

Abstract
Patients with long-term type 1 and type 2 diabetes mellitus (DM) can develop skeletal complications or “diabetic osteopathy”. These include osteopenia, osteoporosis and an increased incidence of low-stress fractures. In this context, it is important to evaluate whether current anti-diabetic treatments can secondarily affect bone metabolism. Adenosine monophosphate-activated protein kinase (AMPK) modulates multiple metabolic pathways and acts as a sensor of the cellular energy status; recent evidence suggests a critical role for AMPK in bone homeostasis. In addition, AMPK activation is believed to mediate most clinical effects of the insulin-sensitizer metformin. Over the past decade, several research groups have investigated the effects of metformin on bone, providing a considerable body of pre-clinical (in vitro, ex vivo and in vivo) as well as clinical evidence for an anabolic action of metformin on bone. However, two caveats should be kept in mind when considering metformin treatment for a patient with type 2 DM at risk for diabetic osteopathy. In the first place, metformin should probably not be considered an anti-osteoporotic drug; it is an insulin sensitizer with proven macrovascular benefits that can secondarily improve bone metabolism in the context of DM. Secondly, we are still awaiting the results of randomized placebo-controlled studies in humans that evaluate the effects of metformin on bone metabolism as a primary endpoint.
Keywords: Diabetes mellitus, Osteoporosis, Bone fractures, Metformin, AMP-activated kinase
Core tip: Patients with long-term type 1 and type 2 diabetes mellitus (DM) can develop skeletal complications. These include osteopenia, osteoporosis and increased incidence of low-stress fractures. In this context, it is important to evaluate whether current anti-diabetic treatments can secondarily affect bone metabolism. Over the past decade, several research groups have investigated the effects of metformin on bone, providing a considerable body of pre-clinical (in vitro, ex vivo and in vivo) as well as clinical evidence for an anabolic action of metformin on bone. This could be particularly relevant when considering treatment options for DM in the context of diabetic osteopathy.
INTRODUCTION
Diabetes mellitus (DM) is a highly prevalent global disease associated with long-term microvascular and macrovascular complications. Over the past 30 years, an increasing body of experimental and clinical evidence has reported the association of type 1 and type 2 DM with osteopenia, osteoporosis and an increased incidence of low-stress fractures, in what has been called diabetic osteopathy[1]. Many adult patients with type 1 DM show mild osteopenia, with a decrease in bone mineral density (BMD) of around 10%[2] that would be expected to double the risk of non-vertebral fragility fractures[3]. However, the incidence of low-stress fractures in type 1 DM is 7-12 times that of age-matched non-diabetic individuals[4,5]. On the other hand, patients with type 2 DM tend to have normal or even moderately elevated BMD. Although this would be expected to reduce their incidence of osteoporotic fractures, they actually show a 2-fold increase in hip, extremity and vertebral fractures[3-7]. Taken together, these clinical observations are considered to be evidence for a significant decrease in bone quality of patients with both types of DM[8] that would explain their increase in low-stress fractures.
Several mechanisms have been proposed to explain diabetic osteopathy, such as disturbed glucose metabolism, tissue (bone) and systemic low-grade inflammation, changes in the secretory pattern of growth factors and/or cytokines, increased oxidative stress and excess accumulation of advanced glycation end products (AGEs). In particular, excess accumulation of AGEs in bone extracellular matrix (ECM) occurs as a function of aging and duration of Diabetes, and has been found to impair the mechanical properties of bone[9]. Poorly compensated DM elevates circulating reactive oxygen species (ROS), glucose and/or carbonyl stress, which can induce excess AGEs formation on bone ECM, reducing bone strength and post-yield properties. Additionally, collagen-AGEs interact with the receptor for AGEs (RAGE) expressed by osteoblasts and osteoclasts, inhibiting their functionality and decreasing bone turnover. This induces an even greater accumulation of AGEs in bone that contributes to diabetic osteopathy, and can increase fracture risk[1].
Treatment of patients with DM can include either an absolute requirement for exogenous insulin (type 1 DM), or relative requirement of glucose-lowering medication such as insulin and/or oral drugs (type 2 DM). Oral glucose-lowering agents fall into different classes that include (but are not limited to) insulin secretagogues, insulin sensitizers, incretin-based treatments and inhibitors of renal proximal tubule glucose reabsorption. Each class operates through distinct biological pathways and has certain advantages as well as disadvantages. Recently, several commonly prescribed oral medications for type 2 DM have been found to secondarily affect bone metabolism, in some cases modifying the incidence of fragility fractures[10]. Depending on their specific skeletal effects these drugs could either help to prevent diabetic osteopathy, or contribute to worsen this complication of DM.
Members of the thiazolidinediones (TZD) family of insulin-sensitizers such as rosiglitazone have been shown to be detrimental for bone health. In rodents, TZD increase the adipocytic commitment of mesenchymal stem cells (MSC) while decreasing their osteogenic potential, via a decrease in the Runx2/PPARγ ratio. This increases bone marrow adiposity and promotes bone loss[11]. In the ADOPT clinical trial[12], results showed a higher risk of fracture in diabetic women, but not men, on rosiglitazone monotherapy.
Post-prandial incretin secretion is believed to play a physiological role linking nutrient ingestion to suppression of bone resorption and stimulation of bone formation. Thus, incretin-based treatments would be expected to show anabolic effects on bone, as has been suggested in a recent meta-analysis[13]. However, this may not be so in certain cases such as the DPP4 inhibitor saxagliptin, which has been found to impair MSC osteogenic potential and bone micro-architecture in rodent models[14].
Inhibitors of the sodium glucose cotransporter 2 such as dapagliflozin and canagliflozin, that decrease plasma glucose and body weight by impairing proximal tubule glucose reabsorption, have recently been associated with alterations in mineral metabolism and with an increase in bone fractures. This undesirable effect is probably due to the fact that these drugs can induce hyperphosphataemia and an increase in fibroblast growth factor-23 and para-thyroid hormone levels[15].
Metformin is an insulin-sensitizing biguanide that was developed several decades ago; however, it is still the most widely used oral anti-diabetic medication, particularly since the United Kingdom Prospective Diabetes Study demonstrated its efficacy for reducing macrovascular complications in obese type 2 DM patients[16]. Although the exact mechanism of action for metformin is still incompletely understood, in various tissues and organs it improves glucose metabolism via activation of the ubiquitously expressed AMP-activated protein kinase (AMPK)[17,18]. AMPK subunit expression and activation is tissue-specific, with the α1 subunit accounting for most of bone AMPK[19]. Over the last 10 years, pre-clinical and clinical evidence has accumulated pointing to an anabolic effect of metformin on bone, in part due to AMPK activation.
AMPK-A KEY ENERGY SENSOR
AMPK is a Ser/Thr protein kinase that modulates multiple metabolic pathways and acts as a sensor of the cellular energy status[20]. AMPK is a heterotrimer of three subunits. The α subunit holds the catalytic domain with a Ser/Thr kinase domain (KD). It contains a Thr172 residue, critical for the kinase activity. AMPK-α also contains a regulatory domain, which interacts with the KD of the unphosphorylated inactive form. The β subunit contains a carbohydrate-binding module and acts as a scaffold for the assembly of α and γ subunits. It also defines the subcellular localization of AMPK as well as its substrate specificity. The γ subunit contains four cystathionine-β-synthase (CBS) domains, which bind adenine nucleotides. Expression of all three subunits is required for AMPK activity. The mechanism of activation first requires the reversible phosphorylation of the α subunit; then after AMP binding to the CBS domain of γ subunit, an allosteric stimulatory effect occurs. Several upstream kinases (AMPKK) are involved in the phosphorylation of AMPK, such as liver kinase B1 (LKB1) and Ca2+/calmodulin-dependent protein kinase kinase beta (CaMKKβ). Activation of AMPK can occur by two mechanisms: One is mediated by an increase of AMP/ATP ratio (for instance during exercise), so this mechanism coordinates anabolic and catabolic pathways to equilibrate nutrient supply with energy expenditure[21]. Several compounds can activate AMPK by this mechanism: 5-Aminoimidazole-4-carboxamide ribonucleotide (AICAR), H2O2, MAPK inhibitors, TZD, leptin, adiponectin and α-lipoic acid[22]. In the second mechanism, AMPK activation can be independent of AMP/ATP ratio and involves alternative AMPK regulation. Agents such as peroxynitrite, metformin, estradiol, low glucose levels and several membrane receptor agonists can induce AMPK activation by this way.
