

Abstract
Oral cancer is the sixth most common cause of death from cancer with an estimated 400,000 deaths worldwide and a low (50%) 5-year survival rate. The most common form of oral cancer is oral squamous cell carcinoma (OSCC). OSCC is highly inflammatory and invasive, and the degree of inflammation correlates with tumor aggressiveness. The G protein-coupled receptor protease-activated receptor-2 (PAR-2) plays a key role in inflammation. PAR-2 is activated via proteolytic cleavage by trypsin-like serine proteases, including kallikrein-5 (KLK5), or by treatment with activating peptides. PAR-2 activation induces G protein-α-mediated signaling, mobilizing intracellular calcium and Nf-κB signaling, leading to the increased expression of pro-inflammatory mRNAs. Little is known, however, about PAR-2 regulation of inflammation-related microRNAs. Here, we assess PAR-2 expression and function in OSCC cell lines and tissues. Stimulation of PAR-2 activates Nf-κB signaling, resulting in RelA nuclear translocation and enhanced expression of pro-inflammatory mRNAs. Concomitantly, suppression of the anti-inflammatory tumor suppressor microRNAs let-7d, miR-23b, and miR-200c was observed following PAR-2 stimulation. Analysis of orthotopic oral tumors generated by cells with reduced KLK5 expression showed smaller, less aggressive lesions with reduced inflammatory infiltrate relative to tumors generated by KLK5-expressing control cells. Together, these data support a model wherein KLK5-mediated PAR-2 activation regulates the expression of inflammation-associated mRNAs and microRNAs, thereby modulating progression of oral tumors.
Keywords: inflammation, kallikrein, microRNA (miRNA), NF-κB, protease, serine protease, oral cancer, protease-activated receptor
Introduction
Squamous cell carcinoma of the oral cavity (OSCC)2 is a highly inflammatory disease that ranks as the 6th most frequently diagnosed cancer worldwide, with a poor 5-year survival rate of about 50% (1). Early diagnosis of OSCC is challenging, predominantly because early oral cancers and premalignant lesions are often subtle and asymptomatic. Although many patients present for diagnosis with stage III or IV disease, premalignant lesions in the oral mucosa often precede invasive OSCC (2). Furthermore, unlike most solid tumors that are monoclonal in origin, multiple distinct foci of dysplastic cells may be present in the oral mucosa. These epithelial dysplasia are categorized as mild, moderate, severe, or carcinoma in situ based on the histologic abnormalities present in the oral epithelium (3). Studies show that ∼12–36% of epithelial dysplasia progress to carcinoma (4, 5); however, current approaches do not enable accurate identification of premalignant lesions likely to undergo malignant transformation.
The link between inflammation and cancer is now well established, and inflammatory mediators are present in the microenvironment of virtually all solid tumors (6–10). Many studies have associated proteinase-activated receptor-2 (PAR-2) with both inflammation and tumor progression (11–15); however, the expression of PAR-2 in OSCC has not been evaluated. PAR-2 is a G protein-coupled receptor that is activated by trypsin-like serine proteinases. Proteolytic cleavage of the extracellular amino terminus generates a “tethered ligand” that binds to the receptor, initiating Gα protein signaling. PAR-2 can also be activated by PAR2-activating peptides that mimic the tethered ligand amino acid sequence and generate all of the hallmarks of an inflammatory response (11, 12).
Kallikrein 5 (KLK5) is a secreted serine protease that is involved in regulated skin desquamation during epidermal differentiation (16). We have previously reported that KLK5 is up-regulated at the mRNA and protein levels in oral cancer cell lines and human OSCC tissues (17) and catalyzes cleavage of the cell-cell adhesion molecule desmoglein-1, promoting loss of junctional integrity and enhanced invasive activity in vitro (18). Activity of KLK5 is tightly controlled via expression of the proteinase inhibitor lympho-epithelial Kazal-type inhibitor (LEKTI, encoded by SPINK5). In Netherton syndrome, loss-of-function mutations in LEKTI enable unopposed KLK5 activity, leading to stratum corneum detachment and defective skin barrier function (19, 20). Expression of LEKTI is also lost in OSCC, indicative of unregulated KLK5 activity (21, 22). An additional substrate of KLK5 is PAR-2. KLK5 cleavage of the receptor triggers a PAR-2 signaling cascade and activation of Nf-κB target genes, including the pro-inflammatory mediators TNFα, IL8, ICAM1, and TSLP (16).
PAR-2 regulation of pro-inflammatory mediators in both tumor cells and immune cells is effected principally through the Nf-κB signaling pathway (16, 23–25). Downstream of PAR-2 signaling, phosphorylation of RelA by IKKβ and nuclear translocation of RelA-containing Nf-κB dimers activate transcription of an array of pro-inflammatory cytokines and chemokines such as IL8 (CXCL8). IL8 is consistently up-regulated in OSCC, is produced by tumor cells as well as cells in the tumor microenvironment, is a reliable biomarker for the disease, and is associated with angiogenesis, invasion, metastasis, and poor survival in OSCC and other cancers (26–31). Although PAR-2-mediated regulation of pro-inflammatory mRNAs is well established, regulation of microRNA expression by PAR-2 activation has not been evaluated. In this study, we examined KLK5 or peptide-mediated activation of PAR-2 in oral cells and tissues and the resulting effect on mRNA/microRNA expression and tumor progression.
Materials and Methods
Reagents and Antibodies
TNF-α was obtained from Shenandoah Biotechnology (Warwick, PA.); sc-514 was purchased from EMD Millipore Corp. (Billerica, MA); SB 747651A was obtained from Axon MedChem (The Netherlands); pertussis toxin was purchased from Enzo Life Sciences (Farmingdale, New York); fura-2/AM and fluorophore-conjugated secondary antibodies were obtained from Life Technologies, Inc.; PAR-2-activating peptide (SLIGRL-NH2) was purchased from GenScript (Piscataway, NJ); anti-RelA antibody sc-372, anti-PAR-2 antibody sc-13504, and anti-GAPDH antibody sc-47724 were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX); anti-GAPDH-peroxidase was purchased from Sigma; and anti-kallikrein-5 was obtained from Abcam (Cambridge, MA). miRNA microarray MIHS-105Z, SYBR Green kit for miRNA, and all miRNA assays were purchased from Qiagen (Gaithersburg, MD). Active recombinant human KLK5 was obtained from R&D Systems (Minneapolis, MN). Specific activity of the protease is guaranteed by the manufacturer based on enzymatic cleavage of t-butyloxycarbonyl-Val-Pro-Arg-7-amino-4-methylcoumarin.
Culture of Cell Lines
Cell lines OKF6/T, SCC1, and SCC25 were obtained from Dr. J. Rheinwald (Brigham & Women's Hospital, Harvard Institutes of Medicine). OKF6/T cells are derived from normal oral keratinocytes immortalized with the telomerase catalytic subunit human TERT (32). These cells retain keratinocyte growth controls and differentiate normally in culture. SCC1 and SCC25 cell lines were established from squamous cell carcinoma of the oral cavity (33). These cell lines do not stratify in organotypic culture, display growth factor-independent growth, and are tumorigenic in nude mice. In our studies, the SCC1-initiated tumors produce pulmonary metastases in nude mice (34). SCC25-initiated tumors invade tongue muscle, nerve bundles, and blood and lymphatic vessels (35, 36); however, distant metastasis is not observed prior to sacrifice for ethical considerations due to difficulty with feeding. Generation of an SCC25 cell line with reduced KLK5 expression (KLK5 knockdown, designated SCC25-KLK5-KD) and empty vector control cells (SCC25-Vec) was previously described (18). KLK5 mRNA expression in KLK5-KD cells is reduced by 90% relative to corresponding vector controls, as determined by qPCR, and loss of KLK5 protein was shown by immunocytochemistry (18). SCC25-derived cell lines were routinely maintained in DMEM/F-12 1:1 media (Corning) containing 10% fetal calf serum and supplemented with 100 units/ml penicillin, 100 μg/ml streptomycin, and 0.2 μg/ml puromycin. Immortalized normal oral keratinocytes (OKF6/T cell line) were maintained in keratinocyte-SFM (Life Technologies, Inc.) supplemented with 100 units/ml penicillin, 100 μg/ml streptomycin, 0.2 ng/ml epidermal growth factor (EGF), 25 μg/ml bovine pituitary extract, and 0.4 mm CaCl2. The SCC1 cell line was maintained in Eagle's minimal essential medium with Earle's salts and l-glutamine (Corning) containing 10% fetal calf serum supplemented with 100 units/ml penicillin and 100 μg/ml streptomycin. For studies requiring serum-free media, cells were washed three times with PBS, and OSCC cells were transferred to media without fetal calf serum, whereas OKF6/T cells were transferred to media without bovine pituitary extract or EGF. Cell lines were routinely passaged at 70% confluence and did not experience extremes of pH. For passage, cells were washed with PBS and trypsinized with 0.25% trypsin, 2.2 mm EDTA (2.5 ml/10-cm plate) for 4 min at 37 °C. Trypsinization was stopped by the addition of 5 ml of medium containing FBS or appropriate media.
