

Abstract
The recapitulation of primary tumour heterogenity and the existence of a minor sub-population of cancer cells, capable of initiating tumour growth in xenografts on serial passages, led to the hypothesis that cancer stem cells (CSCs) exist. CSCs are present in many tumours, among which is breast cancer. Breast CSCs (BCSCs) are likely to sustain the growth of the primary tumour mass, as well as to be responsible for disease relapse and metastatic spreading. Consequently, BCSCs represent the most significant target for new drugs in breast cancer therapy. Both the hypoxic condition in BCSCs biology and pro-inflammatory cytokine network has gained increasing importance in the recent past. Breast stromal cells are crucial components of the tumours milieu and are a major source of inflammatory mediators. Recently, the anti-inflammatory role of some nuclear receptors ligands has emerged in several diseases, including breast cancer. Therefore, the use of nuclear receptors ligands may be a valid strategy to inhibit BCSCs viability and consequently breast cancer growth and disease relapse.
Keywords: Cancer stem cells, Hypoxia, Inflammation, Nuclear receptors, Retinoids, Peroxisome proliferator-activator receptors
Core tip: This review examines the roles of breast cancer stem cells (BCSC) in the eliminate breast cancer disease. BCSCs represent the most significant target for new drugs in breast cancer therapy. The use of nuclear receptors ligands may be a valid strategy to inhibit BCSCs viability and consequently breast cancer growth and disease relapse.
INTRODUCTION
The new hypothesis: Cancer stem cells
Several studies in the past years have shown that particular stem cells can have a significant role in cancer formation. These cells were identified in the hematopoietic system, central nervous system and mammary glands. These cells are a rare cell population of “tumour initiators”, with particular biological characteristics[1-4]. These stem cells have the ability to self-renew and to develop into all the cells that form the tumour mass and are called cancer stem cells (CSCs)[5]. In the CSCs hypothesis, cancer derives from normal stem cells that are transformed into tumour cells[6]. Adult stem cells are characterized as long-living with a low proliferative rate and are exposed for prolonged periods to agents that can induce damage and can accumulate mutations that result in neoplastic transformation[7]. Therefore, this condition implies the adoption of a new model to explain the carcinogenesis. Contrary to the “stochastic” model of tumorigenesis, for which the neoplastic transformation would result from random mutations incurred by a healthy cell that, consequently, undergoes clonal expansion. The CSCs hypothesis argues that the tumour begins from a stem cell, probably due to a dysregulation of the pathways involved in self-renewal[6]. However, these mechanisms are not exclusive and we can consider that other mechanisms can participate in the genesis and tumour progression, contributing to the heterogeneity of the tumour.
Breast cancer stem cells
The existence of CSCs in tumours of the mammary gland has been widely demonstrated by several studies, based mainly on transplants. The hypothesis of the origin of the breast cancer stem cells (BCSCs) was confirmed by the finding that only a minority of human breast cancer cells have the ability to induce new tumours when transplanted into immunocompromised mice (NOD/SCID)[8,9]. The presence of BCSCs indicates the onset of a breast tumour and they are distinguishable from other cancer cells by expression of specific membrane markers such as CD44 and an Epithelial Specific Antigen and by the absence or low expression of CD24 protein (CD44 and CD24 are adhesion molecules). Therefore, BCSCs are isolated from the tumour mass as CD44+/CD24- by FACS analysis. Approximately 200 cells characterized by this phenotype induced tumour growth in NOD/SCID mice while 20000 cells with a different phenotype did not have this capability. CD44+/CD24- breast cancer cells can generate cells of the same phenotype and cells phenotypically different, so that the tumour from which they develop in mice repeats the entire heterogeneity of its initial cancer[4]. The mammary gland epithelial components are thought to arise from stem cells that undergo both self-renewal and differentiation. Self-renewal has been shown to be regulated by the Hedgehog, Notch, and Wnt pathways. Deregulation of the self-renewal in stem cells/progenitors might be a key event in mammary carcinogenesis[10]. Different combinations of cell surface markers such as CD44, CD49f, CD24, and CD29 as well as the activity of certain enzymes such as aldehyde dehydrogenase isoform 1 (ALDH1) have been used to identify BCSCs[11].
The new BCSCs hypothesis has important therapeutic implications. BCSCs have many similar characteristics to normal stem cells, such as apoptosis resistance, the capacity to repair DNA damage and multidrug-resistance (MDR). MDR is important to explain the capacity of breast cancer to overcome chemotherapy. BCSCs are characterized by expression of genes encoding ATP-binding cassette (ABC) proteins, which are transmembrane transporters involved in the extrusion of drugs from cancer cells, as ABCC1, ABCG2 and ABCB1. The principal MDR proteins that are expressed in BCSCs are the P-glycoprotein, the multidrug-resistance protein 1 (MDR1) and the cluster differentiation 243 (CD243). Chemotherapy drugs with anti-proliferative effects are less effective on CSCs population, because these cells divide less frequently than cancer cells[6,7]. For these reasons chemotherapy destroy many neoplastic cells but does not affect the minority component of tumour such as BCSCs. Accordingly, BCSCs induce tumour relapse and metastases[8,9]. So a change in chemotherapy strategy is necessary to kill also BCSCs.
The stem cell niche and CSC
Stem cells are localized in a niche that is a local tissue microenvironment. The niche has a limited area where the cells can maintain their peculiarity. Significant progress has been made by the studies on the interactions between the stem cells and the microenvironment in Caenorhabditis elegans and mammals[12,13]. Comparing the stem cell niches in these systems, various common features and functions have emerged. The niche is formed by a group of cells (fibroblasts of the stroma) which have a support function for stem cells, serving as the anchor point for the stem cells and physical adhesion molecules mediate the interactions between the support cells and stem cells (as well as those between the stem cells and the extracellular matrix). The niche generates factors that control number, proliferation and differentiation of stem cells. Normally, it maintains the stem cells in a quiescent state, providing them with the signals that inhibit the growth and proliferation. Only after implementing a stimulus transient activator, stem cells are able to divide in order to participate in tissue regeneration. This suggests that control stem cell dependent signaling mechanisms, resulting from dynamic niche and maintaining the balance between the proliferative and anti-proliferative signals, are the key to the homeostatic regulation of the stem cells[12,13]. When there is a change in the niche and growth and proliferation signals prevail, the stem cell population is exposed to an uncontrolled expansion which can lead to spread CSCs[13].
