SFK
- Src family kinase
- PLCγ
- phospholipase Cγ
- Rho
- Ras homology
- PAK
- p21-activated kinase
- PBD
- p21 binding domain
- MBCD
- methyl β-cyclodextrin
- Akt
- α-serine/threonine-protein kinase
- PBMC
- peripheral blood mononuclear cell.
References
- 1. Ma L. L., Wang C. L., Neely G. G., Epelman S., Krensky A. M., and Mody C. H. (2004) NK cells use perforin rather than granulysin for anticryptococcal activity. J. Immunol. 173, 3357–3365 [DOI] [PubMed] [Google Scholar]
- 2. Jiang K., Zhong B., Gilvary D. L., Corliss B. C., Hong-Geller E., Wei S., and Djeu J. Y. (2000) Pivotal role of phosphoinositide-3 kinase in regulation of cytotoxicity in natural killer cells. Nat. Immunol. 1, 419–425 [DOI] [PubMed] [Google Scholar]
- 3. Jones G. J., Wiseman J. C., Marr K. J., Wei S., Djeu J. Y., and Mody C. H. (2009) In contrast to anti-tumor activity, YT cell and primary NK cell cytotoxicity for Cryptococcus neoformans bypasses LFA-1. Int. Immunol. 21, 423–432 [DOI] [PubMed] [Google Scholar]
- 4. Wiseman J. C., Ma L. L., Marr K. J., Jones G. J., and Mody C. H. (2007) Perforin-dependent cryptococcal microbicidal activity in NK cells requires PI3K-dependent ERK1/2 signaling. J. Immunol. 178, 6456–6464 [DOI] [PubMed] [Google Scholar]
- 5. Amin A. R., Ichigotani Y., Oo M. L., Biswas M. H., Yuan H., Huang P., Mon N. N., and Hamaguchi M. (2003) The PLC-PKC cascade is required for IL-1β-dependent Erk and Akt activation: their role in proliferation. Int. J. Oncol. 23, 1727–1731 [PubMed] [Google Scholar]
- 6. Diaz-Flores E., Goldschmidt H., Depeille P., Ng V., Akutagawa J., Krisman K., Crone M., Burgess M. R., Williams O., Houseman B., Shokat K., Sampath D., Bollag G., Roose J. P., Braun B. S., and Shannon K. (2013) PLC-γ and PI3K link cytokines to ERK activation in hematopoietic cells with normal and oncogenic Kras. Sci. Signal. 6, ra105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Caraux A., Kim N., Bell S. E., Zompi S., Ranson T., Lesjean-Pottier S., Garcia-Ojeda M. E., Turner M., and Colucci F. (2006) Phospholipase C-γ2 is essential for NK cell cytotoxicity and innate immunity to malignant and virally infected cells. Blood 107, 994–1002 [DOI] [PubMed] [Google Scholar]
- 8. Maffucci T., Raimondi C., Abu-Hayyeh S., Dominguez V., Sala G., Zachary I., and Falasca M. (2009) A phosphoinositide 3-kinase/phospholipase Cγ1 pathway regulates fibroblast growth factor-induced capillary tube formation. PLoS One 4, e8285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Djeu J. Y., Jiang K., and Wei S. (2002) A view to a kill: signals triggering cytotoxicity. Clin. Cancer Res. 8, 636–640 [PubMed] [Google Scholar]
- 10. Bustelo X. R., Sauzeau V., and Berenjeno I. M. (2007) GTP-binding proteins of the Rho/Rac family: regulation, effectors and functions in vivo. Bioessays 29, 356–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Malorni W., Quaranta M. G., Straface E., Falzano L., Fabbri A., Viora M., and Fiorentini C. (2003) The Rac-activating toxin cytotoxic necrotizing factor 1 oversees NK cell-mediated activity by regulating the actin/microtubule interplay. J. Immunol. 171, 4195–4202 [DOI] [PubMed] [Google Scholar]
- 12. Vyas Y. M., Mehta K. M., Morgan M., Maniar H., Butros L., Jung S., Burkhardt J. K., and Dupont B. (2001) Spatial organization of signal transduction molecules in the NK cell immune synapses during MHC class I-regulated noncytolytic and cytolytic interactions. J. Immunol. 167, 4358–4367 [DOI] [PubMed] [Google Scholar]
