

Abstract
Mechanisms controlling the metabotropic γ-aminobutyric acid receptor (GABAB) cell surface stability are still poorly understood. In contrast with many other G protein-coupled receptors (GPCR), it is not subject to agonist-promoted internalization, but is constitutively internalized and rapidly down-regulated. In search of novel interacting proteins regulating receptor fate, we report that the ubiquitin-specific protease 14 (USP14) interacts with the GABAB(1b) subunit's second intracellular loop. Probing the receptor for ubiquitination using bioluminescence resonance energy transfer (BRET), we detected a constitutive and phorbol 12-myristate 13-acetate (PMA)-induced ubiquitination of the receptor at the cell surface. PMA also increased internalization and accelerated receptor degradation. Overexpression of USP14 decreased ubiquitination while treatment with a small molecule inhibitor of the deubiquitinase (IU1) increased receptor ubiquitination. Treatment with the internalization inhibitor Dynasore blunted both USP14 and IU1 effects on the receptor ubiquitination state, suggesting a post-endocytic site of action. Overexpression of USP14 also led to an accelerated degradation of GABAB in a catalytically independent fashion. We thus propose a model whereby cell surface ubiquitination precedes endocytosis, after which USP14 acts as an ubiquitin-binding protein that targets the ubiquitinated receptor to lysosomal degradation and promotes its deubiquitination.
Keywords: bioluminescence resonance energy transfer (BRET), biosensor, deubiquitylation (deubiquitination), G protein-coupled receptor (GPCR), GABA receptor, protein degradation, receptor endocytosis, ubiquitylation (ubiquitination)
Introduction
G protein-coupled receptors (GPCRs)5 are the largest family of cell-surface proteins in the human genome and represent the target of a large proportion of current pharmacological agents. Regulation of GPCR activity in response to different stimuli provides cells with important mechanisms to fine tune the response to natural ligands as well as drugs.
For most GPCRs, desensitization results from agonist-promoted phosphorylation by second messenger-activated and GPCR kinases (GRK) (1), leading to β-arrestin-promoted uncoupling from the G protein and subsequent endocytosis. In contrast, the metabotropic γ-aminobutyric acid receptor (GABAB), an obligatory hetero-oligomer composed of two different 7TM proteins, GABAB(1) and GABAB(2), which provides the metabotropic response to the inhibitory neurotransmitter GABA (2), does not undergo β-arrestin engagement and receptor endocytosis following agonist stimulation. Two GRK isoforms (GRK4 and GRK5) (3) have been proposed to play a role in GABAB desensitization. In the case of GRK4, this effect was found to be independent of its kinase activity, as its regulator of G protein signaling (RGS) domain alone is sufficient for desensitization of the receptor (4). Also, in contrast with most GPCR, GABAB activity is not correlated with the overall phosphorylation state of the receptor. Agonist-promoted protein kinase C (PKC) phosphorylation decreases receptor activity (5) while phosphorylation by adenosine monophosphate activated-kinase (AMPK) on GABAB(2) serine 783 (6) and by protein kinase A on GABAB(2) serine 893 (7) have been linked to receptor sensitization and increased effector coupling. Finally, interaction of the receptor with members of the potassium channel tetramerization domain (KCTD) family can also modulate receptor-effector coupling (8), adding yet another level of regulation.
Removal of agonist-desensitized receptor from the cell surface through endocytosis is a common step for GPCR resensitization or degradation (9). However, GABAB does not undergo agonist-promoted endocytosis (4, 5) but internalizes constitutively at a rapid rate, up to 50% of the receptor being removed from the cell surface in 2 h (10). This phenomenon has been observed in both heterologous expression systems and primary neuron cultures (4, 11). The exact mechanism underlying constitutive internalization is not yet characterized, but it has been suggested that a direct interaction between the GABAB receptor and the clathrin adaptor protein-2 (AP-2) complex could be involved (10). More recently, evidence has surfaced that glutamate receptor activation can control cell surface availability of GABAB (12) through distinct mechanisms. The NMDA receptor promotes PP2A-mediated dephosphorylation of serine 783 on GABAB(2), favoring degradation over recycling of the receptor (13). CamKII phosphorylation of serine 867 on GABAB(1) promotes GABAB endocytosis (14) in response to NMDA receptor activation. AMPAR and mGluRs have also been suggested to have a role in glutamate-mediated GABAB down-regulation (15). None of the downstream cellular machinery linked to this process has been identified yet.
Among the mechanism controlling cell surface stability of GPCRs, ubiquitination has been increasingly shown to play an important role. Activity-dependent ubiquitination of β-arrestin has been linked to the early steps of endocytosis (16) whereas the ubiquitination state of receptors has generally been linked to degradation (17) and in some cases to endocytosis (18). In the case of the β2-adrenergic receptor (β2AR), members of the Nedd4 E3 ubiquitin ligases family have been shown to regulate receptor ubiquitination after recruitment by β-arrestin or arrestin domain-containing protein-3 and -4 (19, 20) while the deubiquitinases USP20 and USP33 were suggested to promote receptor recycling (21). In the case of the CXCR4 chemokine receptor, both ubiquitination by AIP4 (22) and deubiquitination by USP14 (23) are necessary for efficient CXCR4 degradation, in a mechanism that could suggest the importance of a ubiquitination/deubiquitination cycle. The deubiquitinase USP8 has also been shown to promote trafficking and degradation of CXCR4 (24).
Using the second intracellular loop of the GABAB(1) subunit in search of regulatory proteins interacting with the receptor, we identified USP14 in a yeast-two hybrid screen. Considering the role of USP14 as a deubiquitinating enzyme, and the evidence suggesting that GABAB ubiquitination could be linked to down-regulation (15) and endoplasmic reticulum (ER)-associated proteasomal degradation (25), we took advantage of a bioluminescence resonance energy transfer (BRET) assay to measure real-time GABAB ubiquitination in living cells (26) and assessed the role of USP14 in the de-ubiquitination and degradation of the GABAB. The regulation of the receptor's ubiquitination state and degradation by PKC was also assessed.
Experimental Procedures
Materials
HA-GABAB(1a)-CFP, cMyc-GABAB(2)-YFP, and Usp14-YFP were prepared by subcloning of CFP or YFP into pRK5-HA-GABAB(1a), pRK5-cMyc-GABAB(2) and pRK5-Usp14 (cloned from mouse mRNA). pcDNA3-Myc-GABAB(1b), pcDNA3-Myc-GABAB(1b)-Rluc, pcDNA3-HA-GABAB(2) and pcDNA3-HA-GABAB(2)-Rluc constructs have been previously described (5, 27). pEYFP-Ubi and pEYFP-UbiAA were constructed as described in Ref. 26, but the human ubiquitin (Ubi) coding sequence was inserted in pEYFP-C1(Clontech) instead of pGFP2-C1. pcDNA3-USP14b, pcDNA3-USP14b-GFP and pcDNA3-Rluc-USP14b were constructed by PCR from a corresponding cDNA clone (OriGene) and inserted in the pcDNA3 vector. Point mutations were done by PCR to create pcDNA3-USP14b-C79A and pcDNA3-USP14b-C79A-GFP as well as all the lysine mutants of GABAB(1b) (pcDNA3-Myc-GABAB(1b)-KA1-Rluc (K887A/K893A), pcDNA3-Myc-GABAB(1b)-KA2-Rluc (K900A/K905A), pcDNA3-Myc-GABAB(1b)-KKAA-Rluc (K887A/K893A/K900A/K905A)), and GABAB(2) (pcDNA3-HA-GABAB(2)-KA3-Rluc (K767A/K771A), HA-GABAB(2)-KA4 (K801A/K807A)) and the ER-retention signal mutant of GABAB(1b) (pcDNA3-Myc-GABAB(1b)-ASRR-Rluc). CXCR4-Rluc (28) and pcDNA3-V2R-Rluc (29) have been previously described. pcDNA3-PAR1-Rluc was a generous gift from Dr. Terry Hébert (McGill University, Montreal, Canada). Dynasore, chloroquine, PMA, and MG132 were purchased from Sigma-Aldrich while GABA was from Calbiochem, IU1 from Cayman Chemical, coelenterazine h from Nanolight Technology and EZ-Link Sulfo-NHS-LC-Biotin from Thermo Scientific. Poly-d-lysine was from MP Biomedicals while poly-l-lysine was from Gibco. All cell culture reagents were from Wisent or Gibco. All others chemicals were from Sigma. Anti-Myc (9E10) was an ascites fluid prepared in house while anti-GFP was from Clontech (632592). Anti-GABAB(2) was a generous gift from GlaxoSmithKline (United Kingdom). Anti-Rluc was obtained from Ray Biotechnologies (130–00005). Anti-actin antibody (sc-1616) was from Santa Cruz Biotechnologies. Anti-USP14 antibody (8159), USP14-siRNA and control siRNA were from Cellular Signaling. Anti-mouse IgG (NA931V) and anti-rabbit IgG (NA934V) secondary antibody coupled to horseradish peroxidase were from GE HealthCare Life Sciences. Pep-il2 (VHTVFTKKEEKKEWRKTLEP-YGRKKRRQRRR) and the random sequence peptide pep-RSP (ISHVCKLFAM-YGRKKRRQRRR) were synthesized by GenScript.