It has recently been demonstrated that AMPK can participate in the control of whole-body energy homeostasis by integrating signals from diverse cellular environments[23]. AMPK participates in several physiological events, such as survival, growth and development. This kinase could also be implicated in pathological conditions such as type 2 DM, insulin resistance, cardiovascular disease and cancer. For instance, AMPK can control epigenetic processes in certain cells to avoid reproductive defects in their subsequent generations[24]; or it can suppress proinflammatory-signalling pathways in adipocytes[25]. It has recently been reported that in patients with insulin resistance, AMPK is depressed in adipose tissue; an effect that is associated with oxidative stress, increased expression of inflammatory cytokines and decreased expression of genes regulating oxidative phosphorylation. On the contrary, AMPK activation increases mitochondrial biogenesis[26]. AMPK can also inhibit the proliferation of muscle stem cells, and induce the differentiation of endothelial cell progenitors[21].
ROLE OF AMPK IN CELL METABOLISM
AMPK functions as an intracellular sensor regulating energy balance in different cell types, and thus can regulate diverse metabolic pathways (Table 1).
Table 1.
Role of AMP-activated protein kinase activation on cell metabolism in different organs
| Organ | Effect | Mechanism of action | Ref. |
| Liver | Inhibition of anabolic pathways | Inhibition of fatty acid synthesis | [27] |
| Inhibition of gluconeogenesis | |||
| Stimulation of ATP synthesis | Stimulation of Mitochondrial oxidative phosphorylation | [27] | |
| Skeletal muscle | Regulation of energy expenditure during exercise | Favours the transition from glycolitic to oxidative skeletal muscle fibers | [28] |
| Regulation of myocitic uptake and oxidation of fatty acids | [21] | ||
| Enhanced glucose uptake via an increase in GLUT4 expression | [21] | ||
| Increase in skeletal muscle regeneration | Regulation of post-injury inflammatory response | [29] | |
| Stem cell reprogramming: Induction of proliferation, differentiation and self-renewal | [21] | ||
| Bone | Increase in osteoblastogenesis | Increases MSC differentiation towards the osteoblastic lineage favouring Runx2 expression | [30,37] |
| Decreases PPARγ expression diminishing MSC differentiation towards the adipocytic phenotype | [36,37] | ||
| Decrease in osteoclastogenesis | Negative regulation of RANKL expression by osteoblasts | [35] |
MSC: Mesenchymal stem cells; ATP: Adenosine triphosphate; GLUT4: Glucose transporter type 4; RANKL: Receptor activator for nuclear factor κB ligand.
In the liver, AMPK acts as a “metabolic master switch”[27]; its activation inhibits energy-consuming pathways and stimulates ATP-producing catabolic pathways. For instance, after AMPK activation in a fasting state, fatty acid synthesis is inhibited while mitochondrial oxidative phosphorylation is stimulated. These effects occur by a reduction in malonyl-CoA content that is mediated by inhibition of Acetyl-CoA carboxylase. In addition, malonyl CoA decarboxylase is activated, thus further increasing fatty acid oxidation. Activation of AMPK also suppresses glucose production (gluconeogenesis), as has been demonstrated in metformin-treated primary hepatocyte cultures[17]. This has been confirmed in mice with a liver-selective deletion of the AMPKα2 gene, which exhibit hyperglycaemia and glucose intolerance in the fasting state[27].
In skeletal muscle, AMPK regulates energy expenditure during exercise in order to optimize and enhance energy production. It participates in the transition from more glycolytic fibres to more oxidative fibres, following exercise training[28]. A role for AMPK in the myocytic uptake and oxidation of fatty acids has also been postulated. In addition, during exercise an increase in ATP turnover is accompanied by enhanced glucose uptake, in turn associated with an increase in myocyte plasma membrane GLUT4 expression[21]. Thus, use of metformin in patients with type 2 DM will increase their AMPK-induced glucose uptake and disposal. AMPK activation has also been postulated to induce skeletal muscle regeneration, by regulating its post-injury inflammatory response[29]. Postnatal skeletal muscle regeneration involves stem cell reprogramming that induces their proliferation, differentiation and/or self-renovation, and activation of the AMPK pathway is believed to regulate these processes[21]. Due to the regulatory effects of AMPK on integrated metabolism, activation of AMPK is considered a therapeutic target for hyperglycaemic states. For instance metformin, an anti-diabetic drug that is widely used for treatment of patients with type 2 DM, suppresses hepatic glucose production and decreases plasma glucose levels via activation of AMPK pathways.
The precise effects of AMPK on bone metabolism are incompletely known; however, recent evidence supports an active role for this kinase in bone physiology[30]. Several reports have demonstrated that AMPK modulates bone cell differentiation and function. In AMPKα-deleted animals, a reduction was found in trabecular bone mass[31]. Single α1 or α2 knockout (KO) mice are viable, but the double KO is embryonically lethal[27]. In addition, histomorphometric analysis revealed that AMPKα1 KO mice show an elevated rate of bone remodelling in vivo, associated with increased osteoclastogenesis in vitro.
In vitro experiments have demonstrated that AMPK activation enhances osteogenesis[30,32] while compound C (an AMPK inhibitor) reduces osteoblastic mineralization[30]. In other experiments AICAR (an activator of AMPK) was found to stimulate alkaline phosphatase activity (ALP) and mineral nodule formation by rat calvaria-derived cells, while compound C suppressed these effects[33]. A decrease in AMPK activity has been reported during osteoblastic differentiation; this could be associated with the high-energy requirements of maturing osteoprogenitor cells[34]. Although studies of AMPK action on bone resorption have led to conflicting results, it appears that AMPK is a negative regulator for RANKL and can thus decrease osteoclast-mediated bone resorption[35]. The AMPK pathway may also be involved in regulating the fate of bone marrow stromal cells (MSC) toward the osteoblastic or adipocytic lineage by reciprocally regulating the expression of Runx2 and PPARγ[30]. AMPK has been shown on one hand to induce phosphorylation of β-catenin, suppress PPARγ expression and thus reduce adipogenesis[36]; while on the other hand it increases Runx2 expression and thus osteoblastic differentiation of MSC[37]. This evidence suggests a critical role for AMPK in bone homeostasis.
MOLECULAR MECHANISMS OF METFORMIN ACTION
Metformin has been widely used in the United States since 1995 as an oral anti-diabetic treatment for type 2 DM[22]. Even though its precise mechanism of action is not completely known, metformin is known to activate AMPK[17]; however, AMPK-independent pathways have also been postulated[38].
Metformin can be incorporated into cells by a facilitated mechanism that is mediated by different isoforms of the organic cation transporter. Metformin induces mild and specific inhibition of the mitochondrial respiratory chain complex in hepatocytes and other tissues[39]; it can also inhibit the mitochondrial production of ROS. Inhibition of mitochondrial activity induces a decrease in the cell energy status, which in turn triggers depletion of ATP and a diminished ATP/AMP ratio. This effect then induces phosphorylation and activation of AMPK via LKB1. Metformin also induces an acute inhibition of gluconeogenesis: this can be explained by the decrease in ATP/AMP ratio, which inhibits key enzymes of the gluconeogenic pathway such as fructose-1,6-bisphosphatase. New evidence suggests that this metformin-induced inhibition of glucose production could also be mediated by a down-regulation of gluconeogenic genes via a transcription-independent process. It has been suggested that reduction in energy status, but not AMPK activation, is critical for metformin inhibition of hepatic glucose production[38].