- 13. Aspenström P., Lindberg U., and Hall A. (1996) Two GTPases, Cdc42 and Rac, bind directly to a protein implicated in the immunodeficiency disorder Wiskott-Aldrich syndrome. Curr. Biol. 6, 70–75 [DOI] [PubMed] [Google Scholar]
- 14. Aspenström P. (1999) Effectors for the Rho GTPases. Curr. Opin. Cell Biol. 11, 95–102 [DOI] [PubMed] [Google Scholar]
- 15. Iden S., and Collard J. G. (2008) Crosstalk between small GTPases and polarity proteins in cell polarization. Nat. Rev. Mol. Cell Biol. 9, 846–859 [DOI] [PubMed] [Google Scholar]
- 16. Takenawa T., and Suetsugu S. (2007) The WASP-WAVE protein network: connecting the membrane to the cytoskeleton. Nat. Rev. Mol. Cell Biol. 8, 37–48 [DOI] [PubMed] [Google Scholar]
- 17. Putney J. W. (2002) PLC-γ: an old player has a new role. Nat. Cell Biol. 4, E280–E281 [DOI] [PubMed] [Google Scholar]
- 18. Park B. J., Wannemuehler K. A., Marston B. J., Govender N., Pappas P. G., and Chiller T. M. (2009) Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. AIDS 23, 525–530 [DOI] [PubMed] [Google Scholar]
- 19. Gibson J. F., and Johnston S. A. (2015) Immunity to Cryptococcus neoformans and C. gattii during cryptococcosis. Fungal. Genet. Biol. 78, 76–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hidore M. R., and Murphy J. W. (1986) Natural cellular resistance of beige mice against Cryptococcus neoformans. J. Immunol. 137, 3624–3631 [PubMed] [Google Scholar]
- 21. Lipscomb M. F., Alvarellos T., Toews G. B., Tompkins R., Evans Z., Koo G., and Kumar V. (1987) Role of natural killer cells in resistance to Cryptococcus neoformans infections in mice. Am. J. Pathol. 128, 354–361 [PMC free article] [PubMed] [Google Scholar]
- 22. Hidore M. R., Nabavi N., Sonleitner F., and Murphy J. W. (1991) Murine natural killer cells are fungicidal to Cryptococcus neoformans. Infect. Immun. 59, 1747–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Levitz S. M., Dupont M. P., and Smail E. H. (1994) Direct activity of human T lymphocytes and natural killer cells against Cryptococcus neoformans. Infect. Immun. 62, 194–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li S. S., Kyei S. K., Timm-McCann M., Ogbomo H., Jones G. J., Shi M., Xiang R. F., Oykhman P., Huston S. M., Islam A., Gill M. J., Robbins S. M., and Mody C. H. (2013) The NK receptor NKp30 mediates direct fungal recognition and killing and is diminished in NK cells from HIV-infected patients. Cell Host Microbe 14, 387–397 [DOI] [PubMed] [Google Scholar]
- 25. Bryceson Y. T., Ljunggren H. G., and Long E. O. (2009) Minimal requirement for induction of natural cytotoxicity and intersection of activation signals by inhibitory receptors. Blood 114, 2657–2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kwon H. J., and Kim H. S. (2012) Signaling for synergistic activation of natural killer cells. Immune. Netw. 12, 240–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Oykhman P., Timm-McCann M., Xiang R. F., Islam A., Li S. S., Stack D., Huston S. M., Ma L. L., and Mody C. H. (2013) Requirement and redundancy of the Src family kinases Fyn and Lyn in perforin-dependent killing of Cryptococcus neoformans by NK cells. Infect. Immun. 81, 3912–3922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Keely P. J., Westwick J. K., Whitehead I. P., Der C. J., and Parise L. V. (1997) Cdc42 and Rac1 induce integrin-mediated cell motility and invasiveness through PI(3)K. Nature 390, 632–636 [DOI] [PubMed] [Google Scholar]