Transfection
HEK293T cells were transfected with the indicated plasmids using PEI, as described previously (29). The following day, cells were transferred to a poly-d-lysine-coated 24 well plate (200 000 cells per well). COS7 cells were grown onto poly-l-Lysine-coated coverslips overnight, transiently transfected by calcium phosphate co-precipitation with equal amount of HA-GABAB(1a)-CFP, cMyc-GABAB(2)-YFP, or Usp14-YFP constructs in pRK5 vector.
Yeast Two-hybrid
Sequence corresponding to the GABAB(1) second intracellular loop (GB1i2, protein sequence: TKIWWVHTVFTKKEEKKEWRKTLEPWKLY) was introduced in a pGBT9 bait vector in-frame with the Gal4 DNA-binding domain. The construct was screened against a commercially obtained rat brain cDNA library according to Clontech protocols (Clontech, Palo Alto, CA). The screening efficiency with pGBT9-GB1i2 bait was 5.1 × 106, resulting in the identification of 24 positive colonies. Yeast-mating assays for GB1i2 binding were performed using yeast strains HF7c and Y187, with pGBT9 and pACT2 vectors. One of the positive clones, USP14, contained the cDNA coding for amino acid 239 to the stop codon and the untranslated region up to the poly-A tail. The cDNA coding for the entire open reading frame was cloned from isolated rat brain RNA (reverse transcriptase, PCR amplification, and TA-cloning system with blue-white selection (Invitrogen)) and sequenced.
Fluorescent Microscopy
24 h post-transfection, DMEM (Gibco) was replaced by PBS and the living COS7 cells were placed under an inverted fluorescent microscope Zeiss Axiovert 200 m. The fluorescent pictures were processed by MetaMorph software (Molecular Devices).
Ubiquitination Measurement by BRET
To measure ubiquitination, a BRET-based assay monitoring the addition of ubiquitin-YFP to GABAB subunits linked to Rluc was performed as previously described (26). Briefly, a ubiquitin mutant form of Ubi (mono-Ubi-YFP) in which lysines 48 and 63, the two major sites for polyubiquitination, were mutated to alanine to avoid steric hindrance, quenching or other interference phenomena due to the presence of multiple YFP that could lead to a blunted BRET signal. A ubiquitin-YFP lacking the two terminal glycines required for covalent ubiquitination (UbiAA-YFP) was used as a negative control. Treatments were done directly in 6- or 12-well plates for the indicated time. Medium was then removed, cells washed twice in PBS and resuspended in PBS. 100,000 cells were then transferred to 96-well white plates. BRET measurements were done as described in Ref. 29. In short, cells were treated for 2 min with 5 μm coelenterazine h before reading in a Berthold Mithras LB940 equipped with acceptor (530 ± 20 nm) and donor (480 ± 20 nm) filters. netBRET was calculated as the ratio of the signal detected in the 530 ± 20 nm window (corresponding to the YFP emission) divided by the signal detected in the 480 ± 20 nm window (corresponding to the Rluc emission) when both Rluc and YFP partners were expressed minus the same ratio when only the Rluc partner was expressed. The specific ubiquitination BRET was calculated by subtracting the nonspecific netBRET measured when UbiAA-YFP was expressed as the energy acceptor from the netBRET measured when MonoUbi-YFP was expressed as the energy acceptor.
Degradation Assay using Biotin Labeling and Streptavidin Precipitation
Biotin labeling was modified from Ref. 10. HEK293 cells expressing Myc-tagged GABAB(1b) and HA-tagged GABAB(2) were treated with EZ-Link Sulfo-NHS-LC-Biotin (0.5 mg/ml) in buffer A (25 mm HEPES, pH 7.4, 119 mm NaCl, 5 mm KCl, 2 mm CaCl2, 2 mm MgCl2, 30 mm glucose) at 4 °C for 30 min, then washed three times with ice-cold buffer A. Cells were then either kept on ice (control) or treated with the appropriate compound at 37 °C for the indicated chase time. Cells were solubilized in RIPA buffer (50 mm Tric-HCl pH 7.4, 150 mm NaCl, 2.5 mm EDTA, 1% Nonidet P-40, 0.5% sodium deoxycholate, 1 mm DTT, 5 μg/ml leupeptin, 5 μg/ml soybean trypsin inhibitor, 10 μg/ml benzamidine) and biotin-labeled receptors purified by pull-down with streptavidin-Sepharose beads and subjected to SDS-PAGE and Western blotting with anti-Myc (9E10) mouse antibody. Quantification of the receptors was done by chemiluminescence using a LAS-3000 from Fujifilm and analyzed using Quantity One software from Bio-Rad.
Internalization Assay by ELISA
Two days following transfection, media was removed and cells were washed in ice-cold buffer A. Cells were then incubated with 9E10 anti-Myc antibody (1:1000 in buffer A containing 0.2% BSA) for 1 h on ice, then washed three times in cold buffer A. Cells were then treated or not (vehicle) at 37 °C for 1 h. Following incubation, cells were washed in cold buffer A and incubated with secondary antibody (anti-mouse IgG coupled to horseradish peroxidase) for 45 min. Cells were then washed three times, and the colorimetric ELISA signal was measured after adding SIGMAFASTTM.
Immunoprecipitation and Western Blotting
Immunoprecipitations (IPs) were done as described in (5) using RIPA buffer Anti-Myc antibody was added in a 1:400 dilution. After incubation for 16 h, beads were washed three times with RIPA buffer containing 350 mm NaCl. Remaining complexes were dissolved in Laemmli buffer before being subjected to SDS-PAGE and Western blotting using anti GABAB(2), 9E10 anti-Myc, anti-GFP, and anti-Rluc antibodies. To detect USP14 expression after siRNA transfection, cells were lysed and subjected to Western blotting as described above using anti USP14. Actin was used as internal control for normalization.
Statistical Analysis
All statistical analysis were done using GraphPad Prism 5.0. Analysis of variance (ANOVA) followed by post hoc Dunnett or Tukey tests were used for multiple comparisons; Dunnett tests being used to compare multiple conditions to a single control, whereas Tukey tests were used to identify the difference within a group. Student's t tests were performed for pairwise comparison.
Results
Identification of USP14 as a Novel GABAB Interactor
Using the evolutionary conserved second cytoplasmic loop of GABAB(1b) as a bait in a yeast two-hybrid screen, we identified a rat cDNA encoding part of the catalytic domain of the Ubiquitin-Specific Protease 14 (USP14) (Fig. 1A). The interaction between USP14 and GABAB(1b) in the context of the functional heterodimer was confirmed by co-immunoprecipitation studies in HEK293T cells. As shown in Fig. 1B, GFP-USP14 was detected by Western blot analysis following receptor immunoprecipitation. The appearance of USP14 as a doublet (with two bands separated by ≈3 kDa) may result from a post-translational modification or degradation of a fraction of USP14. Although the precise reason is unknown, similar migration profiles have been previously observed for USP14 (23, 30). Considering that the interacting USP14 cDNA found in the yeast-two-hybrid screen encodes its catalytic domain, we tested whether the interaction was dependent on the deubiquitinase activity of USP14. For this purpose, we took advantage of a mutant form of USP14 (USP14-C79A) which is catalytically inactive (31). Immunoprecipitation revealed that both WT- and USP14-C79A interacted to the same extent with the receptor, suggesting that USP14 deubiquitinase activity is not required for its interaction with GABAB(1).
FIGURE 1.

GABAB interacts with USP14 through its GABAB(1) subunit. A, schematic representation of the GABAB(1) second intracellular loop used as bait and the USP14 region recovered in a rat brain cDNA yeast two-hybrid screening. B, HEK293T cells expressing a combination of Myc-GABAB(1b), HA-GABAB(2), Rluc-USP14, and/or Rluc-USP14-C79A were solubilized and immunoprecipitated (IP) using an anti-myc antibody. Total (input, 5% of IP) and IP samples were separated on SDS-PAGE and proteins detected by Western blotting (WB) using the indicated antibodies (anti-myc for GABAB(1b), anti GABAB(2) for GABAB(2), and anti-Rluc for USP14). C, HEK293T cells expressing a combination of Myc-GABAB(1b), HA-GABAB(2) and/or GFP-USP14 were treated for 2 h without (vehicle) or with 10 μm of either Pep-il2 or Pep-RSP, then solubilized and immunoprecipitated (IP) using an anti-myc antibody. Total (input, 5% of IP) and IP samples were separated on SDS-PAGE and proteins detected by Western blotting (WB) using the indicated antibodies (anti-myc for GABAB(1b), anti GABAB(2) for GABAB(2), and anti-GFP for USP14). The results shown are representative of three independent experiments.