In addition to its effects in the liver, metformin can also affect other organs via multiple molecular mechanisms. One important action of metformin is its reduction of endothelial activation and of atherogenesis[16]. Metformin decreases intracellular ROS production in endothelial cells by inhibiting both NADPH oxidation and the respiratory chain complex[40], and this effect appears to be independent of AMPK activation.
METFORMIN MEETS BONE
Over the past decade, several research groups have investigated the effects of metformin on bone. The results of these studies are discussed in detail below. They have provided a considerable body of pre-clinical (in vitro, ex vivo and in vivo) as well as clinical evidence for an anabolic action of metformin on bone, which could be particularly relevant when considering treatment options for DM in the context of diabetic osteopathy.
In vitro effects of metformin on bone cells
Metformin has been found to modulate the physiology of osteoblasts (Figure 1) and osteoclasts, as well as influencing the phenotypic progression of bone MSC.
Figure 1.
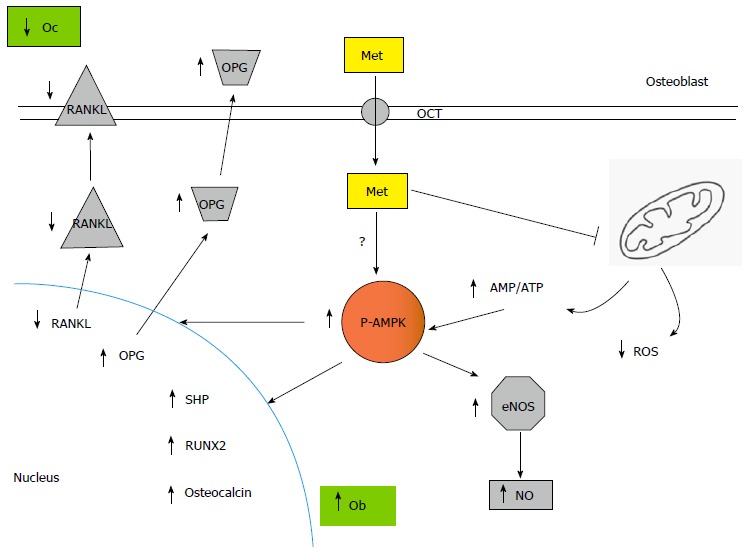
Metformin actions via osteoblasts are pro-osteogenic and anti-resorptive. Metformin is incorporated into osteoblasts, where it inhibits intracellular ROS production and induces AMPK phosphorylation/activation. This increases eNOS activity and NO production, promoting osteoblast proliferation. In addition, activated AMPK up regulates osteoblastic differentiation and mineralization via expression of Runx2 and SHP, while decreasing osteoclastic recruitment and bone-resorbing activity through a reduction in osteoblastic RANKL/OPG ratio. ROS: Reactive oxygen species; AMPK: AMP-activated protein kinase; OPG: Osteoprotegerin; RANKL: Reduced receptor activator of nuclear factor-κB ligand; SHP: Small heterodimer partner; RUNX2: Runt-related transcription factor 2; AMP: Adenosine monophosphate; ATP: Adenosine triphosphate; Met: Metformin.
Cortizo et al[41] were the first to describe an in vitro effect of metformin on bone-derived cells, showing that it dose-dependently increased osteoblastic proliferation, differentiation and mineralization. This effect was mediated by activation of extracellular-regulated kinases and by induction of NO synthases. Several researchers have corroborated these results[19,42-44], additionally showing that the osteogenic in vitro action of metformin on osteoblasts is dependent on activation of the AMPK signalling pathway and subsequent bone morphogenetic protein-2 production. In an interesting mechanistic study, Jang et al[45] found that metformin increased the osteoblastic transcription of small heterodimer partner (SHP) and osteocalcin genes, an effect that was inhibited either by a dominant negative form of AMPK or by compound C. They also found that metformin-induced SHP gene expression was mediated by upstream stimulatory factor-1 (USF-1), that AMPK activation increased the expression of Runx2 and that SHP interacts physically and forms a complex with Runx2 on the osteocalcin gene promoter in osteoblastic cells. Thus, metformin appears to enhance osteoblast differentiation through the transactivation of Runx2 via the AMPK/USF-1/SHP regulatory cascade[45]. In another study, Mai et al[46] found that metformin dose-dependently stimulated osteoprotegerin (OPG) and reduced receptor activator of nuclear factor-κB ligand (RANKL) mRNA and protein expression by cultured osteoblastic cells, a potentially anti-osteoclastogenic effect that they were able to block by inhibition of AMPK[46].
Since one of the proposed mechanisms for diabetic osteopathy is hyperglycaemia-mediated accumulation of AGEs on bone collagen, and AGEs can decrease osteoblastic maturation and survival via binding to their receptor RAGE, Schurman et al[47] investigated whether this process could be modulated in vitro by metformin. They found that metformin was able to prevent the increase in apoptosis, caspase 3 activity, inhibition of ALP and alterations in intracellular oxidative stress induced by AGEs in osteoblastic cells, via a metformin-dependent down regulation in the osteoblastic expression of RAGE[47]. In another study, Zhen et al[33] evaluated whether metformin could prevent the anti-proliferative effect of high-glucose exposure on primary osteoblasts in culture. They found that incubation with metformin decreased the high-glucose-induced intracellular ROS production and apoptosis, and that it additionally induced an osteogenic effect on osteoblasts that was mediated by an increase in Runx2 and IGF-1 gene expression[33]. These results have been recently confirmed by other investigators[48].
Three different studies have investigated the in vitro effects of metformin on stromal cells isolated from bone marrow (MSC). In the first report, Gao et al[49] found that metformin increased Runx2 and decreased PPARγ expression, and consequently stimulated ALP and mineralization while inhibiting the intracellular accumulation of lipid droplets. These results suggest that metformin could influence the reciprocal relationship between osteoblastic and adipogenic differentiation of MSC, tipping the balance towards osteogenesis. Molinuevo et al[37] further demonstrated that metformin could induce an in vitro dose-dependent increase in MSC osteogenic potential (ALP, type 1 collagen secretion and mineralization). They also found that metformin dose-dependently prevents rosiglitazone-induced intracellular lipid accumulation by MSC[37]. In another study, Sedlinsky et al[50] demonstrated that the in vitro osteogenic effect of metformin on MSC is AMPK-dependent, and that it can be completely blocked by the AMPK inhibitor compound C.
Metformin has also been found to modulate in vitro osteoclast recruitment, differentiation and bone-resorbing activity in some[35,46,51] but not all[52] published reports. OPG and RANKL are predominantly secreted by osteoblasts and play critical roles in osteoclast physiology. As stated above, in vitro experiments have shown that metformin increases OPG and reduces RANKL mRNA and protein expression by osteoblasts, which is potentially anti-osteoclastogenic. Additionally, when a macrophage cell line was incubated with the supernatant of osteoblasts treated with metformin, this reduced the formation of tartrate resistant acid phosphatase (TRAP)-positive multi-nucleated osteoclasts[46]. In another interesting in vitro study, AMPK was found to be expressed by bone marrow pre-osteoclasts and as such is a regulatory target for osteoclast differentiation and resorptive activity. Pharmacological inhibition of pre-osteoclastic AMPK with compound C increased the RANKL-induced formation of TRAP-positive multinucleated cells and their resorptive activity on dentine discs, via downstream activation of p38, JNK, NF-κB, Akt, CREB, c-Fos, and NFATc1. On the contrary, metformin dose-dependently suppressed formation of TRAP-positive multinucleated cells and dentine resorption[35]. In unpublished results using indirect immunofluorescence, we have found a metformin-induced increase and sub-cellular redistribution in phosphorylated (activated) AMPK of multinucleated osteoclasts obtained from osteoblast-macrophage co-cultures (Figure 2), which could be mediating the effects of metformin in this cell type.