Inflammation and breast cancer stroma
The idea that inflammation could play a role in carcinogenesis was born in 1863, when Rudolf Virchow noted the presence of leukocytes in neoplastic tissues. After this observation, more and more data have demonstrated that malignancy may begin at sites of infection or chronic inflammation and approximately 25% of all cancers are associated with such conditions. In fact, although the inflammation represents a defensive response adaptive to infection or injury and is, under normal conditions, a self-limiting process which culminates in the repair of damaged tissues, an inadequate resolution induces chronic diseases or cancer[14]. Chronic inflammation is involved in all stages of carcinogenesis (initiation, promotion and progression). Inflammation induces an excessive production of reactive oxygen species that could cause genomic instability and mutation and consequently a tumour[14]. Stromal cells as fibroblasts, that are around the tumour, and inflammatory cells, as macrophage, that are infiltrated in the tumour, help to create an environment favoring the increase of inflammation[15]. Fibroblasts are among the most abundant cell types in solid tumours and are especially important in breast, pancreas, colon and prostate cancer[16]. In physiological conditions, fibroblasts have a low proliferative rate and have a constant production of extracellular matrix (ECM). ECM has anchoring function for the epithelial cells maintaining the integrity of structural epithelium. In the carcinoma in situ, the stroma is not compromised because it remains separated from the tumour cells through the basement membrane integrity. With the acquisition of infiltrating characteristics, some tumour cells manage to cross the basal lamina whose breaking mimics a traumatic insult to the tissue, causing changes in non-epithelial cell types. Fibroblasts are activated and become tumour associated fibroblasts (TAF) thus contributing to the growth and expansion of tumour in several ways: they produce proteins such as matrix metalloproteinases (MMPs) that have proteolytic activity on the components of the extracellular matrix[17]; release high levels of stromal-derived factor-1 (SDF-1) which attracts endothelial progenitor cells in the tumour mass (thus promoting angiogenesis); directly promote the growth of cancer cells through interaction with their receptor CXCR4[18]; release some growth factors such as epidermal growth factor (EGF) and transforming growth factor β (TGFβ) and release a wide range of inflammatory cytokines[16,19]. Macrophages resident in the stroma and monocytes that act together with tumour chemotactic factors, undergo changes that lead them to favor tumour growth. Thus, tumour associated macrophages (TAM) support tumour angiogenesis through the secretion of pro-angiogenic factors as the vascular endothelial growth factor (VEGF), the interleukin-1β (IL1β) and the angiogenin (Ang). TAM facilitate the migration of cancer cells through the release of tumor necrosis factor-α (TNFα), MMPs (such as the MMP9) and other proteases such as tissue plasminogen activator[20,21]. Moreover, TAM produce factors, such as EGF, that directly promote the growth of cancer cells[22] and have a role in facilitating the invasion of neoplastic cells[23]. Finally, TAF can activate macrophages that produce cytokines to maintain an inflamed microenvironment[24,25]. Inflammatory cytokines, including SDF-1, interleukin-1 (IL-1), IL-6 and IL-8, may affect tumour growth by regulating of CSCs population[26]. In particular, it has been demonstrated that IL-6 can induce the acquisition of malignant characteristics in multicellular spheroids called mammosphere (MS), formed from stem cells and progenitors of the mammary gland; such aggregates were obtained in vitro in conditions of non-adherence from MCF-7 breast cancer cell line (MCF-MS) or obtained from breast surgical specimens (normal and tumour, N-MS and T-MS respectively)[27]. High levels of IL-6 mRNA were detected in T-MS, however IL-6 can stimulate the growth and self-renewal in both T-MS and N-MS. In particular, IL-6 induce overexpression of Notch-3 and of its ligand Jagged1, both implicated in the maintenance of stem cells in an undifferentiated state. It has been demonstrated that the pathway IL-6/Notch-3 increases the expression of the protein carbonic anhydrase IX (CAIX), from which depends the survival of MCF7-MS in hypoxic environment, as well as an increase of their invasive potential. IL-6 acting through different signal transduction pathways involving protein kinases such as mitogen activated protein kinase (MAPK) or the phosphatidylinositol-triphosphate kinase (PI3K) and having the ability to directly activate STAT transcription factors (such as STAT3) via the kinase JAK2 which is associated its receptor, leads to a number of responses that favor the proliferation, inhibits apoptosis and increases the invasive capacity of tumour cells[28,29]. IL-6 induces the activation of the transcription factor NF-κβ that can code for several cytokines[27]. Other studies show that the TNFα, the major inducer of NF-κβ, involves an increase in the formation of MCF-7-MS cells through up-regulation of the Slug gene, a regulator of stem mammary tumour phenotype[30]. TNFα induces, after 10 d of treatment, the acquisition of typical characteristics of BCSCs (CD44+/CD24-) in not transformed mammary epithelial cell line MCF-10A; this effect is accompanied by the reduction of the E-cadherin expression and an increase of mesenchymal markers expression such as vimentin and smooth-muscle actin-α (αSMA)[31,32]. These considerations lead to the hypothesis that the survival of the BCSCs is dependent on the activation of NF-κβ, in turn resulting from the stimulation exerted, for example, by pro-inflammatory cytokines, as confirmed by the effect of inhibition of the proliferation of MCF7-MS due to the use of selective inhibitors of the NF-κβ, as parthenolide[33].
Hypoxia and BCSCs
Hypoxia plays a key role in carcinogenesis. Solid tumours are characterized by poorly vascularized regions and can progress under hypoxic conditions. Hypoxia is a condition that generally is found within the stem cell niche, which requires low concentrations of oxygen in order to minimize the damage that the eventual oxidation of the DNA could generate[34]. Hypoxia is also involved in the maintenance of an undifferentiated cell, thus playing a crucial factor in the stem cells condition and for this reason; could potentially contribute to the generation and or to support the CSCs. In hypoxic condition, there is an induction of the octamer-binding transcription factor 4 (Oct4) and Notch1 expression in CSCs, two proteins that are involved in the self-renewal and differentiation pathways[35,36]. Hypoxia induces Shc gene expression in BCSCs, a gene that coding for p66Shc protein, involved in cellular response to oxidative stress, which induce the up-regulation of Notch-3 and its ligand Jagged-1. Interestingly, there is a correlation of Snail expression with histological grade and lymph node status in breast carcinomas[37]. Snail coding for CAIX protein is a molecule that is overexpressed in hypoxic condition and in BCSCs[38]. Cancer cells in the hypoxic tumour niche overexpressed the hypoxic inducible factor (HIF). HIF is a heterodimeric transcription factor consisting of an α-subunit (HIF-1α or HIF-2α), and a β-subunit (HIF-1β), expressed constitutively. HIF-1 affects a variety of malignant features, such as hypoxic cancer cell survival, via the regulation of a large number of genes, including CAIX[27]. The oxygen-dependent HIF activity is mediated by a series of enzymes containing iron (Fe2+), belonging to the superfamily of 2-oxoglutarate-dependent dioxygenase that are oxygen sensitive. Members of this family are the prolyl hydroxylase domain-containing protein (PHD), as PHD1, PHD2, PHD3 and the factor inhibiting HIF. In normoxic conditions, the HIF-1α subunit is characterized by a very short half-life. In hypoxic condition, HIF-1α translocates into the nucleus and leads to gene activation by binding to a specific sequence (59-RCGTG-39) called Hypoxic Responsive Element (HRE), through the recruitment of coactivators CBP/300[39]. HIF-1α causes a metabolic change that allows cancer cells to adapt to poorly oxygenated environments: It results in the use of glycolysis at the expense of oxidative phosphorylation, even in aerobic conditions, with a decrease in mitochondrial respiration and an increased lactate. This phenomenon is called “Warburg effect” and is frequently found in cancer[40]. HIF induces a metabolic “shift” via transcriptional activation of genes involved in glucose turnover, including those coding for glucose transporters, glycolytic enzymes and enzymes involved in the production of lactate and in the metabolism of pyruvate[41,42]. HIF induces the VEGF expression and reduce anti-angiogenic factors, such as thrombospondin[43]. A recent study has shown that, in several cancer cell lines (breast, lung, cervical and ovarian), HIF-1α increases cell invasion[44]. Since HIF-1α induces the transmembrane protein CAIX expression, through various mechanisms CAIX can increase the invasive potential of cancer cells[45]. Moreover HIF controls the expression of LOX (lysyl oxidase) and the cytokine receptor CXCR4 expression that are essential for metastasis induction[46]. Finally, HIF reduces E-cadherin expression and induces the epithelial-mesenchymal transition[47-49].
NUCLEAR RECEPTORS
Nuclear hormone receptors (NRs) include receptors for steroid hormones such as estrogen receptors (ERs) and progesterone receptors (PRs), receptors for the thyroid hormone (TRs), receptors for vitamin D (VDRs), retinoic acid receptors (RARs), retinoid X receptors (RXRs) and a number of receptors that respond to intermediary metabolites, among which there are the peroxisome proliferator-activator receptors (PPARs) activated by fatty acids and prostaglandins[50-52]. The members of this superfamily act as transcription factors activated by ligands and have a conserved structure[53]. NRs are characterized by the presence of two conserved domains: (1) A central DNA-binding domain (DBD) which interacts with the core motif, that have specific DNA sequences called “response elements”(monomeric NR recognize a single core motif, while dimeric NR complexes interact with repeated occurrences of this core motif); and (2) A C-terminal ligand-binding domain, which determines specific NRs properties and is highly variable between the different receptors. NRs are characterized by a flexible linker region between the two previous domains. NRs have a carboxy-terminal E-domain that is responsible for the ligand binding, dimerization, and contain an inducible transactivation function dependent on ligand (AF-2). Finally, the N-terminal terminal A/B-domain of the NR molecule contains a constitutive activation function independent on ligand (AF-1). NR can be activated by specific ligands that can modulate gene transcription and induce differentiation and anti-proliferative effects in cancer cells in several tumours[54].
