

Abstract
Positron emission tomography measures the activity of radioactively labeled compounds which distribute and accumulate in central nervous system regions in proportion to their metabolic rate or blood flow. Specific circuits such as the dopaminergic nigrostriatal projection can be studied with ligands that bind to the pre-synaptic dopamine transporter or post-synaptic dopamine receptors (D1 and D2). Single photon emission computerized tomography (SPECT) measures the activity of similar tracers labeled with heavy radioactive species such as technetium and iodine. In essential tremor, there is cerebellar hypermetabolism and abnormal GABAergic function in premotor cortices, dentate nuclei and ventral thalami, without significant abnormalities in dopaminergic transmission. In Huntington’s disease, there is hypometabolism in the striatum, frontal and temporal cortices. Disease progression is accompanied by reduction in striatal D1 and D2 binding that correlates with trinucleotide repeat length, disease duration and severity. In dystonia, there is hypermetabolism in the basal ganglia, supplementary motor areas and cerebellum at rest. Thalamic and cerebellar hypermetabolism is seen during dystonic movements, which can be modulated by globus pallidus deep brain stimulation (DBS). Additionally, GABA-A receptor activity is reduced in motor, premotor and somatosensory cortices. In Tourette’s syndrome, there is hypermetabolism in premotor and sensorimotor cortices, as well as hypometabolism in the striatum, thalamus and limbic regions at rest. During tics, multiple areas related to cognitive, sensory and motor functions become hypermetabolic. Also, there is abnormal serotoninergic transmission in prefrontal cortices and bilateral thalami, as well as hyperactivity in the striatal dopaminergic system which can be modulated with thalamic DBS. In Parkinson’s disease (PD), there is asymmetric progressive decline in striatal dopaminergic tracer accumulation, which follows a caudal-to-rostral direction. Uptake declines prior to symptom presentation and progresses from contralateral to the most symptomatic side to bilateral, correlating with symptom severity. In progressive supranuclear palsy (PSP) and multiple system atrophy (MSA), striatal activity is symmetrically and diffusely decreased. The caudal-to-rostral pattern is lost in PSP, but could be present in MSA. In corticobasal degeneration (CBD), there is asymmetric, diffuse reduction of striatal activity, contralateral to the most symptomatic side. Additionally, there is hypometabolism in contralateral parieto-occipital and frontal cortices in PD; bilateral putamen and cerebellum in MSA; caudate, thalamus, midbrain, mesial frontal and prefrontal cortices in PSP; and contralateral cortices in CBD. Finally, cardiac sympathetic SPECT signal is decreased in PD. The capacity of molecular imaging to provide in vivo time courses of gene expression, protein synthesis, receptor and transporter binding, could facilitate the development and evaluation of novel medical, surgical and genetic therapies in movement disorders.
Keywords: Positron emission tomography, Single photon emission computerized tomography, Movement disorders, Essential tremor, Huntington’s disease, Dystonia, Tourette’s syndrome, Parkinson’s disease, Parkinsonism
Core tip: By evaluating changes in regional brain perfusion, glucose metabolism and neurotransmitter systems, molecular imaging has shed light onto the etiology, pathophysiology, diagnosis, progression and therapeutic options of movement disorders, including the identification and individualization of potential neuromodulation targets. Continuing progress in the design of positron emission tomography and single photon emission computerized tomography systems, such as new detector materials and image reconstruction algorithms, higher performance technology, and improved availability will contribute to a wider range of applications. In particular, the combined use of genetic therapy and molecular imaging could provide opportunities for the design and evaluation of novel therapies at early stages of the disease.
INTRODUCTION
Movement disorders manifest with excess or paucity of movements (i.e., hyper- or hypokinetic disorders). The most common hyperkinetic disorders are tremor, chorea, dystonia, tics and myoclonus. In the hypokinetic group, the parkinsonian syndromes are the most frequent. Many of these conditions are related to dysfunction of the basal ganglia, the cerebellum and/or their connections with primary or associative motor cortices. In these regions, neuronal alterations usually begin at the genetic, molecular and cellular levels, later progressing to neurotransmitter and network dysfunction.
Structural imaging with techniques such as magnetic resonance imaging (MRI) could demonstrate anatomical abnormalities in motor circuits. These defects are usually identified at advanced stages when continued neurodegeneration has led to volume loss (atrophy). MRI can also identify ischemia, neoplasm, infection, demyelination or abnormal substance deposition as the etiology of abnormal movements.
In contrast, molecular imaging techniques could provide neurochemical information useful to learn about the pathophysiology of the disorder, to diagnose the conditions at early stages, to monitor disease progression, as well as to plan and assess the response to medical and surgical interventions, including the identification of potential targets for neuromodulation. In this review, we describe the use of positron emission tomography (PET) and single-photon emission computerized tomography (SPECT) in movement disorders.
BASICS OF PET AND SPECT
PET evaluates neurobiological processes at the molecular level by using nanomolar concentrations of radioactively labeled compounds without producing significant disturbances in the biological system being assessed. The essential components of PET are: (1) the production of positron-emitting species in a cyclotron such as fluorine [18F] (half-life = 109.8 min), carbon [11C] (half-life = 20.3 min) and oxygen [15O] (half-life = 2 min); (2) the radiopharmaceutical procedure to attach the positron-emitting species to a physiological substrate such as glucose or amino acids; (3) the introduction of this tracer into the human body and its distribution in central nervous system (CNS) regions based on uptake and metabolic characteristics; (4) the detection and indirect measurement of its positron-emitting activity (high-energy gamma-rays) in a positron tomograph camera; (5) the construction of the corresponding images; and (6) the tracer-kinetic model for the interpretation of temporal changes in the regional distribution, accumulation and clearance of that positron-emitting activity[1-3].
2-[18F]-fluoro-2-deoxy-D-glucose (FDG) is the most widely used tracer in human PET studies. The same carrier transports both FDG and glucose across the blood-brain barrier (BBB) into the CNS. Both compounds then enter the glycolysis cycle and are phosphorylated by hexokinase. Unlike glucose-6-phosphate, FDG-6-phosphate is not a substrate for further metabolism and remains trapped in the tissue for at least 1 h. Accordingly, FDG-6-phosphate will accumulate in the tissue proportionally to its metabolic rate[1-3]. Additionally, PET can estimate regional cerebral blood flow (CBF) with the use of [15O]-labeled tracers such as [15O]H2O. Consequently, several “metabolic signatures” corresponding to different movement disorders have been obtained with PET.
Neurotransmitter systems can also be evaluated with PET. Particularly, the dopaminergic nigrostriatal circuit has been extensively studied using [18F]-labeled L-3,4-dihydroxiphenylalanine (FDOPA) and ligands that selectively bind to the pre-synaptic dopamine transporter (DAT) or the post-synaptic dopamine receptors (D1 and D2)[4-6]. With these PET techniques, neurochemical deficits have been identified that uniquely characterize a wide variety of movement disorders.
In SPECT, low-energy gamma-rays emitted by probes labeled with heavy radioactive species such as technetium [99mTc] (half-life = 6 h) and iodine [123I] (half-life = 13.3 h) are directly detected in a gamma camera, which has reduced sensitivity as compared with PET. However, the longer half-lives of SPECT tracers enable transportation from distance and even performance of several studies, in contrast to PET studies, which usually depend on an adjacent cyclotron. Thus, SPECT is usually more available than PET. For example, regional brain perfusion has been investigated extensively with the SPECT tracer [99mTc]hexamethyl propylene amine oxime (HMPAO).
MOLECULAR IMAGING OF TREMOR
Essential tremor (ET) is a common disorder characterized by bilateral, symmetric postural and kinetic tremor, more pronounced in the hands. It is thought to be related to abnormalities in the connections between the inferior olive, dentate nucleus, red nucleus, thalamus and motor cortices (“tremor-network”). However, there is controversy between neurodegeneration, dysfunction of GABAergic systems and/or abnormal tremor-network oscillations to explain the origin of ET. In fact, PET studies have demonstrated increased bilateral cerebellar metabolic activity during both tremor and at rest, as well as its suppression with ethanol consumption[7,8]. Also, PET has shown abnormal GABAergic function in premotor cortices, dentate nuclei and ventral thalami with the use of the GABA-A receptor antagonist [11C]flumazenil[9] (Table 1). Interestingly, a PET study has shown increased CBF in the supplementary motor area (SMA) ipsilateral to ventral intermediate thalamic deep brain stimulation (DBS), suggesting a stimulating rather than inactivating effect[10]. Further investigation with PET will likely continue increasing our understanding of ET.
Table 1.
Summary of molecular imaging findings in common movement disorders
| Movement disorder |
Metabolism and/or perfusion |
Nigrostriatal dopaminergic activity |
Other abnormal systems | ||
| Increased | Decreased | Pre-synaptic | Post-synaptic | ||
| Tremor: Essential tremor | Cerebellar, brainstem, thalamic, motor cortices (“tremor-network”) | - | Normal | Normal | GABAergic: Reduced in cerebellum, thalami and premotor |
| Chorea: Huntington’s disease | - | Striatum, frontal and temporal | Reduced | Reduced | - |
| Dystonia | Rest: Basal ganglia, cerebellar, sensorimotor Dystonia: Thalamic, cerebellar | Focal dystonia: Contralateral primary motor cortex | Focal and dopa-responsive dystonia: Normal DAT | Reduced D2 Dopa-responsive dystonia: Increased D2, normal D1 | GABA-A: Reduced in motor, premotor and somatosensory cortices |
| Tics: Tourette’s syndrome | Rest: Premotor, sensorimotor Tic: Multiple cognitive and sensorimotor regions | Rest: Striatum, thalamus, limbic | Hyper responsive, correlates with decreased serotonin | Hyper responsive, correlates with decreased serotonin | Serotoninergic: Prefrontal and thalamic GABA-A: Reduced in amygdala and ventral striatum |
| Parkinsonism: Parkinson’s disease | Basal ganglia, thalamus, contralateral to initial/worse symptoms, ipsilateral cerebellum | Parieto-occipital, frontal premotor, contralateral to initial/worse symptoms | Reduced in striatum contralateral to initial/worse symptoms, caudal-to-rostral (putamen-to-caudate) | Normal or increased putaminal D2 if untreated, could normalize with therapy | Cholinergic: Reduced early Noradrenergic, locus coeruleus: Increased early, reduced later Cardiac sympathetic: Reduced |
| Multiple system atrophy | - | Bilateral striatal (putamen) and cerebellum | Diffuse reduction in bilateral striatum, variable caudal-to-rostral | Reduced D2 | - |
| Progressive supranuclear palsy | - | Bilateral mesial frontal, prefrontal, striatal, thalamic, midbrain | Diffuse reduction in bilateral striatum, not caudal-to-rostral | Reduced D2 | - |
| Corticobasal degeneration | - | Cortices contralateral to symptoms | Diffuse striatal reduction contralateral to symptoms, not caudal-to-rostral | - | - |
In addition, molecular imaging including FDOPA PET and DAT SPECT studies have not shown abnormalities in dopamine striatal neurotransmission in ET[11,12]. Thus, evaluation of the dopaminergic system with PET or SPECT could aid in the distinction between ET and tremor-predominant parkinsonism when the clinical diagnosis is equivocal.
