

Abstract
Protein lysine posttranslational modification by an increasing number of different acyl groups is becoming appreciated as a regulatory mechanism in cellular biology. Sirtuins are class III histone deacylases that use NAD+ as a co-substrate during amide bond hydrolysis. Several studies have described the sirtuins as sensors of the NAD+/NADH ratio, but it has not been formally tested for all the mammalian sirtuins in vitro. To address this problem, we first synthesized a wide variety of peptide-based probes, which were used to identify the range of hydrolytic activities of human sirtuins. These probes included aliphatic ϵ-N-acyllysine modifications with hydrocarbon lengths ranging from formyl (C1) to palmitoyl (C16) as well as negatively charged dicarboxyl-derived modifications. In addition to the well established activities of the sirtuins, “long chain” acyllysine modifications were also shown to be prone to hydrolytic cleavage by SIRT1–3 and SIRT6, supporting recent findings. We then tested the ability of NADH, ADP-ribose, and nicotinamide to inhibit these NAD+-dependent deacylase activities of the sirtuins. In the commonly used 7-amino-4-methylcoumarin-coupled fluorescence-based assay, the fluorophore has significant spectral overlap with NADH and therefore cannot be used to measure inhibition by NADH. Therefore, we turned to an HPLC-MS-based assay to directly monitor the conversion of acylated peptides to their deacylated forms. All tested sirtuin deacylase activities showed sensitivity to NADH in this assay. However, the inhibitory concentrations of NADH in these assays are far greater than the predicted concentrations of NADH in cells; therefore, our data indicate that NADH is unlikely to inhibit sirtuins in vivo. These data suggest a re-evaluation of the sirtuins as direct sensors of the NAD+/NADH ratio.
Keywords: acetylation, fatty acid, histone deacetylase (HDAC), nicotinamide adenine dinucleotide (NADH), protein acylation, sirtuin, lysine myristoylation, lysine palmitoylation, nicotinamide adenine dinucleotide (NAD+), post-translational modification (PTM)
Introduction
Acetylation of lysine residues in core histones (H2A, H2B, H3, and H4) in chromatin complexes plays important roles in gene regulation, and enzymes that modify the levels of ϵ-N-acetyllysine (Kac)5 have become important drug targets (1–5). Protein acetylation has furthermore been recognized as a general posttranslational modification with biological implications in numerous pathways, especially in metabolism (6–11). In addition to lysine acetylation, examples of formylation (12), propionylation and butyrylation (13–15), and myristoylation (16–18) have been reported. More recently, the list of acyl groups has been extended considerably with malonyl (19, 20), succinyl (19–21), glutaryl (22), crotonyl (23, 24), α-hydroxyisobutyryl (25), and 3-phosphoglyceryl (26), thus extending the scope of acyl-based posttranslational modifications considerably (10, 27–30).
The degree of acetylation in mammalian cells is regulated by opposing functions of acetyltransferases and hydrolases that catalyze removal of the acetyl groups from ϵ-amino groups of lysine residues. In humans, enzymes from two different classes have been recognized to have the latter capability, namely the 7 NAD+-dependent homologs of Sir2 (silent information regulator 2) enzymes (the sirtuins, SIRT1–7) (31–33) and the 11 Zn2+-dependent histone deacetylases (HDAC1–11) (34). The sirtuins and HDACs act on a wide range of acyllysine substrates. For example, ϵ-N-malonyllysine, ϵ-N-succinyllysine, and ϵ-N-glutaryllysine all appear to be selectively hydrolyzed by SIRT5 (19, 20, 22, 35), whereas ϵ-N-crotonyllysine has been cleaved by HDAC3 (23, 36), SIRT1 (35, 37), and SIRT2 (37) in vitro. Furthermore, a recent study using cross-linking affinity-based probes and SILAC (stable isotope labeling with amino acids in cell culture) suggested that SIRT3 processes ϵ-N-crotonyllysine in cells (38). SIRT6-mediated ϵ-N-myristoyllysine hydrolysis of TNF-α was reported to regulate its secretion (18); SIRT1–3 were shown to have robust fatty acid deacylase activity as well (37, 39, 40); and very recently, ϵ-N-lipoyllysine and ϵ-N-biotinyllysine were also reported to be hydrolyzed by SIRT4 (41).
Unlike the Zn2+-dependent HDACs, the sirtuins uniquely use NAD+ as a co-substrate in their deacylation reactions (42). Because of this unique enzymatic requirement, sirtuin activity has been linked to the energetic status of the cell, and the mammalian sirtuins have been described as sensors of the NAD+/NADH ratio (43–47). However, the sensitivity of the latest list of reported mammalian sirtuin deacylation reactions to NADH has not been formally tested. To do this, we first had to establish the full array of sirtuin-mediated lysine deacylation reactions in our assays. Given that there is such a large variety of reported sirtuin activities and that in some cases these reports are conflicting, it was imperative that we perform a comprehensive analysis to confirm previously reported sirtuin activities in our assays. Thus, we first established a range of sirtuin-mediated lysine deacylation reactions using three different in vitro assays. We then tested the sensitivity of a selection of these reactions to NADH. The data presented here have important implications for the fundamental understanding of sirtuin activity as well as the wide scope of biology they regulate.
Experimental Procedures
Materials
SIRT1 (aa 193–741 with N-terminal GST tag, >60% purity), SIRT2 (aa 50–356 with C-terminal His tag, >90% purity), SIRT4 (aa 25–314 with N-terminal GST tag, >70% purity), SIRT5 (full-length with N-terminal GST tag, >57% purity), SIRT6 (full-length with N-terminal GST tag, >64% purity), SIRT7 (full length with C-terminal FLAG tag, >50% purity) HDAC1 (full-length with C-terminal His tag and C-terminal FLAG tag, >62% purity), HDAC2 (full-length with C-terminal His tag, >86% purity), HDAC3·NCoR2 (full-length HDAC3 with C-terminal His tag in complex with NCoR2 (aa 395–489) with N-terminal GST-tag, >90% purity), HDAC4 (aa 627–1085 with C-terminal GST tag, >89% purity), HDAC5 (aa 657–1123 with C-terminal His tag, >90% purity, HDAC8 (full-length with C-terminal His tag, >90% purity), and HDAC9 (aa 604–1066 with C-terminal His tag, >76% purity) were purchased from BPS Biosciences (San Diego, CA); HDAC7 (aa 383–end with N-terminal His tag, >50% purity) was purchased from Millipore (Temecula, CA); SIRT3 (aa 101–399 with N-terminal His tag; >90% purity) was purchased from Cayman Chemical (Ann Arbor, MI); SIRT5 (aa 37–310 with N-terminal His tag, >90% purity) was purchased from Enzo Life Sciences (Farmingdale, NY). Purities were based on SDS-PAGE and Coomassie Blue staining according to the supplier, and all enzyme concentrations given are based on stock concentrations according to the supplier. Assay buffer was prepared as described in the Biomol International product sheets BML-KI-143 (Tris/Cl (50 mm), NaCl (137 mm), KCl (2.7 mm), MgCl2 (1 mm), pH 8.0) with or without the addition of BSA (1.0 mg/ml) for fluorescence and HPLC-MS experiments, respectively. Trypsin (10,000 units/mg, l-1-tosylamido-2-phenylethyl chloromethyl ketone-treated from bovine pancreas, T1426) was from Sigma-Aldrich (Steinheim, Germany). NADH (as a hydrate of the disodium salt, N8129) was from Sigma-Aldrich, and the stock solution (250 mm, in degassed MilliQ water) was prepared based on the absorbance at 340 nm (ϵ340 = 6220 m−1·cm−1). All chemicals and solvents were of analytical grade and were used without further purification as obtained from commercial suppliers.
Chemical Synthesis
Fluorogenic substrate series 1–3 were synthesized using a combination of solution-phase and solid-phase peptide synthesis (see the supplemental information and Fig. 1A for details). Octamer and dodecamer substrate series 4 and 5 were prepared by standard Fmoc (N-(9-fluorenyl)methoxycarbonyl) solid-phase peptide synthesis on Chem-Matrix® resin with a Rink amide linker using HATU as the coupling reagent. Compounds 4i and 4j were prepared by HATU-mediated acylation of the unprotected peptide (4n) with myristic acid and palmitic acid, respectively, to give the desired substrates as the major products. Compound 5i was prepared by HATU-mediated acylation of the resin-bound, appropriately protected peptide with myristic acid, and compounds 5p and 5q were synthesized by acylation of the resin-bound, appropriately protected peptide with the desired anhydride.
Fluorescence-based HDAC and Sirtuin Deacylase Assays
All reactions were performed in black low binding 96-well microtiter plates (Corning half-area wells), with duplicate series in each assay, and each assay was performed at least twice. Control wells without enzyme were included in each plate. All reactions were performed in assay buffer, with appropriate concentrations of substrates and inhibitors obtained by dilution from 10–250 mm stock solutions in either water or DMSO and an appropriate concentration of enzyme obtained by dilution of the stock provided by the supplier. All plates were analyzed using a PerkinElmer Life Sciences Enspire plate reader with excitation at 360 nm and detecting emission at 460 nm. Fluorescence measurements (relative fluorescence units) were converted to [AMC] concentrations based on an [AMC]-fluorescence standard curve, and all data analysis was performed using GraphPad Prism.
Fluorogenic Sirtuin Substrate Screening
The initial screening for substrate deacylation activity was performed with end point fluorophore cleavage by trypsin. For a final volume of 25 μl/well, acyl substrates (Ac-TARKacyl-AMC (1a–m) or Ac-QPKKacyl-AMC (2a–k) 50 μm) and NAD+ (500 μm) were added to each well, followed by a solution of sirtuin enzyme (SIRT1, 333 nm; SIRT2, 222 nm; SIRT3, 250 nm; SIRT4, 270 nm; SIRT5, 211 nm; SIRT6, 313 nm; SIRT7, 400 nm). The reaction was incubated at 37 °C for 60 min, and then 25 μl of a solution of trypsin and nicotinamide (5.0 mg/ml and 4 mm, respectively; final concentration 2.5 mg/ml and 2 mm, respectively) was added, and the assay development was allowed to proceed for 90 min at room temperature before fluorescence analysis. The data were analyzed to afford [AMC] relative to control wells.