Retinoic acid receptors and retinoid x receptors
Nuclear receptors retinoic acid receptors (RARs) and retinoid x receptors (RXRs) mediate the effects of retinoids. Retinoids are a class of compounds that includes natural metabolites of vitamin A (retinol) and its synthetic analogues. The natural retinoids are produced in vivo by oxidation of retinol, a two-step process that leads to the formation of all-trans-retinaldehyde due to the action of alcohol dehydrogenase, followed by oxidized retinaldehyde due to the action of the enzyme dehydrogenase. In the reaction all-trans-retinoic acid (ATRA) is produced, which is then metabolized by CYP26 to produce hydroxylated metabolites[51]. There are three receptor subtypes, encoded by different genes, called RARα, β, γ and RXRα, β, γ. RARs subtypes can bind with high affinity not only ATRA as well as 9-cis retinoic acid (9cRA), the product of isomerization of ATRA, that is able to interact with RXRs, a feature that sets it apart from trans retinoic acid isoforms that do not have this possibility[55]. Following the activation induced by the ligand, the RARs form heterodimers with RXRs (RAR-RXR) that lead to gene transcription by binding to specific DNA sequences in the promoter of target genes, those corresponding to the Retinoic Acid Response Element (RARE), while homodimers formed by RXRs (RXR-RXR) bind to sequences denominated RXRE (Retinoic X Response Element)[52]. The RXRs are the only nuclear receptors that are capable to form both homodimers (RXR-RXR) and heterodimers (NR-RXR), constituting factors required for efficient DNA binding of many other members of the NR superfamily, including RARs and PPARs precisely[52]. These considerations underscore the importance of RXRs ligands because they can mediate effects affecting many biological processes. The NRs partner of RXRs receptors can be “permissive”, as PPARs. The heterodimer that is formed can be activated independently from agonists of one or other receptors or, synergistically, by both. RXRs may be “non-permissive”, when the heterodimer cannot be activated by RXRs agonists alone, necessitating the presence of a ligand for the receptor partners (in the case of the dimer RAR-RXR is necessary a RARs ligand as ATRA)[51]. Various studies targetting the identification of a natural endogenous ligand for RXRs did not produce the desired results because the molecules proposed for this role (9cRA, phytanic acid, docosahexaenoic acid) have not demonstrated a selectivity only for binding to RXRs; for this reason synthetic compounds that bind only to RXRs (called rexinoids) could be essential to better understand the role of these receptors[52].
Retinoids and breast cancer
Retinoids are widely used to treat dermatological diseases. Retinoids have recently received considerable attention for the prevention and treatment of cancer due to their role in cell differentiation and their anti-proliferative, pro-apoptotic and anti-oxidant effects[56]. Epidemiological studies show that a low intake of vitamin A leads to a higher risk of developing cancer. Altered expression of RARs and RXRs is associated with malignant transformation both in animal tissues and in cultured cells[57]. Furthermore, in animal models retinoids reduce cancer of skin, lung, breast, bladder, ovary and prostate. In humans, retinoids can reverse epithelial precancerous lesions, induce differentiation of myeloid cells, and have an important role in the lung, liver and breast cancer prevention[58]. Moreover, retinoids regulate stem cell differentiation[59]. Retinoic acid is used today in various diseases: ATRA is the principal retinoid investigated in clinical trials for the treatment of lymphoma, leukemia, melanoma, lung cancer, cervix, kidney, neuroblastoma, and glioblastoma. Its clinical use has more effect in the treatment of the acute promyelocytic leukemia (APL). Since 1995, the FDA approved ATRA to APL treatment[60]. 9cRA differs from ATRA for its ability to activate both RAR and RXR. In addition, 9cRA activates different nuclear receptors such as PPARs, FXRs, PXRs and VDRs through RXR heterodimerization. In preclinical studies, 9cRA is effective in the prevention of prostate cancer and breast cancer and was also approved by the FDA for the topical treatment of cutaneous lesions of Kaposi's sarcoma[61]. The natural retinoid 13-cis retinoic acid (13cRA), binds both receptors RARs and RXRs, has anti-inflammatory activity and is in clinical development for different types of cancer, including cancer of the thyroid[62]. Preclinical and clinical studies have shown the anti-tumoural effects of retinoids in breast cancer. It has been observed that 9cRA inhibits proliferation and induces differentiation and apoptosis in the breast cancer cell line MCF-7 cells. Recently, it has been demonstrated that retinoid have a role also in the regulation of BCSCs self-renewal and differentiation; ATRA reduces BCSCs proliferation demonstrated by ALDH assay[11]. However, clinical studies have shown that natural retinoids can have side effects such as the hypervitaminosis A. It has been demonstrated that retinoids selective for RARs have chemopreventive activity with side effects, while selective RXRs retinoids (called rexinoids) suppress mammary tumorigenesis without side effects[63]. Since hypertriglyceridemia can be induced by rexinoids, recent research has investigated new rexinoids that have anti-tumoural effects without side effects. Among these there is (2E,4E,6Z,8Z)-8-(3’,4’-Dihydro-1’(2H)-naphthalen-1’-ylidene)-3,7-dimethyl-2,3,6 octatrienoinic acid (UAB30) that is currently undergoing clinical evaluation as a novel breast cancer prevention agent[64]. Furthermore, some patients may experience relapses cancer because cancer cells become resistant to retinoids therapies. For these reasons, the synergic use of multiple molecules as NRs ligands with other molecules at lower doses might be a good strategy to block breast cancer growth, while inducing less side effects in patients. Immunotherapy with the use of retinoids and T cell has proved effective in the treatment of neuroblastoma and 13cRA+interferon-α2a significantly increases the survival of patients with metastatic renal cell carcinoma[65]. Lee and co-workers have shown that administration of ATRA increased the effectiveness of EGCG at a low concentration. Indeed, ATRA increased the synthesis of a EGCG molecular targets, the 67 kDa laminin receptor (LR67), which plays a key role in cell adhesion and in the breast metastatic process[66]. ATRA is a regulator of epithelial mesenchymal transition (EMT) that is a determinant of the breast cancer cell invasion and metastatic behaviour. It has shown that in HER2-positive SKBR3 and UACC812 cells, there is an amplification of the ERBB2 and RARA genes and ATRA activated a RARα-dependent epithelial differentiation program. Moreover, ATRA blocked Notch-1 up-regulation by EGF and/or heregulin-β1 and switches TGFβ from an EMT-inducing and pro-migratory determinant to an anti-migratory mediator[67]. ATRA can reduce the MS-forming ability of a subset of breast cancer cells, which correlates with induction of apoptosis, reducing SOX2 expression and inducing of its antagonist CDX2. The SOX2/CDX2 ratio has prognostic relevance in BCSCs[68]. K-Ras mutant BCSCs was resistant to ATRA, which was reversed by MAPK inhibitors. Thus, ATRA can be used in combination to reduce BCSC proliferation[68]. Interestingly, also the combination ATRA and doxorubicin can differentiate and kill the BCSCs. Differentiation of CSCs into non-CSCs can reduce their self-renewal capacity and increase their sensitivity to chemotherapy in a synergistic manner[69].