- 29. Shirakawa F., Tanaka Y., Eto S., Suzuki H., Yodoi J., and Yamashita U. (1986) Effect of interleukin 1 on the expression of interleukin 2 receptor (Tac antigen) on human natural killer cells and natural killer-like cell line (YT cells). J. Immunol. 137, 551–556 [PubMed] [Google Scholar]
- 30. James A. M., Hsu H. T., Dongre P., Uzel G., Mace E. M., Banerjee P. P., and Orange J. S. (2013) Rapid activation receptor- or IL-2-induced lytic granule convergence in human natural killer cells requires Src, but not downstream signaling. Blood 121, 2627–2637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huston S. M., Li S. S., Stack D., Timm-McCann M., Jones G. J., Islam A., Berenger B. M., Xiang R. F., Colarusso P., and Mody C. H. (2013) Cryptococcus gattii is killed by dendritic cells, but evades adaptive immunity by failing to induce dendritic cell maturation. J. Immunol. 191, 249–261 [DOI] [PubMed] [Google Scholar]
- 32. Falasca M., Logan S. K., Lehto V. P., Baccante G., Lemmon M. A., and Schlessinger J. (1998) Activation of phospholipase Cγ by PI 3-kinase-induced PH domain-mediated membrane targeting. EMBO J. 17, 414–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Buhl A. M., Osawa S., and Johnson G. L. (1995) Mitogen-activated protein kinase activation requires two signal inputs from the human anaphylatoxin C5a receptor. J. Biol. Chem. 270, 19828–19832 [DOI] [PubMed] [Google Scholar]
- 34. Tassi I., and Colonna M. (2005) The cytotoxicity receptor CRACC (CS-1) recruits EAT-2 and activates the PI3K and phospholipase Cγ signaling pathways in human NK cells. J. Immunol. 175, 7996–8002 [DOI] [PubMed] [Google Scholar]
- 35. Islam A., Li S. S., Oykhman P., Timm-McCann M., Huston S. M., Stack D., Xiang R. F., Kelly M. M., and Mody C. H. (2013) An acidic microenvironment increases NK cell killing of Cryptococcus neoformans and Cryptococcus gattii by enhancing perforin degranulation. PLoS Pathog. 9, e1003439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shutes A., Onesto C., Picard V., Leblond B., Schweighoffer F., and Der C. J. (2007) Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J. Biol. Chem. 282, 35666–35678 [DOI] [PubMed] [Google Scholar]
- 37. Ferri N., Corsini A., Bottino P., Clerici F., and Contini A. (2009) Virtual screening approach for the identification of new Rac1 inhibitors. J. Med. Chem. 52, 4087–4090 [DOI] [PubMed] [Google Scholar]
- 38. Welch H. C., Coadwell W. J., Stephens L. R., and Hawkins P. T. (2003) Phosphoinositide 3-kinase-dependent activation of Rac. FEBS Lett. 546, 93–97 [DOI] [PubMed] [Google Scholar]
- 39. Pfeifer M., Grau M., Lenze D., Wenzel S. S., Wolf A., Wollert-Wulf B., Dietze K., Nogai H., Storek B., Madle H., Dörken B., Janz M., Dirnhofer S., Lenz P., Hummel M., Tzankov A., and Lenz G. (2013) PTEN loss defines a PI3K/AKT pathway-dependent germinal center subtype of diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. U.S.A. 110, 12420–12425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Filbert E. L., Le Borgne M., Lin J., Heuser J. E., and Shaw A. S. (2012) Stathmin regulates microtubule dynamics and microtubule organizing center polarization in activated T cells. J. Immunol. 188, 5421–5427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Srinivasan S., Wang F., Glavas S., Ott A., Hofmann F., Aktories K., Kalman D., and Bourne H. R. (2003) Rac and Cdc42 play distinct roles in regulating PI(3,4,5)P3 and polarity during neutrophil chemotaxis. J. Cell Biol. 160, 375–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Murphy J. W., Hidore M. R., and Nabavi N. (1991) Binding interactions of murine natural killer cells with the fungal target Cryptococcus neoformans. Infect. Immun. 