

Abstract
The aggregation of amyloid β protein (Aβ) is a fundamental pathogenic mechanism leading to the neuronal damage present in Alzheimer disease, and soluble Aβ oligomers are thought to be a major toxic culprit. Thus, better knowledge and specific targeting of the pathways that lead to these noxious species may result in valuable therapeutic strategies. We characterized some effects of the molecular chaperone clusterin, providing new and more detailed evidence of its potential neuroprotective effects. Using a classical thioflavin T assay, we observed a dose-dependent inhibition of the aggregation process. The global analysis of time courses under different conditions demonstrated that clusterin has no effect on the elongation rate but mainly interferes with the nucleation processes (both primary and secondary), reducing the number of nuclei available for further fibril growth. Then, using a recently developed immunoassay based on surface plasmon resonance, we obtained direct evidence of a high-affinity (KD = 1 nm) interaction of clusterin with biologically relevant Aβ1–42 oligomers, selectively captured on the sensor chip. Moreover, with the same technology, we observed that substoichiometric concentrations of clusterin prevent oligomer interaction with the antibody 4G8, suggesting that the chaperone shields hydrophobic residues exposed on the oligomeric assemblies. Finally, we found that preincubation with clusterin antagonizes the toxic effects of Aβ1–42 oligomers, as evaluated in a recently developed in vivo model in Caenorhabditis elegans. These data substantiate the interaction of clusterin with biologically active regions exposed on nuclei/oligomers of Aβ1–42, providing a molecular basis for the neuroprotective effects of the chaperone.
Keywords: Alzheimer disease, amyloid β (Aβ), Caenorhabditis elegans, chaperone, kinetics, surface plasmon resonance (SPR), clusterin, kinetics of fibril formation
Introduction
Alzheimer disease (AD)2 is a devastating neurodegenerative disease. The majority of AD cases belong to the sporadic form of AD, which tends to occur later in life. One in 20 people over 65 and one in five over 85 have AD. Because the elderly population will increase 3- to 4-fold by 2050 in highly industrialized regions and increasing age is the strongest risk factor in developing the disease, AD will become a huge social and economic burden (1).
One of the main pathological hallmarks of AD is the presence of amyloid deposits (plaques), mainly made of amyloid β protein (Aβ), in the brain parenchyma. Aβ is formed from sequential cleavage of the amyloid β protein precursor, with the most common species containing either 40 (Aβ1–40) or 42 (Aβ1–42) amino acids. The aggregation of Aβ peptides, Aβ1–42 in particular, is thought to be a fundamental pathogenic mechanism leading to the neuronal damage present in AD. The kinetics of amyloid fibril formation from the soluble Aβ monomers are characterized on a macroscopic level by a lag phase, a growth phase, and a final plateau. Oligomeric intermediates, which have been found to be more toxic than the end-stage fibrillar amyloid species (2), are formed during the first stages. Even though the mechanism of toxicity is not yet understood, generic structural features of oligomers may influence toxicity. Recent data (3, 4) suggest that the exposure of hydrophobic motifs is the main conformational determinant of Aβ oligomer toxicity, whereas size and secondary structures may be less important (3). These findings suggest that better knowledge and specific targeting of the pathways that lead to these toxic oligomeric species may result in valuable therapeutic strategies.
Several classes of molecules, e.g. small molecules, peptides, antibodies, and other proteins, have been reported to influence the aggregation of Aβ peptides. One important class of natural inhibitors is molecular chaperones (5–7).
Molecular chaperones are endogenous proteins that assist the folding/unfolding and assembly/disassembly of proteins or other macromolecular structures (7, 8). Particularly, they have the potential to suppress the formation of aggregates and promote the clearance of misfolded species (7, 8). A key role is played by secreted glycoproteins with ATP-independent chaperone activity, which selectively recognize misfolded/aggregated proteins, preventing inappropriate accumulation of misfolded and aggregated proteins in the extracellular space (7, 8). One of these chaperones is clusterin (also known as apolipoprotein J), whose gene locus is the third strongest known genetic risk factor for late-onset AD (9). Clusterin is a multifunctional, highly glycosylated, heterodimeric protein expressed by a wide variety of tissues and is found in many extracellular fluids (7, 8). It inhibits stress-induced amorphous protein aggregation and the fibrillar aggregation of many amyloidogenic proteins and peptides (7, 10–14). Interestingly, it has been suggested that clusterin preferentially binds to hydrophobic regions exposed on misfolded/aggregated proteins regardless of their identity (8, 15), favoring their degradation or rendering them less toxic because of coverage of hydrophobic patches (12). In vitro data show, in particular, that clusterin hinders the kinetics of Aβ1–42 fibril formation (11), which might be due to the formation of stable complexes with small Aβ1–40/Aβ1–42 oligomers (16, 17).
In this study we further investigated the impact of clusterin on Aβ1–42 fibril formation and oligomerization. We used a thioflavin T (ThT) assay in a recently described experimental approach suitable for determining the dominating underlying molecular process (18, 19) to follow the effects of clusterin on the kinetics of fibril formation. Cumulative evidence suggests that protein aggregation might be dominated by one of the following general classes of polymerization mechanisms (18, 20, 21): inherently slow formation of aggregates from peptide monomers (homogenous primary nucleation); fibril breakage (monomer-independent secondary nucleation); and surface-catalyzed nucleation, where aggregates are formed from monomers on fibril surfaces (monomer-dependent secondary nucleation). To directly substantiate the interaction of clusterin with Aβ1–42 oligomers and evaluate the consequences of such interaction on the formation and toxicity of oligomers, we used two assays recently developed in our lab to specifically recognize biologically relevant soluble oligomers of synthetic Aβ1–42 (22–24). Thus, the transiently formed oligomeric assemblies were recognized by a SPR-based immunoassay, whereas the toxicity of these aggregated species was assessed by a behavioral assay in Caenorhabditis elegans, which recently has been used to detect toxic soluble assemblies of various amyloidogenic proteins (23, 25).
Experimental Procedures
Aβ1–42 Preparation
Depsi-Aβ1–42 was synthesized in-houseas described previously (26). In comparison with the highly aggregating Aβ1–42, the more soluble depsi form has a much lower propensity for spontaneous aggregation (26, 27). The final concentration of depsi-Aβ1–42 in the purified solutions was determined with UV absorption, using the theoretical molar extinction coefficient ϵ(214 nm)Aβ1–42 = 76848 m−1 cm−1 (28).