Fluorogenic HDAC Substrate Screening
The screening for substrate deacylation activity was performed with end point fluorophore cleavage by trypsin. For a final volume of 25 μl/well, acyl substrate (Ac-QPKKacyl-AMC (2a–k) (10 μm)) was added, followed by a solution of HDAC enzyme (10 nm). The reaction was incubated at 37 °C for 60 min, after which a solution of trypsin (25 μl, 5.0 mg/ml, final concentration 2.5 mg/ml) was added, and the assay development was allowed to proceed for 90 min at room temperature before fluorescence analysis. The data were analyzed to afford [AMC] relative to control wells.
Adenosine Diphosphate Ribose (ADPR) and Nicotinamide (NAM) Concentration-Response Experiments
Using the protocol described above with a 30-min reaction time, hydrolase activities of SIRT1–3 and SIRT6 against substrates 1f, 1i, and 3b and SIRT5 against 3p and 3q were evaluated under varying ADPR or NAM concentrations. The sirtuin enzyme (SIRT1, 100 nm; SIRT2, 400 μm; SIRT3, 300 nm; SIRT6, 600 nm; or SIRT5, 200 nm) was incubated with the relevant substrate (300 μm), NAD+ (1000 μm), and ADPR or NAM (1–0.064 mm in a 5-fold dilution series, or 0.0 mm) in a total volume of 25 μl of assay buffer. The data were fitted to the concentration-response equation (Hill slope = −1) using GraphPad Prism.
HPLC-MS-based Assays
All reactions were performed in black low binding 96-well microtiter plates (Corning half-area wells), with each assay performed at least twice. After incubating sirtuin, substrates, and inhibitor (when applicable) for 30 min at 37 °C, a sample of the reaction mixture (10 μl) was taken out and quenched by the addition of buffer/MeOH/HCOOH (50:96:4, 30 μl). The samples were analyzed by HPLC-MS on a Waters Acquity ultra-HPLC-MS system equipped with a diode array detector. A gradient with eluent I (0.1% HCOOH in water (v/v)) and eluent II (0.1% HCOOH in acetonitrile (v/v)) rising linearly 0–10% during t = 0.00–2.10 min followed by 10–60% of eluent II during t = 2.10–4.30 min was applied at a flow rate of 0.6 ml/min. The obtained chromatograms at either 280 nm (where the indole chromophore of tryptophan of substrate series 5 has its absorption maximum) or 326 nm (where the coumarin moiety of substrate series 1 and 3 has its absorption maximum) were used to determine reaction progression, by determining area under the curve of the relevant peaks using Waters MassLynx.
Sirtuin Deacylase Activity
The hydrolase activities of SIRT1–3 against substrate 5b, SIRT1–3 and -6 against substrate 5i, and SIRT5 against substrates 5p and 5q were evaluated. Sirtuin enzyme (SIRT1, 50 nm for 5b and 100 nm for 5i; SIRT2, 200 nm for 5b and 5i; SIRT3, 400 nm for 5b and 600 nm for 5i; SIRT5, 400 nm for 5p and 5q; and SIRT6, 6.00 μm for 5i) was incubated with the relevant dodecapeptide substrate (100 μm), NAD+ (500 μm), and NADH (0.0, 0.5, 5.0, or 50 mm) in a total volume of 25 μl of assay buffer.
NADH Concentration-Response Experiments
Using the protocol described above, hydrolase activities of SIRT1–3 against substrates 1f, 1i, and 3b and SIRT5 against 3p and 3q were evaluated under varying NADH concentrations. The sirtuin enzyme (SIRT1, 400 nm; SIRT2, 2.00 μm; SIRT3, 2.76 μm; SIRT6, 1.88 μm; or SIRT5, 200 nm) was incubated with the relevant substrate (300 μm (SIRT1–3 and SIRT6) or 100 μm (SIRT5)), NAD+ (1000 μm (SIRT1–3 and SIRT6) or 500 μm (SIRT5), and NADH (10–0.01 mm in a 3.16-fold dilution series, or 0.0 mm) in a total volume of 25 μl of assay buffer. The data were fitted to the concentration-response equation (Hill slope = −1) using GraphPad Prism.
Steady-state Rate Inhibition Experiments
Using the protocol described above, hydrolase activities of SIRT1 against substrate 3b and SIRT5 against substrate 3p were determined under varying substrate-NADH and NAD+-NADH concentrations. Sirtuin enzyme (SIRT1, 400 nm; SIRT5, 650 nm) was incubated with substrate (3b (400–12.5 μm, 2-fold dilution or 300 μm) or 3p (400–12.5 μm, 2-fold dilution or 100 μm)), NAD+ (500 μm or 5.0–0.156 mm, 2-fold dilution), and NADH (10–0.156 mm, 2-fold dilution or 0.0 mm). The data were fitted to the Michaelis-Menten equation using GraphPad Prism. Double-reciprocal (Lineweaver-Burk) plots were constructed by fitting the obtained data to a series of first order equations with shared intersection using GraphPad Prism.
[32P]NAD+-based Assays
Based on a method described previously (19), reactions were performed in a total volume of 10 μl in buffer (Tris/Cl (50 mm), NaCl (50 mm), MgCl2 (4 mm), DTT (0.5 mm), pH 9.0) with 0.5 μCi of [32P]NAD+ (PerkinElmer Life Sciences, NEG023X; 800 Ci/mmol) and 50 μm acylated peptide substrate (substrate series 4). The reactions were incubated with 1.5 μg of SIRT1–3 or SIRT6 (BPS Bioscience) for 1 h at 37 °C. At the end of the incubation, 2 μl of each reaction was spotted onto silica gel thin layer chromatography plates (Whatman Partisil LK6D) and eluted with EtOH-aqueous NH4HCO3 (1 m) (70:30 (v/v)). The plates were then air-dried and exposed to an Eastman Kodak Co. storage phosphorimaging screen SD230 (GE Healthcare), and the signal was detected using a STORM820 PhosphorImager (GE Healthcare).
Computational Peptide Substrate Docking
Molecular modeling studies of sirtuin active sites and docking of peptide substrates were conducted using Discovery Studio version 3.5 (Accelrys Software, Inc., San Diego, CA). The crystal structure coordinates for SIRT1 (Protein Data Bank code 4KXQ) (49), SIRT2 (3ZGV) (50), SIRT3 (3GLR) (51), and SIRT6 (3ZG6) (18) were downloaded from the Protein Data Bank. Structures were prepared and subjected to energy minimization utilizing the conjugate gradient minimization protocol with a CHARMm force field and the generalized Born implicit solvent model with simple switching (52) and converged to a root mean square gradient of <0.001 kcal/mol. The crystal structures of SIRT3 and SIRT6 contained co-crystallized peptides with Kac and ϵ-N-myristoyllysine residues, respectively. An additional myristoyl modification was built onto the co-crystallized Kac residue in the SIRT3 structure and likewise minimized as described above. Figures were generated in Discovery Studio with the proteins represented by a solvent-based molecular surface using a 1.2-Å probe and colored by surface hydrophobicity over a smooth gradient from hydrophilic (blue) to neutral (white) to hydrophobic (brown).
Molecular Dynamics Simulations
Molecular dynamics studies of NAD+ binding sites and docking of NAD+ and NADH were conducted in Desmond (Desmond Molecular Dynamics System version 4.4 (D.E. Shaw Research, New York) and Maestro-Desmond Interoperability Tools version 4.4 (Schrödinger, New York)). The crystal structure coordinates for SIRT1/EX527/NAD+ (4I5I) (54) and SIRT3/FZN/ADPR (3GLT) (51) were used to construct the models for molecular dynamics simulations. SIRT1 ternary complexes were built by replacing the inhibitor EX527 with the substrate AceCS2 (from 3GLR) and modifying NAD+ to NADH. SIRT3 complexes were obtained by replacing the ADPR ligand with NAD+ (from 3RIY) or NADH (modified from NAD+) and the FZN ligand with an AceCS2 peptide (from 3GLR) (51), giving systems with the dinucleotide bound in the AC site or modified to accommodate the dinucleotide bound in the alternative AB site. Each system was neutralized by adding sodium ions and solvated with a cubic TIP3P water box having at least 12-Å thickness. Additional sodium and chloride ions were introduced to achieve a salt concentration of 0.15 m (physiological conditions). The resulting systems (51,000–77,000 atoms) were then treated using either the default presimulation protocol (SIRT1) or a modified presimulation protocol where the relaxation time was extended from 1 ps to 1 ns with gradual increase of the temperature from 100 to 300 K. A production run was then carried out for either 250 ns (AC structures) or 500 ns (AB structures) at 300 K using the newly released OPLS3 force field (55). We found that OPLS3 performed better than OPLS2005 in replicating pyramidalization of the pyridyl nitrogen of NADH found using DFT (density functional theory) calculations (56, 57). Periodic boundary conditions with the Nose-Hoover chain thermostat (58, 59) and the Martyna-Tobias-Klein barostat (60) were used, with sampling of coordinates and energies at 10-ps intervals. Three replicates were performed for each system.
Results
Design and Synthesis of Fluorogenic Substrate Series
To investigate the sensitivity of sirtuins to NADH, we first established the range of NAD+-dependent deacylase activities. Three well established assays are consistently used to measure sirtuin deacylation: an AMC-based fluorogenic assay, an HPLC-MS-based assay, and an assay monitoring the conversion of 32P-labeled NAD+ to 32P-labeled O-acyl-ADP-ribose. The AMC-based fluorogenic assay takes advantage of the different electronic properties of N-acylated versus free AMC. AMC is quenched when conjugated but fluoresces in its free form. Peptides containing an ϵ-N-acyllysine residue with an AMC group at the C terminus (Fig. 1B) were used as substrates in which cleavage of the ϵ-N-amide bond by a selected sirtuin was followed by a trypsin-mediated liberation of AMC, to indirectly detect ϵ-N-amide bond cleavage by fluorescence readout. If the ϵ-N-amide bond is not cleaved by the sirtuin, trypsin is unable to liberate AMC, and the fluorophore remains quenched. This assay has been used to measure sirtuin deacetylation as well as deacylation (61).