59, 1476–1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Billadeau D. D., Brumbaugh K. M., Dick C. J., Schoon R. A., Bustelo X. R., and Leibson P. J. (1998) The Vav-Rac1 pathway in cytotoxic lymphocytes regulates the generation of cell-mediated killing. J. Exp. Med. 188, 549–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Faure S., Salazar-Fontana L. I., Semichon M., Tybulewicz V. L., Bismuth G., Trautmann A., Germain R. N., and Delon J. (2004) ERM proteins regulate cytoskeleton relaxation promoting T cell-APC conjugation. Nat. Immunol. 5, 272–279 [DOI] [PubMed] [Google Scholar]
- 45. Porter J. C., Bracke M., Smith A., Davies D., and Hogg N. (2002) Signaling through integrin LFA-1 leads to filamentous actin polymerization and remodeling, resulting in enhanced T cell adhesion. J. Immunol. 168, 6330–6335 [DOI] [PubMed] [Google Scholar]
- 46. Brown A. C., Dobbie I. M., Alakoskela J. M., Davis I., and Davis D. M. (2012) Super-resolution imaging of remodeled synaptic actin reveals different synergies between NK cell receptors and integrins. Blood 120, 3729–3740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Inabe K., Ishiai M., Scharenberg A. M., Freshney N., Downward J., and Kurosaki T. (2002) Vav3 modulates B cell receptor responses by regulating phosphoinositide 3-kinase activation. J. Exp. Med. 195, 189–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bokoch G. M., Vlahos C. J., Wang Y., Knaus U. G., and Traynor-Kaplan A. E. (1996) Rac GTPase interacts specifically with phosphatidylinositol 3-kinase. Biochem. J. 315, 775–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yang H. W., Shin M. G., Lee S., Kim J. R., Park W. S., Cho K. H., Meyer T., and Heo W. D. (2012) Cooperative activation of PI3K by Ras and Rho family small GTPases. Mol. Cell. 47, 281–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Meng X., Krokhin O., Cheng K., Ens W., and Wilkins J. A. (2007) Characterization of IQGAP1-containing complexes in NK-like cells: evidence for Rac 2 and RACK1 association during homotypic adhesion. J. Proteome Res. 6, 744–750 [DOI] [PubMed] [Google Scholar]
- 51. Guo F., Cancelas J. A., Hildeman D., Williams D. A., and Zheng Y. (2008) Rac GTPase isoforms Rac1 and Rac2 play a redundant and crucial role in T-cell development. Blood 112, 1767–1775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pende D., Parolini S., Pessino A., Sivori S., Augugliaro R., Morelli L., Marcenaro E., Accame L., Malaspina A., Biassoni R., Bottino C., Moretta L., and Moretta A. (1999) Identification and molecular characterization of NKp30, a novel triggering receptor involved in natural cytotoxicity mediated by human natural killer cells. J. Exp. Med. 190, 1505–1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Perez-Villar J. J., Whitney G. S., Sitnick M. T., Dunn R. J., Venkatesan S., O'Day K., Schieven G. L., Lin T. A., and Kanner S. B. (2002) Phosphorylation of the linker for activation of T-cells by Itk promotes recruitment of Vav. Biochemistry 41, 10732–10740 [DOI] [PubMed] [Google Scholar]
- 54. Lou Z., Jevremovic D., Billadeau D. D., and Leibson P. J. (2000) A balance between positive and negative signals in cytotoxic lymphocytes regulates the polarization of lipid rafts during the development of cell-mediated killing. J. Exp. Med. 191, 347–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Morgan M. M., Labno C. M., Van Seventer G. A., Denny M. F., Straus D. B., and Burkhardt J. K. (2001) Superantigen-induced T cell:B cell conjugation is mediated by LFA-1 and requires signaling through Lck, but not ZAP-70. J. Immunol. 167, 5708–5718 [DOI] [PubMed] [Google Scholar]