MOLECULAR IMAGING OF CHOREA
Huntington’s disease (HD) is a fatal neurodegenerative condition caused by an abnormally increased number of CAG repeats in the huntingtin gene located in chromosome 4p. Usually between the age of 40-50 years, a significant loss of medium spiny GABAergic neurons in the caudate, putamen and cerebral cortices results in progressive behavioral symptoms, chorea and cognitive dysfunction. The pathological processes underlying this degeneration likely precede symptom-onset by several years.
As the affected GABAergic striatal neurons contain the majority of dopamine receptors in the striatum, decreases in D1 and D2 receptors accompany the extent of cell loss[13]. Actually, initial PET imaging in HD focused on alterations of these receptors using the D1 receptor ligand [11C]SCH 23390 and the D2 receptor ligand [11C]raclopride. These studies have demonstrated that HD progression is accompanied by significant reductions in both D1 and D2 binding[14]. Striatal D2 binding decreases by approximately 5% per year, and this reduction correlates with trinucleotide repeat length, disease duration and severity[15-18]. In patients with HD, dopaminergic uptake is also reduced in pre-synaptic striatal and extrastriatal neurons[16,17].
FDG PET studies have shown hypometabolism in striatum, frontal and temporal cortices in HD patients and asymptomatic carriers preceding neuronal loss[19] (Table 1). In fact, HD carriers who became symptomatic had lower caudate metabolism compared to those who remained asymptomatic after 5 years[20]. PET imaging has also demonstrated increased levels of activated microglia in the striatum of HD carriers, which correlated with the probability of disease-onset and clinical severity[21].
MOLECULAR IMAGING OF DYSTONIA
Dystonia is characterized by sustained or intermittent involuntary contractions of agonist and antagonist muscles resulting in abnormal, repetitive movements and postures that are typically patterned and twisting. Dystonia is associated with abnormal inhibition, processing and plasticity in basal ganglia and sensorimotor networks.
FDG PET studies of large cohorts of patients comparing movement-related and movement-free images obtained during wake and sleep states respectively, have allowed for the development of “metabolic signatures” of dystonia. In DYT1 dystonia, a generalized dystonia caused by a GAG deletion in chromosome 9, hypermetabolism was found in the premotor cortices, basal ganglia, pons and midbrain[22]. The movement-free hypermetabolic network was identified in the basal ganglia, SMA and cerebellum, whereas movement-related hypermetabolism was found in the cerebellum and thalamus (Table 1). In addition, asymptomatic gene carriers were found to have the movement-free but not the movement-related abnormalities[23]. Afterwards, patients with focal dystonia (blepharospasm)[24] and DYT6 dystonia[25] were surprisingly found to have similar movement-free metabolic patterns, suggesting that this topography is not genotype-specific. In another study, DYT1 and DYT6 patients manifesting dystonia were found to have hypermetabolism in the bilateral SMAs and parietal association cortices. In contrast, DYT1 asymptomatic carriers were found to have hypermetabolism in the putamen, anterior cingulate and cerebellum, whereas DYT6 carriers had hypometabolism in the putamen and hypermetabolism in the temporal cortices[26]. Additionally, PET studies with [11C]raclopride have demonstrated that symptomatic and asymptomatic DYT1 and DYT6 individuals have decreased D2 striatal uptake[27,28]. Abnormalities of GABAergic networks have also been postulated in the pathophysiology of dystonia. Particularly, the inhibitory modulation of afferent signals arriving to the somatosensory regions is dysfunctional. In fact, reductions in GABA-A receptor activity in motor, premotor, primary and secondary somatosensory cortices has been revealed in an [11C]flumazenil PET study of patients with dystonia[29]. These results suggest that hereditary dystonia is a neurodevelopmental disorder of sensorimotor integration (“motor preparation”) networks, with resulting excessive output of existing postural control systems[30-32].
The effects of globus pallidus (GP) DBS in patients with dystonia have been assessed by measuring regional cerebral blood flow with [15O]H2O PET. These studies have shown hyperactivity in the thalamus, dorsolateral prefrontal cortex, medial and superior frontal gyri, which could be modulated with GP-DBS[33,34]. Remarkably, long-term GP-DBS may correct these abnormalities, such that continued stimulation may ultimately prove unnecessary[35]. Finally, FDG PET could also be used to monitor and even individualize treatment with other neuromodulation techniques in patients with dystonia[36,37].
DOPA-responsive dystonia (DRD) is a childhood-onset dystonia related to mutations in genes that encode enzymes important for the production of dopamine. DRD patients have been found to have increased [11C]raclopride D2 binding in the striatum as compared to PD patients and healthy controls[38]. In addition, increased striatal dopamine D2 availability with unchanged D1 and DAT binding have been found in patients with DRD[39] (Table 1). The results of these studies might reflect reduced competition and tracer displacement by lack of endogenous dopamine and/or a compensatory response to the dopamine deficiency.
In focal dystonias, post-synaptic striatal D2 binding reduction along with normal pre-synaptic DAT binding have been evidenced in PET and SPECT studies[40,41] (Table 1). Furthermore, patients with focal limb dystonia (“writer’s cramp”) were found to have different patterns of striatal dopamine release during motor tasks involving the dystonic limb as compared to asymptomatic tasks[42]. Botulinum toxin injection, currently considered the first-line treatment of focal dystonia, failed to improve the abnormal activation of the contralateral primary motor cortex in patients with “writer’s cramp” despite clinical improvement[43]. Yet, it may induce central plasticity through modulation of afferent muscle spindle inputs[44].
MOLECULAR IMAGING OF TICS
Tourette’s syndrome (TS) is a childhood-onset neuropsychiatric disorder defined by persistence of motor and phonic tics, as well as complex behavioral disturbances usually including obsessive-compulsive disorder (OCD).
Molecular imaging techniques have suggested abnormal function of cortico-striatal-thalamo-cortical circuits in TS. FDG PET has demonstrated hypermetabolism in premotor and sensorimotor cortices, as well as hypometabolism in the striatum, thalamus and limbic cortices including orbitofrontal and hippocampal regions in the resting state. During the tics, there is hypermetabolism in the anterior cingulate, inferior parietal, medial and lateral premotor cortices, primary motor cortices including Broca’s area, cerebellum, insula, thalamus and the striatum[45,46] (Table 1). Moreover, OCD symptoms correlated with a different pattern characterized by hypometabolism in anterior cingulate and dorsolateral prefrontal cortices, and hypermetabolism in primary motor cortices and precuneus[46]. These results support abnormal hyperactivity of the systems involved in processing sensory information and motor planning in TS.
Given the clinical improvement observed with the use of D2 antagonists, TS has been linked to dysregulation of dopaminergic transmission. In fact, striatal pre-synaptic dopamine release after amphetamine administration was studied in adult TS patients using [11C]raclopride PET. Finally, abnormal regulation of phasic dopamine responses resulted in hyper responsive dopaminergic system activation in TS, suggesting an imbalance between tonic and phasic dopaminergic responses[47].
Anomalies in serotoninergic transmission in the dorsolateral prefrontal cortices and bilateral thalami were evidenced in a study using alpha-[11C]methyl-L-tryptophan ([11C]AMT) PET to study tryptophan metabolism in children with TS[48]. Increased caudate serotonin synthesis has been shown in another study[49]. Furthermore, a strong correlation between phasic dopamine release and low levels of serotonin was found after evaluating patients with TS using [11C]raclopride (D2 ligand), [11C]WIN (DAT antagonist), [11C]McN (5-HT2a receptor antagonist) and [11C]MDL (SERT antagonist) as tracers[50]. The modulating effects of thalamic DBS in the hyperactive basal ganglia dopaminergic system of patients with TS have been evidenced by [18F]Fallypride PET[51,52]. In addition, the GABAergic system of patients with TS has been studied with [11C]flumazenil PET. A consistent decrease of GABA-A receptor activity was found in the amygdala, ventral striatum, insula and thalamus; with increased activity in the cerebellum, substantia nigra and periaqueductal gray[53].
MOLECULAR IMAGING OF PARKINSONISM
Most CNS dopaminergic neurons are located in the hypothalamus, substantia nigra and ventral tegmental area. They project to the hypophysis (tubero-hypophyseal pathway), striatum (nigrostriatal pathway), frontal, limbic and olfactory regions (mesocortical and mesolimbic pathways). The main dopaminergic system involved in motor control arises from the substantia nigra, a midbrain structure divided into a dorsolateral pars compacta (SNc) that projects mostly to the putamen, and a ventromedial pars reticulata (SNr) that projects mainly to the thalamus.