To assess the specificity of USP14 binding to GABAB(1b), we created a membrane-permeable TAT-fused peptide comprising the intracellular 20 amino acids (688 to 707) of the 29-mer sequence from the yeast two-hybrid bait peptide originating from the GABAB(1b) second intracellular loop (pep-il2; Fig. 1A). Pre-treatment of cells with pep-il2 completely abolished the co-immunoprecipitation of GFP-USP14 with the functional GABAB heterodimer whereas a TAT-fused random sequence peptide was without effect (Fig. 1C), confirming the interaction site to the second intracellular loop of GABAB(1b).
Probing GABAB Ubiquitination
To investigate the potential role of USP14 in the regulation of GABAB ubiquitination, we first assessed whether GABAB ubiquitination can be measured in living cells using a BRET-based ubiquitination assay (26). BRET titration curves were obtained using receptor subunits genetically fused to Renilla Luciferase (Rluc) and a genetically engineered ubiquitin (monoUbi) construct linking its N terminus to an enhanced Yellow Fluorescent Protein (YFP) (26). To assess the extent of receptor ubiquitination, an increasing amount of monoUbi-YFP was co-expressed with a constant amount of either GABAB(1b)-Rluc or GABAB(2)-Rluc with the associated untagged partner subunit. A mutated ubiquitin missing the C-terminal glycine residues that cannot be conjugated to its substrate and linked to YFP (UbiAA-YFP) was used as a negative control.
As shown in Fig. 2, increasing Ubi-YFP led to a hyperbolic increase of the BRET signal with either GABAB(1b)-Rluc (Fig. 2A) or GABAB(2)-Rluc (Fig. 2C) when expressed in the presence of their untagged cognate partner subunit (GABAB(2) and GABAB(1b), respectively). Although random collision between energy donor and acceptors and non-covalent binding of Ubi can contribute to the BRET signal, the saturability of the signal suggests the occupancy of specific ubiquitination sites. The selectivity of the signal is further supported by the observation that the UbiAA-YFP negative control led to much weaker signals that generally increased linearly with increasing concentration of UbiAA-YFP, as could be expected from random collision, and was therefore used as background BRET values (32). The specific ubiquitination signals (inset of Fig. 2, A–C) were therefore calculated by subtracting the background BRET (UbiAA-YFP signal) from the total signal observed. Specific signals were detected, whether the GABAB(1b) or GABAB(2) were tagged with luciferase within the heterodimer. The expression of GABAB(1b)-Rluc alone, which is retained in the ER in the absence of GABAB(2) (Fig. 2E), led to a weaker specific ubiquitination signal (Fig. 2B), suggesting that a significant fraction of the ubiquitination signal originates from plasma membrane receptors. Consistent with this notion, a larger ubiquitination BRET signal is observed when using a mutant form of the GABAB(1) (GABAB(1b)-ASRR) that can reach the plasma membrane in the absence of GABAB(2), as a result of a mutation of GABAB(1b) ER-retention signal (33) (Fig. 2B). Specific ubiquitination was also observed when GABAB(2), which can easily access the cell surface, was expressed alone (Fig. 2D). To study ubiquitination of the functional heterodimer that can reach plasma membrane, the GABAB(1b)-Rluc/GABAB(2) pair was used for the rest of the study.
FIGURE 2.

BRET titration curves of GABAB homo- and heterodimer with MonoUbi-YFP or UbiAA-YFP. BRET signal, total fluorescence, and total luminescence were measured in HEK293T cells transfected with constant amount of Myc-GABAB(1b)-Rluc and HA-GABAB(2) (A), wild-type (WT) Myc-GABAB(1b)-Rluc or Myc-GABAB(1b)-ASRR-Rluc (B), Myc-GABAB(1b) and HA-GABAB(2)-Rluc (C) or HA-GABAB(2)-Rluc (D) and increasing amount of either MonoUbi-YFP or UbiAA-YFP. The data obtained in two independent experiments were pooled and used to generate the curves. The specific ubiquitination BRET curves (inset A–D) were obtained by subtracting the UbiAA-YFP curve from the MonoUbi-YFP curve. E, fluorescent microscopy of COS7 cells expressing either HA-GABAB(1a)-CFP (left panel) or both HA-GABAB(1a)-CFP and cMyc-GABAB(2)-YFP (right panel). F, table describing the point mutations in each GABAB(1b) and GABAB(2) constructs used. G and H, BRET signal were measured in HEK293T cells expressing the WT and mutants version of Myc-GABAB(1b)-Rluc (G) or HA-GABAB(2) (H), along with the WT GABAB partner subunit and either MonoUbi-YFP or UbiAA-YFP. UbiAA-YFP signal was subtracted from MonoUbi-YFP signal to generate specific ubiquitination BRET signal. The results are presented as the mean ± S.E. of three independent experiments performed in quadruplicates. (*, p < 0.05; **, p < 0.01.)
To investigate the residues of both GABAB subunits potentially involved in receptor ubiquitination, we mutated four lysines that had previously been found to be ubiquitinated in a brain ubiquitome screen (Lys-887 and Lys-905 on GABAB(1b), and Lys-767 and Lys-801 on GABAB(2) (34) and their proximal lysines (Lys-893A and Lys-900 on GABAB(1b), and Lys-771 and Lys-807 on GABAB(2)) to prevent ubiquitination site shifting (Fig. 2F). As shown in Fig. 2G, mutants Myc-GABAB(1b)-KA1 (K887A/K893A) and Myc-GABAB(1b)-KA2 (K900A/K905A) showed a significant (p < 0,05) decrease (15 ± 3% and 16 ± 3%, respectively) in receptor ubiquitination, and the combination of the four mutations in Myc-GABAB(1b)-KKAA led to a further reduction in BRET signal (27 ± 2%), indicating that the mutated residues represent potential sites of ubiquitination on the GABAB(1b) subunit. On the GABAB(2) subunit, the previously characterized K767A/K771A mutant (25) (HA-GABAB(2)-KA3) showed a 11 ± 3% decrease in BRET signal, while the HA-GABAB(2)-KA4 (K801A/K807A) mutant showed no significant difference (Fig. 2H). These data indicate that some of the previously identified sites contribute to the basal ubiquitination signal but that additional lysines are also involved.
Stimulation of the receptor with GABA was without effect on the ubiquitination signal (Fig. 3A). Similarly, no effect was observed upon treatment with baclofen or the GABAB inverse agonist CGP54626 (35) (data not shown), indicating that neither agonist-promoted nor constitutive activation of the receptor had a direct effect on its ubiquitination level. Since GABAB activity has long been known to be down-regulated by PKC (36), we assess the effect of the PKC activator, PMA (phorbol-13-myristate-12-acetate), on receptor ubiquitination. Treatment with PMA led to a transient increase in the ubiquitination signal (163 ± 14%) that peaked at 4 h post-treatment and returned to basal level at 24 h (Fig. 3A). This increase was blocked by pre-treatment with the PKC inhibitor Bisindolylmaleimide I (Fig. 3A, inset), indicating a specific PKC-mediated effect.
FIGURE 3.

PMA increases ubiquitination and accelerates lysosome-mediated degradation of GABAB. A, HEK293T cells expressing Myc-GABAB(1b)-Rluc, HA-GABAB(2) and either MonoUbi-YFP or UbiAA-YFP were treated or not (vehicle) for the indicated time with either 1 mm GABA, 1 μm PMA and/or 100 nm bisindolylmaleimide I (inset, all 2 h). UbiAA-YFP signal was subtracted from MonoUbi-YFP signal to derive the specific BRET signal. B–D, HEK293T cells expressing Myc-GABAB(1b) and HA-GABAB(2) were labeled with EZ-Link Sulfo-NHS-LC-Biotin and either kept on ice (control) or switch at 37 °C with vehicle (B) or 100 nm PMA (C) for the indicated time. After cell solubilization, biotin-labeled receptors were purified by pull-down with streptavidin-Sepharose beads and detected by Western blot (WB) with anti-Myc (9E10) mouse antibody. Receptors remaining after the chase period were quantified (D) and t1/2 was calculated (inset). E–F, as described for B–C, but the temperature switch at 37 °C was done only for 2 h in the presence of vehicle, 200 μm chloroquine or 5 μg/ml MG132. The results shown (B, C, E) are representative of at least three independent experiments or are the mean ± S.E. of at least five (A) or at least three (D, F) independent experiments performed in quadruplicates. (*, p < 0.05; **, p < 0.01; ***, p < 0.001.)