FIGURE 1.

ϵ-N-Acyllysine posttranslational modifications and synthesized sirtuin substrates. A, ϵ-N-Acyllysine posttranslational modifications from the literature. B, substrates prepared for this study. H3, core histone H3; H4, core histone H4; p53, tumor suppressor p53. Khex, ϵ-N-hexanoyllysine; Koct, ϵ-N-octanoyllysine; Kdec, ϵ-N-decanoyllysine; Klau, ϵ-N-lauroyllysine; Kpal, ϵ-N-palmitoyllysine; Ktfa, ϵ-N-trifluoroacetyllysine.
We prepared and evaluated two series of novel acylated probes (series 1 and 2, Fig. 1B). As ϵ-N-acyl modifications, we included formyl, acetyl (Kac), propionyl, butyryl, hexanoyl, octanoyl, decanoyl, lauroyl, myristoyl, palmitoyl, and crotonyl in each tetrapeptide series and lipoyl and biotinoyl in series 1. Substrates 1a–m, containing amino acids 6–9 of core histone H3, were chosen because H3 Lys-9 acetylation has been reported to affect modulation of telomeric chromatin under regulation by SIRT6 (62) and because SIRT6 was reported to deacylate H3 Lys-9 substrates in vitro (18). Due to the reported regulation of TNF-α trafficking by SIRT6-mediated demyristoylation of Lys-20 in this protein (18), we initially considered constructing a series based on amino acids 17–20 of TNF-α (LPKK). However, based on preliminary synthetic efforts, we found that handling and purification of the long chain acyllysine-containing targets were non-trivial due to their amphiphilic nature and low solubility in assay buffer. Previous reports have solved this challenge by extending the peptide to a hexamer (61), but we instead turned our attention to a sequence containing amino acids 317–320 of p53. This sequence offered improved solubility and allowed us to construct our second substrate series, 2a–k (Fig. 1B). Residue Lys-320 is a well known acetylation site (6), and p53-mediated apoptosis has been shown to be regulated by the acetylation state of this residue, which in turn is mediated, at least in part, by the histone acetyltransferase PCAF (KAT2B) (63–65) and sumoylated HDAC2 (66).
Testing the Range of Sirtuin Deacylase Activities
Initially, to ensure efficient and reliable development conditions, trypsin-mediated release of the fluorophore from non-acylated control compounds was investigated. Whereas non-acylated control substrate 3n is readily cleaved by trypsin to release the fluorophore (200 μm deacylated substrate is fully converted by trypsin (200 ng/μl) in less than 30 s (i.e. kcat > 1 s−1 (data not shown))), the additional trypsin cleavage sites in series 1 and 2 (the vicinal arginine or lysine, respectively) resulted in slow fluorophore release by initial formation of H-K-AMC, which proved to be a poorer substrate for trypsin (kcat = 2.2 × 10−3 ± 0.2 × 10−3 s−1 (data not shown)). Full cleavage of deacylated intermediates was therefore ensured by increased trypsin concentration (2500 ng/μl) and prolonged development time (90 min) in the deacylase activity assays.
We then profiled the deacylation activity of the seven human sirtuins (Fig. 2A) using both substrate series and human Zn2+-dependent HDACs belonging to class I and class IIa (HDAC1, -2, -3, and -8 and HDAC4, -5, -7, and -9, respectively; Fig. 2A) using substrate series 2 only.
FIGURE 2.

Sirtuin deacylase activities. A, deacylation data obtained as end point readings of released fluorophore ([AMC]final) after a 1-h incubation with each substrate at 37 °C (SIRT: 200–400 nm enzyme, 50 μm substrate, 500 μm NAD+; HDAC: 10 nm enzyme, 10 μm substrate), followed by development for 90 min at 25 °C (2.5 mg/ml trypsin and 2 mm nicotinamide for the sirtuins). Sirtuin deacylation data are represented as mean from two individual experiments performed in duplicate, and the values were normalized to control wells without enzyme added. B, HPLC traces (A280) of the hydrolysis of Kac and ϵ-N-myristoyllysine (Kmyr) by SIRT1–3 (40–600 nm enzyme, 100 μm substrate (5b and 5i), 500 μm NAD+). *, unidentified impurity. C, [32P]NAD+ consumption assay using octapeptides 4b, 4i, 4j, and 4n. Incubation with SIRT1–3 and SIRT6 in the presence of 32P-labeled NAD+ followed by separation by thin layer chromatography (TLC) and visualization by autoradiography. Kfor, ϵ-N-formyllysine; Kpro, ϵ-N-propionyllysine; Kbut, ϵ-N-butyryllysine; Khex, ϵ-N-hexanoyllysine; Koct, ϵ-N-octanoyllysine; Kdec, ϵ-N-decanoyllysine; Klau, ϵ-N-lauroyllysine; Kpal, ϵ-N-palmitoyllysine; Kcr, ϵ-N-crotonyllysine; Klip, ϵ-N-lipoyllysine; Kbio, ϵ-N-biotinoyllysine; Ktfa, ϵ-N-trifluoroacetyllysine.
Corroborating recent findings (18, 37, 39, 40, 61, 67, 68), we found that SIRT1–3 and SIRT6 all displayed deacylase activities against a wide range of substrates (Fig. 2A). In contrast to a previous report (37), on the other hand, we did not observe significant levels of deacylation against any substrates by SIRT4, SIRT5, or SIRT7. However, this may be due to our use of AMC-linked pseudopeptides instead of longer peptide sequences as well as the lower enzyme loadings applied in our experiments. In all experiments, enzyme concentrations were equal to those known to furnish robust deacetylation activity for SIRT1–3 in this assay. Thus, our fluorogenic assay revealed selective deacylation of modifications with longer hydrocarbon chains (hexanoyl to palmitoyl) by SIRT6. Interestingly, SIRT1 and SIRT2 appeared to be more potent demyristoylases than SIRT6 when compared using the same substrates and equimolar enzyme amounts. SIRT3 exhibited demyristoylase activity similar to that of SIRT6 based on initial screening results. SIRT2 even appeared more potent against longer chains than against acetyl in both fluorogenic substrate series, which is also in agreement with recently reported results (68).
None of the class IIa HDACs exhibited significant activity against the tested substrates, consistent with previous studies (69–72) where robust deacylase activity has only been demonstrated for peptides functionalized with the non-physiological trifluoroacetyl group (e.g. using substrate 3o (72), Fig. 2A). Class I HDAC1–3, which have previously been demonstrated to exhibit robust deacetylase activity, also displayed deacylation activity against modifications longer than Kac (Fig. 2A). All three isoforms were capable of deacylating the closely related ϵ-N-formyllysine and ϵ-N-propionyllysine, but prominent deacylation of ϵ-N-butyryllysine and ϵ-N-hexanoyllysine was also observed for HDAC3. Interestingly, HDAC1 showed measurable activity against the ϵ-N-hexanoyllysine- to ϵ-N-lauroyllysine derivatives, which warrant further investigation, in particular because the enzyme concentrations chosen for HDACs were based on typical values applied for inhibitor screening, which are ∼20–40-fold lower than those used for sirtuin reactions.
Confirmation of Deacylase Activities Using Non-fluorogenic Peptides
Next, we sought to confirm the enzymatic deacylation activities using additional assays, which would eliminate possible false positives related to the fluorophore (73). Thus, we turned to an HPLC-MS-based assay used previously to monitor sirtuin deacylation activity (18). We prepared octa- and dodecapeptide sequences based on histone H3 (4b,i,j,n and 5b,i,n, respectively; supplemental Fig. 1B) with Trp residues added C-terminally to enable UV detection and tested for deacetylase and demyristoylase activity of SIRT1–3 and SIRT6 using HPLC to monitor the conversion of an acylated peptide (Fig. 2B). Identities of acylated and deacylated peptides were confirmed using mass spectrometry (Fig. 3). We observed robust deacetylase as well as demyristoylase activity of SIRT1–3 and demyristoylase activity exclusively for SIRT6 as in the fluorogenic assays.
FIGURE 3.
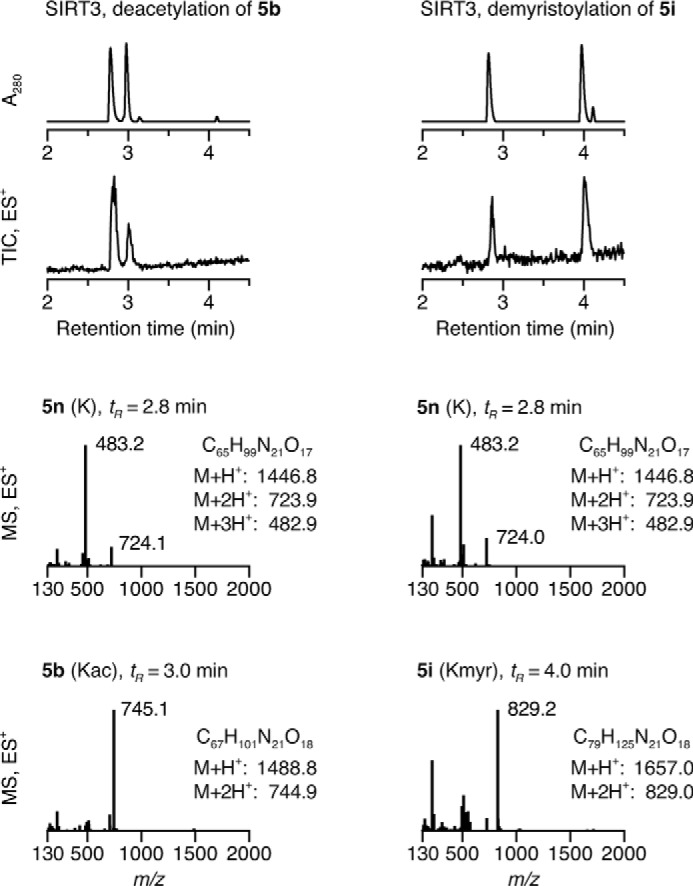
HPLC-MS data of SIRT3-mediated deacylation. UV (A280) and TIC (ES+) chromatograms and mass spectra at relevant time points allowing identification of acylated peptide substrates (5b and 5i) and deacylated peptide product (5n). Kmyr, ϵ-N-myristoyllysine.