Figure 2.
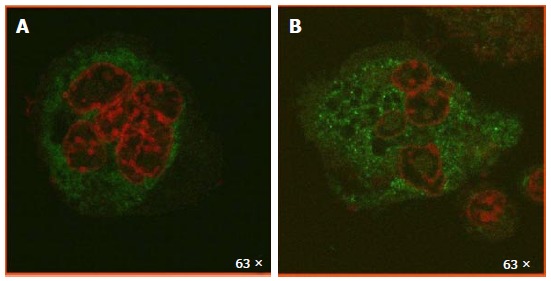
Metformin induces an increase and redistribution of activated AMP-activated protein kinase in multinucleated osteoclasts. UMR106 osteoblasts and Raw 264.7 macrophages were co-cultured for 7 d, in the absence (A) or presence (B) of 500 mol/L metformin. Cells were fixed, permeabilized and incubated with an anti-phosphorylated AMPK antibody, followed by a FITC-conjugated secondary antibody (green). Nuclei were counterstained with propidium iodide (red). Cells were visualized with a Leica TSC SP5 AOBS confocal microscope. Metformin induced an increase in activated AMPK with a punctillate and predominantly cytoplasmic distribution. AMPK: AMP-activated protein kinase.
All in all, these in vitro results point to a global bone-anabolic effect of metformin: Tipping the phenotypic balance of bone MSC towards osteoblastogenesis, increasing the bone-forming capacity of osteoblasts, and decreasing the recruitment and bone-resorbing activity of osteoclasts (Figure 3). These findings are further supported by in vivo and ex vivo (pre-clinical) as well as clinical evidence, pointing to an osteogenic action of metformin in the context of DM.
Figure 3.
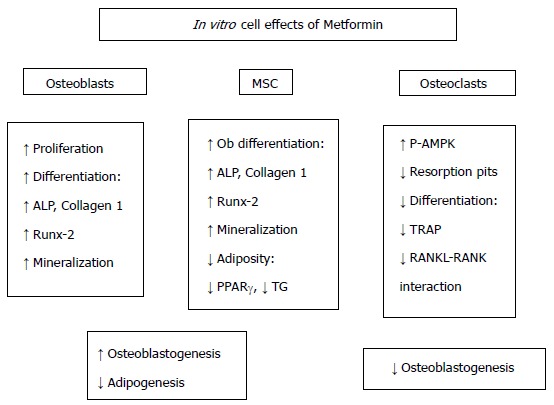
Effects of metformin on bone-derived cells. The results of several in vitro studies show that metformin modulates the phenotypic balance of bone marrow stromal cells (MSC) away from adipogenesis and towards osteoblastogenesis. In addition, metformin increases in vitro the bone-forming capacity of osteoblasts, while decreasing the recruitment and bone-resorbing activity of osteoclasts. ALP: Alkaline phosphatase; TG: Triglycerides; TRAP: Tartrate-resistant acid phosphatase; RANK: Receptor activator for nuclear factor κB; RANKL: RANK ligand.
In vivo and ex vivo effects of metformin on bone metabolism: Animal models
Most studies using animal models (but not all) have shown beneficial actions of metformin on bone metabolism and on bone lesion repair (Figure 4).
Figure 4.
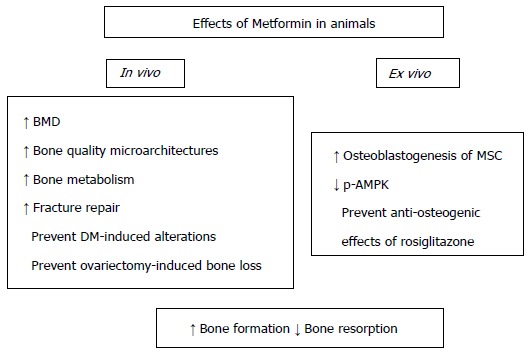
Actions of metformin on bone metabolism - animal studies. Orally administered metformin promotes the osteogenic potential of bone MSC, increases the quality of bone tissue (improving its micro-architecture and mineral density) and facilitates the repair of bone lesions. In addition, metformin may prevent experimental diabetic osteopathy as well as ovariectomy-induced bone loss. MSC: Marrow stromal cells; BMD: Bone mineral density; p-AMPK: Phosphorylated AMP-activated kinase.
Molinuevo et al[37] demonstrated an ex vivo osteogenic effect of metformin: i.e., that bone MSC obtained from rats after a 2-wk treatment with oral metformin, exhibit increased osteogenic potential (Runx2 expression, ALP activity, type 1 collagen production, osteocalcin synthesis, and mineral nodule deposition) vs MSC obtained from non-treated animals. In addition, these metformin-induced effects were found to be secondary to AMPK activation. In this study, metformin treatment also stimulated the repair of a minimal parietal lesion in vivo, both in diabetic and non-diabetic rats.
As stated above, rosiglitazone is a TZD that induces deleterious effects on osteoblast differentiation[53] and on osteocyte survival[54], diverting MSC differentiation toward the adipocyte lineage. In view of the opposite effect that has been demonstrated for metformin on these cell types, Sedlinsky et al[50] investigated the effect of a 2-wk metformin/rosiglitazone combined oral treatment of rats on long-bone metaphyseal microarchitecture, minimal parietal lesion repair and MSC osteogenic potential. Compared to untreated controls, rosiglitazone monotherapy decreased femoral metaphysis trabecular area, osteoblastic and osteocytic density, and TRAP activity associated with epiphyseal growth plates. In addition, it greatly diminished bone repair. It also decreased the ex vivo osteogenic potential of MSC, inducing an increase in PPARγ and a decrease in Runx2 expression, as well as a decrease in phosphorylated (active) AMPK. Metformin/rosiglitazone co-treatment prevented all the in vivo (bone repair and diaphyseal microarchitecture) and ex vivo anti-osteogenic effects of rosiglitazone monotherapy, with a reversion back to control levels of PPARγ, Runx2 and AMPK phosphorylation in MSC[50].
In another study, the skeletal (femoral) effects of rosiglitazone were compared to those of metformin in insulin-resistant female C57BL6J ob/ob mice. The metformin-treated group showed higher BMD, higher trabecular bone volume/total bone volume, higher osteoid width and mineral apposition, lower trabecular spacing and lower bone marrow adiposity, when compared with the rosiglitazone-treated group[55].
Gao et al[56] studied the in vivo effect of oral metformin on bone mass in ovariectomized rats. They found that metformin dose-dependently reverted ovariectomy-induced bone loss, showing an improvement in BMD measured by DEXA, and in bone microarchitecture measured both by micro CT and by bone histology. By real-time PCR of MSC, they found a metformin-dependent increase in Runx2 and Lrp5 (co-receptor for Wnt) expression, both of which are involved in osteoblastic proliferation and differentiation[56].
In another report, Jeyabalan et al[57] studied either ovariectomized C57BL/6 mice or young Wistar rats to evaluate the effect of oral metformin on bone metabolism and fracture repair, respectively. In both models, metformin did not modify bone microarchitecture or cellular activity in vivo as evaluated by micro-CT and bone histomorphometry. In addition, metformin had no significant effect on the repair of a midshaft femoral fracture in Wistar rats[57].
Mai et al[46] further investigated the effects on bone of an oral metformin treatment in ovariectomized (OVX) adult rats. They found that metformin treatment of OVX animals significantly increased total body BMD, enhanced bone mineral content and decreased trabecular separation; supporting the concept that metformin can prevent OVX-induced bone loss. The authors also found that metformin reverted the OVX-associated increase in TRAP-positive osteoclasts of proximal tibiae resorption pits. Metformin treatment also increased serum OPG, and decreased RANKL expression by MSC, in OVX rats. Further in vitro experiments showed that these effects were regulated by AMPK and by its upstream activator CaMKK.