PMA Promotes Faster Degradation and Internalization of GABAB
Since ubiquitination has been linked to the degradation of several membrane proteins, including a few GPCRs (16, 37, 38), we assessed the effect of PMA treatment on the degradation of GABAB using HEK293 cells co-expressing Myc-GABAB(1b) and HA-GABAB(2). Cells were biotinylated at 4 °C and receptor levels were assessed following incubation at 37 °C for up to 4 h using a streptavidin pull-down followed by Western blotting of either GABAB(1b) or GABAB(2). Under basal conditions, the half-life of the plasma membrane-targeted receptor was found to be 2.4 ± 0.6 h (Fig. 3, B and D); the half-life observed being the same whether we monitored GABAB(1b) or GABAB(2) immuno-reactivity (data not shown). Treatment of cells with PMA led to a significant (p < 0.05) increase in the rate of degradation (half-life = 1.0 ± 0.3 h), suggesting a PKC-mediated acceleration of receptor degradation (Fig. 3, C and D). To investigate whether this cell surface disappearance results from proteasomal or lysosomal degradation, we treated cells with the lysosomal acidification inhibitor chloroquine or the proteasomal inhibitor MG132. MG132 had no effect whereas chloroquine almost completely inhibited receptor degradation (Fig. 3, E and F).
Given that GABAB has been shown to have a rapid rate of constitutive endocytosis that contributes to degradation (11), the decrease in receptor-half-life detected could result either from an accelerated rate of internalization or a greater targeting of endocytosed receptors to lysosomal degradation. To distinguish between these two possibilities, we assessed the effect of PMA on internalization. For this purpose, we labeled GABAB heterodimer with Myc-targeted antibody at 4 °C and induced constitutive internalization by switching the temperature to 37 °C, thus monitoring only the disappearance from the plasma membrane. Two hours after temperature switch, 24 ± 4% of the receptor had been internalized under basal condition (Fig. 4A). Treatment with GABA did not increase the extent of internalization, confirming earlier reports that the receptor does not undergo agonist-promoted internalization (4, 39). In contrast, treatment with PMA led to a significant (p < 0.05) increase of endocytosis (33 ± 4%), suggesting that the PMA-accelerated degradation resulted at least in part from an increased internalization.
FIGURE 4.

Internalization inhibitor Dynasore blocks PMA induced internalization and promote constitutive and PMA induced ubiquitination. A, HEK293T cells expressing Myc-GABAB(1b)-Rluc and HA-GABAB(2) were labeled with anti-Myc 9E10 antibody for 1 h on ice. Internalization was then induced at 37 °C for 2 h in the absence (vehicle) or presence of 1 mm GABA, 1 μm PMA, and/or 50 μm Dynasore, while control cells were kept on ice. Receptor amount still at the cell surface were measured by ELISA. The results are presented as the mean ± S.E. of at least four independent experiments performed in quadruplicates. B, HEK293T cells expressing Myc-GABAB(1b)-Rluc, HA-GABAB(2) and either MonoUbi-YFP or UbiAA-YFP were treated or not (vehicle) for 2 h with 50 μm Dynasore and/or 1 μm PMA. UbiAA-YFP signal was subtracted from MonoUbi-YFP signal (specific BRET). Inset shows the PMA induced BRET: PMA signal minus no-PMA added in the absence (vehicle) or presence of Dynasore. The results are presented as the mean ± S.E. of at least three to five independent experiments performed in quadruplicates. (*, p < 0.05; **, p < 0.01; ***, p < 0.001)
To probe whether the ubiquitination of the receptor occurs at the cell surface or in the endosomes following internalization, we assessed the effect of a general blocker of dynamin-dependent endocytosis, Dynasore, on both receptor internalisation and ubiquitination. Dynasore significantly (p < 0.01) blunted both constitutive (54 ± 7% inhibition) and PMA-induced (63 ± 9% inhibition) internalization (Fig. 4A). The Dynasore-mediated block in endocytosis was accompanied by a statistically significant (p < 0.001) 29 ± 3% increase in the basal ubiquitination (Fig. 4B), suggesting that at least part of the modification occurs before internalization of the receptor. Co-treatment with both PMA and Dynasore led to an additive increase in BRET signal (Fig. 4B), indicating that a substantial part of the PMA-promoted increase in ubiquitination also occurs at the plasma membrane.
USP14 Regulates GABAB Ubiquitination and Degradation
To assess the potential role of USP14 in GABAB regulation, we first determined the effect of the deubiquitinase overexpression on the ubiquitination state of the receptor. Co-expression of USP14 with the GABAB receptor led to a 25 ± 4% decrease in receptor ubiquitination whereas the catalytically inactive mutant form of the deubiquitinase, USP14-C79A, did not affect the basal level of ubiquitination (Fig. 5A), indicating that USP14 catalyzed GABAB deubiquitination. Furthermore, both treatment with IU1, a recently discovered inhibitor of USP14 (31), and Pep-il2, the cell-permeable USP14/GABAB(1b) interaction blocking peptide, led to a significant (p < 0.01) increase in GABAB ubiquitination (Fig. 5, B and C). The observation that IU1 (Fig. 5B) increased GABAB ubiquitination in cells overexpressing or not USP14 and that Pep-il2 (Fig. 5C) also inhibited the ubiquitination in cells endogenously expressing USP14 suggests that this enzyme is responsible for the endogenous constitutive deubiquitination of the receptor through a direct protein-protein interaction. To assess whether USP14 acts as a general deubiquitination enzyme of GPCRs, we tested the effect of USP14 overexpression on 3 additional receptors. As previously reported (23), USP14 reduced CXCR4 ubiquitination (Fig. 5D). Similarly, the ubiquitination level of the type-2 vasopressin receptor was also decreased by the enzyme whereas it did not alter PAR-1 ubiquitination, suggesting some level of selectivity in its GPCR substrates.
FIGURE 5.

USP14 deubiquitinates GABAB and accelerates its degradation rate. A–C, HEK293T cells were transfected with Myc-GABAB(1b)-Rluc, HA-GABAB(2), either MonoUbi-YFP or UbiAA-YFP along with pcDNA3, USP14 (A, B) or USP14-C79A (A). For B, cells were treated for three or sixteen hours with 100 μm IU1. For C, cells were treated or not (vehicle) for 2 h with 10 μm Pep-il2 or Pep-RSP. BRET signals were measured and the specific BRET ubiquitination signal calculated. D, HEK293T cells were transfected with Myc-GABAB(1b)-Rluc (GB1b) and HA-GABAB(2) (GB2), CXCR4-Rluc, V2R-Rluc or Par1-Rluc, along with MonoUbi-YFP or UbiAA-YFP, and either pcDNA3 or USP14. Inset shows the percentage of USP14 inhibition: USP14 minus pcDNA3 divided by the pcDNA3 for each condition. The results are presented as the mean ± S.E. of at least three independent experiments performed in triplicates. (*, p < 0.05; **, p < 0.01; ***, p < 0.001.)
Given the proposed roles of ubiquitination on the internalization and degradation of membrane proteins, we assessed the effect of USP14 on these processes for the GABAB. Co-expression of USP14 with GABAB lead to a significant acceleration of the degradation rate of the receptor, reducing the half-life of the receptor from 3 h to less than 1 h (Fig. 6, A, B, G). Surprisingly, the catalytically inactive USP14-C79A, which could not promote deubiquitination of GABAB (Fig. 5A), accelerated receptor degradation to the same extent as the WT form of the enzyme (Fig. 6, C and G), suggesting that the USP14-promoted degradation is independent of its deubiquitinase activity. Consistent with this notion, treatment of cells with the USP14 inhibitor IU1 did not significantly affect degradation (Fig. 6, D and G), also suggesting that the deubiquitinase activity of USP14 does not contribute to GABAB degradation. In contrast, treatment with the Pep-il2 blocking peptide significantly slowed down receptor disappearance whereas treatment with the Pep-RSP had no significant difference (Fig. 6, E–G), indicating the importance of the USP14/GABAB protein-protein interaction in the USP14-mediated receptor degradation. Contrary to the PMA treatment, which increased both the degradation and the endocytosis of the receptor, neither USP14 overexpression nor IU1 treatment affected the extent of internalization (Fig. 6H), indicating that USP14-promoted GABAB degradation is independent of the receptor internalization rate, suggesting a post-endocytic mechanism.
FIGURE 6.

USP14 accelerates the degradation rate of GABAB independently of its catalytic activity. A–G, HEK293T cells expressing Myc-GABAB(1b), HA-GABAB(2) and either pcDNA3 (A, D--F), USP14 (B) or USP14-C79A (C) were labeled with EZ-Link Sulfo-NHS-LC-Biotin and treated or not (vehicle, A–C) with either 100 μm IU1 (D), 10 μm Pep-RSP (E), or 10 μm Pep-il2 (F) for the indicated time, as described in Fig. 3. The receptors remaining after the chase period were quantified and half-life calculated (G). H, HEK293T cells expressing Myc-GABAB(1b)-Rluc, HA-GABAB(2) and either pcDNA3 or USP14 were labeled with anti-Myc 9E10 antibody for one hour on ice. Internalization was induced by temperature switch to 37 °C for 2 h in the absence (vehicle) or presence of 100 μm IU1. Control cells were kept on ice. Receptor amount still at the cell surface were measured by ELISA. The results shown are representative of three independent experiments (A–F) or are the mean ± S.E. of at least three independent experiments performed in triplicates (G, H). (*, p < 0.05.)