Histology and Immunohistochemistry
Tissue microarrays were constructed containing de-identified cores of premalignant oral lesions from Temple University (collected by ER Sauter). The premalignant TMA contained 203 cases, including mild (110 cores), moderate (70 cores), and severe dysplasia (23 cores) as well as carcinoma in situ (24 cores). Human TMAs, OR481and T273, purchased from United States Biomax Ltd., Rockville, MD, contained 24 cores of grade I-II tongue OSCC. In microarrayed tissue sections, not every core was usable in every section. Prior to immunohistochemical staining, endogenous peroxidase activity was quenched with 3.3% hydrogen peroxide in methanol for 30 min. Antigen retrieval was enhanced by microwaving in 10 mm sodium citrate, pH 6.0. Nonspecific binding was blocked with 3% normal horse serum in PBS for 30 min. Sections were incubated with primary antibody (1:25 to 1:200 dilution, as indicated) at 4 °C overnight in 1% BSA in PBS. Staining was detected using an avidin-biotin horseradish peroxidase system (Vectastain Universal Elite ABC kit, Cat. PK-6200, Vector Laboratories, Burlingame, CA), with positive cells staining brown using diaminobenzidine chromogen and hydrogen peroxide substrate (twp-component DAB pack HK542-XAK, BioGenex, San Ramon, CA). Slides were counterstained with Gill's III hematoxylin and then saturated lithium carbonate. Tissue sections were dehydrated through graded ethanol and mixed xylenes and mounted onto coverslips with mounting medium (Surgipath Micromount, Leica Biosystems, Richmond, IL). Staining was designated as absent, weak, moderate, or strong by a pathologist (Z. S.). Immunohistochemical positivity was recorded as a percentage of cells staining with moderate-strong immunoreactivity per ×40 field, with enumeration at 200 cells/sample.
Murine tongue tumor sections (described below) were cut into thin sections (4 μm), incubated at 65 °C, de-paraffinized with xylene, rehydrated in a series of ethanol washes, and stained using hematoxylin and eosin (H&E) using standard procedures. Sections cut from the approximate middle of each tumor were H&E-stained, scanned with an Aperio Slidescanner, and area quantified using Aperio ImageScope software. Lymphocytes were enumerated (by Z. S. and J. J. J.) from H&E-stained sections for each high power field containing or immediately adjacent to tumor nests to provide a correlate to systemic inflammation. Results were presented as counts per high power field. Immunostaining for host (murine) mast cell tryptase with a mouse monoclonal anti-tryptase antibody was performed using the Vector M.O.M. (mouse-on-mouse) kit (Vector Laboratories catalog no. BMK-2202) according to the manufacturer's specifications. Tryptase-positive mast cells were quantified using the hot spot method, starting with the high power field with the most positivity and comparing only fields with similar tumor burden. Results were presented as counts per high power field.
Flow Cytometry Analysis
Cells were collected at 70% confluence and trypsinized briefly with 0.25% trypsin, 2.21 mm EDTA (2.5 ml per 10-cm plate) for 4 min at 37 °C. Trypsinization was stopped by the addition of 5 ml of the appropriate media. After centrifugation, primary antibody (or vehicle) was applied in serum-free media for 30 min at room temperature with occasional gentle mixing. Cells were washed three times in PBS and then incubated with the corresponding fluorescein isothiocyanate-conjugated secondary antibody (Molecular Probes, catalog. no. GM488, Eugene, OR; 1:500 dilution) for 30 min in the dark at room temperature. Cells were washed three times in PBS and resuspended in media for the assay. Fluorescence analysis was done on a Beckman FC500 flow cytometer (Beckman Coulter, Hialeah, FL). Control experiments contained only the appropriate secondary antibody.
Real Time Calcium Imaging
To evaluate cellular response to PAR-2 agonists (50 μm SLIGRL-NH2 or 3.23 μm KLK5), real time calcium imaging was used. The PAR-2-activating peptide SLIGRL-NH2 has been shown to be specific for activation of PAR-2 over PAR-1 and PAR-4 at the concentrations used in this study (20–50 μm) (37, 38). Cell lines (SCC1 and OKF6/T) were grown to 70% confluence on poly-l-lysine-coated 14-mm glass-bottom microwell dishes (Mat Tek, Ashland, MA). The cells were washed three times with artificial cerebrospinal fluid (140 mm NaCl, 5 mm KCl, 2 mm CaCl2, 10 mm Hepes, 24 mm glucose, pH 7.2) and incubated with 1 μm fura-2-acetoxymethyl ester at room temperature for 30–45 min. After incubation, the cells were washed three times in artificial cerebrospinal fluid and incubated for 15–30 min in the same solution to allow de-esterification of intracellular acetoxymethyl esters. The dish was mounted onto an imaging chamber and placed on the stage of a Nikon Eclipse TE 2000-S microscope (Nikon Instruments, Melville, NY). Using the ×40 objective, individual cells were selected using the NIS-Elements AR 3.0 software (Nikon) and exposed to alternating 340 and 380 nm light at 2-s intervals from a xenon lamp using a Lambda LS shutter. Images were acquired with a Cascade II 512 camera (Photometrics, Tucson, AZ) to obtain a stable baseline prior to addition of the PAR-2 agonist and monitoring of the ratiometric trace for each individual cell. The calcium response was calculated by subtracting the baseline [Ca2+]i from the peak value. De-sensitization experiments were performed by incubating cells with PAR-2 agonist peptide SLIGRL-NH2 (50 μm) prior to addition of KLK5 (3.23 μm) and monitoring the calcium response as described above.
Western Blotting Analysis
To examine PAR-2 protein levels in cell lines, cells were grown to 70% confluence and harvested in lysis buffer (modified RIPA buffer, with an added protease inhibitor mixture tablet), and protein concentration of samples was determined using a detergent-compatible protein assay kit (Bio-Rad). Proteins were separated by SDS-PAGE and then transferred onto a polyvinylidene di-fluoride (PVDF) microporous membranes (Millipore). After blocking nonspecific binding to membranes in 3% BSA in TBST for 1 h at room temperature, membranes were incubated with primary anti-PAR-2 antibodies for 3 h at room temperature or overnight at 4 °C and then with HRP-conjugated secondary antibodies. Immunoreactivity was determined by SuperSignal West Dura Extended Duration Substrate kit (Thermo Scientific). Blots were stripped with 0.4 n glycine, pH 2.8, for 30 min, washed three times in TBST, then blocked again, and probed with antibodies against GAPDH to ascertain equal protein loading. Images were acquired with Fuji Film LAS-4000 luminescent image analyzer. Western blots were performed in triplicate.
Immunofluorescence
Cells were plated on 22-mm2 glass coverslips in 6-well plates to 50% confluence to ensure areas of separated cell clusters, washed three times in PBS, and serum-starved overnight. Cells were treated with 100 ng/ml TNFα (1.9 nm, positive control for Nf-κB activation and RelA nuclear translocation), with 20 μm SLIGRL-NH2, or with 3.23 μm KLK5 for 30–120 min. Cells were washed once with PBS, and then fixed in 4% paraformaldehyde in PBS containing 0.12 m sucrose for 20 min at room temperature and permeabilized for 5 min in 0.3% Triton in PBS. Cells were rinsed twice in PBS and blocked with 10% BSA in PBS for 1 h. Primary antibody in 1% BSA was applied for 1 h at room temperature (1:100 dilution). Cells were washed three times in PBS followed by application of the secondary antibody (Alexa-Fluor 488) for 30 min (1:500 dilution). Cells were washed three times in PBS and rinsed one time with water, and coverslips were allowed to air-dry protected from light. Coverslips were inverted onto 10 μl of Vectashield containing DAPI on glass slides. Fluorescence was examined on an inverted AMG EVOS All-In-One digital microscope.