Parkinsonism is a syndrome characterized by bradykinesia plus rigidity, resting tremor or postural instability. It encompasses several conditions with overlapping clinical features including secondary parkinsonism, alpha-synucleinopathies such as Parkinson’s disease (PD) and multiple systems atrophy (MSA), as well as tauopathies such as progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD). The most common parkinsonian syndrome is PD, a progressive neurodegenerative condition in which there is abnormal accumulation of alpha-synuclein in SNc neurons and subsequent dopamine deficiency in the nigrostriatal system. In addition to parkinsonism, PD has important non-motor manifestations including cognitive, behavioral and autonomic disturbances.
Unlike many other neurotransmitters, dopamine has biological attributes that make it amenable to imaging with PET. Given that it cannot cross the BBB, dopaminergic neurons normally synthesize dopamine by decarboxylation of its immediate precursor, LDOPA, which can cross the BBB but is typically synthesized in situ by hydroxylation of tyrosine.
The first approach to imaging of the nigrostriatal dopaminergic system integrity was based on the assessment of pre-synaptic striatal dopamine synthesis capacity. Given the short half-life (20 min) and rapid procedures needed for [11C]DOPA synthesis, LDOPA was labeled with [18F][54]. After several studies, [18F]6-fluoro-L-DOPA (FDOPA) became the preferred tracer for measurements of LDOPA distribution and uptake in the CNS[55,56].
FDOPA is transported across the BBB into the brain by the large neutral amino acid transporter, with similar kinetics to LDOPA[57]. Yet, the rate of FDOPA metabolism by peripheral catechol-O-methyl transferase (COMT) is about one fourth of that of LDOPA[58], which is favorable for brain PET studies, but should be considered during studies aiming to measure or monitor treatment with LDOPA. Once localized in the pre-synaptic terminals of striatal dopaminergic neurons, FDOPA is converted to [18F]fluorodopamine (FDA) by the cerebral aromatic amino acid decarboxylase (AAAD) and subsequently stored in synaptic vesicles. Striatal FDOPA activity is increased if preceded by the administration of a peripheral decarboxylase inhibitor and is decreased after treatment with reserpine, a drug that releases catecholamines from pre-synaptic vesicles. This was the first time the regional distribution, localization and pharmacological manipulation of a neurotransmitter were imaged[59]. Later, the first human FDOPA PET study demonstrated a similar pattern of dopaminergic localization[60]. Although FDOPA can be taken up by noradrenergic and serotoninergic neurons, the striatal uptake of FDOPA has been used as a correlate of nigrostriatal dopaminergic pre-synaptic integrity (Figure 1).
Figure 1.
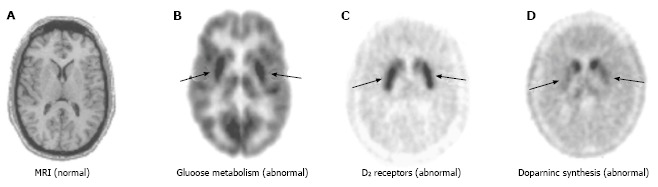
Imaging of early Parkinson’s disease. A: MRI of the striatum does not indicate any significant anatomical abnormalities; B: PET reveals selective regional alterations in diverse neurochemical processes: FDG PET shows putaminal hypermetabolism relative to the caudate (approximately 10%); C: [18F]fluoroethylspiperone PET shows approximately 15% greater D2 receptor density in the putamen as compared to the caudate; D: FDOPA PET shows significant reduction in dopamine synthetic capacity (80%) in the putamen but not in the caudate. Arrows in B, C, and D indicate putamen. MRI: Magnetic resonance imaging; PET: Positron emission tomography; FDG: 2-[18F]-fluoro-2-deoxy-D-glucose; FDOPA: [18F]-labeled L-3,4-dihydroxiphenylalanine. (Originally published in the JNM. Phelps ME. PET: the merging of biology and imaging into molecular imaging. J Nucl Med 2000; 41: 661-681. © by the Society of Nuclear Medicine and Molecular Imaging, Inc.)
Another approach is to evaluate dopamine terminal density with tracers that bind to the DAT, a pre-synaptic membrane protein responsible for the reuptake of released dopamine. The cocaine congener, N-[3-[18F]fluoropropyl]-2β-carbomethoxy-3β-(4-iodophenyl)nortropane [18F]FPCIT, which has high binding affinity for the DAT, is the most common PET tracer used for this purpose (Figure 2). Several other tracers have been synthesized for SPECT, such as [99mTc]TRODAT-1, beta-[123I]CIT and [123I]FPCIT (DaTScan™) (Figure 3). Although kinetic properties and DAT selectivity vary between tracers, they all provide similar information about pre-synaptic dopaminergic function. Of note, DATs can undergo compensatory or pharmacological regulation, thus the use of DAT ligands is controversial particularly when used to monitor disease progression or the effect of medical therapy. The pre-synaptic vesicular monoamine transporter type 2 (VMAT-2) is less subject to these regulatory changes but is expressed in all monoaminergic neurons. Since most striatal VMAT-2 activity occurs in dopamine terminals, PET studies labeling VMAT-2 with [11C]dihydrotetrabenazine (DTBZ) or [18F]fluoropropyl-DTBZ might also help evaluate dopaminergic nerve terminal function[61].
Figure 2.
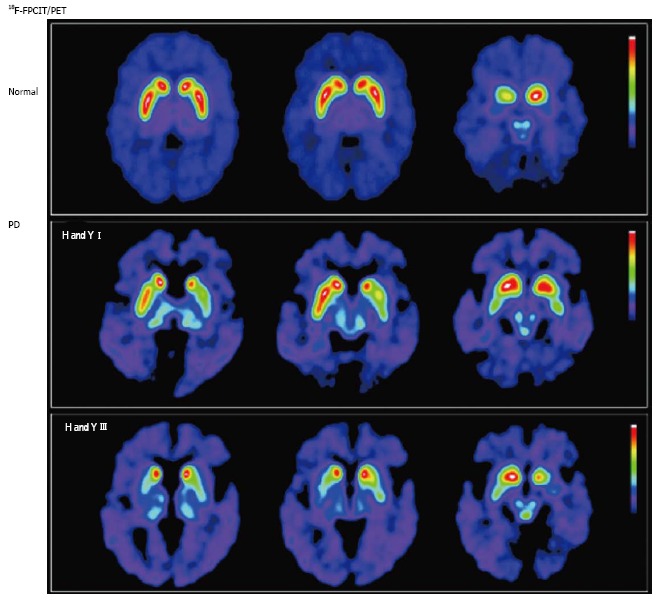
Nigrostriatal dopaminergic system degeneration in Parkinson’s disease. PET images show striatal uptake of [18F]FPCIT, a ligand with high affinity for pre-synaptic dopamine transporters, for a normal volunteer (top row), and 2 patients with Parkinson’s disease (PD) [middle row: Hoehn and Yahr (H and Y) stage I; bottom row: H and Y stage III]. Striatal [18F]FPCIT uptake is mainly reduced in the putamen contralateral to the most symptomatic side in early PD (middle row), progressing to bilateral reduction in a caudal-to-rostral pattern (bottom row). (Originally published in the JNM. Katzumata K, Dhawan V, Chaly T, et al. Dopamine transporter imaging with fluorine-18-FPCIT and PET. J Nucl Med 1998; 39: 1521-1530. © by the Society of Nuclear Medicine and Molecular Imaging, Inc.)
Figure 3.
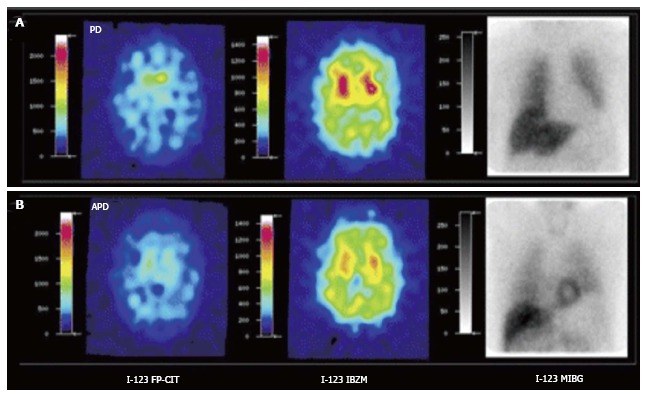
[123I]FPCIT, [123I]IBZM and cardiac SPECT imaging [[123I]MIBG] of Parkinson’s disease (A) and atypical parkinsonism (B). Striatal [123I]FPCIT is decreased in both PD and APD. Striatal D2 receptor binding is normal in PD but decreased in APD. Myocardial sympathetic SPECT signal is decreased in PD but normal in APD. PD: Parkinson’s disease; APD: Atypical parkinsonism; SPECT: Single photon emission computerized tomography. (Originally published in the JNM. Südmeyer M, Antke C, Zizek T, et al. J Nucl Med 2011; 52: 733-740. © by the Society of Nuclear Medicine and Molecular Imaging, Inc.)
Post-synaptic striatal dopaminergic function can be assessed with tracers that bind to D1 and D2. For example, [18F]Fluoroethylspiperone is a PET tracer (Figure 1) and [123I]IBZM is a SPECT tracer (Figure 3), both of which can bind to D2 receptors. D1 and D2 receptors have different functions based on their mechanisms of adenylyl cyclase stimulation and inhibition, respectively[62]. Since receptor binding is subject to competition with endogenous dopamine, molecular tracers with low D2 affinity can be used to estimate the amount of dopamine (i.e., increased synaptic dopamine leads to decreased D2 activity). Furthermore, changes after therapeutic interventions can be assessed with these low receptor affinity ligands. Lastly, high affinity probes can help evaluate extrastriatal D2 binding[63].
The capability to detect localized subregional biochemical deficits in the nigrostriatal system has provided insights into the etiology, pathophysiology, differential diagnosis and therapy for patients with parkinsonian syndromes, particularly PD.