Aβ1–42 was then obtained from the depsi-peptide by a “switching” procedure involving a change in pH (26, 29, 30). We previously demonstrated (23) that freshly prepared Aβ1–42 solution obtained in this manner allows the preparation of reproducible, seed-free stock solutions of monomeric Aβ1–42 (26). This is routinely checked by circular dichroism, size exclusion chromatography, and SPR (26, 29). To prepare oligomer-containing solutions, freshly prepared Aβ1–42 was diluted in 10 mm PBS (pH 7.4) to a final concentration of 100 μm and incubated at 37 °C under quiescent conditions for 2 h (7). The appearance of oligomeric species is monitored by size exclusion chromatography and by the appearance of a pseudo-irreversible SPR binding signal when the solution is flowed onto the anti-Aβ antibody 4G8 (23) (also see below).
Kinetics of Fibril Formation
Aggregation kinetics were followed using an in situ ThT fluorescence assay (31) based on the increase of the fluorescence signal of ThT when bound to β sheet-rich structures (32). Different concentrations of Aβ1–42 were incubated, with and without plasma-derived native human clusterin (Biovendor), under quiescent conditions at 37 °C in microplate wells (Microplate Corning 3881, 96-well, low-binding, Corning Inc. Life Sciences, Acton, MA) in the presence of 20 μm ThT (100 μl solution/well). ThT fluorescence was measured every 2.5 min using an M200 Infinity plate reader (Tecan Italia Srl, Cernusco Sul Naviglio, Italy). The dye was excited at 440 nm, and the emission was measured at 495 nm. Comparison of the time course of aggregation by ThT and turbidity assays (with and without ThT in the reaction mixture) indicates that the presence of ThT does not influence the polymerization reaction (Fig. 1).
FIGURE 1.
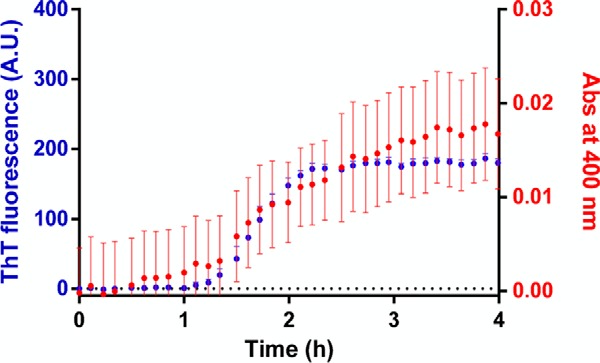
Kinetics of Aβ1–42 fibril formation in the absence and presence of ThT. Aβ1–42 was dissolved in phosphate buffer (pH 7.4), at a final concentration of 5 μm in the presence of 20 μm ThT for the ThT fluorescence assay (blue) or in the absence of ThT for the turbidity assay (red). The 96-microwell plate was loaded onto a plate reader. During incubation at 28 °C, ThT fluorescence and the absorbance at 400 nm were measured. Although the turbidity assay showed a much higher variability than the ThT assay, the time courses were similar (note the similar lag phases and the similar elongation rates). Each value is the mean ± S.D. of three replicates. A. U., arbitrary units; Abs, absorbance.
We followed the protocol described recently by Meisl et al. (19) using the online platform AmyloFit to analyze the ThT data and to determinate the microscopic assembly processes dominating the aggregation kinetics. The analysis is based on the integrated rate laws describing the evolution of total fibril mass in the presence of primary and, possibly, secondary nucleation events. The kinetic models that can be selected depend on the reaction orders nc and n2, describing the dependences of the primary and secondary pathways, respectively, on the initial monomer concentration. To determine these reaction orders, we first obtained the half-times of transition, i.e. the times corresponding to half the maximum ThT signal. Since the initial Aβ1–42 monomer concentration and the half-time of transition are related by a scaling law (half-time of transition ∝ (initial monomer concentration)γ), the slope of the log-log plot corresponds to the scaling exponent γ, which is approximately −nc/2 in the case of primary nucleation and −(n2+1)/2 in the case of secondary nucleation (18). The calculated parameters nc or n2 or both are then used to constrain and fit the proper kinetic model to the experimental reaction curves. The parameters obtained from global fitting are combinations of microscopic rate constants. For instance, in the case of primary nucleation, it is k+kn, whereas, for secondary nucleation, they are k+kn and k+k2, where kn, k+, k−, and k2 are, respectively, the primary nucleation rate, elongation rate, fragmentation rate, and secondary nucleation rate constants. This small number of free parameters allows the determination of the dominant aggregation mechanism.
Analysis of Aβ1–42 Oligomers by SPR
SPR studies were carried out with the ProteOn XPR36 protein interaction array system (Bio-Rad) based on SPR technology. The anti-Aβ antibody 4G8 (Covance, Princeton, NJ) was immobilized on GLC sensor chips (Bio-Rad) using amine-coupling chemistry, as described previously (33). The final immobilization levels were about 5000 resonance units (1 resonance unit = 1 pg protein/mm2). A “reference” surface, using the same immobilization procedure but without addition of the antibody, was always prepared in parallel. After rotation of the microfluidic system (33), oligomer-enriched Aβ1–42 solutions (see above) diluted to a final Aβ1–42 concentration of 1 μm were flowed over the chip surfaces for 2–5 min at a flow rate of 30 μl/ml, subsequently returning to running buffer (see below). According to our previous data, Aβ monomers were completely dissociated from 4G8 after 20 min in running buffer, when most oligomers are still pseudo-irreversibly bound to the antibody (23). Clusterin was either flowed for 10 min over the 4G8-bound oligomers or added to oligomer-containing solutions 10 min before injection over 4G8.
The running buffer, also used to dilute the samples, was 10 mm PBS containing 150 mm NaCl and 0.005% Tween 20 (PBST). All of these assays were performed at 25 °C. The sensorgrams (time course of the SPR signal in resonance units) were normalized to a baseline value of 0. The signal observed on the immobilized antibodies was corrected by subtracting the nonspecific response observed on the reference surface.
Toxicity Test: Pharyngeal Pumping Assay in C. elegans
The procedure described by Stravalaci et al. (23) was used. Briefly, N2 nematodes at the L3-L4 larval stage were collected with M9 buffer, centrifuged, and washed twice with 5 mm PBS (pH 7.4) to eliminate bacteria. 100 μm Aβ1–42, 10 mm H2O2, or 100 μg/ml of the light chain protein H7 (25) was incubated for 2 h at 37 °C in the absence and presence of clusterin (30–300 nm) and diluted 20 times. Worms (100/100 μl) were fed these solutions for 2 h at 37 °C in the absence of Escherichia coli to avoid any potential interference with the peptides. Control worms were incubated with 5 mm PBS (pH 7.4) (vehicle) or clusterin alone.
After 2 h of orbital shaking, the worms were transferred onto nematode growth medium plates seeded with OP50 E. coli. The pharyngeal pumping was scored 2 h later by counting the number of times the terminal bulb of the pharynx contracted over a 1-min interval (pumps per minute).