Real Time Quantitative PCR for mRNA and miRNA
The following primer sets were employed for comparative quantitation of mRNA: IL8 forward, 5′-GAGGGTTGTGGAGAAGTTTTTG-3′, and reverse, 5′-CTGGCATCTTCACTGATTCTTG-3′; IL1A forward, 5′-GTCTCTGAATCAGAAATCCTTCTATC-3′, and reverse, 5′-CATGTCAAATTTCACTGCTTCATCC-3′; MMP9 forward, 5′-GATGCGTGGAGAGTCGAAAT-3′, and reverse, 5′-CACCAAACTGGATGACGATG-3′; endogenous control gene phosphoglycerate kinase (PGK1) forward, 5′-GGGCTGCATCACCATCATAGG-3′, and PGK1 reverse, 5′-GAGAGCATCCACCCCAGGAAG-3′. Oligonucleotides were custom-synthesized by IDT (Coralville, Iowa). Cell lines were grown in 6-well plates to 70% confluence, treated overnight with the appropriate inhibitor or vehicle in serum-free media, and then treated with SLIGRL-NH2 (20 μm, or vehicle control) in the continued presence of inhibitor. For inhibitor studies, cells were pretreated for 16 h with 100 μm Nf-κB inhibitor sc-514, 20 μm Nf-κB inhibitor SB747651A, 1 μg/ml (8.5 nm) general Gα inhibitor pertussis toxin, or the appropriate vehicle in serum-free medium. Cells were then stimulated with SLIGRL-NH2 (20 μm) at time points from 30 min to 6 h. RNA was extracted with TRIzol reagent (Invitrogen), and reverse transcription was performed with 5 μg of the total RNA from each cell population using the RT2 First Strand kit or 1 μg of total RNA using the miScript II RT kit (Qiagen) with Hi-Flex buffer. Real time PCR was performed with SYBR Green Master Mix (Applied Biosystems, Foster City, CA). PCR cycling conditions were 95 °C for 10 min followed by 40 cycles of 95 °C for 15 s and 55 °C for 1 min. Melt curve cycling consisted of 81 30-s cycles beginning at 55 °C and increasing by 0.5 to 95 °C. Each sample was analyzed in triplicate for each PCR measurement. Melting curves were checked to ensure specificity. Relative quantification of mRNA expression was calculated using the standard curve method with the endogenous housekeeping gene PGK level as normalizer and control sample as calibrator, or using the ΔΔCT method where the efficiency of amplification of the target gene was similar (within 10%) to the efficiency of the endogenous control gene (PGK1). The % inhibition of expression was calculated based on the up-regulation of IL8 by SLIGRL-NH2 alone as a control.
Extracted RNA was also evaluated for candidate microRNAs. Primer assays were purchased for candidate microRNAs and several putative normalizer genes from Qiagen (Gaithersburg, MD). A PCR array, the human inflammatory response and autoimmunity miScript miRNA PCR array, was also obtained from Qiagen. Reverse transcription was performed with 1 μg of RNA isolated above using the miScript II RT kit using the Hi-Spec buffer. Real time PCR was performed with the miScript SYBR Green PCR kit (Qiagen). PCR cycling conditions were 95 °C for 15 min followed by 40 cycles of 94 °C for 15 s, 55 °C for 30 s, and 70 °C for 30 s. Melt curve cycling was identical to the mRNA procedure listed above. Relative quantification was calculated as above with RNU6 and RNU44 as normalizers. For inhibitor studies, the % inhibition was calculated based on the effect of treatment with SLIGRL-NH2 alone. The mRNA and microRNA panel experiments were performed in triplicate. The results with the array and the panel were internally consistent. The microRNA qPCR experiments with inhibitors were conducted in duplicate. The results with the SLIGRL-NH2 positive controls (without inhibitors) were also consistent with previous results.
Murine Orthotopic OSCC Model
To evaluate the effect of reduced KLK5 expression on tumor progression, an orthotopic murine model of OSCC of the tongue, which produces tumors that closely resemble human OSCC, was utilized (17, 35, 36). Briefly, 6-week-old male athymic nu/nu mice (n = 10 per cohort) were anesthetized using 2.5% isofluorane, and a 1-ml syringe with a 25-gauge needle was used to inject 30 μl of SCC25-KLK5-KD cells (18) or SCC25-Vec cells (0.8 × 106 cells) into the lateral border of the tongue just anterior to the junction of the anterior ⅔ and posterior ⅓ of the tongue. After 9 weeks, mice were sacrificed, and tissues were fixed in paraformaldehyde, embedded in paraffin, and evaluated as described above. Tumor surface area was quantified by analysis of scanned H&E sections using Aperio ImageScope software. Data were exported to Excel for further analysis. The experiment was repeated in duplicate. All procedures were conducted with approval of the IACUC, University of Notre Dame.
Results
Expression of PAR-2 and KLK5 in Pre-malignant and Malignant Oral Lesions
To examine PAR-2 expression in pre-malignant and malignant human oral tissues, TMAs were evaluated by immunohistochemistry. PAR-2 expression was heterogeneous, with examples of weak and strong staining shown in Fig. 1, A and B, for premalignant and Fig. 1, C and D, for malignant oral tissues. As PAR-2 is also expressed by inflammatory cells, a contribution from these cells cannot be ruled out; however, extensive visible inflammatory influx in the TMA core sections was not evident by H&E staining (data not shown). As quantified in Table 1, PAR-2 staining is moderately enhanced in malignant oral tissues relative to pre-malignant lesions. KLK5 was also expressed in premalignant and malignant oral lesions (Fig. 1, E–H; Table 1). Similar to our previous results (17, 18), KLK5 expression is also enhanced in OSCC (Table 1).
FIGURE 1.
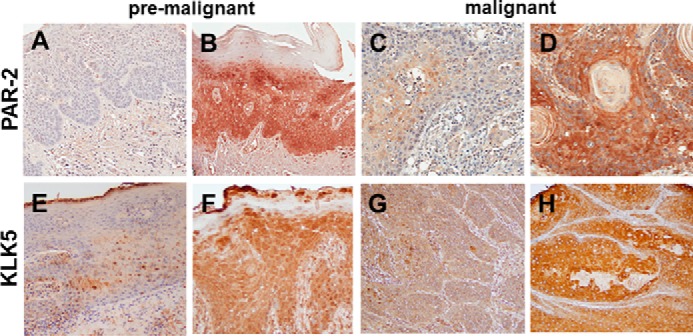
Immunohistochemical analysis of PAR-2 and KLK5 expression in pre-malignant oral lesions and OSCC. A and B, PAR-2 expression in pre-malignant oral lesions. A, 24% of lesions exhibited negative or low PAR-2 expression; B, 76% demonstrated moderate to high PAR-2 expression. PAR-2 antibody was used at a 1:50 dilution. Staining was detected using an avidin-biotin horseradish peroxidase system. C and D, PAR-2 expression in oral cancer. C, 24% of lesions exhibited negative or low PAR-2 expression; D, 76% demonstrated moderate to high PAR-2 expression. Antibody dilutions as in A. E and F, KLK5 expression in pre-malignant lesions. E, 37% of lesions exhibited negative or low KLK5 expression; F, 63% demonstrated moderate to high KLK5 expression. KLK5 antibody was used at a 1:50 dilution. G and H, KLK5 expression in oral cancer. Note that KLK5 expression in OSCC was previously published in Pettus et al. (17). In that study, 22% of lesions exhibited negative or low KLK5 expression, with 78% staining positive or strong positive. Magnification, ×200.
TABLE 1.
PAR-2 and KLK5 expression in human oral tissues
Microarrayed human oral tissues were immunostained for PAR-2 or KLK5 as described under “Materials and Methods,” and staining was scored as absent, weak, moderate, or strong. The average percentage of cells scored as moderate/strong is shown. Mild, moderate, and severe refer to levels of dysplasia; CIS means carcinoma in situ; stage I-II indicates oral squamous cell carcinoma stage I-II.
| Pre-malignant |
Malignant, stage I–II | ||
|---|---|---|---|
| Mild/moderate | Severe/CIS | ||
| PAR-2 | 75% | 56% | 84% |
| n = 122 | n = 38 | n = 22 | |
| KLK5 | 65% | 55% | 83% |
| n = 180 | n = 47 | n = 24 | |
PAR-2 Activation and Nf-κB Signaling in OSCC Cell Lines
To complement human tissue staining, PAR-2 expression was examined in OSCC cell lines and compared with tert-immortalized oral keratinocytes (OKF6/T). Examination of OSCC and immortalized oral mucosal cell lines by both Western blotting and flow cytometry showed enhanced expression and surface localization of PAR-2 in OSCC cell lines (Fig. 2, A–E). To determine whether surface PAR-2 was active, cells were treated with the PAR-2-activating peptide SLIGRL-NH2. Robust PAR-2 activation was triggered by the activating peptide SLIGRL-NH2 in OSCC cell lines relative to immortalized oral keratinocytes as demonstrated by enhanced calcium mobilization (Fig. 2, F and G). Similarly, incubation of OSCC cells with KLK5 also enhanced calcium mobilization (Fig. 2H). Desensitization experiments (Fig. 2I), in which cells were treated with SLIGRL-NH2 prior to addition of KLK5, demonstrated that the KLK5-induced calcium response was due to PAR-2 activation. As PAR-2 regulation of pro-inflammatory mediators is effected principally through the Nf-κB signaling pathway, nuclear localization of RelA was examined following PAR-2 activation. Incubation of OSCC cell lines with either SLIGRL-NH2 or KLK5 induced RelA nuclear translocation (Fig. 3, A–F). Positive controls included incubation of cells with TNFα (1.9 nm), which induced robust RelA nuclear translocation (Fig. 3B).