The new rexinoid IIF can kill BCSCs
In our laboratory, we have investigated the antitumoural effects of ATRA when binded to RARs while with the RXRs ligand, we used the synthetic rexinoid 6-OH-11-O-hydroxyphenanthrene (IIF), a new derivative of retinoic acid, capable of binding selectively to RXR and mainly activating the form RXR-γ[70]. Several in vitro studies show that IIF can be used as an anticancer agent: This rexinoid showed a greater anti-proliferative effect than ATRA and 9cRA in leukemic cell line HL-60, which induces apoptosis[71]. IIF induces differentiation in different tumour cell lines, such as colon carcinoma and neuroblastoma[72]. In the glioblastoma mouse model IIF reduces tumour growth and invasion through the inhibition of MMPs, such as MMP-2 and MMP-9, in combination with increased expression of their inhibitors (TIMP-1 and TIMP-2)[73]. IIF has anti-inflammatory effects in colon cancer by suppressing the expression of cyclooxygenase-2 (COX-2), the inducible form of COX, responsible for the prostaglandin production that is overexpressed in many tumours[73]. Recently, we have demonstrated that ATRA and IIF reduce the inflammation-dependent survival in MS generated from human tumour specimens (T-MS) and from the breast cancer cell line MCF-7 (MCF7-MS), but not in MS derived from normal mammary glands (N-MS). The effect depends on the inhibition of the inflammatory pathway NF-κB/IL-6 which is wired in T-MS. ATRA and IIF, blocking NF-κB axis, reduced expression of genes involved in the maintenance of a tumour stem cell phenotype (such as Slug, Notch-3, Jagged-1) and was accompanied by an increased expression of markers of differentiation such as ERα and keratin-18[74]. A promising strategy is the combination of IIF with natural substances, such as Epigallocatechin-3-gallate (EGCG), that have a cytotoxic effect against breast cancer cells. In a recent study, we demonstrated that the combination of IIF and EGCG had a higher activity than the individual administration. IIF and EGCG can have a common signaling pathway that induces apoptosis by reducing epidermal growth factor receptor activation and its downstream kinase AKT-1[75].
PPAR receptors and their agonists
RXRs receptors can form heterodimers with PPARs receptors. The latter mediate the effects of many synthetic compounds called peroxisome proliferators (PPs-peroxisome proliferators). The PPs influence both the number and the size of the peroxisomes, responsible for various functions within the cell (β-oxidation of fatty acids and cholesterol metabolism). Even PPARs there exist three isoforms (α, β, γ), encoded by different genes and characterized by different tissue localization. They operate as sensors for fatty acids and their derivatives, checking therefore, important pathways concerning lipids and energy metabolism[76]. PPARα is expressed at high levels in organs with significant catabolism of fatty acids. PPARβ has the broadest expression pattern, and the levels of expression depend on the extent of cell proliferation and differentiation. Finally, PPARγ is expressed as two isoforms, of which PPARγ2 is found in the adipose tissues, whereas PPARγ1 has a broader expression pattern and is expressed at high levels in cancer tissue[76]. RXRs dimerization and the presence of coactivators are necessary for PPARs activation as transcription factor[77,78]. There is a wide range of endogenous and exogenous ligands that can interact with PPARs, leading to have different responses. Among PPARs endogenous ligands there are arachidonic acid, eicosapentaenoic acid and prostaglandin J2, while, among exogenous ligands there are the synthetic compounds called thiazolidinediones (TZDs): Pioglitazone (PGZ), rosiglitazone and troglitazone[54]. The TZDs are used in the treatment of type 2 diabetes because they decrease insulin resistance; they increase glucose uptake in peripheral tissues and reduce hepatic production. Some studies show, however, that TZDs could be successfully used also against tumours. In breast cancer, for example, tumour cells often express high levels of PPARs and it was demonstrated that TZDs are able to induce differentiation and inhibit tumour proliferation both in vivo (nude mice) and in vitro (mammary tumour cell line MCF-7); these effects are increased when combined with retinoids[79,80]. It was also noted that treatment with TZD leads to a reduction in the number of breast cancer cells in S phase and an increase of cells in phase G0-G1. Furthermore, TZD and retinoids induced apoptosis in 30%-40% of breast cancer cells through the inhibition of Bcl-2 expression[80]. Among the anticancer mechanisms mediated by PPARs ligands, in addition to the induction of pro-apoptotic proteins and stabilization of cell cycle, the inhibition of the expression or activity of various cytokines and transcription factors involved in inflammatory pathways (as TNFα, IL-1, IL-4, NF-β) could help to slow the growth of transformed cells[54]. Interfering with inflammatory pathway is an ability shown by some ligands of PPARα; fenofibrate and GW7647 (synthetic agonists). For example, they can significantly reduce the levels of pro-inflammatory cytokines such as IL-1, the expression of TNFα, COX-2 and an inducible form of the enzyme nitric oxide synthase in murine microglia BV-2 exposed to radiation. This effect is due to the inhibition of translocation of the NF-κB-p65 subunit or the inhibition of phosphorylation c-jun, a subunit of the transcription factor AP-1, both involved in inflammatory mechanisms[81]. MnSOD expression is significantly amplified in the aggressive breast carcinoma basal subtype. Interestingly, PPARγ activation repressed MnSOD expression and increased chemosensitivity, and inhibited tumour growth in MDA-MB-231 and BT549 breast cancer cell lines[82]. PPARs are also reported to be involved in the modulation of the EMT process in CSCs initiation and in the regulation of CSCs functions[83]. Some data show that activation of PPARα could induce cancer and result in the induction of inflammatory responses. If the stimulation of the PPARs, for example with the TZD, involves the inhibition of neoplastic growth and the induction of differentiation, activation of PPARα significantly increases the proliferation of tumour cells, as demonstrated for the breast cancer cell lines MDA-MB-231 and MCF-7[84]. This stark contrast between these isoforms of PPAR is highlighted by studies that show the effects mediated by an agonist of PPARα, WY-14643. Chronic administration of PP in rats and mice leads to development of hepatocellular carcinoma; as a result of repeated exposure to WY-14643. Mice in which the expression of PPARα is increased, do not develop this type of tumour, as opposed to what happens in wild-type mice for PPARα, demonstrating that the receptor mediates the effects of carcinogenic arising by the stimulation exerted by an agonist[85]. More recently it has been seen that WY-14643 promotes the formation of a MS-tumour (derived from cells of the mammary tumour cell line MCF-7) by stimulating the activation of the NF-κB/IL-6 and, consequently, the expression of genes Slug, Notch-3, Jagged-1, whereas the silencing of PPARα with a specific siRNA reduces tumour-MS formation. Furthermore, PPARα expression is positively correlated with the phenotype of BCSCs obtained from specimens of breast cancer patients[86]. Finally, we have recently demonstrated that IIF potentiates the ability of PGZ to hamper the MS-forming capability of human breast tumours and MCF7 cancer cells, reducing the expression of CSCs regulatory genes (Notch3, Jagged1, SLUG, IL-6, Apolipoprotein E, HIF-1α and CAIX). Notably, these effects are not observed in normal-MS obtained from human breast tissue[87]. Recently, Wang et al[88] demonstrated that PPARγ-binding protein upregulates several genes in the de novo fatty acid synthesis network, which is highly active in ERBB2-positive breast cancer cells. ERBB2 is a prognostic marker occurring in 30% of breast cancers and is associated with aggressive disease and poor outcomes. Inhibition of the PPARγ pathway using PPARγ antagonists (GW9662 and T0070907) reduces the ALDH-positive population and tumour-MS formation in ERBB2-positive breast cancer cells[88].
Vitamin D and BSCSs
Vitamin D-3 exerts most of its cellular effects via its nuclear receptor, the vitamin D-3 receptor (VDR), that heterodimerizes with the RXRs. The VDR-RXR complex binds vitamin D responsive elements (VDRE) in gene promoters and regulates transcription of target genes[89]. It has been reported in literature that vitamin D is a potential preventive/therapeutic agent against CSCs. Several proteins, such as Notch, Hedgehog, Wnt and TGF-β, are modulated by vitamin D in CSCs as well as in normal stem cell[90]. Interestingly, MS derived from BRCA1-silenced MCF7 or MDA-MB-231 breast cancer cells were no longer sensitive to the growth inhibitory effects of vitamin D, 1α, 25-dihydroxyvitamin D 3 (1,25D). Since, the active form of vitamin D is a potent inhibitor of BCSCs growth through the down-regulation of BRCA1 expression, which is the most frequently mutated tumour suppressor gene in breast cancer[91]. Treatment with 1α25(OH)2D3 or BXL0124 (two vitamin D compounds) repressed markers associated with the breast stem cell-like phenotype, such as CD44, CD49f, c-Notch1 and NF-κB. Furthermore, 1α25(OH)2D3 and BXL0124 reduced the expression of pluripotency markers, OCT4 and KLF-4 in BCSCs[92]. However, some authors have shown that MS were relatively insensitive to treatment with 125D compared to more differentiated breast cancer cells; instead combined treatment of 125D and DET- NONOate induce a significant decrease in the overall size of MS and reduced breast tumour volume in nude mice[93]. Combination therapy using 125D with drugs specifically targeting key survival pathways in BCSCs could be a best strategy to overcome aggressive breast cancer.