To confirm the importance of endogenous USP14 toward GABAB deubiquitination and degradation, we knocked-down the expression of the deubiquitinase using siRNA (Fig. 7A). This knock-down led to a significant increase in GABAB ubiquitination (p < 0,01) and a reduced rate of degradation (increased receptor half-life) (p < 0,05) compared with a control siRNA (Fig. 7, B–E).
FIGURE 7.

USP14 siRNA knockdown diminishes GABAB ubiquitination and increase its half-life. A, HEK293T cells were transfected with either 100 nm USP14 siRNA, 100 nm control siRNA or pcDNA3, then solubilized and separated on SDS-PAGE followed by Western blotting (WB) using the indicated antibodies. The results shown are representative of three independent experiments. B, HEK293T cells were transfected with Myc-GABAB(1b)-Rluc, HA-GABAB(2), either MonoUbi-YFP or UbiAA-YFP along with pcDNA3, 100 nm USP14 siRNA, or 100 nm control siRNA. BRET signals were measured and the specific ubiquitination signal calculated. C–E, HEK293T cells expressing Myc-GABAB(1b), HA-GABAB(2) and 100 nm control siRNA (C) or 100 nm USP14 siRNA (D) were labeled with EZ-Link Sulfo-NHS-LC-Biotin and were treated as described in Fig. 3. The receptors remaining after the chase period were quantified (E) and the half-life calculated (E, inset). (*, p < 0.05; **, p < 0.01.)
USP14 Acts as a Post-endocytosis Regulator of GABAB
To determine whether USP14 acts pre- or post-endocytosis, its deubiquitination activity was tested in the presence and absence of the internalization inhibitor Dynasore. As shown in Fig. 8A, treatment with Dynasore led to a time-dependent decrease of the deubiquitination promoted by USP14 overexpression, indicating a post-endocytosis action of USP14. Indeed, overexpressing USP14 resulted, as expected, in an overall reduction of the ubiquitination state of the receptor. However, inhibiting endocytosis with Dynasore significantly blunted the effect of USP14, the reduction in GABAB ubiquitination promoted by USP14 overexpression going from 21% to 9% following a 16h Dynasore treatment (Fig. 8A, inset). Also consistent with the post-endocytotic action of USP14, the increased GABAB ubiquitination observed following inhibition of USP14 by UI1 was blunted from 23% to 8% following Dynasore treatment (Fig. 8B, inset). Together, these data clearly suggest that most of the deubiquitination action of USP14 occurs following endocytosis since a pre-endocytic action of USP14 would have been potentiated, not blunted by inhibiting endocytosis. This is also consistent with a primarily cytoplasmic localization of USP14 (Fig. 8C). Of notice, the deubiquitinating activity of USP14 was not affected by inhibition of lysosomal degradation with chloroquine (Fig. 8D). The observation that chloroquine treatment increased ubiquitination signal in the presence or absence of overexpressed USP14, is consistent with the lysosomal degradation of the ubiquitinated receptor.
FIGURE 8.
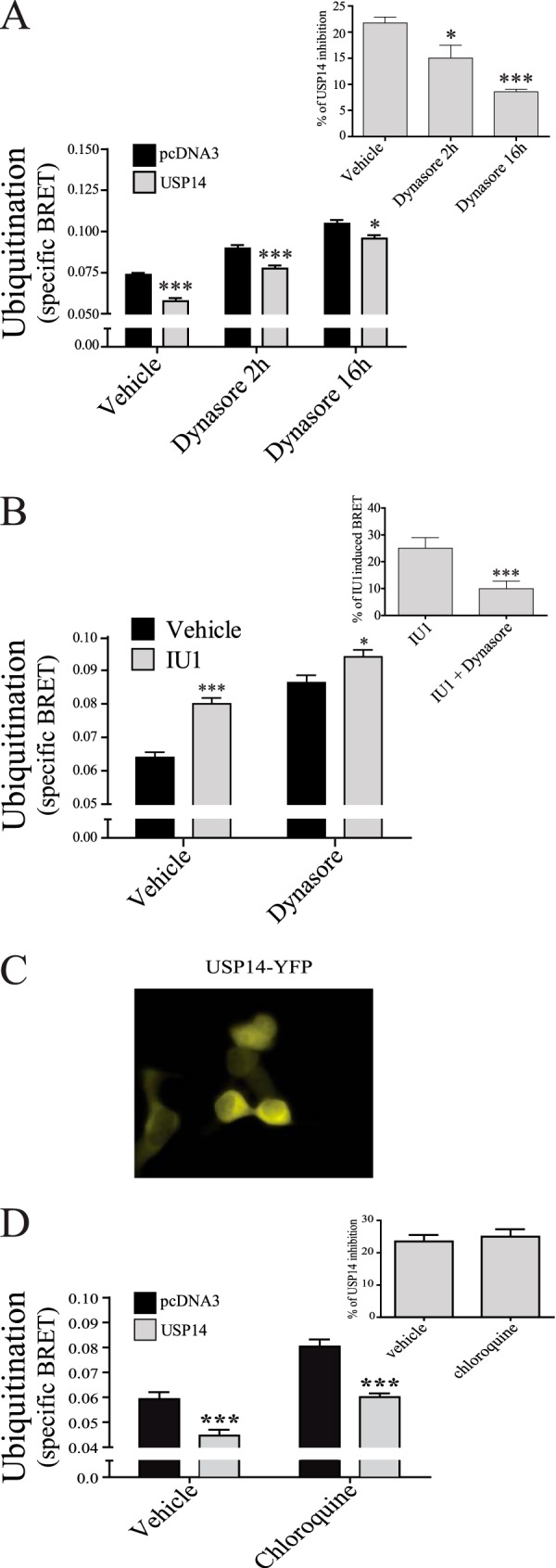
USP14 deubiquitination is decreased by internalization inhibitor Dynasore. HEK293T cells were transfected with Myc-GABAB(1b), HA-GABAB(2), MonoUbi-YFP or UbiAA-YFP, and either pcDNA3 or USP14. A, cells were treated or not (vehicle) for two or sixteen hours with 50 μm Dynasore and specific BRET ubiquitination signal calculated. Inset shows the percentage of inhibition of USP14 and calculated as in Fig. 5C. B, cells were treated or not (vehicle) for 2 h with 50 μm Dynasore, with or without IU1. Inset shows the percentage of IU1 induced BRET: IU1 treated minus vehicle dividing by vehicle condition. C, fluorescent microscopy illustrating the cytoplasmic distribution of USP14-YFP in COS7 cells co-expressing HA-GABAB(1a)-CFP, cMyc-GABAB(2). D, cells transfected as in A were treated or not (vehicle) for 2 h with 200 μm chloroquine and specific BRET ubiquitination signal calculated. Inset shows the percentage of inhibition of USP14 and calculated as in Fig. 5C. The results are presented as the mean ± S.E. of at least three independent experiments performed in triplicates. (*, p < 0.05; ***, p < 0.001.)
Discussion
In the present study, we showed that GABAB is constitutively ubiquitinated in living cells and that this post-translational modification occurs, at least in part, once the receptor has reached the cell surface. Increased ubiquitination promoted by PKC activation is accompanied by an increase in GABAB internalization and accelerated degradation. Deubiquitination of the receptor is catalyzed post-endocytically by the deubiquitinase USP14 that contributes to the targeting of the receptor for lysosomal degradation. In addition, to further our understanding of the constitutive ubiquitination and cell surface regulation of GABAB, our study unravels a new PKC-mediated regulation of this post-translational modification and identifies USP14 as an important regulator of both receptor ubiquitination and degradation.
USP14 is a member of the ubiquitin-specific proteases (USPs)/ubiquitin-specific-processing proteases (UBPs) family of proteases, falling into subfamily of Peptidases C19. This subfamily is, with around 60 members, one of the largest families of peptidases in the human genome (40). These intracellular peptidases remove ubiquitin molecules from poly-ubiquitinated peptides by cleavage of isopeptide bonds, through the hydrolysis of carboxyl group bonds of the extreme C-terminal glycine residue of ubiquitin. The level of sequence conservation varies considerably among the members of the family. USP14 displays diverse sequence identity with the other members of the family, depending on varying regions outside the catalytic core. In the catalytic domain, the conservation of USP14 reaches between 18 and 26% with the other members. Despite the relatively low sequence identity, residues surrounding the catalytic cysteine and histidine are conserved and the overall fold of the catalytic domain seems preserved among these enzymes (41).
As mentioned, in addition to the catalytic domain, USPs also harbor diverse accessory domains that are believed to confer selectivity for the regulation of their catalytic activity, substrate recognition, recruitment of regulatory factors and subcellular localization. USP14 contains a single ubiquitin-like domain on the N terminus that has been proposed to be its main site of interaction with the proteasome (40).