FIGURE 2.

PAR-2 expression and activation in OSCC cell lines. A, Western blot showing elevated PAR-2 protein in SCC1 and SCC25 cell lines relative to immortalized oral keratinocyte line OKF6/T. Cell lysates were electrophoresed on 9% SDS-polyacrylamide gels and electroblotted to Immobilon. Upper panel, blots were probed with murine anti-PAR-2 antibody (1:100 dilution) followed by an HRP-conjugated secondary antibody (1:4000 dilution). Loading controls (lower panel) were probed with mouse anti-GAPDH (1:500) and an HRP-conjugated secondary antibody (1:10,000). The experiment was repeated in triplicate, and a representative blot is shown. B–E, flow cytometry analysis of cell surface PAR-2. B and D, OKF6/T; C and E, SCC1 cell lines were incubated on ice with vehicle (B and C) or anti-PAR-2 antibody (1:100) (D and E) followed by FITC-conjugated secondary antibody (1:500) and evaluated using a Beckman/Coulter FC500 cell sorter. F–I, analysis of PAR-2 activation-induced calcium signaling. Cell lines were loaded with Fura2AM and incubated with the PAR-2 agonist peptide SLIGRL-NH2 (50 μm) or KLK5 (3.23 μm), as indicated. Shown in each trace is a representative single cell Ca2+ response measured as 340/380 fluorescence intensity (F.I.) ratio. The calcium response is the difference in the peak value and baseline value. F, peptide (SLIGRL-NH2, 50 μm) activation of PAR-2 in SCC1 cell line. G, peptide (SLIGRL-NH2, 50 μm) activation of PAR-2 in OKF6/T cell line. H, KLK5 (3.23 μm) activation of PAR-2 in SCC1 cell line. I, de-sensitization experiment showing lack of additional calcium response in SCC1 cells treated with SLIGRL-NH2 (50 μm) followed by KLK5 (3.23 μm).
FIGURE 3.
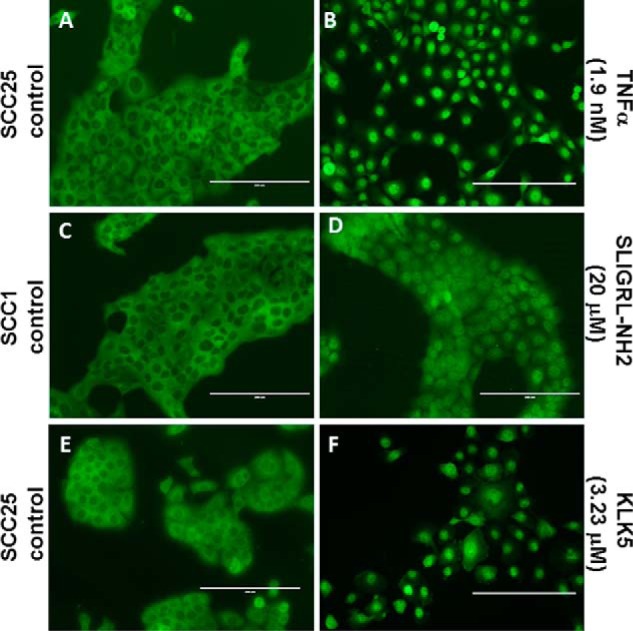
PAR2 induces RelA nuclear translocation. Cells grown on coverslips in serum-free media were untreated (A, C, and E) or treated with TNFα (100 ng/ml, 1.9 nm, 30 min) (B), SLIGRL-NH2 (20 μm, 85 min) (D), or KLK5 (3.23 μm, 60 min) (F). After washing and fixing/permeabilization, cells were treated with antibody to RelA, a FITC secondary, and mounted. Nuclear translocation of RelA was assessed on an inverted AMG EVOS All-In-One digital microscope.
PAR-2 Activation Regulates Both mRNA and MicroRNA Expression
Numerous studies have shown that PAR-2 activation induces Gα-mediated signaling, mobilizing intracellular calcium and Nf-κB signaling, and leading to increased expression of pro-inflammatory mRNAs (11–15). To determine whether PAR-2 activation in OSCC cell lines induces expression of candidate mRNAs known to be associated with inflammation in OSCC, expression of three pro-inflammatory genes known to be regulated downstream of PAR-2 activation was evaluated (27, 39, 40). Interleukin 8 (CXCL8 gene, IL8 protein) is a member of the pro-inflammatory CXC chemokine gene family. Expression levels are elevated in the saliva of oral cancer patients, and high serum IL8 levels correlate with a poor clinical outcome in OSCC (41, 42). The interleukin encoded by the IL1A gene is a potent mediator of inflammation and is involved in epidermal barrier function (43). Expression of IL1A can trigger inflammation leading to tumor formation in differentiated epidermal cells (44). Matrix metalloproteinase 9 (MMP9) is an inflammation-associated marker that may be used to differentiate OSCC from leukoplakia (45). Up-regulation of MMP9 correlates with enhanced inflammation and advanced metastatic OSCC (46). As shown in Fig. 4A, activation of PAR-2 in OSCC cells increased expression of control pro-inflammatory mRNAs encoding IL8, IL1A, and MMP9.
FIGURE 4.
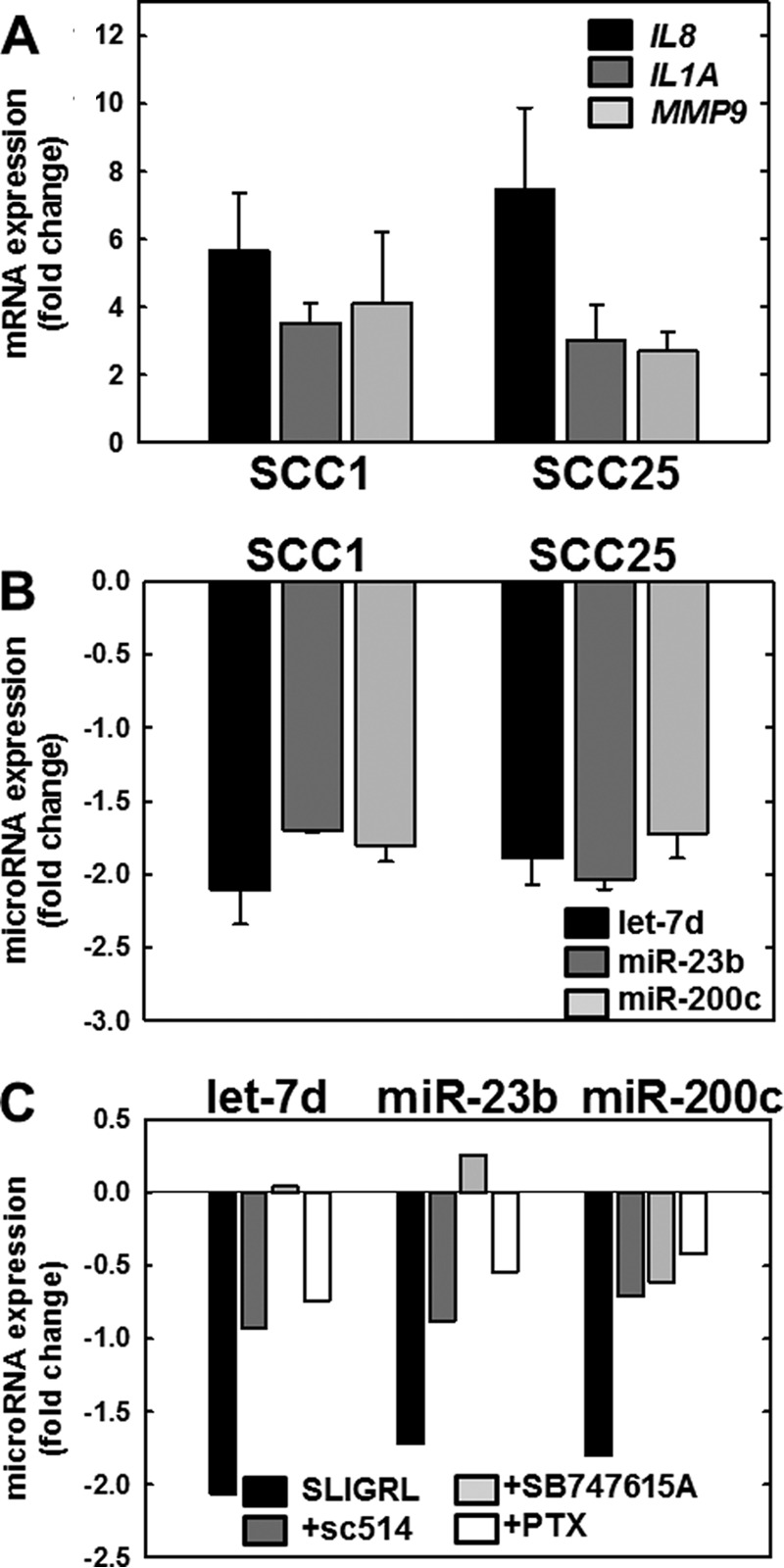
PAR-2 activation regulates expression of mRNAs and microRNAs in OSCC cell lines. A, PAR-2 activation increases pro-inflammatory mRNAs. PAR-2 was activated in SCC1 or SCC25 cell lines, as indicated, by incubation with SLIGRL-NH2 (20 μm, 2 h) followed by immediate RNA isolation and processing for RT-qPCR as described under “Materials and Methods.” Data shown are fold-change in expression of IL8 (black bar), IL1A (dark gray bar), and MMP9 (light gray bar) relative to untreated cells. Experiments were repeated in triplicate; error bars show S.E. B, PAR-2 activation decreases anti-inflammatory microRNAs. PAR-2 was activated in SCC1 or SCC25 cell lines, as indicated, by incubation with SLIGRL-NH2 (20 μm, 2 h) followed by immediate RNA isolation and processing for RT-qPCR as described under “Materials and Methods.” Data shown are fold-change in expression of let-7d (black bar), miR-23b (dark gray bar), and miR-200c (light gray bar) relative to untreated cells. Experiments were repeated in triplicate; error bars show S.E. C, PAR-2-mediated microRNA suppression is reversed by Nf-κB inhibitors. Cells were incubated with vehicle (control, black bar) or pretreated overnight with inhibitors as indicated: sc-514 (100 μm, dark gray bar), SB747651A (20 μm, light gray bar), or pertussis toxin (PTX) (1 μg/ml, 8.5 nm white bar) prior to activation of PAR-2 with SLIGRL-NH2 (20 μm, 2 h) in the continued presence of inhibitor. RNA was isolated immediately and used as template for RT-qPCR. Data shown are fold-change in expression of let-7d, miR-23b, and miR-200c relative to SLIGRL-NH2-activated cells without the addition of an inhibitor. Experiments were performed in duplicate.