Estrogen receptor and BCSCs
It has been reported in literature that many breast cancers express estrogen receptor-α (ERα) and are dependent on estrogens[94]. Tamoxifen is the most widely used in endocrine therapy for ERα positive (ER+) breast cancers during the last 30 years. Unfortunately, up to 40% of metastases from ER+ primary breast cancer do not respond to endocrine therapy. Recent study have demonstrated that tamoxifen was effective in reducing proliferation of ERα positive (ER+) adherent cancer cells, but not their CSCs population[95]. Interestingly, estrogen is essential for the development of the normal breast, but adult mammary stem cells are known to be ERα negative (ER-)[96]. BCSCs sorted derived from ER+ breast cancer tissue and established breast cancer cell lines, have low or absent ER expression[74,96]. However, estrogen stimulated BCSCs activity demonstrated by increased MS-formation through the induction of EGF and Notch receptor signaling pathways[96]. Breast cancer cells develop resistance to endocrine therapies by shifting between ER-regulated and growth factor receptor-regulated survival signaling pathways[97]. However, the roles of BCSCs in antiestrogen resistance and the underlying molecular mechanisms have not been well established. Recent, a novel variant of ERα, called ERα36 (molecular weight of 36 kDa) it has been investigated. ERα36 mediates rapid antiestrogen signaling and is highly expressed in ER+ breast progenitor cells. Antiestrogens increased the percentages of the BCSCs from ER+ breast cancer cell through stimulation of luminal epithelial lineage specific and these BCSCs are more resistant to antiestrogens than the bulk cells. Finally, ERα36 mediated antiestrogen signaling such as the PI3K/AKT that plays an important role in antiestrogen resistance of ER+/BCSCs[98].
CONCLUSION
BCSCs represent the most significant target for new anti-breast cancer drugs. In fact, BCSCs are likely to sustain the growth of the primary tumour mass, as well as to be responsible for disease relapse and metastatic spreading in breast cancer[6-10]. The activity of NF-κB in BCSCs and in the tumour stroma (mainly formed by fibroblasts and inflammatory cells) has been recognized to be of pivotal importance in normal and CSCs survival[99]. It has been proposed that “NF-κB activity addiction” would make CSCs more susceptible to NF-κB inhibitors than their normal counterparts[26,55]. For these reasons the use of molecules, as NRs ligands, capable of inhibiting NF-κB dependent inflammation may be the best strategy to hamper BCSCs growth[74,86]. Recently, we demonstrated that BCSCs have a particular NRs phenotype[86,87] (Figure 1). Therefore, the synergic use of multiple molecules (as ligands of NRs) at lower doses might be a good strategy to kill BCSCs, while inducing fewer side effects in patients. Moreover, the use of NRs ligands in combination with each other (as ligands of PPARs with ligands of RXRs) or with other substances (e.g., EGCG) may be a valid strategy to inhibit BCSCs viability.
Figure 1.
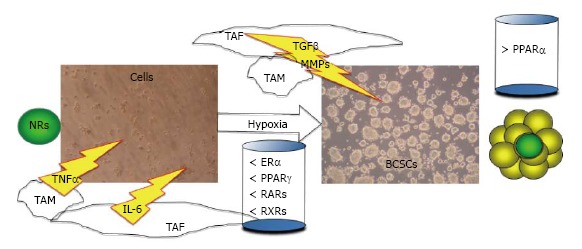
The nuclear receptors phenotype in breast cancer stem cells. In breast cancer, TAF and TAM promote inflammation and invasion through the secretion of cytokines, as IL6, TNFα and TGFβ and the secretion of MMPs, as MMP9. In this hypoxic inflammatory niche, particular stem cells can form the BCSCs as mammospheres. BCSCs are characterized by a particular nuclear receptors phenotype: a lower level of ERα, PPARγ, RARs, RXRs, and a higher level of PPARα is expressed than adherent breast cancer cells. NRs: Nuclear receptors; BCSCs: Breast cancer stem cells; TAF: Tumour associated fibroblast; TAM: Tumour associated macrophage; MMPs: Metalloproteinases; IL6: Interleukin-6; ERα: Estrogen receptor-α; PPARs: Peroxisome proliferator-activator receptors; RARs: Retinoic acid receptors; RXRs: Retinoid x receptors; TNFα: Tumor necrosis factor-α; TGFβ: Trasforming growth factor-β.
Footnotes
Conflict-of-interest statement: Authors declare no conflict of interests for this article.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: November 3, 2015
First decision: December 4, 2015
Article in press: January 29, 2016
P- Reviewer: Liu SH, Wang LS S- Editor: Qiu S L- Editor: A E- Editor: Lu YJ
References
- 1.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 2.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 3.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 4.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jordan CT, Guzman ML, Noble M. Cancer stem cells. N Engl J Med. 2006;355:1253–1261. doi: 10.1056/NEJMra061808. [DOI] [PubMed] [Google Scholar]
- 6.Charafe-Jauffret E, Monville F, Ginestier C, Dontu G, Birnbaum D, Wicha MS. Cancer stem cells in breast: current opinion and future challenges. Pathobiology. 2008;75:75–84. doi: 10.1159/000123845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dontu G, Al-Hajj M, Abdallah WM, Clarke MF, Wicha MS. Stem cells in normal breast development and breast cancer. Cell Prolif. 2003;36 Suppl 1:59–72. doi: 10.1046/j.1365-2184.36.s.1.6.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, Wicha MS. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003;17:1253–1270. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dontu G, Liu S, Wicha MS. Stem cells in mammary development and carcinogenesis: implications for prevention and treatment. Stem Cell Rev. 2005;1:207–213. doi: 10.1385/SCR:1:3:207. [DOI] [PubMed] [Google Scholar]
- 10.Liu S, Dontu G, Wicha MS. Mammary stem cells, self-renewal pathways, and carcinogenesis. Breast Cancer Res. 2005;7:86–95. doi: 10.1186/bcr1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ginestier C, Wicinski J, Cervera N, Monville F, Finetti P, Bertucci F, Wicha MS, Birnbaum D, Charafe-Jauffret E. Retinoid signaling regulates breast cancer stem cell differentiation. Cell Cycle. 2009;8:3297–3302. doi: 10.4161/cc.8.20.9761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Albini A, Cesana E, Noonan DM. Cancer stem cells and the tumor microenvironment: soloists or choral singers. Curr Pharm Biotechnol. 2011;12:171–181. doi: 10.2174/138920111794295756. [DOI] [PubMed] [Google Scholar]
- 13.Bonafè M, Storci G, Franceschi C. Inflamm-aging of the stem cell niche: breast cancer as a paradigmatic example: breakdown of the multi-shell cytokine network fuels cancer in aged people. Bioessays. 2012;34:40–49. doi: 10.1002/bies.201100104. [DOI] [PubMed] [Google Scholar]
- 14.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kundu JK, Surh YJ. Inflammation: gearing the journey to cancer. Mutat Res. 2008;659:15–30. doi: 10.1016/j.mrrev.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 16.Allen M, Louise Jones J. Jekyll and Hyde: the role of the microenvironment on the progression of cancer. J Pathol. 2011;223:162–176. doi: 10.1002/path.2803. [DOI] [PubMed] [Google Scholar]