Results
Clusterin Inhibits Aβ1–42 Fibrillogenesis
We used a ThT assay in a recently described experimental design (18, 19) suitable to dissect the molecular mechanisms underlying the kinetics of Aβ1–42 fibril formation.
Initial studies were carried out to characterize the kinetics in the absence of clusterin, in particular to verify whether the results shown previously with recombinant Aβ1–42 (31) were replicated with our synthetic Aβ1–42. For this purpose, we used the same reducing conditions as indicated in Cohen et al. (31). Seven concentrations of freshly prepared seed-free Aβ1–42, ranging from 1–5 μm, were incubated at 37 °C under quiescent conditions with 20 μm ThT, measuring the fluorescence intensity every 2.5 min for 18 h. Fig. 2 illustrates the main results. Fig. 2A shows the raw data, whereas Fig. 2B shows the data after normalization on the maximum fluorescence intensity (at 18 h) to allow easier visual comparison of the polymerization rates. The half-times of transition (i.e. the time points at which 50% of the maximal ThT signal was reached) were inversely correlated to the concentration of Aβ1–42, with a linear dependence in the log-log plot (Fig. 2C). The slope of this straight line (scaling exponent γ) is −1.04 ± 0.07 (mean ± S.D. from three independent experiments), slightly higher than the value observed previously with recombinant Aβ1–42 (−1.3), which allows the determination of reaction numbers nc ≥ 2 and n2 ≥ 1. The global fitting of all ThT curves with the equations corresponding to the three possible theoretical models primary nucleation (nc = 3) (Fig. 2D), secondary nucleation (nc = n2 = 2) (Fig. 2E), and fibril breakage (nc = 2) (Fig. 2F) allowed us to identify the surface-induced secondary nucleation model as the one that best describes the experimental data (Fig. 2E), as shown previously for recombinant Aβ1–42 (31). The combined microscopic rate constants knk+ and k2k+ were estimated as fitting parameters. Assuming a fibril elongation rate of k+ = 3.00 × 106 m−1s−1 (31, 34), the microscopic rate constants were as follows: primary nucleation rate kn = 1.36 ± 0.53 × 10−4 m−1s−1 and secondary nucleation rate k2 = 7.32 ± 0.71 × 102 m−2s−1 (means ± S.D. from three independent experiments).
FIGURE 2.

Kinetics of Aβ1–42 fibril formation under reducing conditions. Freshly prepared Aβ1–42 was diluted at the indicated concentrations in 20 mm phosphate buffer (pH 8.0) containing 0.02% NaN3 and 200 μm EDTA and incubated with 20 μm ThT at 37 °C under quiescent conditions. Fluorescence intensities were collected every 2.5 min for 18 h. A, raw fluorescent values (mean ± S.D. of three replications). B, after normalization of the data in A on the corresponding maximal ThT value. C, log-log plot of Aβ concentration versus the half-time of transition (the time corresponding to 50% of the maximal ThT signal) for each of three independent experiments. D–F, fitting of the normalized data (only the mean is shown for clarity) with the three different models of microscopic assembly processes (fitting is shown with broken lines).
We then evaluated whether and how clusterin interferes with the aggregation of Aβ1–42 under the same experimental conditions. Fig. 3A shows the results of a representative experiment carried out with 3.6 μm Aβ1–42, showing that clusterin dose-dependently increases the half-time of transition from 2.6 h in the absence of clusterin to 5.6 h in the presence of 30 nm clusterin. The same experiment was repeated twice for different Aβ1–42 concentrations. Fig. 3B shows that clusterin shifted the log-log straight line to the right in a concentration-dependent manner, consistent with the slowdown of the polymerization kinetics, and also decreased the corresponding slopes from −1.04 in the absence of clusterin to −1.31 in the presence of 30 nm clusterin. As above, the global fitting of all ThT curves allowed us to extract the combined microscopic rate constants knk+ and k2k+ (18, 31), which appeared to be reduced by clusterin to a similar extent, approaching 1 order of magnitude at the higher concentration (Fig. 3, C and D). This could be due to either a similar effect of clusterin on both primary (kn) and secondary (k2) nucleation rates or an effect on the elongation rate (k+).
FIGURE 3.

Effects of clusterin on the kinetics of Aβ1–42 fibril formation under reducing conditions. Freshly prepared Aβ1–42 was diluted as in Fig. 2 and incubated with 20 μm ThT and different concentrations of clusterin (CLU) at 37 °C under quiescent conditions. Fluorescence intensities were collected every 2.5 min for 18 h. A, results from a representative experiment using 3.6 μm Aβ1–42. Fluorescence data are reported after normalization on the corresponding maximal ThT value. B, log-log plot of Aβ concentration versus the half-time of transition (the time corresponding to 50% of the maximal ThT signal). The results from two independent experiments are shown, with the corresponding regressions (solid and broken lines). C and D, clusterin-dependent changes of microscopic rate constants as determined by global fitting with the secondary nucleation model. Values are normalized on the rate constants obtained in the absence of clusterin.
Because the influence of clusterin on the primary nucleation events has already been demonstrated (12, 17), we further investigated the effect of clusterin on the secondary nucleation. To clarify this point, we carried out an experiment in which we reduced the influence of the primary nucleation events by adding preformed fibrils to the reaction solution (3.66 μm Aβ1–42 with or without clusterin). Under these conditions, polymerization initiated immediately without any lag phase. Clusterin slowed it down in a concentration-dependent manner, with an increase in half-time of transition from 2 to 6.6 h with 30 nm clusterin (Fig. 4A). Notably, even in the presence of clusterin, the shape of the curve is still hyperbolic, with no appearance of a lag phase. Numerical simulation, assuming the presence of seeds and a decrease in combined microscopic rate constants by 1 order of magnitude (as with 30 nm clusterin), indicates that the experimentally observed shape is expected when only the secondary nucleation is affected, whereas an effect on the elongation rate would have resulted in a sigmoidal shape (Fig. 4B).
FIGURE 4.
Seeded fibril growth of Aβ1–42 under reducing conditions in the presence of clusterin. A, normalized ThT fluorescence data of 3.66 μm freshly prepared Aβ1–42 incubated in the presence of 1% fibril seeds at different clusterin (CLU) concentrations at 37 °C and in the presence of 20 μm ThT under quiescent conditions. Fluorescence intensities were collected every 2.5 min for 30 h. B, numerical simulation of the seeded aggregation of 3 μm solution of Aβ1–42 in the presence of 1% fibril seeds using the kinetic parameter obtained from the kinetic analysis in Fig. 2 (red line), reducing the nucleation-related rate constants kn and k2 by 1 order of magnitude (green line), and reducing the elongation rate k+ by 1 order of magnitude (green broken line).