Finally, we confirmed these enzymatic activities using a [32P]NAD+ consumption assay (37), which monitors the conversion of [32P]NAD+ to O-[32P]acyl-ADP-ribose by separation using thin layer chromatography and visualization by autoradiography. Again, we observed that long chain acyllysine modifications were hydrolyzed by a mechanism utilizing NAD+ as a co-substrate, as expected for sirtuin-mediated amide bond hydrolysis (Fig. 2C). In addition, this assay further supported the findings that SIRT1–3 cleaved both acetyl and myristoyl modifications, whereas SIRT6 cleaved myristoyl only. Together, these series of assays provide very strong evidence for several different enzymatic activities of the sirtuin deacylases.
Effects of NAD+/NADH Ratio on Deacylase Activity
Next, we investigated the potential regulation of deacylation events by NADH. The NAD+/NADH redox couple is intrinsically linked to cellular metabolism, and because sirtuins are NAD+-dependent enzymes, changes in the NAD+/NADH ratio could potentially affect sirtuin activity via altered NAD+ concentrations or by putative NADH-mediated sirtuin inhibition. Thus, we found it relevant to compare effects of NADH levels on these novel sirtuin deacylase activities with the effects on deacetylation. Because the Zn2+-dependent HDACs do not use NAD+ as a co-substrate, they were not included in this investigation; indeed, the activity of HDAC1 and -2 has been reported to be insensitive to NADH (74).
We first tested the effects of NADH in the AMC-based assay. However, we observed nearly complete spectral overlap of both UV absorbance and fluorescence of NADH and AMC (Fig. 4A). Thus, fluorogenic AMC-based assays are not suitable for measuring effects of NADH on sirtuin activity. Therefore, we used the HPLC-MS-based assay to test the influence of NADH against SIRT1–3 and SIRT6 (5b or 5i; Fig. 4B), as well as on SIRT5-mediated hydrolysis of succinylated and glutarylated peptide substrates (5n or 5o; Fig. 4B). All enzyme and substrate combinations investigated were inhibited at 50 mm NADH (Fig. 4C). At 0.5 and 5.0 mm NADH, deacetylation by SIRT2 and SIRT3 and demyristoylation by SIRT2 and SIRT6 showed little or no inhibition (Fig. 4C). In contrast, deacylation by SIRT1 and SIRT5 was inhibited at these lower concentrations (Fig. 4C).
FIGURE 4.

NADH inhibition of sirtuin activity. A, absorbance spectra of AMC, NAD+, and NADH as well as fluorescence spectra of AMC, NADH, and a combined AMC-NADH sample. The sum of the individual AMC and NADH signals is also included. B, HPLC traces (A280) of the hydrolysis of Kac or ϵ-N-myristoyllysine (Kmyr) by SIRT1 and SIRT2 and ϵ-N-succinyllysine (Ksuc) or ϵ-N-glutaryllysine (Kglu) by SIRT5 (100 μm peptide substrate, 500 μm NAD+) at different NADH concentrations. C, fractional conversion for the hydrolysis of ϵ-N-acetyllysine, ϵ-N-myristoyllysine, ϵ-N-succinyllysine, and ϵ-N-glutaryllysine by SIRT1–3, SIRT5, and SIRT6 (100 μm peptide substrate, 500 μm NAD+), based on area under the relevant peaks.
The differential effects of NADH prompted further investigation of its inhibitory activity on SIRT1–3, SIRT6, and SIRT5. To increase the sensitivity of the HPLC-MS-based assay, we used substrate series 1 and 3, because the chromatographic separation allows us to exploit the coumarin moiety as a chromophore without overlap of NADH absorbance. Using this system, we tested the inhibitory activity of NADH on the deacylase activity of SIRT1–3 and 6 using substrates 3b, 1f, and 1i and of SIRT5 using substrates 3p and 3q (Fig. 5). Under these conditions, all of the investigated deacylation reactions were inhibited by NADH with IC50 values between 1.3 ± 0.4 and 68 ± 9 mm (Fig. 5 and Table 1). We also tested the inhibitory activities of ADPR and NAM under the same substrate concentrations. Because neither ADPR nor NAM absorb or fluoresce at relevant wavelengths, the AMC-based fluorescence assay was used. ADPR was found to be a poor inhibitor, with IC50 values from 1.1 ± 0.1 mm to >10 mm, except for SIRT1-mediated deacetylation (IC50 = 0.22 ± 0.03 mm) and SIRT6-mediated deoctanoylation and demyristoylation (IC50 = 74 ± 15 and 89 ± 29 μm, respectively), whereas nicotinamide, in agreement with the literature, was found to be more potent, with IC50 values ranging from 28 ± 5 to 180 ± 30 μm.
FIGURE 5.

Concentration response of NADH, ADPR, and NAM. Residual activity of SIRT1–3 and SIRT6-mediated hydrolysis of Kac (LGKac), ϵ-N-octanoyllysine (TARKoct), and ϵ-N-myristoyllysine (TARKmyr) (300 μm peptide substrate (3b, 1f, and 1i), 1000 μm NAD+) as well as SIRT5-mediated hydrolysis of ϵ-N-succinyllysine (LGKsuc) and ϵ-N-glutaryllysine (LGKglu) (100 μm peptide substrate (3p and 3q), 500 μm NAD+) at different NADH, ADPR, and NAM concentrations. Conversions are based on relative areas under the relevant peaks in the UV chromatograms.
TABLE 1.
IC50values for NADH, ADPR, and NAM
Inhibition constants of SIRT1–3-, SIRT6-, and SIRT5-mediated hydrolysis of relevant substrates (300 μm peptide substrate (3b, 1f, and 1i), 1000 μm NAD+, or 100 μm peptide substrate (3p and 3q), 500 μm NAD+), calculated from concentration-response experiments shown in Fig. 5.
| NADH | ADPR | NAM | |
|---|---|---|---|
| mm | mm | μm | |
| SIRT1 | |||
| 3b | 1.3 ± 0.2 | 0.22 ± 0.03 | 180 ± 30 |
| 1f | 3.0 ± 0.1 | 1.9 ± 0.4 | 84 ± 11 |
| 1i | 1.3 ± 0.4 | 1.1 ± 0.3 | 68 ± 6 |
| SIRT2 | |||
| 3b | 4.8 ± 0.2 | 4.9 ± 3.0 | 41 ± 6 |
| 1f | 32 ± 2 | 3.9 ± 0.5 | 44 ± 5 |
| 1i | 21 ± 8 | 8.0 ± 3.5 | 35 ± 4 |
| SIRT3 | |||
| 3b | 3.3 ± 0.4 | 6.0 ± 3.7 | 72 ± 15 |
| 1f | 68 ± 9 | 3.9 ± 0.6 | 76 ± 7 |
| 1i | 8.9 ± 1.9 | 1.1 ± 0.1 | 53 ± 7 |
| SIRT6 | |||
| 3b | NDa | ND | ND |
| 1f | 9.3 ± 0.5 | 0.074 ± 0.014 | 150 ± 40 |
| 1i | 1.8 ± 0.3 | 0.089 ± 0.029 | 120 ± 30 |
| SIRT5 | |||
| 3p | 3.8 ± 0.2 | >10b | 28 ± 5 |
| 3q | 2.4 ± 0.3 | 4.3 ± 2.2 | 99 ± 30 |
a ND, not determined due to the prohibitively high concentration of enzyme needed.
b The IC50 value could not be determined from the concentration-response curve (84% residual activity at 1 mm, the highest concentration tested).
To determine the inhibitory mechanism of NADH, we measured initial velocities of SIRT1-mediated deacetylation of substrate 3b and SIRT5-mediated desuccinylation of substrate 3p at constant NAD+ concentrations and varying concentrations of substrate and NADH as well as at constant acyl substrate concentrations and varying concentrations of NAD+ and NADH (Fig. 6, A and B). Lineweaver-Burk (1/rate versus 1/[NAD+]) plots of all four enzyme-inhibitor experiments indicated a non-competitive inhibition mechanism for both substrates (peptide and NAD+) for SIRT1 and SIRT5. Initial velocities were fitted to the Michaelis-Menten equation to obtain Km, kcat, Kis, and Kii values (Table 2). The derived Kii and Kis constants are composites of multiple equilibria in the bisubstrate deacylation reaction, rendering it difficult to separate contributions to individual rate constants. However, the overall Kii and Kis values were in the same range, which also suggests non-competitive mechanisms with respect to both peptide substrate and NAD+. We therefore decided to perform molecular dynamics simulations to attempt to rationalize these results.
FIGURE 6.

Steady-state rate inhibition experiments. A, Michaelis-Menten and Lineweaver-Burk plots of NADH inhibition of SIRT1-mediated deacetylation using substrate 3b (LGKac) at varying substrate or NAD+ concentrations. B, Michaelis-Menten and Lineweaver-Burk plots of NADH inhibition of SIRT5-mediated desuccinylation of substrate 3p (LGKsuc) at varying substrate or NAD+ concentrations.
TABLE 2.