MicroRNAs are also involved in inflammation; however, regulation of inflammation-related microRNAs by PAR-2 has not been investigated. We therefore investigated a panel of inflammation-related microRNAs and a commercially available qPCR array of inflammation- and immunity-related microRNAs. Our results showed that the anti-inflammatory microRNAs (miRNAs) let-7d, miR-23b, and miR-200c are consistently suppressed in OSCC cell lines after treatment with SLIGRL-NH2 (Fig. 4B). Pre-treatment with pertussis toxin, an inhibitor of Gα signaling, reversed PAR-2-mediated down-regulation of let-7d, miR-23b, and miR-200c (Fig. 4C, white bars). Similarly, inhibitors of Nf-κB signaling, including sc-514 (which inhibits IKKβ phosphorylation of RelA at Ser-536) and SB 747651A (which prevents MSK1-mediated RelA phosphorylation at Ser-276), also reversed PAR-2-mediated suppression of all three microRNAs (Fig. 4C, dark gray and light gray bars, respectively).
Down-regulation of KLK5 Expression Reduces Tumor Progression and Inflammation
To evaluate the effect of KLK5 expression on tumor progression, an orthotopic murine tongue xenograft model was utilized (17, 35, 36). Cells with diminished KLK5 expression (designated SCC25-KLK5-KD) (18) or vector controls (SCC25-Vec) were injected into the lateral border of the base of the tongue and allowed to grow for 9 weeks. Examination of the resulting tumors by H&E staining showed that SCC25-Vec cells formed moderately differentiated SCC. Some tumors were poorly circumscribed with invasive cords (Fig. 5, A–C) with evident vascular and perineural invasion (data not shown). By contrast, SCC25-KLK5-KD cells formed smaller, well circumscribed, and well differentiated tumors with numerous keratin pearls and with fewer examples of vascular or perineural invasion (Fig. 5, D–F). Tumors formed from SCC25-KLK5-KD cells were significantly smaller than tumors formed from SCC25-Vec cells (Fig. 5G). As our data showed that PAR-2 activation enhanced pro-inflammatory mRNAs and reduced expression of anti-inflammatory miRNAs, inflammatory cell recruitment to the oral tumor microenvironment was assessed. Tumors initiated with SCC25-KLK5-KD cells had significantly less lymphatic infiltrate relative to tumors initiated with SCC25-Vec cells (Fig. 6, A–C). Tryptase-positive mast cells were also significantly reduced in tumors from SCC25-KLK5-KD cells relative to SCC25-Vec cells (Fig. 6, D–F).
FIGURE 5.
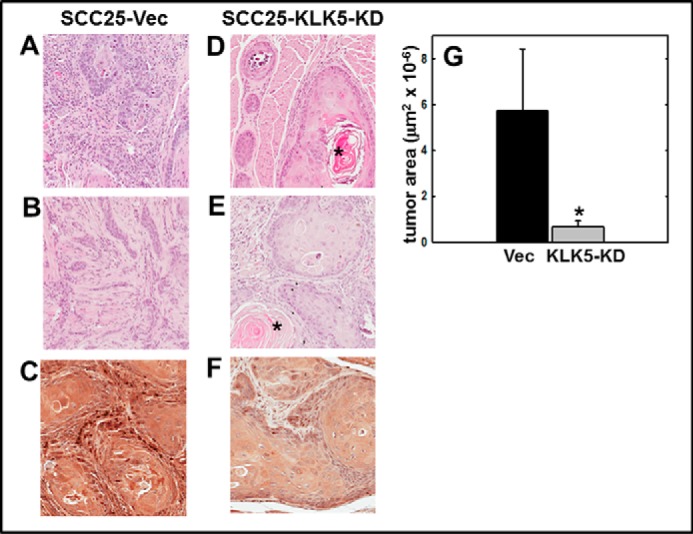
KLK5 silencing inhibits tumor growth and progression in an orthotopic murine OSCC xenograft model. Cells (0.8 × 106 in 30 μl of PBS) with diminished KLK5 expression (designated SCC25-KLK5-KD) or vector controls (SCC25-Vec) were injected into the lateral border of the base of the tongue and allowed to grow for 9 weeks. Following sacrifice, tumors were harvested, processed for histology, and stained with H&E. A and B, representative tumors formed from SCC25-Vec control cells are poorly circumscribed with invasive cords of tumor cells, characteristic of moderately differentiated OSCC. Magnification, ×200. C, section of SCC25-Vec tumor immunostained with anti-KLK 5 antibody (1:200) and peroxidase-conjugated secondary antibody. Magnification, ×400. D and E, representative tumors formed from SCC25-KLK5-KD cells with silenced KLK5 expression are well circumscribed with pushing tumor margins and keratin pearls (*), characteristic of well differentiated OSCC. Magnification, ×200. F, representative section of SCC25-KLK5-KD tumor immunostained with anti-KLK5 antibody. Magnification, ×400. G, quantitation of tumor area. Tumors formed from SCC25-Vec cells were ∼8-fold larger than SCC25-KLK5-KD tumors (p = 0.002).
FIGURE 6.

KLK5 silencing reduces tumor inflammatory infiltrate. A–C, inflammatory cells were enumerated from H&E-stained sections for each high power field containing or immediately adjacent to tumor nests. Results were presented as counts per high power field. Magnification A and B, ×200. *, p = 0.001. D–F, tumor sections were evaluated for mast cell infiltration by staining for mast cell tryptase using murine anti-tryptase (1:100) and the Vector M.O.M. (mouse-on-mouse) kit according to the manufacturer's specifications. Tryptase-positive mast cells were quantified as described under “Materials and Methods.” Magnification C and D, ×200. *, p = 0.004.
Discussion
Chronic inflammation is an established risk factor for cancer development, and inflammatory cells are a component of the tumor microenvironment in virtually all solid tumors. Increased presence of inflammatory cells and inflammatory mediators contributes to the complex cross-talk between the tumor and its microenvironment, regulating proliferation, angiogenesis, and metastasis. Many pro-inflammatory mRNAs, including those encoding cytokines and chemokines, are regulated downstream of PAR-2 through activation of the transcription factor Nf-κB (16, 22–25). We have shown previously that the secreted serine proteinase KLK5 is overexpressed in OSCC (17). In this study, we show that KLK5 and PAR-2 expression are elevated in pre-malignant and malignant oral tissues. Interestingly, expression of the KLK5 inhibitor LEKTI (encoded by SPINK5) is lost in human OSCC (21, 22), indicative of unregulated KLK5 activity. KLK5-catalyzed activation of PAR-2 results in calcium mobilization and activation of Nf-κB signaling. The downstream effects on expression of pro-inflammatory mRNAs and suppression of anti-inflammatory microRNAs may modify the tumor microenvironment and alter tumor progression. This is supported by data showing that tumors initiated by cells with reduced KLK5 expression were less aggressive and exhibited reduced inflammatory cell influx relative to KLK5-expressing controls. Our data support a model wherein KLK5-mediated PAR-2 activation regulates the expression of inflammation-associated mRNAs and microRNAs, thereby modulating progression of oral tongue tumors.