In a rat model of partially insulin-deficient nicotinamide/streptozotocin-induced DM, tibia histomorphometry showed a diabetes-induced decrease in trabecular bone volume, osteocyte density, growth plate height and osteoclast (TRAP positive) activity in the primary spongiosa, as well as an increase in bone marrow adiposity. MSC from diabetic animals showed a decrease in their osteoblastic potential, an increase in adipocytic commitment, a reduction in their Runx2/PPARγ ratio and an increased expression of the AGEs receptor RAGE. A 2-wk oral treatment with metformin prevented all these Diabetes-induced alterations in bone micro-architecture and MSC osteogenic potential, and also induced a down-regulation of RAGE expression by MSC[58].
Clinical evidences of metformin effects on bone
There are few published clinical studies reporting the skeletal effects of metformin. In addition, randomized placebo-controlled studies in humans that evaluate the effects of metformin on bone metabolism as a primary end point are so far unavailable. The results of published clinical reports are summarized in Table 2.
Table 2.
Clinical evidences of metformin effects on bone
| Study design | Study population | n | Outcome | Ref. |
| Case control study | All subjects with bone fracture in Denmark (year 2000), vs 3-fold controls | 124655 fracture patients 373962 control | Fracture risk = 0.81 (95%CI: 0.70-0.93)1 (for metformin) | [59] |
| Cohort Study | Rochester residents first meeting Diabetes glycaemic criteria (1970-1994) | 1964 diabetic patients | Fracture risk = 0.7 (95%CI: 0.6-0.96)2 (for metformin) | [6] |
| Case control study | A study nested within a cohort of 1945 diabetic Tuscany outpatients (1998-2004) | 83 fracture patients 249 control | Fracture risk = 0.60 (95%CI: 0.34-1.08)3 (for metformin) | [60] |
| Double-blind, randomized, controlled clinical trial | Recently diagnosed, drug-naïve patients with type 2 diabetes, treated for a median of 4 yr with rosiglitazone, metformin, or glyburide | Rosiglitazone: n = 1456; Metformin: n = 1454; Glyburide: n = 1441 | Nº Fractures (%): Rosiglitazone 60 (9.30) Metformin 30 (5.08)4 Glyburide 21 (3.47)4 | [12] |
| Double-blind, randomized, controlled clinical trial | Recently diagnosed, drug-naïve patients with type 2 diabetes, treated for a median of 4 yr with RSG, MET, or GLY | Paired baseline and 12-mo stored serum samples from 1605 patients | In women, CTX increased by 6.1% with RSG, decreased by 1.3% with MET (P = 0.03) In men, CTX was unchanged on RSG (-1.0%) and fell with MET -12.7% (P = 0.001) | [61] |
| Randomized, parallel group, double-blind, multicentre study | Drug naïve, male and female patients who had an established clinical diagnosis of type 2 diabetes mellitus | 688 patients equally randomized to RSG/MET or MET | BMD at week 80: Lumbar = (-2.2) (95%CI: -3.5, -0.9) Total hip = (-1.5) (95%CI: -2.3, -0.7)5 | [62] |
| Prospective randomized study with active comparator study | Forty postmenopausal diabetic women recruited from Tanta University Hospitals | 20 patients on metformin and 20 on sitagliptin, for 12 wk | BMD was unchanged in both groups at week 12 Bone turnover markers remained unchanged from baseline in MET | [63] |
| Prospective randomized double-blind, double-dummy with active comparator | Men with uncomplicated type 2 diabetes mellitus, aged 45-65 yr | 71 men were randomized to PIO once daily or MET twice daily | Sclerostin levels at week 24 increased by 11% in PIO-treated patients and decreased by 1.8% in MET-treated patients (P = 0.018) | [64] |
Relative risk of any fracture interpreted as OR with 95%CI for several variables in the population of Denmark (National Health Registry, year 2000);
Multivariate HR for the development of any new fracture among 1964 Rochester, MN, United States residents after recognition of diabetes mellitus in 1970-1994;
Exposure for at least 36 mo to hypoglycemic treatments in case subjects and matched control subjects, interpreted as OR with 95%CI;
P < 0.01 for comparison of fracture risk in women with rosiglitazone (unadjusted, contingency χ2 test);
Percentage of change in BMD at week 80, comparing RSG/MET vs MET. RSG: Rosiglitazone; MET: Metformi; GLY: Glyburide; PIO: Pioglitazone; BMD: Bone mineral density; OR: Odds ratio; HR: Hazard ratios.
Several epidemiological studies have reported the effects of diabetes and antidiabetic agents on bone fracture risk. In 2005, Vestergaard et al[59] published a Danish population-based study evaluating risk of fractures and its relationship with T1DM and T2DM and anti-diabetic agents. They found that both T1DM and T2DM patients had a significant increase in bone fracture risk, and that the use of metformin was associated with a significantly decreased risk for fracture at any site.
Melton et al[6] conducted another population-based study in Rochester, United States, to evaluate fracture risk factors in T2DM patients. They found that patients had an increased risk of hip fracture after 10 years of DM, and that use of biguanides such as metformin was protective even after adjusting for other risk factors (HR, 0.7; 95%CI: 0.6-0.96).
Monami et al[60] conducted a case-control study, nested within a retrospective cohort, comparing 83 case subjects with a history of bone fractures and 249 control subjects, in all cases exposed to insulin, insulin secretagogues or metformin treatment for the past 10 years, in order to assess the risk for bone fractures associated with exposure to insulin or different oral hypoglycaemic agents. This study was unable to demonstrate a reduction in bone fractures associated with metformin treatment, but showed an increased rate of fractures in patients on insulin treatment, probably related to worse diabetes control or to hypoglycaemic episodes. Nevertheless, the authors acknowledged that the lack of a statistically significant fracture reduction associated with metformin treatment was probably related to an insufficient sample size.
The ADOPT study (A Diabetes Outcome Progression Trial), that compared the glycaemic effects of rosiglitazone, metformin and glyburide, showed that among the adverse effects of rosiglitazone was an increased risk of fracture in women. At the same time they showed that metformin had a lower risk of fracture, both in women and men, for every skeletal site assessed[12]. In an add-on report to the ADOPT study, C-telopeptide levels (CTX, a bone resorption marker) were found to be reduced by metformin treatment and increased in rosiglitazone-treated patients, suggesting that changes in bone resorption may be partly responsible for the differences in fracture risk observed for both treatments[61].
A randomized, parallel group, double-blind, multicentre study comparing the efficacy and safety of rosiglitazone/metformin co-treatment (RSG/MET) vs metformin monotherapy (MET) was conducted in order to assess glycaemic control and BMD after 80 wk of treatment in drug-naïve T2DM patients. Although the RSG/MET combination was superior to MET in achieving significant reductions in glycated haemoglobin and fasting plasma glucose, RSG/MET was associated with a significantly lower BMD in comparison with MET at week 80 in the hip and lumbar spine[62].
In another study, Hegazy[63] evaluated the possible anti-osteoporotic effect of metformin vs sitagliptin in 40 post-menopausal diabetic women. They were randomly divided into two groups, one receiving 500 mg metformin twice a day, and the other 100 mg sitagliptin once a day, for 12 wk. In the metformin-treated group, serum ALP and urinary D-piridinoline (DPD) were not significantly different from baseline; conversely in the sitagliptin group, serum ALP and urinary DPD decreased significantly after 12 wk, although BMD was unchanged in both groups.