Given that mutation that abolished the catalytic activity of USP14 did not prevent the interaction between the deubiquiting enzyme and the receptor nor its ability to promote the lysosomal-dependent degradation of GABAB, the role played by each of these domains on the binding and degradation of GABAB remains to be determined. Whether USP family members act similarly on GABAB or other GPCRs also remains a largely unanswered question, although USP8 has been shown to promote CXCR4 trafficking and degradation (24).
A role for PKC in the regulation of GABAB had been previously suggested, as PKC has been shown to phosphorylate GABAB (5) and down-regulate synaptic activity of the receptor (36). Although the role of PKC-mediated phosphorylation in GABAB ubiquitination remains to be investigated, phosphorylation of other substrates, such as the gap junction protein connexin-43 (42) and the organic anion transporter-1 (43) by PKC has been shown to promote their ubiquitination, resulting in their degradation and internalization, respectively. In the case of GABAB, we found that inhibition of endocytosis led to an increased level of both constitutive and PMA-induced ubiquitination, indicating that the PKC-promoted modification occurred at least in part at the plasma membrane and preceded endocytosis. Combined with our observation that PMA treatment led to an elevation of both ubiquitination and endocytosis, these results are consistent with the findings for other membrane proteins that PMA increases ubiquitination and promotes endocytosis (44). In contrast, receptor ubiquitination at the plasma membrane was found to inhibit the constitutive internalization of another GPCR, PAR1, whereas agonist-promoted deubiquitination favored its endocytosis (18). It should be emphasized, however, that PAR1 may represent a special case since this receptor is activated by proteolytic cleavage of its N terminus, resulting in a continuous activation that requires degradation for its inactivation. Further studies will be needed to determine whether ubiquitination directly favors internalization of GABAB through the recruitment of endocytosis machinery elements.
We have identified six potential residues (Lys-887, Lys-893, Lys-900, and Lys-905 on GABAB(1b); Lys-767 and Lys-771 on GABAB(2)) involved in ubiquitination of GABAB. Three of those residues (Lys-887 and Lys-905 on GABAB(1b) and Lys-767 on GABAB(2)) had been found to be ubiquitinated in the human brain (34) and both Lys-767 and Lys-771 have been linked to proteasomal degradation of GABAB through a mechanism involving Rpt6 (25, 45). Lys-893 and Lys-900, however, represent novel ubiquitination sites revealed by our study. Whether these sites are responsible for the cell surface ubiquitination or the ubiquitination associated with the ERAD system remains to be investigated. Proteasomal degradation has been previously shown to affect the expression of GABAB but was linked to the ER and forward trafficking of the receptor (25). Our findings identify yet an additional and distinct cell-membrane and post-endocytic role for ubiquitination and USP14.
The observation that stimulation of GABAB by its agonists GABA and baclofen did not affect the extent of receptor ubiquitination contrasts with what was observed for several GPCRs, such as the β2AR (16, 19) and CXCR4 (38). In those cases, agonist-promoted β-arrestin recruitment brings E3 ubiquitin ligases to the receptors, promoting ubiquitination. In contrast, GABAB stimulation has not been found to promote β-arrestin recruitment and internalization (4, 39), nor to promote receptor down-regulation (11), consistent with the lack of agonist-stimulated ubiquitination observed. The process leading to the constitutive ubiquitination of GABAB and the identity of the E3 ligase involved remains to be investigated. However, our data clearly identify USP14 as a deubiquitinase that regulates the ubiquitination state of GABAB. This action of USP14 was blocked by an inactivating mutation of its catalytic domain, indicating that this diminution is due to deubiquitination of the receptor and not to a decrease in de novo receptor ubiquitination. The USP14 deubiquitination was significantly blunted when the endocytic process was inhibited by Dynasore, indicating that the deubiquitination occurred following the internalization of the receptor. While Dynasore may have off-target actions, its effect on deubiquitination is consistent with the observation that USP14 overexpression led to an accelerated rate of GABAB lysosomal degradation, while siRNA-promoted knock-down of USP14 led to a decreased rate of receptor removal. Such a role for USP14 in the post-endocytic processing of GPCRs has been observed for the CXCR4. Indeed, knock-down of USP14 expression was found to block receptor degradation following stimulation with its agonist CXCL12 (23).
In contrast to its proposed role in the deubiquitination-promoted degradation of CXCR4 (23), the USP14-accelerated rate of degradation of GABAB observed in our study was found to be independent of its catalytic activity. The inactive enzyme showed a similar ability to decrease the receptor's half-life as its wild type counterpart. This was dependent on its interaction with GABAB, as the cell-permeable Pep-il2 peptide promoted an increase receptor half-life, most likely by preventing interaction with endogenous USP14. These results suggest that USP14 action on the GABAB receptor stability results from direct interaction with the receptor and is not a by-product of the deubiquitination. Such a non-catalytic action of USP14 has been proposed for its role on the activation of the 20S proteasome. Indeed, direct interaction between USP14 and the ubiquitin chain was found to promote the opening of the proteasome, leading to the degradation of the deubiquitinase cargo (46, 47). The data obtained herein for the GABAB are however at variance with this canonical role of USP14 as a proteasomal-associated deubiquitinase (31, 46, 47), since we found the USP14-promoted GABAB degradation to be lysosomal- and not proteasomal-dependent. It is presently unknown whether, as is the case for its proteasomal role (47), USP14 could act both as a substrate targeting receptor and degradation activator at the lysosome. It is noteworthy that USP14 has also been suggested to be involved in ERAD-associated proteasomal degradation of GABAB, as IU1 treatment was found to decrease receptor trafficking to the cell surface (25), indicating that USP14-mediated deubiquitination can have distinct actions on GABAB steady-state levels. Again, this is distinct from the catalytic-independent GABAB degradation-promoting activity of USP14 described in the present study.
Taken together, our results suggest a novel mechanism controlling GABAB cell surface expression that involves receptor ubiquitination and the binding of USP14 to promote receptor degradation. The proposed model (Fig. 9) shows cell surface ubiquitination of the GABAB receptor occurring constitutively or promoted by PMA. Upon endocytosis, USP14 acts as a catalytically-independent ubiquitin-binding protein to promote endosomal sorting of highly ubiquitinated GABAB toward lysosomal degradation. The USP14-promoted deubiquitination of GABAB is not needed for its sorting and degradation-promoting activity, as illustrated by the ability of the catalytically-inactive USP14-C79A to lead to lysosomal degradation. The deubiquitinase activity in this model may however be important for ubiquitin recycling, as previously suggested for USP14 (46). Our data suggest that increased ubiquitination of GABAB can coexist with faster degradation (ie: when USP14-C79A is overexpressed). There is therefore no need to invoke the action of another deubiquitinase before lysosomal degradation but we cannot exclude the contribution of such another enzyme. In such a model, non-ubiquitinated receptor would not bind to USP14 and would be targeted toward the recycling pathway, allowing reinsertion of the receptor in the plasma membrane and leading to the resensitization of the cells to GABA stimulation. Interestingly, glutamate has been shown to promote internalization (14) and down-regulation of GABAB (15) that involves a balance between receptor recycling and degradation (49) in which ubiquitination plays an important role, mirroring multiple elements from our proposed model. Possible activation of PKC by glutamate or others physiological stimuli could provide a general mechanism of regulation of GABAB activity, as we have observed that stimulation of endogenous muscarinic receptors with carbachol also leads to increased GABAB ubiquitination (data not shown). It is also tempting to speculate that the constitutively active isoform of PKC, PKMζ, involved in long-term potentiation maintenance could act in part by down-regulating GABAB activity (50). Further studies are needed to examine the possible links between various physiological stimuli and the USP14-promoted degradation of GABAB.
FIGURE 9.

Proposed model of GABAB ubiquitination and USP14 mediated post-endocytosis degradation. Cell surface GABAB receptors undergo constitutive or PKC-mediated ubiquitination and constitutive internalization. Non-ubiquitinated receptors do not effectively engage USP14 and are sorted toward recycling (1), while ubiquitinated receptors bind USP14 in a catalytically independent manner, through its ubiquitin-binding domain, and are targeted toward lysosomal degradation (2). The catalytically-deficient USP14 (C79A) leads to receptor lysosomal degradation without ubiquitin recycling (3 and 5). However, the catalytically active USP14 deubiquitinates the receptors during trafficking, allowing ubiquitin recycling while the receptor is degraded (4 and 5).
The proposed role of USP14 in receptor regulation may have important physiological consequences. Indeed, a transgenic mouse model with an invalidated USP14 gene (USP14 knock-out), also known as the ataxia mice, shows profound defects in synaptic transmission (51). These defects have been shown to result in part from an increased GABAA cell surface expression and activity that is due to altered endosomal sorting and decreased receptor degradation (52), suggesting a similar role of USP14 on GABAA and GABAB. Also, the ataxia mice show a reduction in the size of the releasable vesicle pool that results in altered neurotransmitter release (53). The GABAB receptor being directly involved in regulating vesicle priming at the release site (54), it is tempting to speculate that USP14 knock-down promotes increased level of synaptic GABAB receptor through reduced degradation, leading to an amplified GABAB activity and reduced releasable vesicle priming. Consistent with this hypothesis, the decreased size of the releasable vesicle pool was rescued by expression of the catalytically inactive USP14 mutant (48), mirroring our finding of a catalytic-independent role of USP14.