One of the pro-inflammatory mediators regulated by PAR-2 activation is the chemokine interleukin 8 (IL8). Previous studies have shown that IL8 is significantly up-regulated in OSCC and is a biomarker associated with proliferation, angiogenesis, migration, invasion, metastasis, and poor prognosis (26–31). IL-8 contributes to cross-talk between the tumor and its microenvironment via induced differentiation of CD163-positive M2 macrophages from monocytes, which produce IL-10. Although it is clear that PAR-2 activation culminates in the transcription of pro-inflammatory mRNAs in OSCC, including IL8, IL1A, and MMP9, PAR-2 regulation of inflammation-related microRNAs in OSCC is a novel finding of this study. Here, we demonstrate that PAR-2 activation suppresses three anti-inflammatory microRNAs: let-7d, miR-23b, and miR-200c. let-7d has been shown to target pro-inflammatory mRNAs encoding IL6, IL13, and TLR4 (47–49). miR-23b limits tissue inflammation via suppression of Nf-κB activation and inflammatory cytokine expression by targeting TAB2, TAB3, and IKKα (50). miR-200c directly targets many pro-inflammatory genes, including IL-8, VEGFA, and VEGFR2. Additionally, miR-200c also targets IKKβ (IKBKB), thereby decreasing Nf-κB signaling and inhibiting scores of pro-inflammatory Nf-κB-target genes (51–53). Furthermore, as let-7d, miR-23b, and miR-200c are established tumor suppressor genes that are significantly down-regulated in OSCC, our data provide a potential mechanism whereby PAR-2 activation contributes to tumor progression (54–61).
In addition to targeting DICER1, which suppresses proliferation in numerous cell types, knockdown of let-7d promotes epithelial-mesenchymal transition (EMT), leads to expression of TWIST and SNAIL, and promotes invasion in OSCC cells (56). Down-regulation of let-7d is associated with poor survival, disease recurrence, and distant metastasis in oral cancer (62). Similarly, down-regulation of miR-200c correlates with EMT and loss of E-cadherin (61). miR-23b is a known suppressor of cancer metastasis. Putative target genes that are up-regulated in oral cancer include the proliferation and motility gene MARCKS, the RAS oncogene RAP1B, and the growth factor HDGFRP3 (58, 63). miR-23b also suppresses EMT by reduction of vimentin and Snail and up-regulation of E-cadherin (64–66). Thus, down-regulation of miR-23b mediates multiple steps in progression and metastasis, including EMT, tumor growth and survival, cell migration, invasion, and angiogenesis.
PAR-2-mediated up-regulation of IL-8 and suppression of let-7d, miR-23b, and miR-200c were reversed by pretreatment with inhibitors of Gα (pertussis toxin) and Nf-κB (sc-514 and SB747651A) signaling. We have previously shown that SB747651A and sc-514 are potent inhibitors of OSCC invasion (67). The restoration of anti-inflammatory/tumor suppressor microRNAs, inhibition of pro-inflammatory mediators, and the resulting effects on tumor cell behavior suggest the utility of PAR-2 antagonists or Nf-κB inhibitors in the treatment of inflammatory diseases and cancer. Although the inhibition of Nf-κB leads to undesirable effects due to the influence of Nf-κB target genes on numerous other pathways in health and disease, inhibition of PAR-2 may have clinical potential because murine PAR-2 knock-out mice are viable and exhibit very mild phenotypic changes (14). Although complete inhibition of the PAR-2 pathway has yet to be achieved, the most promising antagonist thus far has been the small molecule PAR-2 specific antagonist GB88. GB88 can reversibly inhibit activation of PAR-2 by proteolytic and non-proteolytic agonists at low concentrations, blocking intracellular Ca2+ release, cAMP stimulation, receptor internalization, and pro-inflammatory cytokine release (68). GB88 is also serum-stable and orally active and has already been utilized in a number of animal models of human inflammatory diseases and cancer. The results of this study suggest that evaluation of PAR-2 antagonists such as GB88 for efficacy in OSCC is warranted.
Author Contributions
J. J. J. designed, performed, and analyzed the experiments shown in Figs. 1–6 and drafted the paper. D. L. M. contributed to data analysis and interpretation of data in Fig. 4. R. J. generated the cell lines used in this study. Y. L. provided technical assistance with data collection for Figs. 2 and 4. Z. S. provided pathology support and assistance with cell and tissue staining in Figs. 1, 5, and 6. L. T. provided technical assistance with tissue-based studies in Figs. 1, 5, and 6. R. W. assisted with experiments shown in Fig. 3. R. B. performed and analyzed the experiments shown in Fig. 2. E. R. S. contributed key reagents for the experiments in Fig. 1. M. S. S. conceived and designed the study, supervised the study, assisted with development of methodology, provided data interpretation, and revised the paper. All authors reviewed the results and approved the final version of the manuscript.
This work was supported in part by National Institutes of Health NCI Research Grant RO1CA085870 (to M. S. S.) and NIDCR Grant F31DE021926 (to D. L. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- OSCC
- oral squamous cell carcinoma
- PAR-2
- protease-activated receptor-2
- KLK5
- kallikrein-5
- qPCR
- quantitative PCR
- TMA
- tissue microarray
- miRNA
- microRNA
- EMT
- epithelial-mesenchymal transition.
References
- 1. World Health Organization (2012) GLOBOCAN 2012: Estimated cancer incidence, mortality and prevalence worldwide in 2012 (globocan.iarc.fr/Pages/fact_sheets_cancer.aspx)
- 2. Neville B. W., and Day T. A. (2002) Oral cancer and precancerous lesions. CA Cancer J. Clin. 52, 195–215 [DOI] [PubMed] [Google Scholar]
- 3. Kramer I. R., Lucas R. B., Pindborg J. J., and Sobin L. H. (1978) Definition of leukoplakia and related lesions: an aid to studies on oral precancer. Oral Surg. Oral Med. Oral Pathol. 46, 518–539 [PubMed] [Google Scholar]
- 4. Lumerman H., Freedman P., and Kerpel S. (1995) Oral epithelial dysplasia and the development of invasive squamous cell carcinoma. Oral Surg. Oral Med. Oral Pathol. Oral Radiol Endod. 79, 321–329 [DOI] [PubMed] [Google Scholar]
- 5. Vedtofte P., Holmstrup P., Hjorting-Hansen E., and Pindborg J. J. (1987) Surgical treatment of premalignant lesions of the oral mucosa. Int. J. Oral Maxillofac. Surg. 16, 656–664 [DOI] [PubMed] [Google Scholar]
- 6. Coussens L. M., and Werb Z. (2002) Inflammation and cancer. Nature. 420, 860–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mantovani A., Allavena P., Sica A., and Balkwill F. (2008) Cancer-related inflammation. Nature 454, 436–444 [DOI] [PubMed] [Google Scholar]
- 8. Rao S. K., Pavicevic Z., Du Z., Kim J. G., Fan M., Jiao Y., Rosebush M., Samant S., Gu W., Pfeffer L. M., and Nosrat C. A. (2010) Pro-inflammatory genes as biomarkers and therapeutic targets in oral squamous cell carcinoma. J. Biol. Chem. 285, 32512–32521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Candido J., and Hagemann T. (2013) Cancer-related inflammation. J. Clin. Immunol. 33, S79–S84 [DOI] [PubMed] [Google Scholar]
- 10. Diakos C. I., Charles K. A., McMillan D. C., and Clarke S. J. (2014) Cancer-related inflammation and treatment effectiveness. Lancet Oncol. 15, e493–503 [DOI] [PubMed] [Google Scholar]
- 11. Vergnolle N., Wallace J. L., Bunnett N. W., and Hollenberg M. D. (2001) Protease-activated receptors in inflammation, neuronal signaling and pain. Trends Pharmacol. Sci. 22, 146–152 [DOI] [PubMed] [Google Scholar]
- 12. Rothmeier A. S., and Ruf W. (2012) Protease-activated receptor 2 signaling in inflammation. Semin. Immunopathol. 34, 133–149 [DOI] [PubMed] [Google Scholar]
- 13. Adams M. N., Ramachandran R., Yau M. K., Suen J. Y., Fairlie D. P., Hollenberg M. D., and Hooper J. D. (2011) Structure, function and pathophysiology of protease-activated receptors. Pharmacol. Ther. 130, 248–282 [DOI] [PubMed] [Google Scholar]