- 17.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 18.Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 19.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 20.Romer J, Nielsen BS, Ploug M. The urokinase receptor as a potential target in cancer therapy. Curr Pharm Des. 2004;10:2359–2376. doi: 10.2174/1381612043383962. [DOI] [PubMed] [Google Scholar]
- 21.Coffelt SB, Hughes R, Lewis CE. Tumor-associated macrophages: effectors of angiogenesis and tumor progression. Biochim Biophys Acta. 2009;1796:11–18. doi: 10.1016/j.bbcan.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 22.O’Sullivan C, Lewis CE, Harris AL, McGee JO. Secretion of epidermal growth factor by macrophages associated with breast carcinoma. Lancet. 1993;342:148–149. doi: 10.1016/0140-6736(93)91348-p. [DOI] [PubMed] [Google Scholar]
- 23.Wyckoff JB, Wang Y, Lin EY, Li JF, Goswami S, Stanley ER, Segall JE, Pollard JW, Condeelis J. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007;67:2649–2656. doi: 10.1158/0008-5472.CAN-06-1823. [DOI] [PubMed] [Google Scholar]
- 24.Gyorki DE, Asselin-Labat ML, van Rooijen N, Lindeman GJ, Visvader JE. Resident macrophages influence stem cell activity in the mammary gland. Breast Cancer Res. 2009;11:R62. doi: 10.1186/bcr2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ehninger A, Trumpp A. The bone marrow stem cell niche grows up: mesenchymal stem cells and macrophages move in. J Exp Med. 2011;208:421–428. doi: 10.1084/jem.20110132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korkaya H, Liu S, Wicha MS. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J Clin Invest. 2011;121:3804–3809. doi: 10.1172/JCI57099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sansone P, Storci G, Giovannini C, Pandolfi S, Pianetti S, Taffurelli M, Santini D, Ceccarelli C, Chieco P, Bonafé M. p66Shc/Notch-3 interplay controls self-renewal and hypoxia survival in human stem/progenitor cells of the mammary gland expanded in vitro as mammospheres. Stem Cells. 2007;25:807–815. doi: 10.1634/stemcells.2006-0442. [DOI] [PubMed] [Google Scholar]
- 28.Hodge DR, Hurt EM, Farrar WL. The role of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer. 2005;41:2502–2512. doi: 10.1016/j.ejca.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 29.Asgeirsson KS, Olafsdóttir K, Jónasson JG, Ogmundsdóttir HM. The effects of IL-6 on cell adhesion and e-cadherin expression in breast cancer. Cytokine. 1998;10:720–728. doi: 10.1006/cyto.1998.0349. [DOI] [PubMed] [Google Scholar]
- 30.Storci G, Sansone P, Mari S, D’Uva G, Tavolari S, Guarnieri T, Taffurelli M, Ceccarelli C, Santini D, Chieco P, et al. TNFalpha up-regulates SLUG via the NF-kappaB/HIF1alpha axis, which imparts breast cancer cells with a stem cell-like phenotype. J Cell Physiol. 2010;225:682–691. doi: 10.1002/jcp.22264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhat-Nakshatri P, Appaiah H, Ballas C, Pick-Franke P, Goulet R, Badve S, Srour EF, Nakshatri H. SLUG/SNAI2 and tumor necrosis factor generate breast cells with CD44+/CD24- phenotype. BMC Cancer. 2010;10:411. doi: 10.1186/1471-2407-10-411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Agiostratidou G, Hulit J, Phillips GR, Hazan RB. Differential cadherin expression: potential markers for epithelial to mesenchymal transformation during tumor progression. J Mammary Gland Biol Neoplasia. 2007;12:127–133. doi: 10.1007/s10911-007-9044-6. [DOI] [PubMed] [Google Scholar]
- 33.Zhou J, Zhang H, Gu P, Bai J, Margolick JB, Zhang Y. NF-kappaB pathway inhibitors preferentially inhibit breast cancer stem-like cells. Breast Cancer Res Treat. 2008;111:419–427. doi: 10.1007/s10549-007-9798-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keith B, Simon MC. Hypoxia-inducible factors, stem cells, and cancer. Cell. 2007;129:465–472. doi: 10.1016/j.cell.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, Ruas JL, Poellinger L, Lendahl U, Bondesson M. Hypoxia Requires Notch Signaling to Maintain the Undifferentiated Cell State. Dev Cell. 2005;9:617–628. doi: 10.1016/j.devcel.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 36.Covello KL, Kehler J, Yu H, Gordan JD, Arsham AM, Hu CJ, Labosky PA, Simon MC, Keith B. HIF-2alpha regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006;20:557–570. doi: 10.1101/gad.1399906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blanco MJ, Moreno-Bueno G, Sarrio D, Locascio A, Cano A, Palacios J, Nieto MA. Correlation of Snail expression with histological grade and lymph node status in breast carcinomas. Oncogene. 2002;21:3241–3246. doi: 10.1038/sj.onc.1205416. [DOI] [PubMed] [Google Scholar]
- 38.Cabarcas SM, Mathews LA, Farrar WL. The cancer stem cell niche--there goes the neighborhood? Int J Cancer. 2011;129:2315–2327. doi: 10.1002/ijc.26312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Henze AT, Acker T. Feedback regulators of hypoxia-inducible factors and their role in cancer biology. Cell Cycle. 2010;9:2749–2763. doi: 10.4161/cc.9.14.12591. [DOI] [PubMed] [Google Scholar]
- 40.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 41.Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol. 2003;23:9361–9374. doi: 10.1128/MCB.23.24.9361-9374.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Semenza GL. HIF-1 inhibitors for cancer therapy: from gene expression to drug discovery. Curr Pharm Des. 2009;15:3839–3843. doi: 10.2174/138161209789649402. [DOI] [PubMed] [Google Scholar]
- 43.Dvorak HF. Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J Clin Oncol. 2002;20:4368–4380. doi: 10.1200/JCO.2002.10.088. [DOI] [PubMed] [Google Scholar]
- 44.Pennacchietti S, Michieli P, Galluzzo M, Mazzone M, Giordano S, Comoglio PM. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell. 2003;3:347–361. doi: 10.1016/s1535-6108(03)00085-0. [DOI] [PubMed] [Google Scholar]
- 45.Shin HJ, Rho SB, Jung DC, Han IO, Oh ES, Kim JY. Carbonic anhydrase IX (CA9) modulates tumor-associated cell migration and invasion. J Cell Sci. 2011;124:1077–1087. doi: 10.1242/jcs.072207. [DOI] [PubMed] [Google Scholar]
- 46.Erler JT, Bennewith KL, Nicolau M, Dornhöfer N, Kong C, Le QT, Chi JT, Jeffrey SS, Giaccia AJ. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature. 2006;440:1222–1226. doi: 10.1038/nature04695. [DOI] [PubMed] [Google Scholar]
- 47.Imai T, Horiuchi A, Wang C, Oka K, Ohira S, Nikaido T, Konishi I. Hypoxia attenuates the expression of E-cadherin via up-regulation of SNAIL in ovarian carcinoma cells. Am J Pathol. 2003;163:1437–1447. doi: 10.1016/S0002-9440(10)63501-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Esteban MA, Tran MG, Harten SK, Hill P, Castellanos MC, Chandra A, Raval R, O’brien TS, Maxwell PH. Regulation of E-cadherin expression by VHL and hypoxia-inducible factor. Cancer Res. 2006;66:3567–3575. doi: 10.1158/0008-5472.CAN-05-2670. [DOI] [PubMed] [Google Scholar]