In conclusion, our study reveals a new PKC-mediated regulatory mechanism controlling GABAB ubiquitination, endocytosis and degradation and identifies USP14 as a deubiquitinating enzyme that regulates the post-endocytic ubiquitination state and degradation of the GABAB with potential implications for synaptic transmission.
Author Contributions
N. L. conceived, performed, and analyzed all the experiments described in Figs. 1 to 8 (except the yeast-two hybrid experiments) and wrote the paper. M. K. conceived, performed, and analyzed the experiments related to the yeast two hybrid screen, identifying USP14 as an interactor of GABAB, and the fluorescence microscopy as well as generated constructs that were used in the study. L. P. coordinated the study and wrote the paper. J. B. coordinated the study and wrote the paper. M. B. conceived and coordinated the study and wrote the paper. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Pavel Osten for yeast two hybrid methodology and Dr. Monique Lagace for her expert assistance throughout these studies and her critical reading of the manuscript.
These studies were supported by the Canadian Institutes of Health Research (CIHR) (MOP 10501) and the Czech Science Foundation (GACR P303/12/2408). The authors declare that they have no conflicts of interest with the contents of this article.
- GPCR
- G protein-coupled receptors
- GABAB
- metabotropic γ-aminobutyric acid receptor
- USP14
- ubiquitin-specific protease 14
- BRET
- bioluminescence resonance energy transfer
- ER
- endoplasmic reticulum
- PMA
- phorbol 12-myristate 13-acetate
- GRK
- GPCR kinases
- PKC
- protein kinase C
- AMPK
- adenosine monophosphate activated-kinase
- KCTD
- potassium channel tetramerization domain
- AP-2
- clathrin adaptor protein-2
- β2AR
- β2-adrenergic receptor
- Rluc
- Renilla Luciferase
- YFP
- yellow fluorescent protein
- Ubi
- human ubiquitin
- PAR1
- protease-activated receptor 1.
References
- 1. Luttrell L. M., and Lefkowitz R. J. (2002) The role of {beta}-arrestins in the termination and transduction of G-protein-coupled receptor signals. J. Cell Sci. 115, 455–465 [DOI] [PubMed] [Google Scholar]
- 2. White J. H., Wise a, Main M. J., Green A., Fraser N. J., Disney G. H., Barnes A. A., Emson P., Foord S. M., and Marshall F. H. (1998) Heterodimerization is required for the formation of a functional GABA(B) receptor. Nature 396, 679–682 [DOI] [PubMed] [Google Scholar]
- 3. Kanaide M., and Uezono Y. (2007) Desensitization of GABAB receptor signaling by formation of protein complexes of GABAB2 subunit with GRK4 or GRK5. J. Cell Physiol. 210, 237–245 [DOI] [PubMed] [Google Scholar]
- 4. Perroy J., Adam L., Qanbar R., Chénier S., and Bouvier M. (2003) Phosphorylation-independent desensitization of GABA(B) receptor by GRK4. EMBO J. 22, 3816–3824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pontier S. M., Lahaie N., Ginham R., St-Gelais F., Bonin H., Bell D. J., Flynn H., Trudeau L.-E., McIlhinney J., White J. H., and Bouvier M. (2006) Coordinated action of NSF and PKC regulates GABAB receptor signaling efficacy. EMBO J. 25, 2698–2709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kuramoto N., Wilkins M. E., Fairfax B. P., Revilla-Sanchez R., Terunuma M., Tamaki K., Iemata M., Warren N., Couve A., Calver A., Horvath Z., Freeman K., Carling D., Huang L., Gonzales C., Cooper E., Smart T. G., Pangalos M. N., and Moss S. J. (2007) Phospho-dependent functional modulation of GABA(B) receptors by the metabolic sensor AMP-dependent protein kinase. Neuron 53, 233–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Couve A., Thomas P., Calver A. R., Hirst W. D., Pangalos M. N., Walsh F. S., Smart T. G., and Moss S. J. (2002) Cyclic AMP-dependent protein kinase phosphorylation facilitates GABA(B) receptor-effector coupling. Nat. Neurosci. 5, 415–424 [DOI] [PubMed] [Google Scholar]
- 8. Schwenk J., Metz M., Zolles G., Turecek R., Fritzius T., Bildl W., Tarusawa E., Kulik A., Unger A., Ivankova K., Seddik R., Tiao J. Y., Rajalu M., Trojanova J., Rohde V., Gassmann M., Schulte U., Fakler B., and Bettler B. (2010) Native GABA(B) receptors are heteromultimers with a family of auxiliary subunits. Nature 465, 231–235 [DOI] [PubMed] [Google Scholar]
- 9. Hanyaloglu A. C., and von Zastrow M. (2008) Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu. Rev. Pharmacol. Toxicol. 48, 537–568 [DOI] [PubMed] [Google Scholar]
- 10. Grampp T., Sauter K., Markovic B., and Benke D. (2007) γ-Aminobutyric acid type B receptors are constitutively internalized via the clathrin-dependent pathway and targeted to lysosomes for degradation. J. Biol. Chem. 282, 24157–24165 [DOI] [PubMed] [Google Scholar]
- 11. Grampp T., Notz V., Broll I., Fischer N., and Benke D. (2008) Constitutive, agonist-accelerated, recycling and lysosomal degradation of GABA(B) receptors in cortical neurons. Mol. Cell Neurosci. 39, 628–637 [DOI] [PubMed] [Google Scholar]
- 12. Vargas K. J., Terunuma M., Tello J. A., Pangalos M. N., Moss S. J., and Couve A. (2008) The availability of surface GABA B receptors is independent of γ-aminobutyric acid but controlled by glutamate in central neurons. J. Biol. Chem. 283, 24641–24648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Terunuma M., Vargas K. J., Wilkins M. E., Ramirez O. A., Jaureguiberry-Bravo M., Pangalos M. N., Smart T. G., Moss S. J., and Couve A. (2010) Prolonged activation of NMDA receptors promotes dephosphorylation and alters postendocytic sorting of GABAB receptors. Proc. Natl. Acad. Sci. 107, 13918–13923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guetg N., Aziz S. A., Holbro N., Turecek R., Rose T., Seddik R., Gassmann M., Moes S., Jenoe P., Oertner T. G., Casanova E., and Bettler B. (2010) NMDA receptor-dependent GABAB receptor internalization via CaMKII phosphorylation of serine 867 in GABAB1. Proc. Natl. Acad. Sci. U.S.A. 107, 13924–13929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maier P. J., Marin I., Grampp T., Sommer A., and Benke D. (2010) Sustained glutamate receptor activation down-regulates GABA(B) receptors by shifting the balance from recycling to lysosomal degradation. J. Biol. Chem. 285, 35606–35614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shenoy S. K., McDonald P. H., Kohout T. A., and Lefkowitz R. J. (2001) Regulation of receptor fate by ubiquitination of activated β2-adrenergic receptor and β-arrestin. Science 294, 1307–1313 [DOI] [PubMed] [Google Scholar]
- 17. Sarker S., Xiao K., and Shenoy S. K. (2011) A tale of two sites: How ubiquitination of a G protein-coupled receptor is coupled to its lysosomal trafficking from distinct receptor domains. Commun. Integr. Biol. 4, 528–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wolfe B. L., Marchese A., and Trejo J. (2007) Ubiquitination differentially regulates clathrin-dependent internalization of protease-activated receptor-1. J. Cell Biol. 177, 905–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shenoy S. K., Xiao K., Venkataramanan V., Snyder P. M., Freedman N. J., and Weissman A. M. (2008) Nedd4 mediates agonist-dependent ubiquitination, lysosomal targeting, and degradation of the β2-adrenergic receptor. J. Biol. Chem. 283, 22166–22176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nabhan J. F., Pan H., and Lu Q. (2010) Arrestin domain-containing protein 3 recruits the NEDD4 E3 ligase to mediate ubiquitination of the β2-adrenergic receptor. EMBO Rep. 11, 605–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Berthouze M., Venkataramanan V., Li Y., and Shenoy S. K. (2009) The deubiquitinases USP33 and USP20 coordinate β2 adrenergic receptor recycling and resensitization. EMBO J. 28, 1684–1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Marchese A., Raiborg C., Santini F., Keen J. H., Stenmark H., and Benovic J. L. (2003) The E3 ubiquitin ligase AIP4 mediates ubiquitination and sorting of the G protein-coupled receptor CXCR4. Dev. Cell 5, 709–722 [DOI] [PubMed] [Google Scholar]