Clusterin Recognizes Oligomeric Forms of Synthetic Aβ1–42
We used a recently developed SPR-based immunoassay to directly investigate the interaction of clusterin with oligomeric species (23). This assay is based on the ability of the anti-Aβ antibody 4G8, immobilized on the sensor chip, to bind oligomers in a pseudo-irreversible manner (i.e. with very slow dissociation, likely because of multivalent binding), whereas monomers bind 4G8 with significantly faster dissociation rates, and higher-order aggregates are not detectable. Notably, we demonstrated that the oligomers captured by 4G8 are also bound by the OC antibody (23), an antibody believed to selectively target fibrillar oligomers (35). Fig. 5A shows the steps to determine the binding of clusterin to the oligomers captured by 4G8. At first the oligomer-enriched solution was flowed for 10 min over 4G8 and immobilized on the sensor chip, subsequently returning to running buffer. According to our previous data, Aβ monomers were completely dissociated from 4G8 after 20 min in running buffer, when most oligomers are still pseudo-irreversibly bound to the antibody (23). At this point, the injection of clusterin permitted the evaluation of its affinity for the captured oligomers. BSA was injected in parallel as a control for specificity. Fig. 5A shows that the chaperone, but not BSA, binds to oligomers in a dose-dependent manner, with an estimated KD value of 1.0 nm (Fig. 5B). We also observed some binding of clusterin to 4G8, i.e. in parallel chambers in which oligomers were not present (Fig. 5A). However, it was not possible to use this binding signal to correct that observed in the presence of oligomers, i.e. to obtain the oligomer-dependent signal, because the resulting sensorgrams had unusual shapes (i.e. increase of SPR signal during the dissociation phase). This suggests that clusterin binding to 4G8 is much lower when oligomers are bound to the antibody.
FIGURE 5.
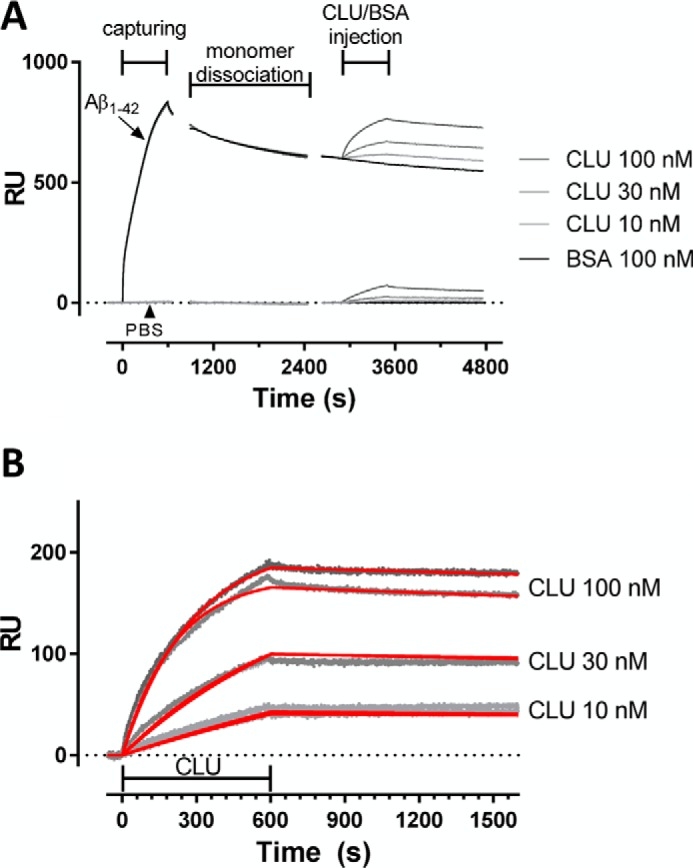
High-affinity binding of clusterin to oligomers. SPR studies showing binding of clusterin (CLU) to 4G8-captured Aβ1–42 oligomers. A, freshly dissolved Aβ1–42 (100 μm) was incubated at 37 °C, sampled after 2 h in 10 mm PBS diluted to 1 μm (pH 7.4), and injected over immobilized 4G8 for 10 min. As a control, PBS alone was injected in parallel. After allowing dissociation of Aβ monomers, clusterin solutions (10, 30, and 100 nm) or BSA (100 nm) were injected for 10 min, followed by 20-min dissociation. B, sensorgrams (i.e. time course of SPR signal in resonance units (RU) versus time) showing the dose-dependent binding of clusterin to Aβ1–42 oligomers after subtraction of the signal observed with BSA (reference). The graph reports the results obtained in two independent sessions. For each of them, the sensorgrams were globally fitted using the Langmuir equation, modeling a simple bimolecular interaction. The corresponding fittings are shown in red. According to this analysis, the estimated KD values were 0.9 and 1.1 nm.
Clusterin Inhibits the Binding of Aβ1–42 Oligomers to 4G8
As a first step in assessing the biological importance of the clusterin-Aβ1–42 interaction, we investigated the ability of the chaperone to counteract the binding of Aβ1–42 oligomers to a third protein (e.g. a putative oligomer receptor). For this, we evaluated whether a short incubation of clusterin with a solution enriched in preformed Aβ oligomers affects binding of the latter to 4G8. We observed that clusterin decreased this binding in a concentration-dependent manner (Fig. 6). The substoichiometric concentration of clusterin effective in this assay (clusterin, Aβ1–42 ratios ≥ 1:1000) is also consistent with an interaction with a subpopulation of Aβ1–42 aggregates.
FIGURE 6.
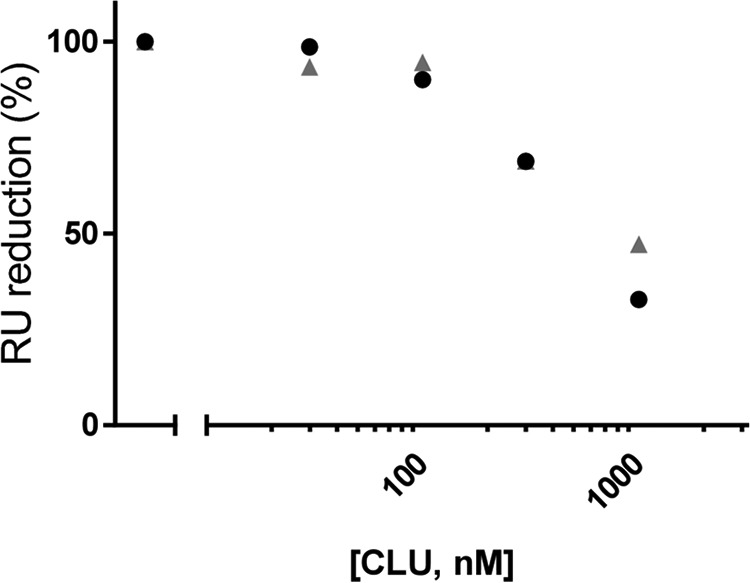
Clusterin inhibits the binding of Aβ1–42 oligomers to 4G8. A volume of 100 μm Aβ1–42 was incubated at 37 °C in PBS (pH 7.4) for 2 h to form oligomers. This solution was subsequently incubated for an additional 10 min in the absence or presence of 30, 100, 300, and 1000 nm clusterin (CLU), diluted 100-fold, and immediately injected over immobilized 4G8. The graph illustrates the binding signals in resonance units (RU) taken after 3 min of injection. Results from two independent experiments are shown.