The effects of pioglitazone and metformin on circulating sclerostin (an osteocyte-derived osteoblast proliferation inhibitor), and biochemical markers of bone turnover were studied in 71 men with T2DM. This group as a whole showed higher serum sclerostin levels than healthy controls. Sclerostin levels were further increased in the sub-set of patients that were treated with pioglitazone, who also showed an increase in serum CTX. On the contrary, metformin-treated patients vs healthy controls showed significantly lower sclerostin levels and unchanged CTX levels[64]. Although sclerostin is a well-established inhibitor of bone formation, recent evidence indicates that it can also promote osteoclastogenesis by stimulating RANKL produced by osteocytes[65], suggesting that pioglitazone could increase bone resorption while decreasing bone formation, and the opposite would occur with metformin.
Finally, metformin was tested for bone-defect healing purposes in a clinical study, adding this biguanide to platelet-rich fibrin in order to treat intrabony defects in patients with chronic periodontitis. The study was designed to evaluate the efficacy of platelet-rich fibrin, 1% metformin gel, or platelet-rich fibrin plus 1% metformin gel, in all cases with open flap debridement, for treatment of intrabony defects in 120 patients with chronic periodontitis. The group treated with platelet-rich fibrin plus 1% metformin gel showed the greatest improvements in clinical parameters, with an increase in percentage radiographic defect depth reduction when compared to metformin alone, platelet-rich fibrin alone or open flap debridement alone[66].
CONCLUSION AND PERSPECTIVES
Patients with long-term T1 DM and T2 DM can develop skeletal complications or “diabetic osteopathy”. These include osteopenia, osteoporosis and an increased incidence of low-stress fractures. In this context, it is important to evaluate whether current anti-diabetic treatments can secondarily affect bone metabolism. Over the past 10 years, many investigators have studied the effects of metformin on bone, providing a considerable body of pre-clinical (in vitro, ex vivo and in vivo) as well as clinical evidence for an anabolic action of metformin on bone. However three reports (one in vitro, one in vivo, one clinical) have been unable to link metformin treatment with bone anabolic processes, underscoring the differences that exist between experimental models in pre-clinical studies, and the low statistical potency inherent in clinical reports that include a relatively small number of patients. In this sense, two caveats should be kept in mind when considering metformin treatment for a patient with T2DM at risk for diabetic osteopathy. In the first place, metformin should probably not be considered an anti-osteoporotic drug; it is an insulin sensitizer with proven macrovascular benefits that can secondarily improve bone metabolism in the context of DM. Secondly, we are still awaiting the results of randomized placebo-controlled studies in humans that evaluate the effects of metformin on bone metabolism as a primary endpoint.
Footnotes
Supported by MINCyT Argentina, No. PICT-2012-0053.
Conflict-of-interest statement: All authors declare that they have no conflicting interests.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: October 3, 2015
First decision: December 4, 2015
Article in press: January 31, 2016
P- Reviewer: Gómez-Sáez JM, Markopoulos AK S- Editor: Song XX L- Editor: A E- Editor: Wu HL
References
- 1.McCarthy AD, Molinuevo MS, Cortizo AM. AGEs and Bone Ageing in Diabetes Mellitus. J Diabetes Metab. 2013;4:276. [Google Scholar]
- 2.Bouillon R. Diabetic bone disease. Calcif Tissue Int. 1991;49:155–160. doi: 10.1007/BF02556109. [DOI] [PubMed] [Google Scholar]
- 3.Hofbauer LC, Brueck CC, Singh SK, Dobnig H. Osteoporosis in patients with diabetes mellitus. J Bone Miner Res. 2007;22:1317–1328. doi: 10.1359/jbmr.070510. [DOI] [PubMed] [Google Scholar]
- 4.Forsén L, Meyer HE, Midthjell K, Edna TH. Diabetes mellitus and the incidence of hip fracture: results from the Nord-Trøndelag Health Survey. Diabetologia. 1999;42:920–925. doi: 10.1007/s001250051248. [DOI] [PubMed] [Google Scholar]
- 5.Nicodemus KK, Folsom AR. Type 1 and type 2 diabetes and incident hip fractures in postmenopausal women. Diabetes Care. 2001;24:1192–1197. doi: 10.2337/diacare.24.7.1192. [DOI] [PubMed] [Google Scholar]
- 6.Melton LJ, Leibson CL, Achenbach SJ, Therneau TM, Khosla S. Fracture risk in type 2 diabetes: update of a population-based study. J Bone Miner Res. 2008;23:1334–1342. doi: 10.1359/JBMR.080323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schwartz AV, Sellmeyer DE, Ensrud KE, Cauley JA, Tabor HK, Schreiner PJ, Jamal SA, Black DM, Cummings SR. Older women with diabetes have an increased risk of fracture: a prospective study. J Clin Endocrinol Metab. 2001;86:32–38. doi: 10.1210/jcem.86.1.7139. [DOI] [PubMed] [Google Scholar]
- 8.Silva MJ, Brodt MD, Lynch MA, McKenzie JA, Tanouye KM, Nyman JS, Wang X. Type 1 diabetes in young rats leads to progressive trabecular bone loss, cessation of cortical bone growth, and diminished whole bone strength and fatigue life. J Bone Miner Res. 2009;24:1618–1627. doi: 10.1359/JBMR.090316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saito M, Marumo K. Collagen cross-links as a determinant of bone quality: a possible explanation for bone fragility in aging, osteoporosis, and diabetes mellitus. Osteoporos Int. 2010;21:195–214. doi: 10.1007/s00198-009-1066-z. [DOI] [PubMed] [Google Scholar]
- 10.Gilbert MP, Pratley RE. The impact of diabetes and diabetes medications on bone health. Endocr Rev. 2015;36:194–213. doi: 10.1210/er.2012-1042. [DOI] [PubMed] [Google Scholar]
- 11.Ali AA, Weinstein RS, Stewart SA, Parfitt AM, Manolagas SC, Jilka RL. Rosiglitazone causes bone loss in mice by suppressing osteoblast differentiation and bone formation. Endocrinology. 2005;146:1226–1235. doi: 10.1210/en.2004-0735. [DOI] [PubMed] [Google Scholar]
- 12.Kahn SE, Haffner SM, Heise MA, Herman WH, Holman RR, Jones NP, Kravitz BG, Lachin JM, O’Neill MC, Zinman B, et al. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med. 2006;355:2427–2443. doi: 10.1056/NEJMoa066224. [DOI] [PubMed] [Google Scholar]
- 13.Monami M, Dicembrini I, Antenore A, Mannucci E. Dipeptidyl peptidase-4 inhibitors and bone fractures: a meta-analysis of randomized clinical trials. Diabetes Care. 2011;34:2474–2476. doi: 10.2337/dc11-1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sbaraglini ML, Molinuevo MS, Sedlinsky C, Schurman L, McCarthy AD. Saxagliptin affects long-bone microarchitecture and decreases the osteogenic potential of bone marrow stromal cells. Eur J Pharmacol. 2014;727:8–14. doi: 10.1016/j.ejphar.2014.01.028. [DOI] [PubMed] [Google Scholar]