Michaelis-Menten parameters and inhibition constants for NADH inhibition
Michaelis-Menten parameters and inhibition constants of SIRT1- and SIRT5-mediated hydrolysis of relevant substrates (3b and 3p, respectively) obtained by fitting to the non-competitive inhibitor equation using GraphPad Prism.
| Km | kcat | Kis | Kii | |
|---|---|---|---|---|
| mm | ×10−2s−1 | mm | mm | |
| SIRT1 | ||||
| 3b | 250 ± 10 | 6.3 ± 0.2 | 0.77 ± 0.06 | 1.2 ± 0.1 |
| NAD+ | 750 ± 70 | 6.3 ± 0.2 | 0.94 ± 0.17 | 4.2 ± 0.9 |
| SIRT5 | ||||
| 3p | 72 ± 4 | 2.8 ± 0.1 | 2.2 ± 0.3 | 4.3 ± 0.5 |
| NAD+ | 200 ± 10 | 1.8 ± 0.1 | 0.65 ± 0.07 | 1.3 ± 0.1 |
Molecular Modeling; Structural Basis for Deacylation and NADH Inhibition
The findings that SIRT1–3 and SIRT6 can remove long-chain acyl groups prompted us to investigate the molecular basis for accommodation of long acyl-amide substrates in the active sites of sirtuins 1–3 and 6. The active sites of SIRT2 and -6 have been recognized to contain a hydrophobic pocket accommodating the long acyl chain of a myristoylated peptide (18, 68) that is distinct from the NAD+ cofactor binding site (75). Based on published x-ray crystal structures, we analyzed the hydrophobicity of the active sites of SIRT1–3 and SIRT6 and found that a hydrophobic pocket was also present in SIRT1 and SIRT3 (Fig. 7A). We also investigated whether SIRT5 can accommodate a long acyl chain. However energetic analysis suggests that this potential interaction is unlikely to occur. The crystal structure of SIRT5 (19) also features a less hydrophobic binding pocket that includes critical arginine and tyrosine residues forming hydrogen bonds with the terminal carboxylic acid moiety of succinylated substrates. We then modeled the predicted interaction of SIRT3 with both the ϵ-N-acetyllysine that was contained within the original crystal structure and that of an ϵ-N-myristoyllysine and found that both acyl groups could be accommodated in the hydrophobic pocket of the enzyme (Fig. 7A). The x-ray crystal structures show high similarity between the catalytic pockets of these sirtuins and provide a potential structural rationale for the observed enzymatic activities.
FIGURE 7.

Molecular modeling structures. A, the SIRT1 and SIRT3 active sites were analyzed for their hydrophobicity to determine the potential for binding long-chain fatty acyl groups. Structures shown are crystal structures of SIRT1 (Protein Data Bank entry 4KXQ) and model structures for active site complexes of SIRT3·Kac and SIRT3·ϵ-N-myristoyllysine (Kmyr) (modified from 3GLR). Conformations shown are the most energetically favorable. Gray areas indicate the position of a slice through the vertical z-plane. B, Conformations obtained after the production run of SIRT1 and SIRT3 in complex with ϵ-N-acetylated peptide substrate and NAD+ or NADH. Substrates are shown with carbon atoms in beige, and non-polar hydrogen atoms have been omitted from the peptide substrate. Isoleucine and aspartate residues forming hydrogen bonds with NAD+ and A, B, and C pockets for NAD+ binding have been labeled.
We then performed molecular dynamics simulations of SIRT1 and SIRT3 in complex with either NAD+ or NADH and a relevant acetylated peptide substrate (Fig. 7B). NAD+ can bind in two different sites, the AB and the AC pockets (51, 76). The ADP-ribose moiety is positioned in the A pocket, whereas the nicotinamide moiety may occupy either the B or the C pocket, with only AC pocket binding leading to deacetylation. In our systems, the absence of peptide substrate compromised protein stability and prevented stable NAD+ binding. This is in agreement with previous findings of an ordered binding (51, 77), where peptide substrate binding closes the binding cleft, stabilizes the complex, and allows NAD+ to bind in a productive conformation. On the other hand, although NADH was initially placed in the AC pocket, the dihydronicotinamide moiety almost immediately (after a 2-ns simulation time) started to move away from the C pocket, leading to an opening of the enzyme structure and an alternative binding of the dihydropyridyl moiety. No direct interactions with the pyridyl nitrogen could be observed for either dinucleotide, in agreement with previous reports showing delocalization of the charge in NAD+ to the full pyridinium system (78). The different binding affinities of SIRT1 and SIRT3 for NAD+ and NADH were also analyzed by the hydrogen bonds established between protein and dinucleotide. Positioning of the nicotinamide moiety in the C pocket and hydrogen bonding to two highly conserved residues (Ile-347/230 and Asp-348/231 in SIRT1/3, through backbone amide carbonyl and side chain carboxylate, respectively) is essential for deacetylation, and both hydrogen bonds form frequently between both enzymes and NAD+ but not NADH during simulations (Fig. 7B). To further evaluate the stability of potential AB pocket binding, we also performed molecular dynamics simulations starting from structures with the nicotinamide part of the dinucleotides positioned in the B pocket with extended simulation time (500 ns). For both SIRT1 and SIRT3, the dihydronicotinamide moiety of NADH moved even further away from the B and C pockets. However, in the case of NAD+, the nicotinamide moiety tended to move closer to the C pocket, i.e. toward the productive binding conformation.
Discussion
We have prepared and evaluated chemical tool compounds containing ϵ-N-acylated lysine residues containing acyl groups ranging from formyl (C1) to palmitoyl (C16) in length as well as dicarboxyl-derived substrates. Using three different assay formats, we established substrate specificities of the human sirtuins (SIRT1–7) in vitro. Based on these enzymatic activities, we then investigated the ability of NADH to inhibit selected sirtuin-mediated deacylation reactions to address the hypothesis that sirtuins may be directly sensing fluctuations in the NAD+/NADH ratio in cells.
SIRT1 and SIRT2 enzymes exhibited more potent deacylase activity than SIRT6 against long chains (>C8; ϵ-N-octanoyllysine), whereas SIRT3 appeared to be equipotent, which indicates that these enzymes may play even more versatile roles in biology than previously realized. Molecular modeling studies based on published x-ray crystal structures (Fig. 7A) suggest that, in addition to SIRT6 (18), SIRT1–3 may also accommodate the ϵ-N-myristoyllysine side chain in accordance with our experimental results as well as the recently published x-ray crystal structures of SIRT2 (68, 75). SIRT5, on the other hand, did not exhibit deacylase activity regardless of acyl chain length and only showed activity against its known substrates, ϵ-N-succinyllysine and ϵ-N-glutaryllysine. This is in agreement with the reported SIRT5 crystal structure (19), which demonstrates a less hydrophobic binding pocket, including critical arginine and tyrosine residues forming hydrogen bonds with the terminal carboxylic acid moiety of the succinylated substrate.
The requirement of NAD+ for sirtuin activity connects this enzyme class to the metabolic state of the cell because the NAD+/NADH redox pair is coupled to energy consumption. A shift from cytosolic fermentation to aerobic mitochondrial metabolism leads to decreasing intracellular levels of NADH and an increased NAD+/NADH ratio (44). Thus, the NAD+/NADH ratio can be affected in vitro by hypoxia (79) or by substrate metabolism, where low glucose leads to an increased NAD+/NADH ratio, and high glucose, lactate, or ethanol leads to a decrease in the NAD+/NADH ratio (43, 44, 80–82). Similarly, changes in the dinucleotide ratio can be accomplished in vivo by exercise (82), by calorie restriction, or by allowing high fat or high ethanol intake (82–84). Because changing the NAD+/NADH ratio has been shown to impact protein acetylation levels (44, 45), sirtuins could be affected by this ratio.
The potential direct regulation of sirtuin deacetylation activity by NADH was initially investigated more than a decade ago. Guarente and co-workers (44) demonstrated inhibition of ySir2 and hSIRT1 (no IC50 values reported) via an NAD+-competitive mechanism. Similarly, Denu and co-workers (85) reported inhibition of ySir2, yHST2, and hSIRT2 (IC50 = 15, 28, and 11 mm, respectively) and also included a very thorough evaluation of the potential effects of in vivo NADH concentrations, concluding that direct NADH inhibition of SIRT2-mediated deacetylation does not contribute significantly to the observed changes in acetylation patterns when changing the NAD+/NADH ratio (85). Elaborating on these results, we here show that NADH also inhibits the more recently discovered long chain deacylase activities of human SIRT1–3 and SIRT6 as well as the dicarboxyl-derived deacylase activities of SIRT5. Using short pseudopeptide substrates, we determined the various IC50 values to be in the low millimolar range, and data obtained using a dodecamer sequence also indicated that millimolar concentrations of NADH are necessary to inhibit any of the tested deacylase activities of human sirtuins. Furthermore, our results suggest that both SIRT1-mediated deacetylase and SIRT5-mediated desuccinylase activities are inhibited via a non-competitive mechanism for both peptide substrate and NAD+. Deacetylation by SIRT1–3 has been reported to follow an ordered bireactant mechanism with initial binding of the acetylated peptide followed by NAD+ binding (51, 77). If NADH binds in the AC pockets after peptide binding, an uncompetitive inhibition mechanism would be expected for peptide substrate and a competitive mechanism for NAD+. A potential random order binding of peptide and NAD+ has been suggested for SIRT6 (86). Under these conditions, a non-competitive inhibition would be expected when varying the peptide substrate concentration, whereas competitive inhibition would be expected when varying the NAD+ concentration. As a result, the observed non-competitive inhibition mechanism with respect to both substrates indicates a more complex binding mode of NADH. An alternative binding mode is also supported by our modeling results, where the dihydropyridine ring is poorly accommodated in the C pocket and instead breaks critical hydrogen bonding, leading to a conformational change of the protein. These findings provide a potential molecular basis for differentiation between the two dinucleotides and could justify the non-competitive inhibition observed in the activity assays.