Etiology and diagnosis
The cause of PD is still unclear. The combination of genetic susceptibility with environmental factors probably plays a significant role. A longitudinal FDOPA PET study of PD monozygotic twins showed that many unaffected twins had decreased tracer uptake, concordant with their affected siblings. When followed for several years, 75% of the twin pairs were clinically or radiologically concordant[64]. This study helped stimulate debate on the genetics of PD. A digenic genotype with complete penetrance was even theorized to explain the pattern of PD inheritance rather than multiple genes or a single gene with incomplete penetrance[65].
PD is characterized by asymmetrical progressive decline in tracer accumulation in the striatum contralateral to the most symptomatic side of the body (Figures 1-3). The reduction of striatal dopaminergic pre-synaptic activity follows a caudal-to-rostral direction, being initially more severe in the posterior striatum (Figure 2). This pattern corresponds with the lateral-to-medial pattern of neurodegeneration in the substantia nigra, with earlier and more severe involvement of the ventrolateral aspect of the SNc[66]. Regional striatal alterations showing larger increases in D2 receptor binding in the putamen than in the caudate can also been detected (Figure 1). As expected, neuropsychologically normal patients with PD can have reduced FDOPA PET activity in the putamen but not in the caudate contralateral to their major motor signs. This reduction can be similar between tremor-predominant and akinetic-rigid patients. To explain these findings, it has been postulated that the putamen is mainly involved in motor control, whereas the caudate participates mostly in cognitive functions[67]. However, most patients in these initial studies had early PD, perhaps before involvement of the caudate could be appreciated. In addition, FDOPA activity can be increased relative to the degree of denervation in early disease, possibly due to compensatory upregulation of AAAD. Moreover, FDG PET studies have shown hypermetabolism in the same areas with decreased FDOPA uptake[68] (Figure 4). Although characteristic, this appearance is not completely specific for PD and might be noted in other parkinsonian syndromes, particularly MSA (Figure 3).
Figure 4.
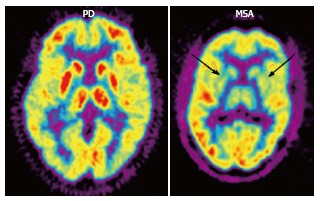
2-[18F]-fluoro-2-deoxy-D-glucose positron emission tomography (FDG PET) in Parkinson’s disease (PD) and multiple system atrophy (MSA). FDG PET shows hypometabolism in bilateral parieto-occipital and prefrontal cortices, as well as hypermetabolism in the basal ganglia and thalamus in PD, as opposed to bilateral striatal and thalamic hypometabolism in MSA (arrows). (Originally published in the JNM. Brooks DJ. J Nucl Med 2010; 51: 596-609. © by the Society of Nuclear Medicine and Molecular Imaging, Inc.)
Differential diagnosis
PSP is an atypical parkinsonism associated with reduction of vertical gaze and axial rather than appendicular rigidity manifesting with premature falls. MSA is defined by autonomic failure and either poor LDOPA-responsive parkinsonism (MSA-P, striatonigral degeneration) or cerebellar dysfunction (MSA-C, olivopontocerebellar atrophy). CBD presents with asymmetric onset of limb bradykinesia, rigidity, dystonia or myoclonus; plus apraxia, cortical sensory deficits or alien limb phenomenon.
The direction of pathologic changes in the substantia nigra of patients with PSP is from medial-to-lateral, corresponding with their early truncal rigidity, but opposite to the direction seen in PD[66,69]. In PSP and MSA, pre-synaptic striatal activity is usually symmetrically and diffusely decreased. The caudal-to-rostral pattern seen in PD is lost in PSP, but could be variably present in MSA (Figure 3). In CBD, there is asymmetric reduction of FDOPA accumulation in the striatum contralateral to the most symptomatic side. Additionally, increased post-synaptic putamen D2 binding that can be normalized with therapy is seen in PD. D2 binding is reduced in PSP and MSA[61] (Table 1, Figure 3).
Although not specific, FDG PET or HMPAO SPECT patterns might also help differentiate between the parkinsonian syndromes. In fact, characteristic hypometabolism has been demonstrated in contralateral parieto-occipital and frontal cortices in PD; bilateral putamen and cerebellum in MSA (Figure 4); caudate, thalami, midbrain, mesial frontal and prefrontal cortices in PSP; and contralateral cortical regions in CBD. In addition, PD patients have hypermetabolism in the basal ganglia, thalami, pons and cerebellum[70] (Table 1, Figure 4). Furthermore, SPECT studies have shown decreased cardiac sympathetic terminals signal indicating denervation in PD as opposed to the atypical parkinsonisms[71] (Figure 3).
Remarkably, up to 20% of patients with a clinical diagnosis of PD can have normal molecular imaging of the nigrostriatal dopaminergic system. This phenomenon has been labeled “symptoms without evidence for dopamine deficiency” (SWEDD)[72]. Most cases of SWEDD are due to clinical misdiagnosis. Yet, a small proportion of SWEDD patients may actually have PD based on clinical progression, LDOPA responsiveness and/or repeated imaging[73]. Although still controversial, these findings imply that normal functional imaging cannot completely exclude early PD[74]. Clearly, additional studies comparing PET and/or SPECT with neuropathology for the diagnosis of PD are needed.
Pathophysiology and disease progression
Molecular imaging studies evaluate dopaminergic activity rather than the number of neurons. Moreover, disability in PD is related to deficiencies in multiple other neurotransmitter systems. Nonetheless, imaging of the nigrostriatal dopaminergic system is one of the best markers to assess PD progression.
Neuroinflammation might precede neuronal death in neurodegenerative disorders. PET studies targeting specific molecules expressed by activated microglia have provided controversial results in PD[75,76]. More consistent findings have been reported in MSA[77], PSP[78], CBD[79] and HD[80]. The challenging imaging of the usual small amounts of intracellular-predominant alpha-synuclein deposits could lead to exciting advances in the future[81].
In PD, the loss of nigrostriatal neurons eventually exceeds an asymptomatic threshold. In fact, striatal uptake prior to symptom presentation has been measured to decline at an annual rate of 9%-12%[82]. Interestingly, molecular studies have demonstrated subclinical dopamine deficits in people exposed to nigral toxins, individuals with family history of dominant or recessive PD, twins of PD patients[64], and patients with non-motor symptoms of PD such as hyposmia, autonomic dysfunction and/or REM sleep behavior disorder (RBD)[83]. In fact, people with these non-motor symptoms and dopaminergic deficits on functional imaging have a higher risk of developing PD[83]. Early identification and long-term clinico-radiological follow-up of these patients could be of great value to study the natural history of PD and to evaluate if, when and how strongly molecular imaging can predict the conversion from prodromal to clinically-evident PD.
FDOPA uptake is increased in the noradrenergic locus coeruleus in early PD but decreases to abnormal levels with disease progression, suggesting early compensatory upregulation. PET studies have also shown early cholinergic dysfunction in PD patients with impaired olfactory function or RBD with or without cognitive dysfunction[84].
[18F]FPCIT PET studies have demonstrated how the pathology in PD progresses from unilateral to bilateral striatal dopamine deficits, and that its magnitude correlates with the severity of symptoms[85] (Figure 2). Interestingly, decreased striatal dopaminergic uptake correlates with metabolic hyperactivity in the STN and GP[86].
Assessment of disease progression requires objective markers to overcome the confounding effects of symptomatic benefit. The evaluation of PD progression with molecular imaging should be carefully interpreted, since tracer metabolism can be altered by compensatory regulation and/or therapeutic interventions.
Treatment
Deprenyl (Selegiline), an irreversible monoamine oxidase type B (MAO-B) inhibitor employed in the treatment of PD, was used to determine that CNS MAO-B distribution is approximately 60% higher in the thalamus and basal ganglia relative to cortical regions and cerebellum. In further studies, 5 mg of deprenyl were administered daily for 7 d in order to obtain maximal binding to MAO-B. Then, the subjects were evaluated with [11C]deprenyl PET. Relative to pre-treatment values, [11C]deprenyl binding after deprenyl administration was reduced by 90%, indicating near complete inactivation of the enzyme. Multiple PET scans during the next two months revealed that MAO-B binding capacity could be restored only by synthesis of new enzyme. It was calculated that the half-time for MAO-B synthesis was approximately 40 d. These results provided the first in vivo measurement of the synthesis rate of a specific protein in the brain. Furthermore, it was demonstrated that the recommended daily dose of deprenyl (5 mg twice daily) exceeded that necessary for therapeutic efficacy, illustrating how PET can be used to determine drug dosage for optimal efficacy in movement disorders[87].
FDOPA PET has been used in several studies to evaluate the effects of potential neuroprotective agents on dopaminergic function[72]. For example, the relative rates of progression of early PD in patients started on ropinirole or LDOPA were compared in a double-blind, randomized study in which FDOPA PET studies were obtained at baseline and 2 years later. The mean reduction in putamen FDOPA uptake was not significantly different for both groups[88].
Molecular studies have shown that LDOPA-induced dopamine release in the striatum increases with the severity and duration of PD, and is of larger magnitude in patients with LDOPA-induced dyskinesias[89,90]. Moreover, greater magnitude but shorter duration of LDOPA-induced dopamine release in responsive patients could predict the future development of motor fluctuations[91]. In addition, changes in D2 receptor density of PD patients have been measured with PET after exposure to dopaminergic pharmacotherapy and/or the compensatory reactions to the ongoing disease process, showing normal or increased D2 binding potential in untreated PD patients, and reduced potential in PD patients with fluctuating responses to LDOPA[92].
Impulse control disorders have a higher incidence in patients with PD treated with dopamine agonists. [11C]raclopride PET was used to compare D2 receptor availability during a gambling task in PD patients with and without pathological gambling. It was found that those with pathological gambling had lower D2 binding at baseline but increased release of dopamine in the ventral striatum during the gambling task, as is seen in those with chemical addictions in response to the drug to which they are addicted[93]. Later, increased release of dopamine in the ventral striatum during reward cues was also demonstrated, implying that PD patients who develop impulse control disorders could represent a susceptible group of individuals unmasked by dopaminergic agonists[94]. Pharmacokinetic and pharmacodynamic studies with PET will likely continue increasing our understanding of the mechanisms of action, efficacy and complications of medical interventions in patients with PD.