- 15.Taylor SI, Blau JE, Rother KI. Possible adverse effects of SGLT2 inhibitors on bone. Lancet Diabetes Endocrinol. 2015;3:8–10. doi: 10.1016/S2213-8587(14)70227-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34) UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352:854–865. [PubMed] [Google Scholar]
- 17.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Musi N, Hirshman MF, Nygren J, Svanfeldt M, Bavenholm P, Rooyackers O, Zhou G, Williamson JM, Ljunqvist O, Efendic S, et al. Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes. 2002;51:2074–2081. doi: 10.2337/diabetes.51.7.2074. [DOI] [PubMed] [Google Scholar]
- 19.Shah M, Kola B, Bataveljic A, Arnett TR, Viollet B, Saxon L, Korbonits M, Chenu C. AMP-activated protein kinase (AMPK) activation regulates in vitro bone formation and bone mass. Bone. 2010;47:309–319. doi: 10.1016/j.bone.2010.04.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sanz P. AMP-activated protein kinase: structure and regulation. Curr Protein Pept Sci. 2008;9:478–492. doi: 10.2174/138920308785915254. [DOI] [PubMed] [Google Scholar]
- 21.Mounier R, Théret M, Lantier L, Foretz M, Viollet B. Expanding roles for AMPK in skeletal muscle plasticity. Trends Endocrinol Metab. 2015;26:275–286. doi: 10.1016/j.tem.2015.02.009. [DOI] [PubMed] [Google Scholar]
- 22.Strack T. Metformin: a review. Drugs Today (Barc) 2008;44:303–314. doi: 10.1358/dot.2008.44.4.1138124. [DOI] [PubMed] [Google Scholar]
- 23.Carling D, Viollet B. Beyond energy homeostasis: the expanding role of AMP-activated protein kinase in regulating metabolism. Cell Metab. 2015;21:799–804. doi: 10.1016/j.cmet.2015.05.005. [DOI] [PubMed] [Google Scholar]
- 24.Xie M, Roy R. AMP-Activated Kinase Regulates Lipid Droplet Localization and Stability of Adipose Triglyceride Lipase in C. elegans Dauer Larvae. PLoS One. 2015;10:e0130480. doi: 10.1371/journal.pone.0130480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bijland S, Mancini SJ, Salt IP. Role of AMP-activated protein kinase in adipose tissue metabolism and inflammation. Clin Sci (Lond) 2013;124:491–507. doi: 10.1042/CS20120536. [DOI] [PubMed] [Google Scholar]
- 26.Ruderman NB, Carling D, Prentki M, Cacicedo JM. AMPK, insulin resistance, and the metabolic syndrome. J Clin Invest. 2013;123:2764–2772. doi: 10.1172/JCI67227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Viollet B, Andreelli F, Jørgensen SB, Perrin C, Flamez D, Mu J, Wojtaszewski JF, Schuit FC, Birnbaum M, Richter E, et al. Physiological role of AMP-activated protein kinase (AMPK): insights from knockout mouse models. Biochem Soc Trans. 2003;31:216–219. doi: 10.1042/bst0310216. [DOI] [PubMed] [Google Scholar]
- 28.Röckl KS, Hirshman MF, Brandauer J, Fujii N, Witters LA, Goodyear LJ. Skeletal muscle adaptation to exercise training: AMP-activated protein kinase mediates muscle fiber type shift. Diabetes. 2007;56:2062–2069. doi: 10.2337/db07-0255. [DOI] [PubMed] [Google Scholar]
- 29.Saclier M, Cuvellier S, Magnan M, Mounier R, Chazaud B. Monocyte/macrophage interactions with myogenic precursor cells during skeletal muscle regeneration. FEBS J. 2013;280:4118–4130. doi: 10.1111/febs.12166. [DOI] [PubMed] [Google Scholar]
- 30.Jeyabalan J, Shah M, Viollet B, Chenu C. AMP-activated protein kinase pathway and bone metabolism. J Endocrinol. 2012;212:277–290. doi: 10.1530/JOE-11-0306. [DOI] [PubMed] [Google Scholar]
- 31.Kang H, Viollet B, Wu D. Genetic deletion of catalytic subunits of AMP-activated protein kinase increases osteoclasts and reduces bone mass in young adult mice. J Biol Chem. 2013;288:12187–12196. doi: 10.1074/jbc.M112.430389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kanazawa I, Yamaguchi T, Yano S, Yamauchi M, Yamamoto M, Sugimoto T. Adiponectin and AMP kinase activator stimulate proliferation, differentiation, and mineralization of osteoblastic MC3T3-E1 cells. BMC Cell Biol. 2007;8:51. doi: 10.1186/1471-2121-8-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhen D, Chen Y, Tang X. Metformin reverses the deleterious effects of high glucose on osteoblast function. J Diabetes Complications. 2010;24:334–344. doi: 10.1016/j.jdiacomp.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 34.Kasai T, Bandow K, Suzuki H, Chiba N, Kakimoto K, Ohnishi T, Kawamoto S, Nagaoka E, Matsuguchi T. Osteoblast differentiation is functionally associated with decreased AMP kinase activity. J Cell Physiol. 2009;221:740–749. doi: 10.1002/jcp.21917. [DOI] [PubMed] [Google Scholar]
- 35.Lee YS, Kim YS, Lee SY, Kim GH, Kim BJ, Lee SH, Lee KU, Kim GS, Kim SW, Koh JM. AMP kinase acts as a negative regulator of RANKL in the differentiation of osteoclasts. Bone. 2010;47:926–937. doi: 10.1016/j.bone.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 36.Zhao J, Yue W, Zhu MJ, Sreejayan N, Du M. AMP-activated protein kinase (AMPK) cross-talks with canonical Wnt signaling via phosphorylation of beta-catenin at Ser 552. Biochem Biophys Res Commun. 2010;395:146–151. doi: 10.1016/j.bbrc.2010.03.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Molinuevo MS, Schurman L, McCarthy AD, Cortizo AM, Tolosa MJ, Gangoiti MV, Arnol V, Sedlinsky C. Effect of metformin on bone marrow progenitor cell differentiation: in vivo and in vitro studies. J Bone Miner Res. 2010;25:211–221. doi: 10.1359/jbmr.090732. [DOI] [PubMed] [Google Scholar]
- 38.Foretz M, Hébrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, Sakamoto K, Andreelli F, Viollet B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. 2010;120:2355–2369. doi: 10.1172/JCI40671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond) 2012;122:253–270. doi: 10.1042/CS20110386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ouslimani N, Peynet J, Bonnefont-Rousselot D, Thérond P, Legrand A, Beaudeux JL. Metformin decreases intracellular production of reactive oxygen species in aortic endothelial cells. Metabolism. 2005;54:829–834. doi: 10.1016/j.metabol.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 41.Cortizo AM, Sedlinsky C, McCarthy AD, Blanco A, Schurman L. Osteogenic actions of the anti-diabetic drug metformin on osteoblasts in culture. Eur J Pharmacol. 2006;536:38–46. doi: 10.1016/j.ejphar.2006.02.030. [DOI] [PubMed] [Google Scholar]
- 42.Kanazawa I, Yamaguchi T, Yano S, Yamauchi M, Sugimoto T. Metformin enhances the differentiation and mineralization of osteoblastic MC3T3-E1 cells via AMP kinase activation as well as eNOS and BMP-2 expression. Biochem Biophys Res Commun. 2008;375:414–419. doi: 10.1016/j.bbrc.2008.08.034. [DOI] [PubMed] [Google Scholar]
- 43.Jang WG, Kim EJ, Lee KN, Son HJ, Koh JT. AMP-activated protein kinase (AMPK) positively regulates osteoblast differentiation via induction of Dlx5-dependent Runx2 expression in MC3T3E1 cells. Biochem Biophys Res Commun. 2011;404:1004–1009. doi: 10.1016/j.bbrc.2010.12.099. [DOI] [PubMed] [Google Scholar]