Clusterin Binds to Biologically Relevant Aβ1–42 Oligomers
We then investigated the ability of clusterin to counteract Aβ oligomer toxicity in vivo using the invertebrate nematode C. elegans, whose pharyngeal behavior is sensitive to sublethal doses of chemical stressors. We previously reported that both rhythmic contraction and relaxation of the pharyngeal muscle in C. elegans, scored as “pumping rate,” were significantly impaired by feeding the nematodes with a solution enriched in synthetic Aβ1–42 oligomers but not monomers or fibrils (23). Here we evaluated whether preincubation of clusterin with Aβ1–42 oligomers prevented this effect (Fig. 7). Consistent with previous observations, Aβ1–42 oligomers significantly reduced the pumping rate of the worms by 17%. This toxic effect was dose-dependently reverted by clusterin. No effect was found using 30 nm clusterin (data not shown), whereas 200 nm clusterin completely abolished the toxic effect of Aβ1–42 oligomers despite also showing a toxic effect per se (Fig. 7). As above, the substoichiometric concentration of clusterin effective in this assay (clusterin, Aβ1–42 ratios ≥ 1:1000) is consistent with an effect because of the interaction with a subpopulation of Aβ1–42 aggregates. Clusterin had no protective effect on the decrease in pumping rate induced by hydrogen peroxide and the immunoglobulin light chain protein H7 (25). The latter is an amyloidogenic protein, although we used a solution containing monomers-dimers only, which are the toxic species in the pumping rate assay (25).
FIGURE 7.
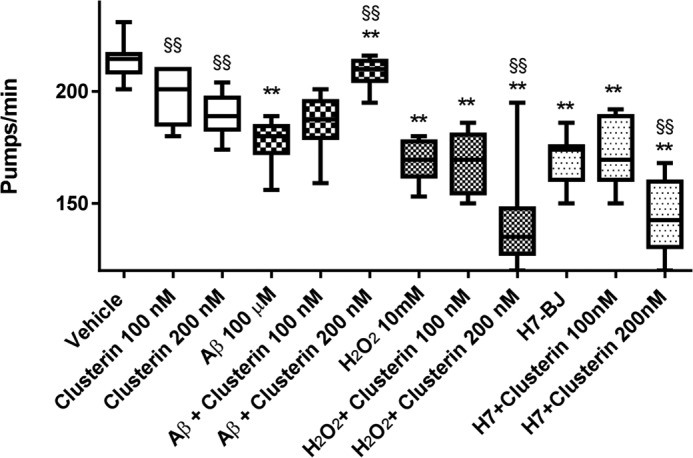
Effect of clusterin on the ability of Aβ1–42 oligomers to reduce the pharyngeal motility of C. elegans. 100 μm Aβ1–42, 10 mm H2O2, or 100 μg/ml of light chain protein H7 was incubated for 2 h at 37 °C in the absence and presence of 100 or 200 nm clusterin and diluted 20 times. Nematodes were fed these solutions for 2 h and then plated on nematode growth medium plates seeded with OP50 E. coli. The pharyngeal pumping rate was scored 2 h after plating. Data are expressed as minimum to maximum box and whisker plots (n = 10 worms/group). Two-way analysis of variance indicated a significant interaction between clusterin and Aβ1–42 (p < 0.01), and between clusterin and H7 (p < 0.05) but not between clusterin and H2O2. **, p < 0.01 versus the corresponding group without stressor; §§, p < 0.01 versus the corresponding group without clusterin (Sidak's multiple comparison test). The figure shows the results of a single study. The antagonism of clusterin on Aβ1–42-induced effects was replicated in another independent experiment, obtaining very similar results.
Discussion
Alzheimer disease is the most common cause of dementia among old people and is recognized as a public health priority. However, no effective therapy is currently available because the vast amount of effort put into the development of disease-modifying drugs has been unsuccessful. Most compounds were designed as inhibitors of Aβ aggregation, although, more recently, soluble toxic Aβ oligomers have been considered as more promising targets for effective therapeutic strategies. However, the reproducibility, reliability, and translatability of data obtained in vitro and in preclinical settings is often limited by the incomplete characterization of the experimental tools and a superficial interpretation of the results. For example, studies with the highly aggregating Aβ1–42 peptide require seed-free starting solutions, but this is not always checked. The biologically relevant oligomers likely represent just a subpopulation of the assemblies present in Aβ1–42 solutions, but most of the studies are usually carried out with heterogeneous mixtures of aggregated species, and the dissection of the microscopic assembly mechanisms affected by putative inhibitors is very important because hindrance of the primary nucleation or elongation would not change or might even dramatically increase the amount of oligomers formed during the aggregation reaction (6).
These points were all considered in this study, aiming to characterize the effects of a member of an important class of endogenous proteins, the molecular chaperones, whose physiological functions also include the recognition of misfolded protein aggregates and the prevention of their toxic effects (36). We chose clusterin because it is an established genetic risk factor for late-onset AD, suggesting a possible important role for this multifunctional globular glycoprotein in the pathogenesis of AD (37). Recent findings also indicate that clusterin binds to Aβ oligomers (12, 17) and prevents Aβ fibril formation (11, 17, 38, 39). However, an overall analysis of its effects is lacking.
The effects of clusterin on Aβ1–42 fibrillation kinetics was evaluated by using a classical in situ ThT assay in a recently proposed experimental design with a rigorous mathematical analysis (18, 19), allowing to extract much more information from the experimental data and to discriminate between the different possible aggregation mechanisms (18). We could thus confirm that the kinetics of fibril formation of synthetic Aβ1–42, like that described for recombinant Aβ1–42, is dominated by a fibril surface-induced secondary nucleation mechanism where nuclei are formed by monomer interaction with the fibril surface (31, 40). Nevertheless, the kinetic nucleation rates kn and k2 are smaller compared with literature data, which could be due to differences in the aggregation propensity of synthetic Aβ1–42 compared with recombinant Aβ1–42 (41). Clusterin interfered with both primary and secondary nucleation steps but not with the elongation rate. These data are consistent with the view that the chaperone interacts with early assemblies, preventing further steps. The molecular chaperones DNAJB6 (5) and Brichos (6) also inhibit primary or secondary nucleation events or both, thus reducing the amount of nuclei available for further fibril growth.