Molecular imaging has also been used to evaluate efficacy of different surgical interventions. For example, in PD patients who received a striatal fetal nigral tissue transplant, a significant increase in FDOPA PET activity was observed. Unfortunately, clinical improvement was dismal, suggesting implant survival without enough or adequate post-synaptic connections[95]. Later, PET imaging has provided evidence that PD patients benefit from surgical interventions such as STN or GP DBS, which reduce the abnormally increased inhibitory output from the basal ganglia to the ventral thalamus[96]. In fact, FDG PET studies have demonstrated that STN DBS suppresses hypermetabolism in the rostral cerebellum, and partly reverses reductions of cortical glucose consumption in limbic and associative regions where basal ganglia connections project, including frontal cortices[97,98].
FUTURE DIRECTIONS
The mission of first defining the impaired molecular mechanisms leading to abnormal motor network dysfunction and movements disorders, and then designing therapies to restore their normal function requires a tremendous effort and integration of different specialties. A fundamental technology in this quest is molecular imaging with PET and SPECT.
In addition to established neuromodulation techniques such as DBS, the use of genetic therapy to alter neurotransmitter activity might be incorporated into therapeutic options in the coming years. These therapies can be quantitatively assessed with specific tracers as mentioned in this review. In fact, the gene for AAAD was inserted into an adeno-associated virus that had been stripped of its replicative functions but was still capable of infecting cells. It was hypothesized that upon injection of the virus into the striatum, the subsequent expression of AAAD in the infected striatal cells would provide a local source of AAAD that could selectively convert LDOPA to dopamine. For the experiment, the subjects first received an intracarotid injection of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a neurotoxin that specifically targets dopaminergic nigral neurons. Afterwards, 6-[18F]fluoro-L-metatyrosine (FMT), an analog of LDOPA, was used as a PET tracer to show unilateral reduction in AAAD activity, indicating that MPTP had produced a unilateral striatal dopamine deficit. Subsequently, the AAAD gene-viral vector was stereotactically injected into the lesioned striatum. Two months later, a robust FMT PET uptake was detected in the striatum that had been injected with the virus containing AAAD[99].
Based on prior studies showing neuroprotective effects of the glial cell-line derived growth factor (GDNF) against MPTP, a solution containing GDNF was stereotactically and unilaterally delivered into the caudate and putamen of vervet monkeys one week prior to systemic administration of methamphetamine. The potential neuroprotective effects of GDNF were then evaluated by PET imaging of the striatum in multiple studies with the tracer [11C]WIN 35428, a selective DAT ligand. The results showed that GDNF pretreatment provided partial neuroprotection from methamphetamine-induced loss of DATs. These results provided evidence that exogenously administered GDNF could provide neuroprotection in the adult non-human primate brain and that a corresponding pharmacotherapy approach could be monitored with PET[100] (Figure 5).
Figure 5.
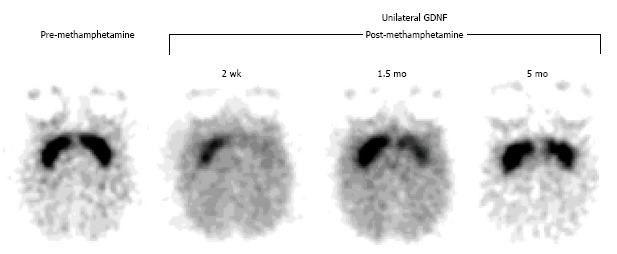
Positron emission tomography assessment of a neuroprotective strategy for methamphetamine-induced neurotoxicity with [11C]WIN 35,428, a selective dopamine transporter ligand. Prior to drug treatment, symmetrical and selective striatal uptake was demonstrated (left, pre-methamphetamine). Glial cell-line derived growth factor (GDNF) was then unilaterally injected into the striatum (left-sided striatum on the images shown). One week later, a neurotoxic methamphetamine (METH) dosage was systemically administered. Subsequent studies showed that the GDNF-pretreated striatum was partially protected from the METH-induced neurotoxicity and that it recovered at a faster rate relative to the contralateral striatum (right, post-methamphetamine). (Originally published in Synapse. Melega WP, Lacan G, Desalles AA, Phelps ME. Long-term methamphetamine-induced decreases of [(11)C]WIN 35,428 binding in striatum are reduced by GDNF: PET studies in the vervet monkey. Synapse 2000; 35: 243-249. © by John Wiley & Sons publications, Inc.)
The capacity of molecular imaging, in particular PET, to provide an in vivo time course of gene expression, protein synthesis, receptor and transporter binding, could enable researchers and clinicians to develop novel therapeutic approaches in humans with movement disorders. These methods should first be evaluated in non-human primate models to validate and optimize drug dosages, targets for injection or modulation, evaluation of treatment safety and efficacy, and more.
CONCLUSION
Molecular imaging has evaluated changes in regional brain perfusion, glucose metabolism and neurotransmitter systems in movement disorders, shedding light into their etiology, pathophysiology, diagnosis, disease progression and therapeutic options, including the identification and individualization of potential neuromodulation targets.
The recognition of asymptomatic and progressive disease by molecular imaging in contrast to the unremarkable data from structural imaging illustrates that significant neurochemical alterations precede and are not necessarily paralleled by morphological changes. In this context, PET and SPECT have provided disease-specific biomarkers that could help determine which individuals are more likely to benefit from different therapeutic modalities.
Continuing progress in the design of PET and SPECT systems, as reflected by new detector materials and image reconstruction algorithms, higher performance computer technology and improved availability of radiopharmaceuticals will all contribute to a wider range of applications in movement disorders. In particular, the combined use of genetic therapy and molecular imaging could provide opportunities for the design and evaluation of novel therapies at early stages of the disease.
Footnotes
Conflict-of-interest statement: The authors declare no potential conflict of interest for this review.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: October 3, 2015
First decision: November 4, 2015
Article in press: January 7, 2016
P- Reviewer: Lin WY, Storto G S- Editor: Qiu S L- Editor: A E- Editor: Lu YJ
References
- 1.Sokoloff L. Cerebral circulation, energy metabolism, and protein synthesis: general characteristics and principles of measurement. In: Phelps M, Mazziotta JC, Heinrich R, Schelbert HR, editors. Positron emission tomography and autoradiography: principles and applications for the brain and heart. New York: Raven Press; 1986. pp. 1–71. [Google Scholar]
- 2.Sokoloff L, Reivich M, Kennedy C, Des Rosiers MH, Patlak CS, Pettigrew KD, Sakurada O, Shinohara M. The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. J Neurochem. 1977;28:897–916. doi: 10.1111/j.1471-4159.1977.tb10649.x. [DOI] [PubMed] [Google Scholar]
- 3.Melega WP, De Salles AAF. Molecular Imaging of the Brain with Positron Emission Tomography. In: Youmans JR, Winn HR, editors. Youmans Neurological Surgery. 6th ed. USA: El Sevier; 2011. pp. 376–392. [Google Scholar]
- 4.Volkow ND, Fowler JS, Gatley SJ, Logan J, Wang GJ, Ding YS, Dewey S. PET evaluation of the dopamine system of the human brain. J Nucl Med. 1996;37:1242–1256. [PubMed] [Google Scholar]
- 5.Blomqvist G, Pauli S, Farde L, Eriksson L, Persson A, Halldin C. Maps of receptor binding parameters in the human brain--a kinetic analysis of PET measurements. Eur J Nucl Med. 1990;16:257–265. doi: 10.1007/BF00842777. [DOI] [PubMed] [Google Scholar]
- 6.Mazière B, Mazière M. Where have we got to with neuroreceptor mapping of the human brain? Eur J Nucl Med. 1990;16:817–835. doi: 10.1007/BF00833018. [DOI] [PubMed] [Google Scholar]
- 7.Wills AJ, Jenkins IH, Thompson PD, Findley LJ, Brooks DJ. Red nuclear and cerebellar but no olivary activation associated with essential tremor: a positron emission tomographic study. Ann Neurol. 1994;36:636–642. doi: 10.1002/ana.410360413. [DOI] [PubMed] [Google Scholar]
- 8.Boecker H, Wills AJ, Ceballos-Baumann A, Samuel M, Thompson PD, Findley LJ, Brooks DJ. The effect of ethanol on alcohol-responsive essential tremor: a positron emission tomography study. Ann Neurol. 1996;39:650–658. doi: 10.1002/ana.410390515. [DOI] [PubMed] [Google Scholar]