- 49.Evans AJ, Russell RC, Roche O, Burry TN, Fish JE, Chow VW, Kim WY, Saravanan A, Maynard MA, Gervais ML, et al. VHL promotes E2 box-dependent E-cadherin transcription by HIF-mediated regulation of SIP1 and snail. Mol Cell Biol. 2007;27:157–169. doi: 10.1128/MCB.00892-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Evans RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240:889–895. doi: 10.1126/science.3283939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Germain P, Chambon P, Eichele G, Evans RM, Lazar MA, Leid M, De Lera AR, Lotan R, Mangelsdorf DJ, Gronemeyer H. International Union of Pharmacology. LX. Retinoic acid receptors. Pharmacol Rev. 2006;58:712–725. doi: 10.1124/pr.58.4.4. [DOI] [PubMed] [Google Scholar]
- 52.Germain P, Chambon P, Eichele G, Evans RM, Lazar MA, Leid M, De Lera AR, Lotan R, Mangelsdorf DJ, Gronemeyer H. International Union of Pharmacology. LXIII. Retinoid X receptors. Pharmacol Rev. 2006;58:760–772. doi: 10.1124/pr.58.4.7. [DOI] [PubMed] [Google Scholar]
- 53.Simons SS, Edwards DP, Kumar R. Minireview: dynamic structures of nuclear hormone receptors: new promises and challenges. Mol Endocrinol. 2014;28:173–182. doi: 10.1210/me.2013-1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Koeffler HP. Peroxisome proliferator-activated receptor gamma and cancers. Clin Cancer Res. 2003;9:1–9. [PubMed] [Google Scholar]
- 55.Konopleva M, Elstner E, McQueen TJ, Tsao T, Sudarikov A, Hu W, Schober WD, Wang RY, Chism D, Kornblau SM, et al. Peroxisome proliferator-activated receptor gamma and retinoid X receptor ligands are potent inducers of differentiation and apoptosis in leukemias. Mol Cancer Ther. 2004;3:1249–1262. [PubMed] [Google Scholar]
- 56.Bushue N, Wan YJ. Retinoid pathway and cancer therapeutics. Adv Drug Deliv Rev. 2010;62:1285–1298. doi: 10.1016/j.addr.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Szanto A, Narkar V, Shen Q, Uray IP, Davies PJ, Nagy L. Retinoid X receptors: X-ploring their (patho)physiological functions. Cell Death Differ. 2004;11 Suppl 2:S126–S143. doi: 10.1038/sj.cdd.4401533. [DOI] [PubMed] [Google Scholar]
- 58.di Masi A, Leboffe L, De Marinis E, Pagano F, Cicconi L, Rochette-Egly C, Lo-Coco F, Ascenzi P, Nervi C. Retinoic acid receptors: from molecular mechanisms to cancer therapy. Mol Aspects Med. 2015;41:1–115. doi: 10.1016/j.mam.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 59.Gudas LJ, Wagner JA. Retinoids regulate stem cell differentiation. J Cell Physiol. 2011;226:322–330. doi: 10.1002/jcp.22417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schenk T, Stengel S, Zelent A. Unlocking the potential of retinoic acid in anticancer therapy. Br J Cancer. 2014;111:2039–2045. doi: 10.1038/bjc.2014.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nagpal S, Cai J, Zheng T, Patel S, Masood R, Lin GY, Friant S, Johnson A, Smith DL, Chandraratna RA, et al. Retinoid antagonism of NF-IL6: insight into the mechanism of antiproliferative effects of retinoids in Kaposi’s sarcoma. Mol Cell Biol. 1997;17:4159–4168. doi: 10.1128/mcb.17.7.4159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Simon D, Körber C, Krausch M, Segering J, Groth P, Görges R, Grünwald F, Müller-Gärtner HW, Schmutzler C, Köhrle J, et al. Clinical impact of retinoids in redifferentiation therapy of advanced thyroid cancer: final results of a pilot study. Eur J Nucl Med Mol Imaging. 2002;29:775–782. doi: 10.1007/s00259-001-0737-6. [DOI] [PubMed] [Google Scholar]
- 63.Wu K, Kim HT, Rodriquez JL, Hilsenbeck SG, Mohsin SK, Xu XC, Lamph WW, Kuhn JG, Green JE, Brown PH. Suppression of mammary tumorigenesis in transgenic mice by the RXR-selective retinoid, LGD1069. Cancer Epidemiol Biomarkers Prev. 2002;11:467–474. [PubMed] [Google Scholar]
- 64.Atigadda VR, Xia G, Deshpande A, Wu L, Kedishvili N, Smith CD, Krontiras H, Bland KI, Grubbs CJ, Brouillette WJ, et al. Conformationally Defined Rexinoids and Their Efficacy in the Prevention of Mammary Cancers. J Med Chem. 2015;58:7763–7774. doi: 10.1021/acs.jmedchem.5b00829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Altucci L, Leibowitz MD, Ogilvie KM, de Lera AR, Gronemeyer H. RAR and RXR modulation in cancer and metabolic disease. Nat Rev Drug Discov. 2007;6:793–810. doi: 10.1038/nrd2397. [DOI] [PubMed] [Google Scholar]
- 66.Tachibana H. Molecular basis for cancer chemoprevention by green tea polyphenol EGCG. Forum Nutr. 2009;61:156–169. doi: 10.1159/000212748. [DOI] [PubMed] [Google Scholar]
- 67.Zanetti A, Affatato R, Centritto F, Fratelli M, Kurosaki M, Barzago MM, Bolis M, Terao M, Garattini E, Paroni G. All-trans-retinoic Acid Modulates the Plasticity and Inhibits the Motility of Breast Cancer Cells: ROLE OF NOTCH1 AND TRANSFORMING GROWTH FACTOR (TGFβ) J Biol Chem. 2015;290:17690–17709. doi: 10.1074/jbc.M115.638510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bhat-Nakshatri P, Goswami CP, Badve S, Sledge GW, Nakshatri H. Identification of FDA-approved drugs targeting breast cancer stem cells along with biomarkers of sensitivity. Sci Rep. 2013;3:2530. doi: 10.1038/srep02530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sun R, Liu Y, Li SY, Shen S, Du XJ, Xu CF, Cao ZT, Bao Y, Zhu YH, Li YP, et al. Co-delivery of all-trans-retinoic acid and doxorubicin for cancer therapy with synergistic inhibition of cancer stem cells. Biomaterials. 2015;37:405–414. doi: 10.1016/j.biomaterials.2014.10.018. [DOI] [PubMed] [Google Scholar]
- 70.Orlandi M, Mantovani B, Ammar K, Avitabile E, Dal Monte P, Bartolini G. Retinoids and cancer: antitumoral effects of ATRA, 9-cis RA and the new retinoid IIF on the HL-60 leukemic cell line. Med Princ Pract. 2003;12:164–169. doi: 10.1159/000070753. [DOI] [PubMed] [Google Scholar]
- 71.Bartolini G, Orlandi M, Papi A, Ammar K, Guerra F, Ferreri AM, Rocchi P. A search for multidrug resistance modulators: the effects of retinoids in human colon carcinoma cells. In Vivo. 2006;20:729–733. [PubMed] [Google Scholar]
- 72.Papi A, Rocchi P, Ferreri AM, Orlandi M. RXRgamma and PPARgamma ligands in combination to inhibit proliferation and invasiveness in colon cancer cells. Cancer Lett. 2010;297:65–74. doi: 10.1016/j.canlet.2010.04.026. [DOI] [PubMed] [Google Scholar]
- 73.Papi A, Tatenhorst L, Terwel D, Hermes M, Kummer MP, Orlandi M, Heneka MT. PPARgamma and RXRgamma ligands act synergistically as potent antineoplastic agents in vitro and in vivo glioma models. J Neurochem. 2009;109:1779–1790. doi: 10.1111/j.1471-4159.2009.06111.x. [DOI] [PubMed] [Google Scholar]
- 74.Storci G, Bertoni S, De Carolis S, Papi A, Nati M, Ceccarelli C, Pirazzini C, Garagnani P, Ferrarini A, Buson G, et al. Slug/β-catenin-dependent proinflammatory phenotype in hypoxic breast cancer stem cells. Am J Pathol. 2013;183:1688–1697. doi: 10.1016/j.ajpath.2013.07.020. [DOI] [PubMed] [Google Scholar]