In cells, the two dinucleotides may be either “free” or bound to protein with two major pools compartmentalized in the cytosol and the mitochondria, respectively. The total (i.e. free + bound) intracellular concentrations of NAD+ and NADH have been reported to be 1–3 mm (44), with an [NAD+]total/[NADH]totalratio of 1.5–10:1 (depending on species, cell type, and metabolic state) (44, 87–89). Studies have estimated that 40–90% of the total NADH may be bound to proteins (79, 90–92). However, the redox state depends on the unbound concentrations of dinucleotides in the different organelles, concentrations that are inherently difficult to measure. Based on indirect measurements of coupled reactions, under assumption of chemical equilibrium, different [NAD+]free/[NADH]free ratios in the cytosol (∼700–200:1) versus the mitochondria (∼10:1) were estimated already in the 1960s (93, 94). Furthermore, different methods have been applied to investigate aspects of the redox state, including [NAD(P)H]free by fluorescence (79, 90), cytosolic [NAD+]free/[NADH]free by NMR (95), and mitochondrial and cytosolic [NADH]free, [NAD+]free, or [NAD+]free/[NADH]freeby genetically encoded biosensors (80, 81, 96). Collectively, these studies estimate cytosolic and mitochondrial [NADH]free to be ∼110 nm and ∼30 μm, respectively (79, 81) and a cytosolic ratio of [NAD+]free/[NADH]free 70–2500:1 (depending on cell type and medium) (80, 95). Thus, the current knowledge of physiological levels of free NAD+ and NADH in the different organelles combined with the observed IC50 values strongly suggests constructive binding of the NAD+ cofactor to the sirtuins and an insignificant direct inhibitory effect of NADH, thereby corroborating the previous conclusions for SIRT2-mediated deacetylation (85) and extending them to include all known sirtuin-mediated deacylation reactions. Alternatively, nicotinamide, formed during the sirtuin deacylation reaction cascade, has been implicated in the regulation of sirtuin activity (43, 97, 98), and its function as a non-competitive inhibitor has been described in detail even in the novel deacylation reactions (75, 77, 99–101), where the inhibitory activity was found to vary with both sirtuin and acyl substrate. Whereas the changes in IC50 values for nicotinamide were more pronounced under the conditions used in the recent report (75), we also observed variation in inhibitory potency of both nicotinamide and ADP-ribose for the different sirtuin-substrate pairs, thereby highlighting potential conformational changes of the active site upon binding of the differently acylated peptides. Interestingly, the highest IC50 values for nicotinamide were found for SIRT1-mediated deacetylation and SIRT6-mediated deacylation, the enzyme-substrate pairs that were the most sensitive to ADPR inhibition. SIRT6-mediated deacylation reactions were especially affected by ADPR. This is a noteworthy observation and in agreement with previous reports that SIRT6 bind ADPR with an affinity of Kd = 4.7 ± 0.5 μm (86). Furthermore, a potential random order binding of peptide and NAD+ has also been suggested for SIRT6 (86), contrary to the other sirtuins, where peptide binding is a prerequisite for stabilization of the NAD+ binding site.
The presented data provide insight into the notion of sirtuins as sensors of the NAD+/NADH ratio. After comprehensively confirming recently reported sirtuin deacylase activities using three different assays, we showed that NADH is only able to inhibit sirtuin activities at non-physiological, millimolar concentrations, both for the previously reported SIRT2-mediated deacetylation and for the recently described long chain deacylase activities. Taken together, our data show that sirtuins are unlikely to be sensors of the NAD+/NADH ratio in vivo. Despite our data showing that the sirtuins are unlikely to sense NADH in vivo, these proteins require NAD+ as a co-substrate and are therefore still considered metabolic sensors. Accordingly, circadian fluctuations in NAD+ concentrations do indeed regulate sirtuin activity (48, 53, 102), but as one of the most important redox couples in the cell, the result of changes in NAD+ and NADH levels (e.g. as a result of calorie restriction) is highly complex and will affect multiple cellular processes, which may in turn affect sirtuin activity and protein acylation levels indirectly.
Author Contributions
A. S. M. devised experiments; performed synthesis, fluorescence-based, and HPLC-MS assays; analyzed data; and wrote the manuscript. C. A., M. D., J. S. L., and S. C. performed synthesis. K. A. A. devised experiments and performed [32P]NAD+ consumption assays. F. K. H. devised experiments, analyzed results, and wrote the manuscript. A. R. C., D. S. B., and P. F. performed molecular modeling. M. D. H. devised experiments, analyzed data, and wrote the manuscript. C. A. O. devised experiments, performed synthesis, analyzed data, and wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank the reviewers for valuable input. We thank Tina Gustafsson (DTU Chemistry) for technical assistance. The University of Colorado Computational Chemistry and Biology Core Facility is supported partly by National Institutes of Health/National Center for Advancing Translational Sciences Colorado Clinical and Translational Science Award Grant UL1TR001082.
This work was supported by Danish Independent Research Council-Technology and Production Sciences Sapere Aude Grants 12-132328 (to A. S. M.) and 11-105487 (to P. F.), Danish Independent Research Council-Natural Sciences Steno Grant 10-080907 (to C. A. O.), the Villum Foundation (to C. A. O.), the Carlsberg Foundation Grants 2013_01_0489 (to P. F.) and 2011_01_0169 and 2013_01_0333 (to C. A. O.). Nvidia generously donated a graphics card for this study to Technical University of Denmark (to P. F.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental information and Fig. 1.
- Kac
- ϵ-N-acetyllysine
- aa
- amino acids
- AMC
- 7-amino-4-methylcoumarin
- ADPR
- adenosine diphosphate ribose
- NAM
- nicotinamide
- HDAC
- histone deacetylase.
References
- 1. Biel M., Wascholowski V., and Giannis A. (2005) Epigenetics: an epicenter of gene regulation: histones and histone-modifying enzymes. Angew. Chem. Int. Ed. Engl. 44, 3186–3216 [DOI] [PubMed] [Google Scholar]
- 2. Minucci S., and Pelicci P. G. (2006) Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 6, 38–51 [DOI] [PubMed] [Google Scholar]
- 3. Kouzarides T. (2007) Chromatin modifications and their function. Cell 128, 693–705 [DOI] [PubMed] [Google Scholar]
- 4. Kazantsev A. G., and Thompson L. M. (2008) Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat. Rev. Drug Discov. 7, 854–868 [DOI] [PubMed] [Google Scholar]
- 5. Haberland M., Montgomery R. L., and Olson E. N. (2009) The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 10, 32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Choudhary C., Kumar C., Gnad F., Nielsen M. L., Rehman M., Walther T. C., Olsen J. V., and Mann M. (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 [DOI] [PubMed] [Google Scholar]
- 7. Zhao S., Xu W., Jiang W., Yu W., Lin Y., Zhang T., Yao J., Zhou L., Zeng Y., Li H., Li Y., Shi J., An W., Hancock S. M., He F., et al. (2010) Regulation of cellular metabolism by protein lysine acetylation. Science 327, 1000–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang Q., Zhang Y., Yang C., Xiong H., Lin Y., Yao J., Li H., Xie L., Zhao W., Yao Y., Ning Z. B., Zeng R., Xiong Y., Guan K. L., Zhao S., et al. (2010) Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327, 1004–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hirschey M. D., Shimazu T., Goetzman E., Jing E., Schwer B., Lombard D. B., Grueter C. A., Harris C., Biddinger S., Ilkayeva O. R., Stevens R. D., Li Y., Saha A. K., Ruderman N. B., Bain J. R., et al. (2010) SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464, 121–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Choudhary C., Weinert B. T., Nishida Y., Verdin E., and Mann M. (2014) The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 15, 536–550 [DOI] [PubMed] [Google Scholar]
- 11. Baeza J., Dowell J. A., Smallegan M. J., Fan J., Amador-Noguez D., Khan Z., and Denu J. M. (2014) Stoichiometry of site-specific lysine acetylation in an entire proteome. J. Biol. Chem. 289, 21326–21338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jiang T., Zhou X., Taghizadeh K., Dong M., and Dedon P. C. (2007) N-Formylation of lysine in histone proteins as a secondary modification arising from oxidative DNA damage. Proc. Natl. Acad. Sci. U.S.A. 104, 60–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen Y., Sprung R., Tang Y., Ball H., Sangras B., Kim S. C., Falck J. R., Peng J., Gu W., and Zhao Y. (2007) Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol. Cell Proteomics 6, 812–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Garrity J., Gardner J. G., Hawse W., Wolberger C., and Escalante-Semerena J. C. (2007) N-Lysine propionylation controls the activity of propionyl-CoA synthetase. J. Biol. Chem. 282, 30239–30245 [DOI] [PubMed] [Google Scholar]