- 56. Pleiman C. M., Hertz W. M., and Cambier J. C. (1994) Activation of phosphatidylinositol-3′ kinase by Src-family kinase SH3 binding to the p85 subunit. Science 263, 1609–1612 [DOI] [PubMed] [Google Scholar]
- 57. Fritsch R., de Krijger I., Fritsch K., George R., Reason B., Kumar M. S., Diefenbacher M., Stamp G., and Downward J. (2013) RAS and RHO families of GTPases directly regulate distinct phosphoinositide 3-kinase isoforms. Cell 153, 1050–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kurosu H., Maehama T., Okada T., Yamamoto T., Hoshino S., Fukui Y., Ui M., Hazeki O., and Katada T. (1997) Heterodimeric phosphoinositide 3-kinase consisting of p85 and p110β is synergistically activated by the βγ subunits of G proteins and phosphotyrosyl peptide. J. Biol. Chem. 272, 24252–24256 [DOI] [PubMed] [Google Scholar]
- 59. Inukai K., Funaki M., Nawano M., Katagiri H., Ogihara T., Anai M., Onishi Y., Sakoda H., Ono H., Fukushima Y., Kikuchi M., Oka Y., and Asano T. (2000) The N-terminal 34 residues of the 55 kDa regulatory subunits of phosphoinositide 3-kinase interact with tubulin. Biochem. J. 346, 483–489 [PMC free article] [PubMed] [Google Scholar]
- 60. Bryceson Y. T., March M. E., Ljunggren H. G., and Long E. O. (2006) Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood 107, 159–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Emmer P. M., Nelen W. L., Steegers E. A., Hendriks J. C., Veerhoek M., and Joosten I. (2000) Peripheral natural killer cytotoxicity and CD56(pos)CD16(pos) cells increase during early pregnancy in women with a history of recurrent spontaneous abortion. Hum. Reprod. 15, 1163–1169 [DOI] [PubMed] [Google Scholar]
- 62. Morse R. H., Séguin R., McCrea E. L., and Antel J. P. (2001) NK cell-mediated lysis of autologous human oligodendrocytes. J. Neuroimmunol. 116, 107–115 [DOI] [PubMed] [Google Scholar]
- 63. Fraison J. B., Guilpain P., Schiffmann A., Veyrac M., Le Moing V., Rispail P., and Le Quellec A. (2013) Pulmonary cryptococcosis in a patient with Crohn's disease treated with prednisone, azathioprine and adalimumab: exposure to chicken manure as a source of contamination. J. Crohns. Colitis. 7, e11–e14 [DOI] [PubMed] [Google Scholar]
- 64. Sethi N. K., Torgovnick J., and Sethi P. K. (2007) Cryptococcal meningitis after Imuran (azathioprine) therapy for autoimmune hepatitis. Eur. J. Gastroenterol. Hepatol. 19, 913–914 [DOI] [PubMed] [Google Scholar]
- 65. Neurath M. (2010) Thiopurines in IBD: what is their mechanism of action? Gastroenterol. Hepatol. (NY) 6, 435–436 [PMC free article] [PubMed] [Google Scholar]
- 66. Marinkovic G., Hibender S., Hoogenboezem M., van Broekhoven A., Girigorie A. F., Bleeker N., Hamers A. A., Stap J., van Buul J. D., de Vries C. J., and de Waard V. (2013) Immunosuppressive drug azathioprine reduces aneurysm progression through inhibition of Rac1 and c-Jun-terminal-N-kinase in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 33, 2380–2388 [DOI] [PubMed] [Google Scholar]
- 67. Daub H., Gevaert K., Vandekerckhove J., Sobel A., and Hall A. (2001) Rac/Cdc42 and p65PAK regulate the microtubule-destabilizing protein stathmin through phosphorylation at serine 16. J. Biol. Chem. 276, 1677–1680 [DOI] [PubMed] [Google Scholar]