We also demonstrated that clusterin binds to Aβ1–42 oligomers with high affinity. The binding constant (KD 1 nm) is 1 order of magnitude smaller than that for the clusterin-related apolipoprotein E (ApoE) when binding to oligomers (42). ApoE, like clusterin, shows some chaperon-like activity, hindering oligomer formation (43) and promoting proteolytic degradation of Aβ (44, 45). Importantly, our SPR assay allowed us to investigate the specific binding of clusterin for a subpopulation of Aβ1–42 oligomers, i.e. those stably captured by the anti-Aβ antibody 4G8 immobilized on an SPR sensor chip.
We previously demonstrated that this is a transient oligomeric population of SDS-labile aggregates, generated during the incubation of synthetic Aβ1–42 and including globular species and short protofibrils, with a main hydrodynamic diameter of 10–30 nm (23). More importantly, different lines of evidence strongly suggest that these 4G8-binding oligomers are biologically active, being able to generate ionic currents in artificial lipid bilayers, and that they inhibit the physiological pharyngeal contractions in C. elegans, an in vivo assay for testing the toxic potential of Aβ oligomers (23). The binding to 4G8 indicates that these oligomers expose the central hydrophobic region of Aβ, implicated in the interactions underlying the formation and elongation of amyloid fibrils (46, 47). Our observation that clusterin prevents the binding of oligomers to 4G8 further substantiates that the chaperone also recognizes and shields these hydrophobic patches (12).
Because the exposure of hydrophobic motifs is considered the main conformational determinant of oligomer toxicity, with size and secondary structures being probably less important (12), it is tempting to speculate that clusterin might exert neuroprotective properties by recognizing this specific population of toxic oligomers with high affinity. Alternatively, clusterin might induce conformational changes toward non-toxic oligomers. Both views are consistent with the in vivo data obtained in C. elegans, showing that clusterin specifically antagonizes the toxic effects of Aβ1–42 oligomers on the pharyngeal pumping rate of the worms. Previous studies reported that clusterin either decreases (11, 12, 36, 48) or enhances (11, 38, 39) the cytotoxicity of Aβ1–42 aggregates. These contradictory results are likely due to the different experimental conditions (11). In our assay, clusterin was protective at a concentration of 200 nm (clusterin, Aβ1–42 = 1:500), which was lower than the concentrations (and ratios) used in most of the previous studies. Moreover, we emphasize that our assay is responsive to short term (2-h) exposure to toxic Aβ1–42 oligomers, a pivotal condition for studying the acute biological effects of these short-lived species (23).
We observed that clusterin dose-dependently reduced the pharyngeal pumping rate of the worms and, surprisingly, that the co-incubation of toxic concentrations of clusterin and Aβ1–42 oligomers completely prevented the toxic effects of both. This mutual inhibition is possibly due to the direct interaction between the two, burying toxicity-associated moieties.
In summary, our data indicate that the molecular chaperone clusterin interacts with biologically active regions, likely hydrophobic patches exposed on oligomers of Aβ1–42, reducing both their further aggregation and toxicity, with high affinity. It has been suggested that the preferential binding of clusterin to misfolded/aggregated proteins, regardless of their identity (8, 15), involves the binding pocket formed by amphipathic α-helical regions (8, 49). Further elucidation of the molecular determinants underlying this naturally occurring interaction may pave the way for the development of new synthetic clusterin mimetics (synthetic chaperones) with potential therapeutic applications in AD and, possibly, other protein misfolding diseases.
Author Contributions
M. B., M. Salmona, and M. G. designed the study and wrote the paper. M. B. conducted most of the experiments and analyzed the results with the assistance of M. Stravalaci for the SPR experiments and A. D. C. for the ThT studies. A. C. and A. R. synthesized and purified Aβ1–42. M. R. and L. D. conducted the C. elegans experiments. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Flamma Spa (Bergamo, Italy) for providing Fmoc amino acids for peptide synthesis. Purified immunoglobulin light chain H7 was provided by Prof. G. Merlini and Dr. P. Rognoni (IRCCS Policlinico San Matteo, Pavia, Italy). C. elegans and OP50 E. coli were provided by the Caenorhabditis Genetics Center, funded by the National Institutes of Health Office Research Infrastructure Programs (P40 OD010440).
This work was supported by the Banca Intesa-San Paolo. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- AD
- Alzheimer disease
- Aβ
- amyloid β
- ThT
- thioflavin T.