- 23. Mines M. A., Goodwin J. S., Limbird L. E., Cui F.-F., and Fan G.-H. (2009) Deubiquitination of CXCR4 by USP14 is critical for both CXCL12-induced CXCR4 degradation and chemotaxis but not ERK ativation. J. Biol. Chem. 284, 5742–5752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Berlin I., Higginbotham K. M., Dise R. S., Sierra M. I., and Nash P. D. (2010) The deubiquitinating enzyme USP8 promotes trafficking and degradation of the chemokine receptor 4 at the sorting endosome. J. Biol. Chem. 285, 37895–37908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zemoura K., Schenkel M., Acuña M. A., Yévenes G. E., Zeilhofer H. U., and Benke D. (2013) Endoplasmic Reticulum-associated Degradation Controls Cell Surface Expression of γ-Aminobutyric Acid, Type B Receptors. J. Biol. Chem. 288, 34897–34905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Perroy J., Pontier S., Charest P., Aubry M., and Bouvier M. (2004) Real-time monitoring of ubiquitination in living cells by BRET. Nat. Methods 1, 203–208 [DOI] [PubMed] [Google Scholar]
- 27. Villemure J.-F., Adam L., Bevan N. J., Gearing K., Chénier S., and Bouvier M. (2005) Subcellular distribution of GABA(B) receptor homo- and hetero-dimers. Biochem. J. 388, 47–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Issafras H., Angers S., Bulenger S., Blanpain C., Parmentier M., Labbé-Jullié C., Bouvier M., and Marullo S. (2002) Constitutive agonist-independent CCR5 oligomerization and antibody-mediated clustering occurring at physiological levels of receptors. J. Biol. Chem. 277, 34666–34673 [DOI] [PubMed] [Google Scholar]
- 29. Breton B., Lagacé M., and Bouvier M. (2010) Combining resonance energy transfer methods reveals a complex between the α2A-adrenergic receptor, Gαi1β1γ2, and GRK2. FASEB J. 24, 4733–4743 [DOI] [PubMed] [Google Scholar]
- 30. Nagai A., Kadowaki H., Maruyama T., Takeda K., Nishitoh H., and Ichijo H. (2009) USP14 inhibits ER-associated degradation via interaction with IRE1α. Biochem. Biophys. Res. Commun. 379, 995–1000 [DOI] [PubMed] [Google Scholar]
- 31. Lee B.-H., Lee M. J., Park S., Oh D.-C., Elsasser S., Chen P.-C., Gartner C., Dimova N., Hanna J., Gygi S. P., Wilson S. M., King R. W., and Finley D. (2010) Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 467, 179–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mercier J.-F., Salahpour A., Angers S., Breit A., and Bouvier M. (2002) Quantitative assessment of β1- and β2-adrenergic receptor homo- and heterodimerization by bioluminescence resonance energy transfer. J. Biol. Chem. 277, 44925–44931 [DOI] [PubMed] [Google Scholar]
- 33. Margeta-Mitrovic M., Jan Y. N., and Jan L. Y. (2001) Function of GB1 and GB2 subunits in G protein coupling of GABA(B) receptors. Proc. Natl. Acad. Sci. U.S.A. 98, 14649–14654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Na C. H., Jones D. R., Yang Y., Wang X., Xu Y., and Peng J. (2012) Synaptic protein ubiquitination in rat brain revealed by antibody-based ubiquitome analysis. J. Proteome Res. 11, 4722–4732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mukherjee R. S., Mcbride E. W., Beinborn M., Dunlap K., and Kopin A. S. (2006) Point mutations in either subunit of the GABAB receptor confer constitutive activity to the heterodimer. Mol. Pharmacol. 70, 1406–1413 [DOI] [PubMed] [Google Scholar]
- 36. Dutar P., and Nicoll R. A. (1988) Pre- and postsynaptic GABAB receptors in the hippocampus have different pharmacological properties. Neuron 1, 585–591 [DOI] [PubMed] [Google Scholar]
- 37. Jacob C., Cottrell G. S., Gehringer D., Schmidlin F., Grady E. F., and Bunnett N. W. (2005) c-Cbl mediates ubiquitination, degradation, and down-regulation of human protease-activated receptor 2. J. Biol. Chem. 280, 16076–16087 [DOI] [PubMed] [Google Scholar]
- 38. Marchese A., and Benovic J. L. (2001) Agonist-promoted ubiquitination of the G protein-coupled receptor CXCR4 mediates lysosomal sorting. J. Biol. Chem. 276, 45509–45512 [DOI] [PubMed] [Google Scholar]
- 39. Fairfax B. P., Pitcher J. A., Scott M. G. H., Calver A. R., Pangalos M. N., Moss S. J., and Couve A. (2004) Phosphorylation and chronic agonist treatment atypically modulate GABAB receptor cell surface stability. J. Biol. Chem. 279, 12565–12573 [DOI] [PubMed] [Google Scholar]
- 40. Clague M. J., Barsukov I., Coulson J. M., Liu H., Rigden D. J., and Urbé S. (2013) Deubiquitylases from genes to organism. Physiol. Rev. 93, 1289–1315 [DOI] [PubMed] [Google Scholar]
- 41. Hu M., Li P., Song L., Jeffrey P. D., Chenova T. A., Wilkinson K. D., Cohen R. E., and Shi Y. (2005) Structure and mechanisms of the proteasome-associated deubiquitinating enzyme USP14. EMBO J. 24, 3747–3756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Leithe E., and Rivedal E. (2004) Ubiquitination and down-regulation of gap junction protein connexin-43 in response to 12-O-tetradecanoylphorbol 13-acetate treatment. J. Biol. Chem. 279, 50089–50096 [DOI] [PubMed] [Google Scholar]
- 43. Li S., Zhang Q., and You G. (2013) Three ubiquitination sites of organic anion transporter-1 synergistically mediate protein kinase C-dependent endocytosis of the transporter. Mol. Pharmacol. 84, 139–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Haglund K., and Dikic I. (2012) The role of ubiquitylation in receptor endocytosis and endosomal sorting. J. Cell Sci. 125, 265–275 [DOI] [PubMed] [Google Scholar]
- 45. Zemoura K., and Benke D. (2014) Proteasomal degradation of γ-aminobutyric acidB receptors is mediated by the interaction of the GABAB2 C terminus with the proteasomal ATPase Rtp6 and regulated by neuronal activity. J. Biol. Chem. 289, 7738–7746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Peth A., Besche H. C., and Goldberg A. L. (2009) Ubiquitinated proteins activate the proteasome by binding to Usp14/Ubp6, which causes 20S gate opening. Mol. Cell 36, 794–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Peth A., Kukushkin N., Bossé M., and Goldberg A. L. (2013) Ubiquitinated proteins activate the proteasomal ATPases by binding to Usp14 or Uch37 homologs. J. Biol. Chem. 288, 7781–7790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Walters B. J., Hallengren J. J., Theile C. S., Ploegh H. L., Wilson S. M., and Dobrunz L. E. (2014) A catalytic independent function of the deubiquitinating enzyme USP14 regulates hippocampal synaptic short-term plasticity and vesicle number. J. Physiol. 592, 571–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kuramoto N., Ito M., Saito Y., Niihara H., Tanaka N., Yamada K.-I., Yamamura Y., Iwasaki K., Onishi Y., and Ogita K. (2013) Dephosphorylation of endogenous GABA(B) receptor R2 subunit and AMPK α subunits which were measured by in vitro method using transfer membrane. Neurochem. Int. 62, 137–144 [DOI] [PubMed] [Google Scholar]
- 50. Yao Y., Kelly M. T., Sajikumar S., Serrano P., Tian D., Bergold P. J., Frey J. U., and Sacktor T. C. (2008) PKMζ maintains late-LTP by NSF/GluR2-dependent trafficking of postsynaptic AMPAR receptors. J. Neurosci. 28, 7820–7827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wilson S. M., Bhattacharyya B., Rachel R. A., Coppola V., Tessarollo L., Householder D. B., Fletcher C. F., Miller R. J., Copeland N. G., and Jenkins N. A. (2002) Synaptic defects in ataxia mice result from a mutation in Usp14, encoding a ubiquitin-specific protease. Nat. Genet. 32, 420–425 [DOI] [PubMed] [Google Scholar]
- 52. Lappe-Siefke C., Loebrich S., Hevers W., Waidmann O. B., Schweizer M., Fehr S., Fritschy J.-M., Dikic I., Eilers J., Wilson S. M., and Kneussel M. (2009) The ataxia (axJ) mutation causes abnormal GABAA receptor turnover in mice. PLoS Genet. 5, e1000631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bhattacharyya B. J., Wilson S. M., Jung H., and Miller R. J. (2012) Altered neurotransmitter release machinery in mice deficient for the deubiquitinating enzyme Usp14. Am. J. Physiol. Cell Physiol. 302, C698–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sakaba T., and Neher E. (2003) Direct modulation of synaptic vesicle priming by GABA(B) receptor activation at a glutamatergic synapse. Nature 424, 775–778 [DOI] [PubMed] [Google Scholar]