- 9.Boecker H, Weindl A, Brooks DJ, Ceballos-Baumann AO, Liedtke C, Miederer M, Sprenger T, Wagner KJ, Miederer I. GABAergic dysfunction in essential tremor: an 11C-flumazenil PET study. J Nucl Med. 2010;51:1030–1035. doi: 10.2967/jnumed.109.074120. [DOI] [PubMed] [Google Scholar]
- 10.Perlmutter JS, Mink JW, Bastian AJ, Zackowski K, Hershey T, Miyawaki E, Koller W, Videen TO. Blood flow responses to deep brain stimulation of thalamus. Neurology. 2002;58:1388–1394. doi: 10.1212/wnl.58.9.1388. [DOI] [PubMed] [Google Scholar]
- 11.Antonini A, Moresco RM, Gobbo C, De Notaris R, Panzacchi A, Barone P, Calzetti S, Negrotti A, Pezzoli G, Fazio F. The status of dopamine nerve terminals in Parkinson’s disease and essential tremor: a PET study with the tracer [11-C]FE-CIT. Neurol Sci. 2001;22:47–48. doi: 10.1007/s100720170040. [DOI] [PubMed] [Google Scholar]
- 12.Isaias IU, Marotta G, Hirano S, Canesi M, Benti R, Righini A, Tang C, Cilia R, Pezzoli G, Eidelberg D, et al. Imaging essential tremor. Mov Disord. 2010;25:679–686. doi: 10.1002/mds.22870. [DOI] [PubMed] [Google Scholar]
- 13.Cross A, Rossor M. Dopamine D-1 and D-2 receptors in Huntington’s disease. Eur J Pharmacol. 1983;88:223–229. doi: 10.1016/0014-2999(83)90009-2. [DOI] [PubMed] [Google Scholar]
- 14.Andrews TC, Weeks RA, Turjanski N, Gunn RN, Watkins LH, Sahakian B, Hodges JR, Rosser AE, Wood NW, Brooks DJ. Huntington’s disease progression. PET and clinical observations. Brain. 1999;122(Pt 12):2353–2363. doi: 10.1093/brain/122.12.2353. [DOI] [PubMed] [Google Scholar]
- 15.Antonini A, Leenders KL, Eidelberg D. [11C]raclopride-PET studies of the Huntington’s disease rate of progression: relevance of the trinucleotide repeat length. Ann Neurol. 1998;43:253–255. doi: 10.1002/ana.410430216. [DOI] [PubMed] [Google Scholar]
- 16.Pavese N, Andrews TC, Brooks DJ, Ho AK, Rosser AE, Barker RA, Robbins TW, Sahakian BJ, Dunnett SB, Piccini P. Progressive striatal and cortical dopamine receptor dysfunction in Huntington’s disease: a PET study. Brain. 2003;126:1127–1135. doi: 10.1093/brain/awg119. [DOI] [PubMed] [Google Scholar]
- 17.Ginovart N, Lundin A, Farde L, Halldin C, Bäckman L, Swahn CG, Pauli S, Sedvall G. PET study of the pre- and post-synaptic dopaminergic markers for the neurodegenerative process in Huntington’s disease. Brain. 1997;120(Pt 3):503–514. doi: 10.1093/brain/120.3.503. [DOI] [PubMed] [Google Scholar]
- 18.Antonini A, Leenders KL, Spiegel R, Meier D, Vontobel P, Weigell-Weber M, Sanchez-Pernaute R, de Yébenez JG, Boesiger P, Weindl A, et al. Striatal glucose metabolism and dopamine D2 receptor binding in asymptomatic gene carriers and patients with Huntington’s disease. Brain. 1996;119(Pt 6):2085–2095. doi: 10.1093/brain/119.6.2085. [DOI] [PubMed] [Google Scholar]
- 19.Ciarmiello A, Cannella M, Lastoria S, Simonelli M, Frati L, Rubinsztein DC, Squitieri F. Brain white-matter volume loss and glucose hypometabolism precede the clinical symptoms of Huntington’s disease. J Nucl Med. 2006;47:215–222. [PubMed] [Google Scholar]
- 20.Ciarmiello A, Giovacchini G, Orobello S, Bruselli L, Elifani F, Squitieri F. 18F-FDG PET uptake in the pre-Huntington disease caudate affects the time-to-onset independently of CAG expansion size. Eur J Nucl Med Mol Imaging. 2012;39:1030–1036. doi: 10.1007/s00259-012-2114-z. [DOI] [PubMed] [Google Scholar]
- 21.Politis M, Pavese N, Tai YF, Kiferle L, Mason SL, Brooks DJ, Tabrizi SJ, Barker RA, Piccini P. Microglial activation in regions related to cognitive function predicts disease onset in Huntington’s disease: a multimodal imaging study. Hum Brain Mapp. 2011;32:258–270. doi: 10.1002/hbm.21008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eidelberg D, Moeller JR, Ishikawa T, Dhawan V, Spetsieris P, Przedborski S, Fahn S. The metabolic topography of idiopathic torsion dystonia. Brain. 1995;118(Pt 6):1473–1484. doi: 10.1093/brain/118.6.1473. [DOI] [PubMed] [Google Scholar]
- 23.Eidelberg D, Moeller JR, Antonini A, Kazumata K, Nakamura T, Dhawan V, Spetsieris P, deLeon D, Bressman SB, Fahn S. Functional brain networks in DYT1 dystonia. Ann Neurol. 1998;44:303–312. doi: 10.1002/ana.410440304. [DOI] [PubMed] [Google Scholar]
- 24.Hutchinson M, Nakamura T, Moeller JR, Antonini A, Belakhlef A, Dhawan V, Eidelberg D. The metabolic topography of essential blepharospasm: a focal dystonia with general implications. Neurology. 2000;55:673–677. doi: 10.1212/wnl.55.5.673. [DOI] [PubMed] [Google Scholar]
- 25.Trost M, Carbon M, Edwards C, Ma Y, Raymond D, Mentis MJ, Moeller JR, Bressman SB, Eidelberg D. Primary dystonia: is abnormal functional brain architecture linked to genotype? Ann Neurol. 2002;52:853–856. doi: 10.1002/ana.10418. [DOI] [PubMed] [Google Scholar]
- 26.Carbon M, Su S, Dhawan V, Raymond D, Bressman S, Eidelberg D. Regional metabolism in primary torsion dystonia: effects of penetrance and genotype. Neurology. 2004;62:1384–1390. doi: 10.1212/01.wnl.0000120541.97467.fe. [DOI] [PubMed] [Google Scholar]
- 27.Asanuma K, Ma Y, Okulski J, Dhawan V, Chaly T, Carbon M, Bressman SB, Eidelberg D. Decreased striatal D2 receptor binding in non-manifesting carriers of the DYT1 dystonia mutation. Neurology. 2005;64:347–349. doi: 10.1212/01.WNL.0000149764.34953.BF. [DOI] [PubMed] [Google Scholar]
- 28.Carbon M, Niethammer M, Peng S, Raymond D, Dhawan V, Chaly T, Ma Y, Bressman S, Eidelberg D. Abnormal striatal and thalamic dopamine neurotransmission: Genotype-related features of dystonia. Neurology. 2009;72:2097–2103. doi: 10.1212/WNL.0b013e3181aa538f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garibotto V, Romito LM, Elia AE, Soliveri P, Panzacchi A, Carpinelli A, Tinazzi M, Albanese A, Perani D. In vivo evidence for GABA(A) receptor changes in the sensorimotor system in primary dystonia. Mov Disord. 2011;26:852–857. doi: 10.1002/mds.23553. [DOI] [PubMed] [Google Scholar]
- 30.Marsden CD. The mysterious motor function of the basal ganglia: the Robert Wartenberg Lecture. Neurology. 1982;32:514–539. doi: 10.1212/wnl.32.5.514. [DOI] [PubMed] [Google Scholar]
- 31.Carbon M, Argyelan M, Habeck C, Ghilardi MF, Fitzpatrick T, Dhawan V, Pourfar M, Bressman SB, Eidelberg D. Increased sensorimotor network activity in DYT1 dystonia: a functional imaging study. Brain. 2010;133:690–700. doi: 10.1093/brain/awq017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Niethammer M, Carbon M, Argyelan M, Eidelberg D. Hereditary dystonia as a neurodevelopmental circuit disorder: Evidence from neuroimaging. Neurobiol Dis. 2011;42:202–209. doi: 10.1016/j.nbd.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Detante O, Vercueil L, Thobois S, Broussolle E, Costes N, Lavenne F, Chabardes S, Lebars D, Vidailhet M, Benabid AL, et al. Globus pallidus internus stimulation in primary generalized dystonia: a H215O PET study. Brain. 2004;127:1899–1908. doi: 10.1093/brain/awh213. [DOI] [PubMed] [Google Scholar]
- 34.Thobois S, Ballanger B, Xie-Brustolin J, Damier P, Durif F, Azulay JP, Derost P, Witjas T, Raoul S, Le Bars D, et al. Globus pallidus stimulation reduces frontal hyperactivity in tardive dystonia. J Cereb Blood Flow Metab. 2008;28:1127–1138. doi: 10.1038/sj.jcbfm.9600610. [DOI] [PubMed] [Google Scholar]
- 35.Hebb MO, Chiasson P, Lang AE, Brownstone RM, Mendez I. Sustained relief of dystonia following cessation of deep brain stimulation. Mov Disord. 2007;22:1958–1962. doi: 10.1002/mds.21616. [DOI] [PubMed] [Google Scholar]
- 36.Lalli S, Piacentini S, Franzini A, Panzacchi A, Cerami C, Messina G, Ferré F, Perani D, Albanese A. Epidural premotor cortical stimulation in primary focal dystonia: clinical and 18F-fluoro deoxyglucose positron emission tomography open study. Mov Disord. 2012;27:533–538. doi: 10.1002/mds.24949. [DOI] [PubMed] [Google Scholar]
- 37.Romito LM, Franzini A, Perani D, Carella F, Marras C, Capus L, Garibotto V, Broggi G, Albanese A. Fixed dystonia unresponsive to pallidal stimulation improved by motor cortex stimulation. Neurology. 2007;68:875–876. doi: 10.1212/01.wnl.0000256816.83036.c9. [DOI] [PubMed] [Google Scholar]
- 38.Styczyńska B, Krzemińska A, Mańkowska H, Sereda B. [Kelevan used as poisoned nutrition in the destruction of Pharaoh’s ants (Monomorium pharaonis L.)] Rocz Panstw Zakl Hig. 1976;27:577–585. [PubMed] [Google Scholar]