References
- 1. Minati L., Edginton T., Bruzzone M. G., and Giaccone G. (2009) Current concepts in Alzheimer's disease: a multidisciplinary review. Am. J. Alzheimers Dis. Other Demen. 24, 95–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bemporad F., and Chiti F. (2012) Protein misfolded oligomers: experimental approaches, mechanism of formation, and structure-toxicity relationships. Chem. Biol. 19, 315–327 [DOI] [PubMed] [Google Scholar]
- 3. Campioni S., Mannini B., Zampagni M., Pensalfini A., Parrini C., Evangelisti E., Relini A., Stefani M., Dobson C. M., Cecchi C., and Chiti F. (2010) A causative link between the structure of aberrant protein oligomers and their toxicity. Nat. Chem. Biol. 6, 140–147 [DOI] [PubMed] [Google Scholar]
- 4. Ladiwala A. R., Litt J., Kane R. S., Aucoin D. S., Smith S. O., Ranjan S., Davis J., Van Nostrand W. E., and Tessier P. M. (2012) Conformational differences between two amyloid β oligomers of similar size and dissimilar toxicity. J. Biol. Chem. 287, 24765–24773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Månsson C., Arosio P., Hussein R., Kampinga H. H., Hashem R. M., Boelens W. C., Dobson C. M., Knowles T. P., Linse S., and Emanuelsson C. (2014) Interaction of the molecular chaperone DNAJB6 with growing amyloid-β 42 (Aβ42) aggregates leads to sub-stoichiometric inhibition of amyloid formation. J. Biol. Chem. 289, 31066–31076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cohen S. I., Arosio P., Presto J., Kurudenkandy F. R., Biverstål H., Dolfe L., Dunning C., Yang X., Frohm B., Vendruscolo M., Johansson J., Dobson C. M., Fisahn A., Knowles T. P., and Linse S. (2015) A molecular chaperone breaks the catalytic cycle that generates toxic Aβ oligomers. Nat. Struct. Mol. Biol. 22, 207–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wyatt A. R., Yerbury J. J., Ecroyd H., and Wilson M. R. (2013) Extracellular chaperones and proteostasis. Annu. Rev. Biochem. 82, 295–322 [DOI] [PubMed] [Google Scholar]
- 8. Wyatt A. R., Yerbury J. J., Dabbs R. A., and Wilson M. R. (2012) Roles of extracellular chaperones in amyloidosis. J. Mol. Biol. 421, 499–516 [DOI] [PubMed] [Google Scholar]
- 9. Harold D., Abraham R., Hollingworth P., Sims R., Gerrish A., Hamshere M. L., Pahwa J. S., Moskvina V., Dowzell K., Williams A., Jones N., Thomas C., Stretton A., Morgan A. R., Lovestone S., et al. (2009) Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat. Genet. 41, 1088–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Humphreys D. T., Carver J. A., Easterbrook-Smith S. B., and Wilson M. R. (1999) Clusterin has chaperone-like activity similar to that of small heat shock proteins. J. Biol. Chem. 274, 6875–6881 [DOI] [PubMed] [Google Scholar]
- 11. Yerbury J. J., Poon S., Meehan S., Thompson B., Kumita J. R., Dobson C. M., and Wilson M. R. (2007) The extracellular chaperone clusterin influences amyloid formation and toxicity by interacting with prefibrillar structures. FASEB J. 21, 2312–2322 [DOI] [PubMed] [Google Scholar]
- 12. Mannini B., Cascella R., Zampagni M., van Waarde-Verhagen M., Meehan S., Roodveldt C., Campioni S., Boninsegna M., Penco A., Relini A., Kampinga H. H., Dobson C. M., Wilson M. R., Cecchi C., and Chiti F. (2012) Molecular mechanisms used by chaperones to reduce the toxicity of aberrant protein oligomers. Proc. Natl. Acad. Sci. U.S.A. 109, 12479–12484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hatters D. M., Wilson M. R., Easterbrook-Smith S. B., and Howlett G. J. (2002) Suppression of apolipoprotein C-II amyloid formation by the extracellular chaperone, clusterin. Eur. J. Biochem. 269, 2789–2794 [DOI] [PubMed] [Google Scholar]
- 14. McHattie S., and Edington N. (1999) Clusterin prevents aggregation of neuropeptide 106–126 in vitro. Biochem. Biophys. Res. Commun. 259, 336–340 [DOI] [PubMed] [Google Scholar]
- 15. Wyatt A. R., Yerbury J. J., and Wilson M. R. (2009) Structural characterization of clusterin-chaperone client protein complexes. J. Biol. Chem. 284, 21920–21927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Matsubara E., Soto C., Governale S., Frangione B., and Ghiso J. (1996) Apolipoprotein J and Alzheimer's amyloid β solubility. Biochem. J. 316, 671–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Narayan P., Orte A., Clarke R. W., Bolognesi B., Hook S., Ganzinger K. A., Meehan S., Wilson M. R., Dobson C. M., and Klenerman D. (2012) The extracellular chaperone clusterin sequesters oligomeric forms of the amyloid-β(1–40) peptide. Nat. Struct. Mol. Biol. 19, 79–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cohen S. I., Vendruscolo M., Dobson C. M., and Knowles T. P. (2012) From macroscopic measurements to microscopic mechanisms of protein aggregation. J. Mol. Biol. 421, 160–171 [DOI] [PubMed] [Google Scholar]
- 19. Meisl G., Kirkegaard J. B., Arosio P., Michaels T. C., Vendruscolo M., Dobson C. M., Linse S., and Knowles T. P. (2016) Molecular mechanisms of protein aggregation from global fitting of kinetic models. Nat. Protoc. 11, 252–272 [DOI] [PubMed] [Google Scholar]
- 20. Ferrone F. (1999) Analysis of protein aggregation kinetics. Methods Enzymol. 309, 256–274 [DOI] [PubMed] [Google Scholar]
- 21. Ferrone F. A. (2006) Nucleation: the connections between equilibrium and kinetic behavior. Methods Enzymol. 412, 285–299 [DOI] [PubMed] [Google Scholar]
- 22. Beeg M., Diomede L., Stravalaci M., Salmona M., and Gobbi M. (2013) Novel approaches for studying amyloidogenic peptides/proteins. Curr. Opin. Pharmacol. 13, 797–801 [DOI] [PubMed] [Google Scholar]
- 23. Stravalaci M., Bastone A., Beeg M., Cagnotto A., Colombo L., Di Fede G., Tagliavini F., Cantù L., Del Favero E., Mazzanti M., Chiesa R., Salmona M., Diomede L., and Gobbi M. (2012) Specific recognition of biologically active amyloid-β oligomers by a new surface plasmon resonance-based immunoassay and an in vivo assay in Caenorhabditis elegans. J. Biol. Chem. 287, 27796–27805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stravalaci M., Beeg M., Salmona M., and Gobbi M. (2011) Use of surface plasmon resonance to study the elongation kinetics and the binding properties of the highly amyloidogenic Aβ(1–42) peptide, synthesized by depsi-peptide technique. Biosens. Bioelectron. 26, 2772–2775 [DOI] [PubMed] [Google Scholar]