- 15. Liu B., Lin Y., Darwanto A., Song X., Xu G., and Zhang K. (2009) Identification and characterization of propionylation at histone H3 lysine 23 in mammalian cells. J. Biol. Chem. 284, 32288–32295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stevenson F. T., Bursten S. L., Locksley R. M., and Lovett D. H. (1992) Myristyl acylation of the tumor necrosis factor α precursor on specific lysine residues. J. Exp. Med. 176, 1053–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stevenson F. T., Bursten S. L., Fanton C., Locksley R. M., and Lovett D. H. (1993) The 31-kDa precursor of interleukin 1α is myristoylated on specific lysines within the 16-kDa N-terminal propiece. Proc. Natl. Acad. Sci. U.S.A. 90, 7245–7249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jiang H., Khan S., Wang Y., Charron G., He B., Sebastian C., Du J., Kim R., Ge E., Mostoslavsky R., Hang H. C., Hao Q., and Lin H. (2013) SIRT6 regulates TNF-α secretion through hydrolysis of long-chain fatty acyl lysine. Nature 496, 110–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Du J., Zhou Y., Su X., Yu J. J., Khan S., Jiang H., Kim J., Woo J., Kim J. H., Choi B. H., He B., Chen W., Zhang S., Cerione R. A., Auwerx J., et al. (2011) Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 334, 806–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xie Z., Dai J., Dai L., Tan M., Cheng Z., Wu Y., Boeke J. D., and Zhao Y. (2012) Lysine succinylation and lysine malonylation in histones. Mol. Cell Proteomics 11, 100–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang Z., Tan M., Xie Z., Dai L., Chen Y., and Zhao Y. (2011) Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 7, 58–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tan M., Peng C., Anderson K. A., Chhoy P., Xie Z., Dai L., Park J., Chen Y., Huang H., Zhang Y., Ro J., Wagner G. R., Green M. F., Madsen A. S., Schmiesing J., et al. (2014) Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 19, 605–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tan M., Luo H., Lee S., Jin F., Yang J. S., Montellier E., Buchou T., Cheng Z., Rousseaux S., Rajagopal N., Lu Z., Ye Z., Zhu Q., Wysocka J., Ye Y., et al. (2011) Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146, 1016–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sabari B. R., Tang Z., Huang H., Yong-Gonzalez V., Molina H., Kong H. E., Dai L., Shimada M., Cross J. R., Zhao Y., Roeder R. G., and Allis C. D. (2015) Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Mol. Cell 58, 203–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dai L., Peng C., Montellier E., Lu Z., Chen Y., Ishii H., Debernardi A., Buchou T., Rousseaux S., Jin F., Sabari B. R., Deng Z., Allis C. D., Ren B., Khochbin S., et al. (2014) Lysine 2-hydroxyisobutyrylation is a widely distributed active histone mark. Nat. Chem. Biol. 10, 365–370 [DOI] [PubMed] [Google Scholar]
- 26. Moellering R. E., and Cravatt B. F. (2013) Functional lysine modification by an intrinsically reactive primary glycolytic metabolite. Science 341, 549–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hirschey M. D. (2011) Old enzymes, new tricks: sirtuins are NAD+-dependent de-acylases. Cell Metab. 14, 718–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Olsen C. A. (2012) Expansion of the lysine acylation landscape. Angew. Chem. Int. Ed. Engl. 51, 3755–3756 [DOI] [PubMed] [Google Scholar]
- 29. Olsen C. A. (2014) An update on lysine deacylases targeting the expanding “acylome”. ChemMedChem 9, 434–437 [DOI] [PubMed] [Google Scholar]
- 30. Jing H., and Lin H. (2015) Sirtuins in epigenetic regulation. Chem. Rev. 115, 2350–2375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Haigis M. C., and Sinclair D. A. (2010) Mammalian sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol. 5, 253–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schemies J., Uciechowska U., Sippl W., and Jung M. (2010) NAD+-dependent histone deacetylases (sirtuins) as novel therapeutic targets. Med. Res. Rev. 30, 861–889 [DOI] [PubMed] [Google Scholar]
- 33. Houtkooper R. H., Pirinen E., and Auwerx J. (2012) Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 13, 225–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gregoretti I. V., Lee Y. M., and Goodson H. V. (2004) Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J. Mol. Biol. 338, 17–31 [DOI] [PubMed] [Google Scholar]
- 35. Madsen A. S., and Olsen C. A. (2012) Substrates for efficient fluorometric screening employing the NAD-dependent sirtuin 5 lysine deacylase (KDAC) enzyme. J. Med. Chem. 55, 5582–5590 [DOI] [PubMed] [Google Scholar]
- 36. Madsen A. S., and Olsen C. A. (2012) Profiling of substrates for zinc-dependent lysine deacylase enzymes: HDAC3 exhibits decrotonylase activity in vitro. Angew. Chem. Int. Ed. Engl. 51, 9083–9087 [DOI] [PubMed] [Google Scholar]
- 37. Feldman J. L., Baeza J., and Denu J. M. (2013) Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. J. Biol. Chem. 288, 31350–31356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bao X. C., Wang Y., Li X., Li X. M., Liu Z., Yang T. P., Wong C. F., Zhang J. W., Hao Q., and Li X. D. (2014) Identification of “erasers” for lysine crotonylated histone marks using a chemical proteomics approach. eLife 10.7554/eLife.02999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Galleano I., Schiedel M., Jung M., Madsen A. S., and Olsen C. A. (2016) A continuous, fluorogenic sirtuin 2 deacylase assay: substrate screening and inhibitor evaluation. J. Med. Chem. 59, 1021–1031 [DOI] [PubMed] [Google Scholar]
- 40. Chiang Y. L., and Lin H. (2016) An improved fluorogenic assay for SIRT1, SIRT2, and SIRT3. Org. Biomol. Chem. 14, 2186–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mathias R. A., Greco T. M., Oberstein A., Budayeva H. G., Chakrabarti R., Rowland E. A., Kang Y., Shenk T., and Cristea I. M. (2014) Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity. Cell 159, 1615–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Imai S., Armstrong C. M., Kaeberlein M., and Guarente L. (2000) Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403, 795–800 [DOI] [PubMed] [Google Scholar]
- 43. Fulco M., Schiltz R. L., Iezzi S., King M. T., Zhao P., Kashiwaya Y., Hoffman E., Veech R. L., and Sartorelli V. (2003) Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Mol. Cell 12, 51–62 [DOI] [PubMed] [Google Scholar]
- 44. Lin S. J., Ford E., Haigis M., Liszt G., and Guarente L. (2004) Calorie restriction extends yeast life span by lowering the level of NADH. Genes Dev. 18, 12–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jang S. Y., Kang H. T., and Hwang E. S. (2012) Nicotinamide-induced mitophagy: event mediated by high NAD+/NADH ratio and SIRT1 protein activation. J. Biol. Chem. 287, 19304–19314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Karamanlidis G., Lee C. F., Garcia-Menendez L., Kolwicz S. C. Jr., Suthammarak W., Gong G., Sedensky M. M., Morgan P. G., Wang W., and Tian R. (2013) Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab. 18, 239–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Marcu R., Wiczer B. M., Neeley C. K., and Hawkins B. J. (2014) Mitochondrial matrix Ca2+ accumulation regulates cytosolic NAD+/NADH metabolism, protein acetylation, and sirtuin expression. Mol. Cell. Biol. 34, 2890–2902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bass J., and Takahashi J. S. (2010) Circadian integration of metabolism and energetics. Science 330, 1349–1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Davenport A. M., Huber F. M., and Hoelz A. (2014) Structural and functional analysis of human SIRT1. J. Mol. Biol. 426, 526–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Moniot S., Schutkowski M., and Steegborn C. (2013) Crystal structure analysis of human Sirt2 and its ADP-ribose complex. J. Struct. Biol. 182, 136–143 [DOI] [PubMed] [Google Scholar]
- 51. Jin L., Wei W., Jiang Y., Peng H., Cai J., Mao C., Dai H., Choy W., Bemis J. E., Jirousek M. R., Milne J. C., Westphal C. H., and Perni R. B. (2009) Crystal structures of human SIRT3 displaying substrate-induced conformational changes. J. Biol. Chem. 284, 24394–24405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Feig M., Onufriev A., Lee M. S., Im W., Case D. A., and Brooks C. L. 3rd (2004) Performance comparison of generalized born and Poisson methods in the calculation of electrostatic solvation energies for protein structures. J. Comput. Chem. 25, 265–284 [DOI] [PubMed] [Google Scholar]
- 53. Nakahata Y., Sahar S., Astarita G., Kaluzova M., and Sassone-Corsi P. (2009) Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science 324, 654–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhao X., Allison D., Condon B., Zhang F., Gheyi T., Zhang A., Ashok S., Russell M., MacEwan I., Qian Y., Jamison J. A., and Luz J. G. (2013) The 2.5 Å crystal structure of the SIRT1 catalytic domain bound to nicotinamide adenine dinucleotide (NAD+) and an indole (EX527 analogue) reveals a novel mechanism of histone deacetylase inhibition. J. Med. Chem. 56, 963–969 [DOI] [PubMed] [Google Scholar]
- 55. Harder E., Damm W., Maple J., Wu C., Reboul M., Xiang J. Y., Wang L., Lupyan D., Dahlgren M. K., Knight J. L., Kaus J. W., Cerutti D., Krilov G., Jorgensen W. L., Abel R., et al. (2016) OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 12, 281–296 [DOI] [PubMed] [Google Scholar]
- 56. Wu Y. D., and Houk K. N. (1991) Theoretical evaluation of conformational preferences of NAD+ and NADH: an approach to understanding the stereospecificity of NAD+/NADH-dependent dehydrogenases. J. Am. Chem. Soc. 113, 2353–2358 [Google Scholar]
- 57. Beis K., Allard S. T., Hegeman A. D., Murshudov G., Philp D., and Naismith J. H. (2003) The structure of NADH in the enzyme dTDP-d-glucose dehydratase (RmlB). J. Am. Chem. Soc. 125, 11872–11878 [DOI] [PubMed] [Google Scholar]
- 58. Nosé S. (1984) A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 81, 511–519 [Google Scholar]
- 59. Hoover W. G. (1985) Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A 31, 1695–1697 [DOI] [PubMed] [Google Scholar]
- 60. Martyna G. J., Tobias D. J., and Klein M. L. (1994) Constant pressure molecular dynamics algorithms. J. Chem. Phys. 101, 4177–4189 [Google Scholar]