- 75.Farabegoli F, Govoni M, Ciavarella C, Orlandi M, Papi A. A RXR ligand 6-OH-11-O-hydroxyphenanthrene with antitumour properties enhances (-)-epigallocatechin-3-gallate activity in three human breast carcinoma cell lines. Biomed Res Int. 2014;2014:853086. doi: 10.1155/2014/853086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Michalik L, Auwerx J, Berger JP, Chatterjee VK, Glass CK, Gonzalez FJ, Grimaldi PA, Kadowaki T, Lazar MA, O’Rahilly S, et al. International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol Rev. 2006;58:726–741. doi: 10.1124/pr.58.4.5. [DOI] [PubMed] [Google Scholar]
- 77.Ehrmann J, Vavrusová N, Collan Y, Kolár Z. Peroxisome proliferator-activated receptors (PPARs) in health and disease. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2002;146:11–14. doi: 10.5507/bp.2002.002. [DOI] [PubMed] [Google Scholar]
- 78.Janani C, Ranjitha Kumari BD. PPAR gamma gene--a review. Diabetes Metab Syndr. 2006;9:46–50. doi: 10.1016/j.dsx.2014.09.015. [DOI] [PubMed] [Google Scholar]
- 79.Mueller E, Sarraf P, Tontonoz P, Evans RM, Martin KJ, Zhang M, Fletcher C, Singer S, Spiegelman BM. Terminal differentiation of human breast cancer through PPAR gamma. Mol Cell. 1998;1:465–470. doi: 10.1016/s1097-2765(00)80047-7. [DOI] [PubMed] [Google Scholar]
- 80.Elstner E, Müller C, Koshizuka K, Williamson EA, Park D, Asou H, Shintaku P, Said JW, Heber D, Koeffler HP. Ligands for peroxisome proliferator-activated receptorgamma and retinoic acid receptor inhibit growth and induce apoptosis of human breast cancer cells in vitro and in BNX mice. Proc Natl Acad Sci USA. 1998;95:8806–8811. doi: 10.1073/pnas.95.15.8806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ramanan S, Kooshki M, Zhao W, Hsu FC, Robbins ME. PPARalpha ligands inhibit radiation-induced microglial inflammatory responses by negatively regulating NF-kappaB and AP-1 pathways. Free Radic Biol Med. 2008;45:1695–1704. doi: 10.1016/j.freeradbiomed.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kumar AP, Loo SY, Shin SW, Tan TZ, Eng CB, Singh R, Putti TC, Ong CW, Salto-Tellez M, Goh BC, et al. Manganese superoxide dismutase is a promising target for enhancing chemosensitivity of basal-like breast carcinoma. Antioxid Redox Signal. 2014;20:2326–2346. doi: 10.1089/ars.2013.5295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang Y, Zhang X, Wang J, Shen Y, Tang X, Yu F, Wang R. Expression and Function of PPARs in Cancer Stem Cells. Curr Stem Cell Res Ther. 2015:Jul 28; Epub ahead of print. doi: 10.2174/1574888x10666150728122921. [DOI] [PubMed] [Google Scholar]
- 84.Suchanek KM, May FJ, Robinson JA, Lee WJ, Holman NA, Monteith GR, Roberts-Thomson SJ. Peroxisome proliferator-activated receptor alpha in the human breast cancer cell lines MCF-7 and MDA-MB-231. Mol Carcinog. 2002;34:165–171. doi: 10.1002/mc.10061. [DOI] [PubMed] [Google Scholar]
- 85.Gonzalez FJ. The peroxisome proliferator-activated receptor alpha (PPARalpha): role in hepatocarcinogenesis. Mol Cell Endocrinol. 2002;193:71–79. doi: 10.1016/s0303-7207(02)00098-9. [DOI] [PubMed] [Google Scholar]
- 86.Papi A, Storci G, Guarnieri T, De Carolis S, Bertoni S, Avenia N, Sanguinetti A, Sidoni A, Santini D, Ceccarelli C, et al. Peroxisome proliferator activated receptor-α/hypoxia inducible factor-1α interplay sustains carbonic anhydrase IX and apoliprotein E expression in breast cancer stem cells. PLoS One. 2013;8:e54968. doi: 10.1371/journal.pone.0054968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Papi A, Guarnieri T, Storci G, Santini D, Ceccarelli C, Taffurelli M, De Carolis S, Avenia N, Sanguinetti A, Sidoni A, et al. Nuclear receptors agonists exert opposing effects on the inflammation dependent survival of breast cancer stem cells. Cell Death Differ. 2012;19:1208–1219. doi: 10.1038/cdd.2011.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang X, Sun Y, Wong J, Conklin DS. PPARγ maintains ERBB2-positive breast cancer stem cells. Oncogene. 2013;32:5512–5521. doi: 10.1038/onc.2013.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Deeb KK, Trump DL, Johnson CS. Vitamin D signalling pathways in cancer: potential for anticancer therapeutics. Nat Rev Cancer. 2007;7:684–700. doi: 10.1038/nrc2196. [DOI] [PubMed] [Google Scholar]
- 90.So JY, Suh N. Targeting cancer stem cells in solid tumors by vitamin D. J Steroid Biochem Mol Biol. 2015;148:79–85. doi: 10.1016/j.jsbmb.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pickholtz I, Saadyan S, Keshet GI, Wang VS, Cohen R, Bouwman P, Jonkers J, Byers SW, Papa MZ, Yarden RI. Cooperation between BRCA1 and vitamin D is critical for histone acetylation of the p21waf1 promoter and growth inhibition of breast cancer cells and cancer stem-like cells. Oncotarget. 2014;5:11827–11846. doi: 10.18632/oncotarget.2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wahler J, So JY, Cheng LC, Maehr H, Uskokovic M, Suh N. Vitamin D compounds reduce mammosphere formation and decrease expression of putative stem cell markers in breast cancer. J Steroid Biochem Mol Biol. 2015;148:148–155. doi: 10.1016/j.jsbmb.2014.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pervin S, Hewison M, Braga M, Tran L, Chun R, Karam A, Chaudhuri G, Norris K, Singh R. Down-regulation of vitamin D receptor in mammospheres: implications for vitamin D resistance in breast cancer and potential for combination therapy. PLoS One. 2013;8:e53287. doi: 10.1371/journal.pone.0053287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Conzen SD. Minireview: nuclear receptors and breast cancer. Mol Endocrinol. 2008;22:2215–2228. doi: 10.1210/me.2007-0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Karthik GM, Ma R, Lövrot J, Kis LL, Lindh C, Blomquist L, Fredriksson I, Bergh J, Hartman J. mTOR inhibitors counteract tamoxifen-induced activation of breast cancer stem cells. Cancer Lett. 2015;367:76–87. doi: 10.1016/j.canlet.2015.07.017. [DOI] [PubMed] [Google Scholar]
- 96.Harrison H, Simões BM, Rogerson L, Howell SJ, Landberg G, Clarke RB. Oestrogen increases the activity of oestrogen receptor negative breast cancer stem cells through paracrine EGFR and Notch signalling. Breast Cancer Res. 2013;15:R21. doi: 10.1186/bcr3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chen C, Baumann WT, Clarke R, Tyson JJ. Modeling the estrogen receptor to growth factor receptor signaling switch in human breast cancer cells. FEBS Lett. 2013;587:3327–3334. doi: 10.1016/j.febslet.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Deng H, Yin L, Zhang XT, Liu LJ, Wang ML, Wang ZY. ER-α variant ER-α36 mediates antiestrogen resistance in ER-positive breast cancer stem/progenitor cells. J Steroid Biochem Mol Biol. 2014;144 Pt B:417–426. doi: 10.1016/j.jsbmb.2014.08.017. [DOI] [PubMed] [Google Scholar]
- 99.Li L, Neaves WB. Normal stem cells and cancer stem cells: the niche matters. Cancer Res. 2006;66:4553–4557. doi: 10.1158/0008-5472.CAN-05-3986. [DOI] [PubMed] [Google Scholar]