- 25. Diomede L., Rognoni P., Lavatelli F., Romeo M., del Favero E., Cantù L., Ghibaudi E., di Fonzo A., Corbelli A., Fiordaliso F., Palladini G., Valentini V., Perfetti V., Salmona M., and Merlini G. (2014) A Caenorhabditis elegans-based assay recognizes immunoglobulin light chains causing heart amyloidosis. Blood 123, 3543–3552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Beeg M., Stravalaci M., Bastone A., Salmona M., and Gobbi M. (2011) A modified protocol to prepare seed-free starting solutions of amyloid-β Aβ1–40 and Aβ1–42 from the corresponding depsipeptides. Anal. Biochem. 411, 297–299 [DOI] [PubMed] [Google Scholar]
- 27. Sohma Y., and Kiso Y. (2006) “Click peptides”: chemical biology-oriented synthesis of Alzheimer's disease-related amyloid β peptide (aβ) analogues based on the “O-acyl isopeptide method”. ChemBioChem 7, 1549–1557 [DOI] [PubMed] [Google Scholar]
- 28. Kuipers B. J., and Gruppen H. (2007) Prediction of molar extinction coefficients of proteins and peptides using UV absorption of the constituent amino acids at 214 nm to enable quantitative reverse phase high-performance liquid chromatography-mass spectrometry analysis. J. Agric. Food Chem. 55, 5445–5451 [DOI] [PubMed] [Google Scholar]
- 29. Balducci C., Beeg M., Stravalaci M., Bastone A., Sclip A., Biasini E., Tapella L., Colombo L., Manzoni C., Borsello T., Chiesa R., Gobbi M., Salmona M., and Forloni G. (2010) Synthetic amyloid-β oligomers impair long-term memory independently of cellular prion protein. Proc. Natl. Acad. Sci. U.S.A. 107, 2295–2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Taniguchi A., Sohma Y., Hirayama Y., Mukai H., Kimura T., Hayashi Y., Matsuzaki K., and Kiso Y. (2009) “Click peptide”: pH-triggered in situ production and aggregation of monomer Aβ1–42. ChemBioChem 10, 710–715 [DOI] [PubMed] [Google Scholar]
- 31. Cohen S. I, Linse S., Luheshi L. M., Hellstrand E., White D. A., Rajah L., Otzen D. E., Vendruscolo M., Dobson C. M., and Knowles T. P. (2013) Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. U.S.A. 110, 9758–9763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. LeVine H., 3rd (1993) Thioflavine T interaction with synthetic Alzheimer's disease β-amyloid peptides: detection of amyloid aggregation in solution. Protein Sci. 2, 404–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bravman T., Bronner V., Lavie K., Notcovich A., Papalia G. A., and Myszka D. G. (2006) Exploring “one-shot” kinetics and small molecule analysis using the ProteOn XPR36 array biosensor. Anal. Biochem. 358, 281–288 [DOI] [PubMed] [Google Scholar]
- 34. Buell A. K., Dhulesia A., White D. A., Knowles T. P., Dobson C. M., and Welland M. E. (2012) Detailed analysis of the energy barriers for amyloid fibril growth. Angew. Chem. Int. Ed. Engl. 51, 5247–5251 [DOI] [PubMed] [Google Scholar]
- 35. Kayed R., Head E., Sarsoza F., Saing T., Cotman C. W., Necula M., Margol L., Wu J., Breydo L., Thompson J. L., Rasool S., Gurlo T., Butler P., and Glabe C. G. (2007) Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol. Neurodegener. 2, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cascella R., Conti S., Tatini F., Evangelisti E., Scartabelli T., Casamenti F., Wilson M. R., Chiti F., and Cecchi C. (2013) Extracellular chaperones prevent Aβ42-induced toxicity in rat brains. Biochim. Biophys. Acta 1832, 1217–1226 [DOI] [PubMed] [Google Scholar]
- 37. Desikan R. S., Thompson W. K., Holland D., Hess C. P., Brewer J. B., Zetterberg H., Blennow K., Andreassen O. A., McEvoy L. K., Hyman B. T., Dale A. M., and Alzheimer's Disease Neuroimaging Initiative Group (2014) The role of clusterin in amyloid-β-associated neurodegeneration. JAMA Neurol. 71, 180–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lambert M. P., Barlow A. K., Chromy B. A., Edwards C., Freed R., Liosatos M., Morgan T. E., Rozovsky I., Trommer B., Viola K. L., Wals P., Zhang C., Finch C. E., Krafft G. A., and Klein W. L. (1998) Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U.S.A. 95, 6448–6453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Oda T., Wals P., Osterburg H. H., Johnson S. A., Pasinetti G. M., Morgan T. E., Rozovsky I., Stine W. B., Snyder S. W., and Holzman T. F. (1995) Clusterin (apoJ) alters the aggregation of amyloid β-peptide (A β 1–42) and forms slowly sedimenting A β complexes that cause oxidative stress. Exp. Neurol 136, 22–31 [DOI] [PubMed] [Google Scholar]
- 40. Ferrone F. A. (2015) Assembly of Aβ proceeds via monomeric nuclei. J. Mol. Biol. 427, 287–290 [DOI] [PubMed] [Google Scholar]
- 41. Finder V. H., Vodopivec I., Nitsch R. M., and Glockshuber R. (2010) The recombinant amyloid-β peptide Aβ1–42 aggregates faster and is more neurotoxic than synthetic Aβ1–42. J. Mol. Biol. 396, 9–18 [DOI] [PubMed] [Google Scholar]
- 42. Petrlova J., Hong H. S., Bricarello D. A., Harishchandra G., Lorigan G. A., Jin L. W., and Voss J. C. (2011) A differential association of Apolipoprotein E isoforms with the amyloid-β oligomer in solution. Proteins 79, 402–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ly S., Altman R., Petrlova J., Lin Y., Hilt S., Huser T., Laurence T. A., and Voss J. C. (2013) Binding of apolipoprotein E inhibits the oligomer growth of amyloid-β peptide in solution as determined by fluorescence cross-correlation spectroscopy. J. Biol. Chem. 288, 11628–11635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jiang Q., Lee C. Y., Mandrekar S., Wilkinson B., Cramer P., Zelcer N., Mann K., Lamb B., Willson T. M., Collins J. L., Richardson J. C., Smith J. D., Comery T. A., Riddell D., Holtzman D. M., et al. (2008) ApoE promotes the proteolytic degradation of Aβ. Neuron 58, 681–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. DeMattos R. B., Cirrito J. R., Parsadanian M., May P. C., O'Dell M. A., Taylor J. W., Harmony J. A., Aronow B. J., Bales K. R., Paul S. M., and Holtzman D. M. (2004) ApoE and clusterin cooperatively suppress Aβ levels and deposition: evidence that ApoE regulates extracellular Aβ metabolism in vivo. Neuron 41, 193–202 [DOI] [PubMed] [Google Scholar]
- 46. Tjernberg L. O., Callaway D. J., Tjernberg A., Hahne S., Lilliehöök C., Terenius L., Thyberg J., and Nordstedt C. (1999) A molecular model of Alzheimer amyloid β-peptide fibril formation. J. Biol. Chem. 274, 12619–12625 [DOI] [PubMed] [Google Scholar]
- 47. Murphy R. M. (2002) Peptide aggregation in neurodegenerative disease. Annu. Rev. Biomed. Eng. 4, 155–174 [DOI] [PubMed] [Google Scholar]
- 48. Boggs L. N., Fuson K. S., Baez M., Churgay L., McClure D., Becker G., and May P. C. (1996) Clusterin (Apo J) protects against in vitro amyloid-β (1–40) neurotoxicity. J. Neurochem. 67, 1324–1327 [DOI] [PubMed] [Google Scholar]
- 49. Bailey R. W., Dunker A. K., Brown C. J., Garner E. C., and Griswold M. D. (2001) Clusterin, a binding protein with a molten globule-like region. Biochemistry 40, 11828–11840 [DOI] [PubMed] [Google Scholar]