- 44.Salai M, Somjen D, Gigi R, Yakobson O, Katzburg S, Dolkart O. Effects of commonly used medications on bone tissue mineralisation in SaOS-2 human bone cell line: an in vitro study. Bone Joint J. 2013;95-B:1575–1580. doi: 10.1302/0301-620X.95B11.31158. [DOI] [PubMed] [Google Scholar]
- 45.Jang WG, Kim EJ, Bae IH, Lee KN, Kim YD, Kim DK, Kim SH, Lee CH, Franceschi RT, Choi HS, et al. Metformin induces osteoblast differentiation via orphan nuclear receptor SHP-mediated transactivation of Runx2. Bone. 2011;48:885–893. doi: 10.1016/j.bone.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 46.Mai QG, Zhang ZM, Xu S, Lu M, Zhou RP, Zhao L, Jia CH, Wen ZH, Jin DD, Bai XC. Metformin stimulates osteoprotegerin and reduces RANKL expression in osteoblasts and ovariectomized rats. J Cell Biochem. 2011;112:2902–2909. doi: 10.1002/jcb.23206. [DOI] [PubMed] [Google Scholar]
- 47.Schurman L, McCarthy AD, Sedlinsky C, Gangoiti MV, Arnol V, Bruzzone L, Cortizo AM. Metformin reverts deleterious effects of advanced glycation end-products (AGEs) on osteoblastic cells. Exp Clin Endocrinol Diabetes. 2008;116:333–340. doi: 10.1055/s-2007-992786. [DOI] [PubMed] [Google Scholar]
- 48.Shao X, Cao X, Song G, Zhao Y, Shi B. Metformin rescues the MG63 osteoblasts against the effect of high glucose on proliferation. J Diabetes Res. 2014;2014:453940. doi: 10.1155/2014/453940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gao Y, Xue J, Li X, Jia Y, Hu J. Metformin regulates osteoblast and adipocyte differentiation of rat mesenchymal stem cells. J Pharm Pharmacol. 2008;60:1695–1700. doi: 10.1211/jpp.60/12.0017. [DOI] [PubMed] [Google Scholar]
- 50.Sedlinsky C, Molinuevo MS, Cortizo AM, Tolosa MJ, Felice JI, Sbaraglini ML, Schurman L, McCarthy AD. Metformin prevents anti-osteogenic in vivo and ex vivo effects of rosiglitazone in rats. Eur J Pharmacol. 2011;668:477–485. doi: 10.1016/j.ejphar.2011.07.033. [DOI] [PubMed] [Google Scholar]
- 51.Son HJ, Lee J, Lee SY, Kim EK, Park MJ, Kim KW, Park SH, Cho ML. Metformin attenuates experimental autoimmune arthritis through reciprocal regulation of Th17/Treg balance and osteoclastogenesis. Mediators Inflamm. 2014;2014:973986. doi: 10.1155/2014/973986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Patel JJ, Butters OR, Arnett TR. PPAR agonists stimulate adipogenesis at the expense of osteoblast differentiation while inhibiting osteoclast formation and activity. Cell Biochem Funct. 2014;32:368–377. doi: 10.1002/cbf.3025. [DOI] [PubMed] [Google Scholar]
- 53.Lecka-Czernik B, Moerman EJ, Grant DF, Lehmann JM, Manolagas SC, Jilka RL. Divergent effects of selective peroxisome proliferator-activated receptor-gamma 2 ligands on adipocyte versus osteoblast differentiation. Endocrinology. 2002;143:2376–2384. doi: 10.1210/endo.143.6.8834. [DOI] [PubMed] [Google Scholar]
- 54.Rzonca SO, Suva LJ, Gaddy D, Montague DC, Lecka-Czernik B. Bone is a target for the antidiabetic compound rosiglitazone. Endocrinology. 2004;145:401–406. doi: 10.1210/en.2003-0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang C, Li H, Chen SG, He JW, Sheng CJ, Cheng XY, Qu S, Wang KS, Lu ML, Yu YC. The skeletal effects of thiazolidinedione and metformin on insulin-resistant mice. J Bone Miner Metab. 2012;30:630–637. doi: 10.1007/s00774-012-0374-0. [DOI] [PubMed] [Google Scholar]
- 56.Gao Y, Li Y, Xue J, Jia Y, Hu J. Effect of the anti-diabetic drug metformin on bone mass in ovariectomized rats. Eur J Pharmacol. 2010;635:231–236. doi: 10.1016/j.ejphar.2010.02.051. [DOI] [PubMed] [Google Scholar]
- 57.Jeyabalan J, Viollet B, Smitham P, Ellis SA, Zaman G, Bardin C, Goodship A, Roux JP, Pierre M, Chenu C. The anti-diabetic drug metformin does not affect bone mass in vivo or fracture healing. Osteoporos Int. 2013;24:2659–2670. doi: 10.1007/s00198-013-2371-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tolosa MJ, Chuguransky SR, Sedlinsky C, Schurman L, McCarthy AD, Molinuevo MS, Cortizo AM. Insulin-deficient diabetes-induced bone microarchitecture alterations are associated with a decrease in the osteogenic potential of bone marrow progenitor cells: preventive effects of metformin. Diabetes Res Clin Pract. 2013;101:177–186. doi: 10.1016/j.diabres.2013.05.016. [DOI] [PubMed] [Google Scholar]
- 59.Vestergaard P, Rejnmark L, Mosekilde L. Relative fracture risk in patients with diabetes mellitus, and the impact of insulin and oral antidiabetic medication on relative fracture risk. Diabetologia. 2005;48:1292–1299. doi: 10.1007/s00125-005-1786-3. [DOI] [PubMed] [Google Scholar]
- 60.Monami M, Cresci B, Colombini A, Pala L, Balzi D, Gori F, Chiasserini V, Marchionni N, Rotella CM, Mannucci E. Bone fractures and hypoglycemic treatment in type 2 diabetic patients: a case-control study. Diabetes Care. 2008;31:199–203. doi: 10.2337/dc07-1736. [DOI] [PubMed] [Google Scholar]
- 61.Zinman B, Haffner SM, Herman WH, Holman RR, Lachin JM, Kravitz BG, Paul G, Jones NP, Aftring RP, Viberti G, et al. Effect of rosiglitazone, metformin, and glyburide on bone biomarkers in patients with type 2 diabetes. J Clin Endocrinol Metab. 2010;95:134–142. doi: 10.1210/jc.2009-0572. [DOI] [PubMed] [Google Scholar]
- 62.Borges JL, Bilezikian JP, Jones-Leone AR, Acusta AP, Ambery PD, Nino AJ, Grosse M, Fitzpatrick LA, Cobitz AR. A randomized, parallel group, double-blind, multicentre study comparing the efficacy and safety of Avandamet (rosiglitazone/metformin) and metformin on long-term glycaemic control and bone mineral density after 80 weeks of treatment in drug-naïve type 2 diabetes mellitus patients. Diabetes Obes Metab. 2011;13:1036–1046. doi: 10.1111/j.1463-1326.2011.01461.x. [DOI] [PubMed] [Google Scholar]
- 63.Hegazy SK. Evaluation of the anti-osteoporotic effects of metformin and sitagliptin in postmenopausal diabetic women. J Bone Miner Metab. 2015;33:207–212. doi: 10.1007/s00774-014-0581-y. [DOI] [PubMed] [Google Scholar]
- 64.van Lierop AH, Hamdy NA, van der Meer RW, Jonker JT, Lamb HJ, Rijzewijk LJ, Diamant M, Romijn JA, Smit JW, Papapoulos SE. Distinct effects of pioglitazone and metformin on circulating sclerostin and biochemical markers of bone turnover in men with type 2 diabetes mellitus. Eur J Endocrinol. 2012;166:711–716. doi: 10.1530/EJE-11-1061. [DOI] [PubMed] [Google Scholar]
- 65.Wijenayaka AR, Kogawa M, Lim HP, Bonewald LF, Findlay DM, Atkins GJ. Sclerostin stimulates osteocyte support of osteoclast activity by a RANKL-dependent pathway. PLoS One. 2011;6:e25900. doi: 10.1371/journal.pone.0025900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pradeep AR, Nagpal K, Karvekar S, Patnaik K, Naik SB, Guruprasad CN. Platelet-rich fibrin with 1% metformin for the treatment of intrabony defects in chronic periodontitis: a randomized controlled clinical trial. J Periodontol. 2015;86:729–737. doi: 10.1902/jop.2015.140646. [DOI] [PubMed] [Google Scholar]