- 39.Rinne JO, Iivanainen M, Metsähonkala L, Vainionpää L, Pääkkönen L, Nagren K, Helenius H. Striatal dopaminergic system in dopa-responsive dystonia: a multi-tracer PET study shows increased D2 receptors. J Neural Transm. 2004;111:59–67. doi: 10.1007/s00702-003-0053-3. [DOI] [PubMed] [Google Scholar]
- 40.Perlmutter JS, Stambuk MK, Markham J, Black KJ, McGee-Minnich L, Jankovic J, Moerlein SM. Decreased [18F]spiperone binding in putamen in idiopathic focal dystonia. J Neurosci. 1997;17:843–850. doi: 10.1523/JNEUROSCI.17-02-00843.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Naumann M, Pirker W, Reiners K, Lange KW, Becker G, Brücke T. Imaging the pre- and postsynaptic side of striatal dopaminergic synapses in idiopathic cervical dystonia: a SPECT study using [123I] epidepride and [123I] beta-CIT. Mov Disord. 1998;13:319–323. doi: 10.1002/mds.870130219. [DOI] [PubMed] [Google Scholar]
- 42.Berman BD, Hallett M, Herscovitch P, Simonyan K. Striatal dopaminergic dysfunction at rest and during task performance in writer’s cramp. Brain. 2013;136(Pt 12):3645–3658. doi: 10.1093/brain/awt282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ceballos-Baumann AO, Sheean G, Passingham RE, Marsden CD, Brooks DJ. Botulinum toxin does not reverse the cortical dysfunction associated with writer’s cramp. A PET study. Brain. 1997;120(Pt 4):571–582. doi: 10.1093/brain/120.4.571. [DOI] [PubMed] [Google Scholar]
- 44.Walsh R, Hutchinson M. Molding the sensory cortex: spatial acuity improves after botulinum toxin treatment for cervical dystonia. Mov Disord. 2007;22:2443–2446. doi: 10.1002/mds.21759. [DOI] [PubMed] [Google Scholar]
- 45.Jeffries KJ, Schooler C, Schoenbach C, Herscovitch P, Chase TN, Braun AR. The functional neuroanatomy of Tourette’s syndrome: an FDG PET study III: functional coupling of regional cerebral metabolic rates. Neuropsychopharmacology. 2002;27:92–104. doi: 10.1016/S0893-133X(01)00428-6. [DOI] [PubMed] [Google Scholar]
- 46.Pourfar M, Feigin A, Tang CC, Carbon-Correll M, Bussa M, Budman C, Dhawan V, Eidelberg D. Abnormal metabolic brain networks in Tourette syndrome. Neurology. 2011;76:944–952. doi: 10.1212/WNL.0b013e3182104106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Singer HS, Szymanski S, Giuliano J, Yokoi F, Dogan AS, Brasic JR, Zhou Y, Grace AA, Wong DF. Elevated intrasynaptic dopamine release in Tourette’s syndrome measured by PET. Am J Psychiatry. 2002;159:1329–1336. doi: 10.1176/appi.ajp.159.8.1329. [DOI] [PubMed] [Google Scholar]
- 48.Behen M, Chugani HT, Juhász C, Helder E, Ho A, Maqbool M, Rothermel RD, Perry J, Muzik O. Abnormal brain tryptophan metabolism and clinical correlates in Tourette syndrome. Mov Disord. 2007;22:2256–2262. doi: 10.1002/mds.21712. [DOI] [PubMed] [Google Scholar]
- 49.Saporta AS, Chugani HT, Juhász C, Makki MI, Muzik O, Wilson BJ, Behen ME. Multimodality neuroimaging in Tourette syndrome: alpha-[11C] methyl-L-tryptophan positron emission tomography and diffusion tensor imaging studies. J Child Neurol. 2010;25:336–342. doi: 10.1177/0883073809339394. [DOI] [PubMed] [Google Scholar]
- 50.Wong DF, Brasić JR, Singer HS, Schretlen DJ, Kuwabara H, Zhou Y, Nandi A, Maris MA, Alexander M, Ye W, et al. Mechanisms of dopaminergic and serotonergic neurotransmission in Tourette syndrome: clues from an in vivo neurochemistry study with PET. Neuropsychopharmacology. 2008;33:1239–1251. doi: 10.1038/sj.npp.1301528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vernaleken I, Kuhn J, Lenartz D, Raptis M, Huff W, Janouschek H, Neuner I, Schaefer WM, Gründer G, Sturm V. Bithalamical deep brain stimulation in tourette syndrome is associated with reduction in dopaminergic transmission. Biol Psychiatry. 2009;66:e15–e17. doi: 10.1016/j.biopsych.2009.06.025. [DOI] [PubMed] [Google Scholar]
- 52.Kuhn J, Janouschek H, Raptis M, Rex S, Lenartz D, Neuner I, Mottaghy FM, Schneider F, Schaefer WM, Sturm V, et al. In vivo evidence of deep brain stimulation-induced dopaminergic modulation in Tourette’s syndrome. Biol Psychiatry. 2012;71:e11–e13. doi: 10.1016/j.biopsych.2011.09.035. [DOI] [PubMed] [Google Scholar]
- 53.Lerner A, Bagic A, Simmons JM, Mari Z, Bonne O, Xu B, Kazuba D, Herscovitch P, Carson RE, Murphy DL, et al. Widespread abnormality of the γ-aminobutyric acid-ergic system in Tourette syndrome. Brain. 2012;135:1926–1936. doi: 10.1093/brain/aws104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reiffers S, Beerling-van der Molen HD, Vaalburg W, Ten Hoeve W, Paans AM, Korf J, Woldring MG, Wynberg H. Rapid synthesis and purification of carbon-11 labelled DOPA: a potential agent for brain studies. Int J Appl Radiat Isot. 1977;28:955–958. doi: 10.1016/0020-708x(77)90060-6. [DOI] [PubMed] [Google Scholar]
- 55.Garnett ES, Firnau G, Nahmias C, Sood S, Belbeck L. Blood-brain barrier transport and cerebral utilization of dopa in living monkeys. Am J Physiol. 1980;238:R318–R327. doi: 10.1152/ajpregu.1980.238.5.R318. [DOI] [PubMed] [Google Scholar]
- 56.Firnau G, Sood S, Pantel R, Garnett S. Phenol ionization in dopa determines the site of methylation by catechol-O-methyltransferase. Mol Pharmacol. 1981;19:130–133. [PubMed] [Google Scholar]
- 57.Barrio JR, Huang SC, Melega WP, Yu DC, Hoffman JM, Schneider JS, Satyamurthy N, Mazziotta JC, Phelps ME. 6-[18F]fluoro-L-dopa probes dopamine turnover rates in central dopaminergic structures. J Neurosci Res. 1990;27:487–493. doi: 10.1002/jnr.490270408. [DOI] [PubMed] [Google Scholar]
- 58.Firnau G, Garnett ES, Chirakal R, Sood S, Nahmias C, Schrobilgen G. [18F]fluoro-L-dopa for the in vivo study of intracerebral dopamine. Int J Rad Appl Instrum A. 1986;37:669–675. doi: 10.1016/0883-2889(86)90260-1. [DOI] [PubMed] [Google Scholar]
- 59.Garnett S, Firnau G, Nahmias C, Chirakal R. Striatal dopamine metabolism in living monkeys examined by positron emission tomography. Brain Res. 1983;280:169–171. doi: 10.1016/0006-8993(83)91187-3. [DOI] [PubMed] [Google Scholar]
- 60.Garnett ES, Firnau G, Nahmias C. Dopamine visualized in the basal ganglia of living man. Nature. 1983;305:137–138. doi: 10.1038/305137a0. [DOI] [PubMed] [Google Scholar]
- 61.Stoessl AJ, Lehericy S, Strafella AP. Imaging insights into basal ganglia function, Parkinson’s disease, and dystonia. Lancet. 2014;384:532–544. doi: 10.1016/S0140-6736(14)60041-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Robertson HA. Dopamine receptor interactions: some implications for the treatment of Parkinson’s disease. Trends Neurosci. 1992;15:201–206. doi: 10.1016/0166-2236(92)90034-6. [DOI] [PubMed] [Google Scholar]
- 63.Farde L. The advantage of using positron emission tomography in drug research. Trends Neurosci. 1996;19:211–214. doi: 10.1016/0166-2236(96)40002-9. [DOI] [PubMed] [Google Scholar]
- 64.Piccini P, Burn DJ, Ceravolo R, Maraganore D, Brooks DJ. The role of inheritance in sporadic Parkinson’s disease: evidence from a longitudinal study of dopaminergic function in twins. Ann Neurol. 1999;45:577–582. doi: 10.1002/1531-8249(199905)45:5<577::aid-ana5>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 65.Hutchinson M, Spanaki C, Lebedev S, Plaitakis A. Genetic basis of common diseases: the general theory of Mendelian recessive genetics. Med Hypotheses. 2005;65:282–286. doi: 10.1016/j.mehy.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 66.Kish SJ, Shannak K, Hornykiewicz O. Uneven pattern of dopamine loss in the striatum of patients with idiopathic Parkinson’s disease. Pathophysiologic and clinical implications. N Engl J Med. 1988;318:876–880. doi: 10.1056/NEJM198804073181402. [DOI] [PubMed] [Google Scholar]
- 67.Nahmias C, Garnett ES, Firnau G, Lang A. Striatal dopamine distribution in parkinsonian patients during life. J Neurol Sci. 1985;69:223–230. doi: 10.1016/0022-510x(85)90135-2. [DOI] [PubMed] [Google Scholar]
- 68.Brooks DJ. PET studies on the early and differential diagnosis of Parkinson’s disease. Neurology. 1993;43:S6–16. [PubMed] [Google Scholar]
- 69.Fearnley JM, Lees AJ. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain. 1991;114(Pt 5):2283–2301. doi: 10.1093/brain/114.5.2283. [DOI] [PubMed] [Google Scholar]
- 70.Teune LK, Bartels AL, de Jong BM, Willemsen AT, Eshuis SA, de Vries JJ, van Oostrom JC, Leenders KL. Typical cerebral metabolic patterns in neurodegenerative brain diseases. Mov Disord. 2010;25:2395–2404. doi: 10.1002/mds.23291. [DOI] [PubMed] [Google Scholar]
- 71.Rascol O, Schelosky L. 123I-metaiodobenzylguanidine scintigraphy in Parkinson’s disease and related disorders. Mov Disord. 2009;24 Suppl 2:S732–S741. doi: 10.1002/mds.22499. [DOI] [PubMed] [Google Scholar]