- 61. Hu J., He B., Bhargava S., and Lin H. (2013) A fluorogenic assay for screening Sirt6 modulators. Org. Biomol. Chem. 11, 5213–5216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Michishita E., McCord R. A., Berber E., Kioi M., Padilla-Nash H., Damian M., Cheung P., Kusumoto R., Kawahara T. L. A., Barrett J. C., Chang H. Y., Bohr V. A., Ried T., Gozani O., and Chua K. F. (2008) SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature 452, 492–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sakaguchi K., Herrera J. E., Saito S., Miki T., Bustin M., Vassilev A., Anderson C. W., and Appella E. (1998) DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 12, 2831–2841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Liu L., Scolnick D. M., Trievel R. C., Zhang H. B., Marmorstein R., Halazonetis T. D., and Berger S. L. (1999) p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol. Cell. Biol. 19, 1202–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Knights C. D., Catania J., Di Giovanni S., Muratoglu S., Perez R., Swartzbeck A., Quong A. A., Zhang X., Beerman T., Pestell R. G., and Avantaggiati M. L. (2006) Distinct p53 acetylation cassettes differentially influence gene-expression patterns and cell fate. J. Cell Biol. 173, 533–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Brandl A., Wagner T., Uhlig K. M., Knauer S. K., Stauber R. H., Melchior F., Schneider G., Heinzel T., and Krämer O. H. (2012) Dynamically regulated sumoylation of HDAC2 controls p53 deacetylation and restricts apoptosis following genotoxic stress. J. Mol. Cell Biol. 4, 284–293 [DOI] [PubMed] [Google Scholar]
- 67. Edrissi B., Taghizadeh K., and Dedon P. C. (2013) Quantitative analysis of histone modifications: formaldehyde is a source of pathological N6-formyllysine that is refractory to histone deacetylases. PLoS Genet. 9, e1003328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Teng Y. B., Jing H., Aramsangtienchai P., He B., Khan S., Hu J., Lin H., and Hao Q. (2015) Efficient demyristoylase activity of SIRT2 revealed by kinetic and structural studies. Sci. Rep. 5, 8529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lahm A., Paolini C., Pallaoro M., Nardi M. C., Jones P., Neddermann P., Sambucini S., Bottomley M. J., Lo Surdo P., Carfí A., Koch U., De Francesco R., Steinkühler C., and Gallinari P. (2007) Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc. Natl. Acad. Sci. U.S.A. 104, 17335–17340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Jones P., Altamura S., De Francesco R., Gallinari P., Lahm A., Neddermann P., Rowley M., Serafini S., and Steinkühler C. (2008) Probing the elusive catalytic activity of vertebrate class IIa histone deacetylases. Bioorg. Med. Chem. Lett. 18, 1814–1819 [DOI] [PubMed] [Google Scholar]
- 71. Schuetz A., Min J., Allali-Hassani A., Schapira M., Shuen M., Loppnau P., Mazitschek R., Kwiatkowski N. P., Lewis T. A., Maglathin R. L., McLean T. H., Bochkarev A., Plotnikov A. N., Vedadi M., and Arrowsmith C. H. (2008) Human HDAC7 harbors a class IIa histone deacetylase-specific zinc binding motif and cryptic deacetylase activity. J. Biol. Chem. 283, 11355–11363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bradner J. E., West N., Grachan M. L., Greenberg E. F., Haggarty S. J., Warnow T., and Mazitschek R. (2010) Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 6, 238–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hubbard B. P., Gomes A. P., Dai H., Li J., Case A. W., Considine T., Riera T. V., Lee J. E., E S. Y., Lamming D. W., Pentelute B. L., Schuman E. R., Stevens L. A., Ling A. J. Y., Armour S. M., et al. (2013) Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science 339, 1216–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Vogelauer M., Krall A. S., McBrian M. A., Li J. Y., and Kurdistani S. K. (2012) Stimulation of histone deacetylase activity by metabolites of intermediary metabolism. J. Biol. Chem. 287, 32006–32016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Feldman J. L., Dittenhafer-Reed K. E., Kudo N., Thelen J. N., Ito A., Yoshida M., and Denu J. M. (2015) Kinetic and structural basis for acyl-group selectivity and NAD+ dependence in sirtuin-catalyzed deacylation. Biochemistry 54, 3037–3050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Avalos J. L., Bever K. M., and Wolberger C. (2005) Mechanism of sirtuin inhibition by nicotinamide: altering the NAD+ cosubstrate specificity of a Sir2 enzyme. Mol. Cell 17, 855–868 [DOI] [PubMed] [Google Scholar]
- 77. Borra M. T., Langer M. R., Slama J. T., and Denu J. M. (2004) Substrate specificity and kinetic mechanism of the Sir2 family of NAD+-dependent histone/protein deacetylases. Biochemistry 43, 9877–9887 [DOI] [PubMed] [Google Scholar]
- 78. Wu Y. D., and Houk K. N. (1993) Theoretical study of conformational features of NAD+ and NADH analogs: protonated nicotinamide and 1,4-dihydronicotinamide. J. Org. Chem. 58, 2043–2045 [Google Scholar]
- 79. Zhang Q., Piston D. W., and Goodman R. H. (2002) Regulation of corepressor function by nuclear NADH. Science 295, 1895–1897 [DOI] [PubMed] [Google Scholar]
- 80. Hung Y. P., Albeck J. G., Tantama M., and Yellen G. (2011) Imaging cytosolic NADH-NAD+ redox state with a genetically encoded fluorescent biosensor. Cell Metab. 14, 545–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhao Y., Jin J., Hu Q., Zhou H.-M., Yi J., Yu Z., Xu L., Wang X., Yang Y., and Loscalzo J. (2011) Genetically encoded fluorescent sensors for intracellular NADH detection. Cell Metab. 14, 555–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Gambini J., Gomez-Cabrera M. C., Borras C., Valles S. L., Lopez-Grueso R., Martinez-Bello V. E., Herranz D., Pallardo F. V., Tresguerres J. A. F., Serrano M., and Viña J. (2011) Free [NADH]/[NAD+] regulates sirtuin expression. Arch. Biochem. Biophys. 512, 24–29 [DOI] [PubMed] [Google Scholar]
- 83. Chen D., Bruno J., Easlon E., Lin S. J., Cheng H. L., Alt F. W., and Guarente L. (2008) Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 22, 1753–1757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kim H. J., Kim J. H., Noh S., Hur H. J., Sung M. J., Hwang J. T., Park J. H., Yang H. J., Kim M. S., Kwon D. Y., and Yoon S. H. (2011) Metabolomic analysis of livers and serum from high-fat diet induced obese mice. J. Proteome Res. 10, 722–731 [DOI] [PubMed] [Google Scholar]
- 85. Schmidt M. T., Smith B. C., Jackson M. D., and Denu J. M. (2004) Coenzyme specificity of Sir2 protein deacetylases: implications for physiological regulation. J. Biol. Chem. 279, 40122–40129 [DOI] [PubMed] [Google Scholar]
- 86. Pan P. W., Feldman J. L., Devries M. K., Dong A., Edwards A. M., and Denu J. M. (2011) Structure and biochemical functions of SIRT6. J. Biol. Chem. 286, 14575–14587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Lin S. J., and Guarente L. (2003) Nicotinamide adenine dinucleotide, a metabolic regulator of transcription, longevity and disease. Curr. Opin. Cell Biol. 15, 241–246 [DOI] [PubMed] [Google Scholar]
- 88. Easlon E., Tsang F., Dilova I., Wang C., Lu S. P., Skinner C., and Lin S. J. (2007) The dihydrolipoamide acetyltransferase is a novel metabolic longevity factor and is required for calorie restriction-mediated life span extension. J. Biol. Chem. 282, 6161–6171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Wilhelm F., and Hirrlinger J. (2011) The NAD+/NADH redox state in astrocytes: independent control of the NAD+ and NADH content. J. Neurosci. Res. 89, 1956–1964 [DOI] [PubMed] [Google Scholar]
- 90. Patterson G. H., Knobel S. M., Arkhammar P., Thastrup O., and Piston D. W. (2000) Separation of the glucose-stimulated cytoplasmic mitochondrial NAD(P)H responses in pancreatic islet β cells. Proc. Natl. Acad. Sci. U.S.A. 97, 5203–5207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Blinova K., Carroll S., Bose S., Smirnov A. V., Harvey J. J., Knutson J. R., and Balaban R. S. (2005) Distribution of mitochondrial NADH fluorescence lifetimes: steady-state kinetics of matrix NADH interactions. Biochemistry 44, 2585–2594 [DOI] [PubMed] [Google Scholar]
- 92. Yu Q., and Heikal A. A. (2009) Two-photon autofluorescence dynamics imaging reveals sensitivity of intracellular NADH concentration and conformation to cell physiology at the single-cell level. J. Photochem. Photobiol. B 95, 46–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Williamson D. H., Lund P., and Krebs H. A. (1967) The redox state of free nicotinamide-adenine dinucleotide in cytoplasm and mitochondria of rat liver. Biochem. J. 103, 514–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Veech R. L., Eggleston L. V., and Krebs H. A. (1969) The redox state of free nicotinamide-adenine dinucleotide phosphate in cytoplasm of rat liver. Biochem. J. 115, 609–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Christensen C. E., Karlsson M., Winther J. R., Jensen P. R., and Lerche M. H. (2014) Non-invasive in-cell determination of free cytosolic [NAD+]/[NADH] ratios using hyperpolarized glucose show large variations in metabolic phenotypes. J. Biol. Chem. 289, 2344–2352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Bilan D. S., Matlashov M. E., Gorokhovatsky A. Y., Schultz C., Enikolopov G., and Belousov V. V. (2014) Genetically encoded fluorescent indicator for imaging NAD+/NADH ratio changes in different cellular compartments. Biochim. Biophys. Acta 1840, 951–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Anderson R. M., Bitterman K. J., Wood J. G., Medvedik O., and Sinclair D. A. (2003) Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature 423, 181–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Revollo J. R., Grimm A. A., and Imai S. (2004) The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J. Biol. Chem. 279, 50754–50763 [DOI] [PubMed] [Google Scholar]
- 99. Bitterman K. J., Anderson R. M., Cohen H. Y., Latorre-Esteves M., and Sinclair D. A. (2002) Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast Sir2 and human SIRT1. J. Biol. Chem. 277, 45099–45107 [DOI] [PubMed] [Google Scholar]
- 100. Jackson M. D., Schmidt M. T., Oppenheimer N. J., and Denu J. M. (2003) Mechanism of nicotinamide inhibition and transglycosidation by Sir2 histone/protein deacetylases. J. Biol. Chem. 278, 50985–50998 [DOI] [PubMed] [Google Scholar]
- 101. Fischer F., Gertz M., Suenkel B., Lakshminarasimhan M., Schutkowski M., and Steegborn C. (2012) Sirt5 deacylation activities show differential sensitivities to nicotinamide inhibition. PLoS One 7, e45098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Ramsey K. M., Yoshino J., Brace C. S., Abrassart D., Kobayashi Y., Marcheva B., Hong H. K., Chong J. L., Buhr E. D., Lee C., Takahashi J. S., Imai S., and Bass J. (2009) Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science 324, 651–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.