- 14. Sales K. U., Friis S., Konkel J. E., Godiksen S., Hatakeyama M., Hansen K. K., Rogatto S. R., Szabo R., Vogel L. K., Chen W., Gutkind J. S., and Bugge T. H. (2015) Non-hematopoietic PAR-2 is essential for matriptase-driven pre-malignant progression and potentiation of ras-mediated squamous cell carcinogenesis. Oncogene. 34, 346–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yau M. K., Liu L., and Fairlie D. P. (2013) Toward drugs for protease-activated receptor 2 (PAR2). J. Med. Chem. 56, 7477–7497 [DOI] [PubMed] [Google Scholar]
- 16. Briot A., Deraison C., Lacroix M., Bonnart C., Robin A., Besson C., Dubus P., and Hovnanian A. (2009) Kallikrein 5 induces atopic dermatitis-like lesions through PAR2-mediated thymic stromal lymphopoietin expression in Netherton syndrome. J. Exp. Med. 206, 1135–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pettus J. R., Johnson J. J., Shi Z., Davis J. W., Koblinski J., Ghosh S., Liu Y., Ravosa M. J., Frazier S., and Stack M. S. (2009) Multiple kallikrein (KLK 5, 7, 8, and 10) expression in squamous cell carcinoma of the oral cavity. Histol. Histopathol. 24, 197–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jiang R., Shi Z., Johnson J. J., Liu Y., and Stack M. S. (2011) Kallikrein-5 promotes cleavage of desmoglein-1 and loss of cell-cell cohesion in oral squamous cell carcinoma. J. Biol. Chem. 286, 9127–9135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Descargues P., Deraison C., Prost C., Fraitag S., Mazereeuw-Hautier J., D'Alessio M., Ishida-Yamamoto A., Bodemer C., Zambruno G., and Hovnanian A. (2006) Corneodesmosomal cadherins are preferential targets of stratum corneum trypsin- and chymotrypsin-like hyperactivity in Netherton syndrome. J. Invest. Dermatol. 126, 1622–1632 [DOI] [PubMed] [Google Scholar]
- 20. Furio L., and Hovnanian A. (2011) When activity requires breaking up: LEKTI proteolytic activation cascade for specific proteinase inhibition. J. Invest. Dermatol. 131, 2169–2173 [DOI] [PubMed] [Google Scholar]
- 21. Gonzalez H. E., Gujrati M., Frederick M., Henderson Y., Arumugam J., Spring P. W., Mitsudo K., Kim H. W., and Clayman G. L. (2003) Identification of 9 genes differentially expressed in head and neck squamous cell carcinoma. Arch. Otolaryngol. Head Neck Surg. 129, 754–759 [DOI] [PubMed] [Google Scholar]
- 22. Jayakumar A., Kang Y., Henderson Y., Mitsudo K., Liu X., Briggs K., Wang M., Frederick M. J., El-Naggar A. K., Bebök Z., and Clayman G. L. (2005) Consequences of C-terminal domains and N-terminal signal peptide deletions on LEKTI secretion, stability, and subcellular distribution. Arch. Biochem. Biophys. 435, 89–102 [DOI] [PubMed] [Google Scholar]
- 23. Macfarlane S. R., Sloss C. M., Cameron P., Kanke T., McKenzie R. C., and Plevin R. (2005) The role of intracellular Ca2+ in the regulation of proteinase-activated receptor-2 mediated nuclear factor κB signalling in keratinocytes. Br. J. Pharmacol. 145, 535–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rallabhandi P., Nhu Q. M., Toshchakov V. Y., Piao W., Medvedev A. E., Hollenberg M. D., Fasano A., and Vogel S. N. (2008) Analysis of proteinase-activated receptor 2 and TLR4 signal transduction: a novel paradigm for receptor cooperativity. J. Biol. Chem. 283, 24314–24325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Buddenkotte J., Stroh C., Engels I. H., Moormann C., Shpacovitch V. M., Seeliger S., Vergnolle N., Vestweber D., Luger T. A., Schulze-Osthoff K., and Steinhoff M. (2005) Agonists of proteinase-activated receptor-2 stimulate upregulation of intercellular cell adhesion molecule-1 in primary human keratinocytes via activation of NF-κB. J. Invest. Dermatol. 124, 38–45 [DOI] [PubMed] [Google Scholar]
- 26. Jian S. L., Hsieh H. Y., Liao C. T., Yen T. C., Nien S. W., Cheng A. J., and Juang J. L. (2013) Gα12 drives invasion of oral squamous cell carcinoma through up-regulation of proinflammatory cytokines. PLoS ONE 8, e66133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rajkumar K., Nandhini G., Ramya R., Rajashree P., Kumar A. R., and Anandan S. N. (2014) Validation of the diagnostic utility of salivary interleukin 8 in the differentiation of potentially malignant oral lesions and oral squamous cell carcinoma in a region with high endemicity. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 118, 309–319 [DOI] [PubMed] [Google Scholar]
- 28. Elashoff D., Zhou H., Reiss J., Wang J., Xiao H., Henson B., Hu S., Arellano M., Sinha U., Le A., Messadi D., Wang M., Nabili V., Lingen M., Morris D., et al. (2012) Prevalidation of salivary biomarkers for oral cancer detection. Cancer Epidemiol, Biomarkers Prev. 21, 664–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fujita Y., Okamoto M., Goda H., Tano T., Nakashiro K., Sugita A., Fujita T., Koido S., Homma S., Kawakami Y., and Hamakawa H. (2014) Prognostic significance of interleukin-8 and CD163-positive cell-infiltration in tumor tissues in patients with oral squamous cell carcinoma. PLoS ONE 9, e110378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Martin D., Galisteo R., and Gutkind J. S. (2009) CXCL8/IL8 stimulates vascular endothelial growth factor (VEGF) expression and the autocrine activation of VEGFR2 in endothelial cells by activating NFkappaB through the CBM (Carma3/Bcl10/Malt1) complex. J. Biol. Chem. 284, 6038–6042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Christofakis E. P., Miyazaki H., Rubink D. S., and Yeudall W. A. (2008) Roles of CXCL8 in squamous cell carcinoma proliferation and migration. Oral Oncol. 44, 920–926 [DOI] [PubMed] [Google Scholar]
- 32. Dickson M. A., Hahn W. C., Ino Y., Ronfard V., Wu J. Y., Weinberg R. A., Louis D. N., Li F. P., and Rheinwald J. G. (2000) Human keratinocytes that express hTERT and also bypass a p16(INK4a)-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol. Cell. Biol. 20, 1436–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rheinwald J. G., and Beckett M. A. (1981) Tumorigenic keratinocyte lines requiring anchorage and fibroblast support cultured from human squamous cell carcinomas. Cancer Res. 41, 1657–1663 [PubMed] [Google Scholar]
- 34. Shi Z., Johnson J. J., Jiang R., Liu Y., and Stack M. S. (2015) Decreased miR-146a is associated with aggressive human oral squamous cell carcinoma. Arch. Oral Biol. 60, 1416–1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Myers J. N., Holsinger F. C., Jasser S. A., Bekele B. N., and Fidler I. J. (2002) An orthotopic nude mouse model of oral tongue squamous cell carcinoma. Clin. Cancer Res. 8, 293–298 [PubMed] [Google Scholar]
- 36. Ghosh S., Koblinski J., Johnson J., Liu Y., Ericsson A., Davis J. W., Shi Z., Ravosa M. J., Crawford S., Frazier S., and Stack M. S. (2010) Urinary-type plasminogen activator receptor/α3β1 integrin signaling, altered gene expression, and oral tumor progression. Mol. Cancer Res. 8, 145–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Oikonomopoulou K., Hansen K. K., Saifeddine M., Vergnolle N., Tea I., Blaber M., Blaber S. I., Scarisbrick I., Diamandis E. P., and Hollenberg M. D. (2006) Kallikrein-mediated cell signaling: targeting proteinase-activated receptors (PARs). Biol. Chem. 387, 817–824 [DOI] [PubMed] [Google Scholar]
- 38. Lee M. C., and Huang S. C. (2008) Proteinase-activated receptor-1 (PAR(1)) and PAR(2) but not PAR(4) mediate contraction in human and guinea-pig gallbladders. Neurogastroenterol. Motil. 20, 385–391 [DOI] [PubMed] [Google Scholar]