- 72.Marek K, Jennings D, Seibyl J. Imaging the dopamine system to assess disease-modifying drugs: studies comparing dopamine agonists and levodopa. Neurology. 2003;61:S43–S48. doi: 10.1212/wnl.61.6_suppl_3.s43. [DOI] [PubMed] [Google Scholar]
- 73.Stoessl AJ. Scans without evidence of dopamine deficiency: the triumph of careful clinical assessment. Mov Disord. 2010;25:529–530. doi: 10.1002/mds.23138. [DOI] [PubMed] [Google Scholar]
- 74.Vlaar AM, van Kroonenburgh MJ, Kessels AG, Weber WE. Meta-analysis of the literature on diagnostic accuracy of SPECT in parkinsonian syndromes. BMC Neurol. 2007;7:27. doi: 10.1186/1471-2377-7-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, Eggert K, Oertel W, Banati RB, Brooks DJ. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis. 2006;21:404–412. doi: 10.1016/j.nbd.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 76.Bartels AL, Willemsen AT, Doorduin J, de Vries EF, Dierckx RA, Leenders KL. [11C]-PK11195 PET: quantification of neuroinflammation and a monitor of anti-inflammatory treatment in Parkinson’s disease? Parkinsonism Relat Disord. 2010;16:57–59. doi: 10.1016/j.parkreldis.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 77.Gerhard A, Banati RB, Goerres GB, Cagnin A, Myers R, Gunn RN, Turkheimer F, Good CD, Mathias CJ, Quinn N, et al. [11C](R)-PK11195 PET imaging of microglial activation in multiple system atrophy. Neurology. 2003;61:686–689. doi: 10.1212/01.wnl.0000078192.95645.e6. [DOI] [PubMed] [Google Scholar]
- 78.Gerhard A, Trender-Gerhard I, Turkheimer F, Quinn NP, Bhatia KP, Brooks DJ. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in progressive supranuclear palsy. Mov Disord. 2006;21:89–93. doi: 10.1002/mds.20668. [DOI] [PubMed] [Google Scholar]
- 79.Gerhard A, Watts J, Trender-Gerhard I, Turkheimer F, Banati RB, Bhatia K, Brooks DJ. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in corticobasal degeneration. Mov Disord. 2004;19:1221–1226. doi: 10.1002/mds.20162. [DOI] [PubMed] [Google Scholar]
- 80.Pavese N, Gerhard A, Tai YF, Ho AK, Turkheimer F, Barker RA, Brooks DJ, Piccini P. Microglial activation correlates with severity in Huntington disease: a clinical and PET study. Neurology. 2006;66:1638–1643. doi: 10.1212/01.wnl.0000222734.56412.17. [DOI] [PubMed] [Google Scholar]
- 81.Bagchi DP, Yu L, Perlmutter JS, Xu J, Mach RH, Tu Z, Kotzbauer PT. Binding of the radioligand SIL23 to α-synuclein fibrils in Parkinson disease brain tissue establishes feasibility and screening approaches for developing a Parkinson disease imaging agent. PLoS One. 2013;8:e55031. doi: 10.1371/journal.pone.0055031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Morrish PK, Sawle GV, Brooks DJ. An [18F]dopa-PET and clinical study of the rate of progression in Parkinson’s disease. Brain. 1996;119(Pt 2):585–591. doi: 10.1093/brain/119.2.585. [DOI] [PubMed] [Google Scholar]
- 83.Stoessl AJ. Positron emission tomography in premotor Parkinson’s disease. Parkinsonism Relat Disord. 2007;13 Suppl 3:S421–S424. doi: 10.1016/S1353-8020(08)70041-5. [DOI] [PubMed] [Google Scholar]
- 84.Müller ML, Bohnen NI. Cholinergic dysfunction in Parkinson’s disease. Curr Neurol Neurosci Rep. 2013;13:377. doi: 10.1007/s11910-013-0377-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kazumata K, Dhawan V, Chaly T, Antonini A, Margouleff C, Belakhlef A, Neumeyer J, Eidelberg D. Dopamine transporter imaging with fluorine-18-FPCIT and PET. J Nucl Med. 1998;39:1521–1530. [PubMed] [Google Scholar]
- 86.Ma Y, Eidelberg D. Functional imaging of cerebral blood flow and glucose metabolism in Parkinson’s disease and Huntington’s disease. Mol Imaging Biol. 2007;9:223–233. doi: 10.1007/s11307-007-0085-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fowler JS, Volkow ND, Logan J, Wang GJ, MacGregor RR, Schyler D, Wolf AP, Pappas N, Alexoff D, Shea C. Slow recovery of human brain MAO B after L-deprenyl (Selegeline) withdrawal. Synapse. 1994;18:86–93. doi: 10.1002/syn.890180203. [DOI] [PubMed] [Google Scholar]
- 88.Rakshi JS, Pavese N, Uema T, Ito K, Morrish PK, Bailey DL, Brooks DJ. A comparison of the progression of early Parkinson’s disease in patients started on ropinirole or L-dopa: an 18F-dopa PET study. J Neural Transm (Vienna) 2002;109:1433–1443. doi: 10.1007/s00702-002-0753-0. [DOI] [PubMed] [Google Scholar]
- 89.Tedroff J, Pedersen M, Aquilonius SM, Hartvig P, Jacobsson G, Långström B. Levodopa-induced changes in synaptic dopamine in patients with Parkinson’s disease as measured by [11C]raclopride displacement and PET. Neurology. 1996;46:1430–1436. doi: 10.1212/wnl.46.5.1430. [DOI] [PubMed] [Google Scholar]
- 90.de la Fuente-Fernández R, Sossi V, Huang Z, Furtado S, Lu JQ, Calne DB, Ruth TJ, Stoessl AJ. Levodopa-induced changes in synaptic dopamine levels increase with progression of Parkinson’s disease: implications for dyskinesias. Brain. 2004;127:2747–2754. doi: 10.1093/brain/awh290. [DOI] [PubMed] [Google Scholar]
- 91.de la Fuente-Fernández R, Lu JQ, Sossi V, Jivan S, Schulzer M, Holden JE, Lee CS, Ruth TJ, Calne DB, Stoessl AJ. Biochemical variations in the synaptic level of dopamine precede motor fluctuations in Parkinson’s disease: PET evidence of increased dopamine turnover. Ann Neurol. 2001;49:298–303. doi: 10.1002/ana.65.abs. [DOI] [PubMed] [Google Scholar]
- 92.Brooks DJ, Ibanez V, Sawle GV, Playford ED, Quinn N, Mathias CJ, Lees AJ, Marsden CD, Bannister R, Frackowiak RS. Striatal D2 receptor status in patients with Parkinson’s disease, striatonigral degeneration, and progressive supranuclear palsy, measured with 11C-raclopride and positron emission tomography. Ann Neurol. 1992;31:184–192. doi: 10.1002/ana.410310209. [DOI] [PubMed] [Google Scholar]
- 93.Steeves TD, Miyasaki J, Zurowski M, Lang AE, Pellecchia G, Van Eimeren T, Rusjan P, Houle S, Strafella AP. Increased striatal dopamine release in Parkinsonian patients with pathological gambling: a [11C] raclopride PET study. Brain. 2009;132:1376–1385. doi: 10.1093/brain/awp054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.O’Sullivan SS, Wu K, Politis M, Lawrence AD, Evans AH, Bose SK, Djamshidian A, Lees AJ, Piccini P. Cue-induced striatal dopamine release in Parkinson’s disease-associated impulsive-compulsive behaviours. Brain. 2011;134:969–978. doi: 10.1093/brain/awr003. [DOI] [PubMed] [Google Scholar]
- 95.Freed CR, Greene PE, Breeze RE, Tsai WY, DuMouchel W, Kao R, Dillon S, Winfield H, Culver S, Trojanowski JQ, et al. Transplantation of embryonic dopamine neurons for severe Parkinson’s disease. N Engl J Med. 2001;344:710–719. doi: 10.1056/NEJM200103083441002. [DOI] [PubMed] [Google Scholar]
- 96.Ceballos-Baumann AO, Boecker H, Bartenstein P, von Falkenhayn I, Riescher H, Conrad B, Moringlane JR, Alesch F. A positron emission tomographic study of subthalamic nucleus stimulation in Parkinson disease: enhanced movement-related activity of motor-association cortex and decreased motor cortex resting activity. Arch Neurol. 1999;56:997–1003. doi: 10.1001/archneur.56.8.997. [DOI] [PubMed] [Google Scholar]
- 97.Hilker R, Voges J, Weisenbach S, Kalbe E, Burghaus L, Ghaemi M, Lehrke R, Koulousakis A, Herholz K, Sturm V, et al. Subthalamic nucleus stimulation restores glucose metabolism in associative and limbic cortices and in cerebellum: evidence from a FDG-PET study in advanced Parkinson’s disease. J Cereb Blood Flow Metab. 2004;24:7–16. doi: 10.1097/01.WCB.0000092831.44769.09. [DOI] [PubMed] [Google Scholar]
- 98.Hilker R. [Functional imaging of deep brain stimulation in idiopathic Parkinson’s disease] Nervenarzt. 2010;81:1204–1207. doi: 10.1007/s00115-010-3027-3. [DOI] [PubMed] [Google Scholar]
- 99.Bankiewicz KS, Eberling JL, Kohutnicka M, Jagust W, Pivirotto P, Bringas J, Cunningham J, Budinger TF, Harvey-White J. Convection-enhanced delivery of AAV vector in parkinsonian monkeys; in vivo detection of gene expression and restoration of dopaminergic function using pro-drug approach. Exp Neurol. 2000;164:2–14. doi: 10.1006/exnr.2000.7408. [DOI] [PubMed] [Google Scholar]
- 100.Melega WP, Lacan G, Desalles AA, Phelps ME. Long-term methamphetamine-induced decreases of [(11)C]WIN 35,428 binding in striatum are reduced by GDNF: PET studies in the vervet monkey. Synapse. 2000;35:243–249. doi: 10.1002/(SICI)1098-2396(20000315)35:4<243::AID-SYN1>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]