- 39. Wojtukiewicz M. Z., Hempel D., Sierko E., Tucker S. C., and Honn K. V. (2015) Protease-activated receptors (PARs)-biology and role in cancer invasion and metastasis. Cancer Metastasis Rev. 34, 775–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Inaba H., Sugita H., Kuboniwa M., Iwai S., Hamada M., Noda T., Morisaki I., Lamont R. J., and Amano A. (2014) Porphyromonas gingivalis promotes invasion of oral squamous cell carcinoma through induction of proMMP9 and its activation. Cell Microbiol. 16, 131–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fujita Y., Okamoto M., Goda H., Tano T., Nakashiro K., Sugita A., Fujita T., Koido S., Homma S., Kawakami Y., Hamakawa H. (2014) Prognostic significance of interleukin 8 and CD163-positive cell infiltration in tumor tissues in patients with oral squamous cell carcinoma. PLoS ONE 9, e110378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li Y., St John M. A., Zhou X., Kim Y., Sinha U., Jordan R. C., Eisele D., Abemayor E., Elashoff D., Park N. H, and Wong D. T. (2004) Salivary transcriptome diagnostics for oral cancer detection. Clin. Cancer Res. 10, 8442–8450 [DOI] [PubMed] [Google Scholar]
- 43. Barland C. O., Zettersten E., Brown B. S., Ye J., Elias P. M., and Ghadially R. (2004) Imiquimod-induced interleukin-1α stimulation improves barrier homeostasis in aged murine epidermis. J. Invest. Dermatol. 122, 330–336 [DOI] [PubMed] [Google Scholar]
- 44. Arwert E. N., Lal R., Quist S., Rosewell I., van Rooijen N., and Watt F. M. (2010) Tumor formation initiated by nondividing epidermal cells via an inflammatory infiltrate. Proc. Natl. Acad. Sci. U.S.A. 107, 19903–19908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chang P. Y., Kuo Y. B., Wu T. L., Liao C. T., Sun Y. C., Yen T. C., and Chan E. C. (2013) Association and prognostic value of serum inflammation markers in patients with leukoplakia and oral cavity cancer. Clin. Chem. Lab. Med. 51, 1291–1300 [DOI] [PubMed] [Google Scholar]
- 46. Tanis T., Cincin Z. B., Gokcen-Rohlig B., Bireller E. S., Ulusan M., Tanyel C. R., and Cakmakoglu B. (2014) The role of components of the extracellular matrix and inflammation on oral squamous cell carcinoma metastasis. Arch. Oral Biol, 59, 1155–1163 [DOI] [PubMed] [Google Scholar]
- 47. Wang X., Cao L., Wang Y., Wang X., Liu N., and You Y. (2012) Regulation of let-7 and its target oncogenes (review). Oncol. Lett. 3, 955–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wagner S., Ngezahayo A., Murua Escobar H., and Nolte I. (2014) Role of miRNA let-7 and its major targets in prostate cancer. Biomed. Res. Int. 2014, 376326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kumar M., Ahmad T., Sharma A., Mabalirajan U., Kulshreshtha A., Agrawal A., and Ghosh B. (2011) Let-7 microRNA-mediated regulation of IL-13 and allergic airway inflammation. J. Allergy Clin. Immunol. 128, e1–10 [DOI] [PubMed] [Google Scholar]
- 50. Zhu S., Pan W., Song X., Liu Y., Shao X., Tang Y., Liang D., He D., Wang H., Liu W., Shi Y., Harley J. B., Shen N., and Qian Y. (2012) The microRNA miR-23b suppresses IL-17-associated autoimmune inflammation by targeting TAB2, TAB3 and IKK-α. Nat. Med. 18, 1077–1086 [DOI] [PubMed] [Google Scholar]
- 51. Rokavec M., Wu W., and Luo J. L. (2012) IL6-mediated suppression of miR-200c directs constitutive activation of inflammatory signaling circuit driving transformation and tumorigenesis. Mol Cell. 45, 777–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chuang T. D., and Khorram O. (2014) miR-200c regulates IL8 expression by targeting IKBKB: a potential mediator of inflammation in leiomyoma pathogenesis. PLoS ONE 9, e95370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shi L., Zhang S., Wu H., Zhang L., Dai X., Hu J., Xue J., Liu T., Liang Y., and Wu G. (2013) MiR-200c increases the radiosensitivity of non-small-cell lung cancer cell line A549 by targeting VEGF-VEGFR2 pathway. PLoS ONE 8, e78344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kozaki K., Imoto I., Mogi S., Omura K., and Inazawa J. (2008) Exploration of tumor-suppressive microRNAs silenced by DNA hypermethylation in oral cancer. Cancer Res. 68, 2094–2105 [DOI] [PubMed] [Google Scholar]
- 55. Childs G., Fazzari M., Kung G., Kawachi N., Brandwein-Gensler M., McLemore M., Chen Q., Burk R. D., Smith R. V., Prystowsky M. B., Belbin T. J., and Schlecht N. F. (2009) Low-level expression of microRNAs let-7d and miR-205 are prognostic markers of head and neck squamous cell carcinoma. Am. J. Pathol. 174, 736–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chang C. J., Hsu C.C., Chang C. H., Tsai L. L., Chang Y. C., Lu S. W., Yu C. H., Huang H. S., Wang J. J., Tsai C. H., Chou M. Y., Yu C. C., and Hu F. W. (2011) Let-7d functions as novel regulator of epithelial-mesenchymal transition and chemoresistant property in oral cancer. Oncol. Rep. 26, 1003–1010 [DOI] [PubMed] [Google Scholar]
- 57. Jakymiw A., Patel R. S., Deming N., Bhattacharyya I., Shah P., Lamont R. J., Stewart C. M., Cohen D. M., and Chan E. K. (2010) Overexpression of dicer as a result of reduced let-7 microRNA levels contributes to increased cell proliferation of oral cancer cells. Genes Chromosomes Cancer 49, 549–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ramdas L., Giri U., Ashorn C. L., Coombes K. R., El-Naggar A., Ang K. K., and Story M. D. (2009) miRNA expression profiles in head and neck squamous cell carcinoma and adjacent normal tissue. Head Neck 31, 642–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pérez-Sayáns M., Pilar G. D., Barros-Angueira F., Suárez-Peñaranda J. M., Fernández A. C., Gándara-Rey J. M., and García-García A. (2012) Current trends in miRNAs and their relationship with oral squamous cell carcinoma. J. Oral Pathol. Med. 41, 433–443 [DOI] [PubMed] [Google Scholar]
- 60. Gorenchtein M., Poh C. F., Saini R., and Garnis C. (2012) MicroRNAs in an oral cancer context–from basic biology to clinical utility. J. Dent. Res. 91, 440–446 [DOI] [PubMed] [Google Scholar]
- 61. Zidar N., Boštjančič E., Gale N., Kojc N., Poljak M., Glavač D., and Cardesa A. (2011) Down-regulation of microRNAs of the miR-200 family and miR-205, and an altered expression of classic and desmosomal cadherins in spindle cell carcinoma of the head and neck–hallmark of epithelial-mesenchymal transition. Hum. Pathol. 42, 482–488 [DOI] [PubMed] [Google Scholar]
- 62. Jamali Z., Asl Aminabadi N., Attaran R., Pournagiazar F., Ghertasi Oskouei S., and Ahmadpour F. (2015) MicroRNAs as prognostic molecular signatures in human head and neck squamous cell carcinoma: a systematic review and meta-analysis. Oral Oncol. 51, 321–331 [DOI] [PubMed] [Google Scholar]
- 63. Ono M., Kosaka N., Tominaga N., Yoshioka Y., Takeshita F., Takahashi R. U., Yoshida M., Tsuda H., Tamura K., and Ochiya T. (2014) Exosomes from bone marrow mesenchymal stem cells contain a microRNA that promotes dormancy in metastatic breast cancer cells. Sci. Signal. 7, ra63. [DOI] [PubMed] [Google Scholar]
- 64. Zhang H., Hao Y., Yang J., Zhou Y., Li J., Yin S., Sun C., Ma M., Huang Y., and Xi J. J. (2011) Genome-wide functional screening of miR-23b as a pleiotropic modulator suppressing cancer metastasis. Nat. Commun. 22, 554. [DOI] [PubMed] [Google Scholar]
- 65. Pellegrino L., Stebbing J., Braga V. M., Frampton A. E., Jacob J., Buluwela L., Jiao L. R., Periyasamy M., Madsen C. D., Caley M. P., Ottaviani S., Roca-Alonso L., El-Bahrawy M., Coombes R. C., Krell J., and Castellano L. (2013) miR-23b regulates cytoskeletal remodeling, motility and metastasis by directly targeting multiple transcripts. Nucleic Acids Res. 41, 5400–5412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Castilla M. Á., Moreno-Bueno G., Romero-Pérez L., Van De Vijver K., Biscuola M., López-García M. Á., Prat J., Matías-Guiu X., Cano A., Oliva E., and Palacios J. (2011) Micro-RNA signature of the epithelial-mesenchymal transition in endometrial carcinosarcoma. J. Pathol. 223, 72–80 [DOI] [PubMed] [Google Scholar]
- 67. Johnson J., Shi Z., Liu Y., and Stack M. S. (2014) Inhibitors of NF-κB reverse cellular invasion and target gene upregulation in an experimental model of aggressive oral squamous cell carcinoma. Oral Oncol. 50, 468–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Suen J. Y., Cotterell A., Lohman R. J., Lim J., Han A., Yau M. K., Liu L., Cooper M. A., Vesey D. A., and Fairlie D. P. (2014) Pathway-selective antagonism of proteinase activated receptor 2. Br. J. Pharmacol. 171, 4112–4124 [DOI] [PMC free article] [PubMed] [Google Scholar]