

Abstract
Dementia is a contemporary global health issue with far reaching consequences, not only for affected individuals and their families, but for national and global socio-economic conditions. The hallmark feature of dementia is that of irreversible cognitive decline, usually affecting memory, and impaired activities of daily living. Advances in healthcare worldwide have facilitated longer life spans, increasing the risks of developing cognitive decline and dementia in late life. Dementia remains a clinical diagnosis. The role of structural and molecular neuroimaging in patients with dementia is primarily supportive role rather than diagnostic, American and European guidelines recommending imaging to exclude treatable causes of dementia, such as tumor, hydrocephalus or intracranial haemorrhage, but also to distinguish between different dementia subtypes, the commonest of which is Alzheimer’s disease. However, this depends on the availability of these imaging techniques at individual centres. Advanced magnetic resonance imaging (MRI) techniques, such as functional connectivity MRI, diffusion tensor imaging and magnetic resonance spectroscopy, and molecular imaging techniques, such as 18F fluoro-deoxy glucose positron emission tomography (PET), amyloid PET, tau PET, are currently within the realm of dementia research but are available for clinical use. Increasingly the research focus is on earlier identification of at risk preclinical individuals, for example due to family history. Intervention at the preclinical stages before irreversible brain damage occurs is currently the best hope of reducing the impact of dementia.
Keywords: Dementia, Alzheimer’s disease, Magnetic resonance imaging, Molecular imaging, Frontotemporal dementia, Lewy body dementia, Vascular dementia
Core tip: Dementia is a clinical diagnosis that cannot be made on imaging. Structural and molecular imaging techniques are useful to identify the likely underlying neuropathology. Neuroimaging techniques, such as computed tomography (CT) and blood flow single photon emission computed tomography (SPECT) are routinely used in clinical practice in all newly diagnosed dementia patients. Structural imaging with CT or magnetic resonance imaging is useful in suspected frontotemporal dementia. Amyloid positron emission tomography imaging has recently been introduced into clinical practice and is likely to be most useful in early onset Alzheimer’s disease. Dopamine transporter imaging with iodine-123-b-carbo-methoxy-3-b-(4-iodophenyltropane) fluropropyl SPECT has been firmly established in clinical practice to support a diagnosis of Lewy body disease. This article is a review of the imaging techniques not only currently in clinical use but also the emerging imaging techniques in research.
INTRODUCTION
Dementia is a syndrome of progressive memory and cognitive decline affecting an individual in his activities of daily life, secondary to irreversible neuronal damage. With 2%-10% of those affected younger than 65 years, this condition is primarily a disease of the aging population[1]. Dementia is not an inevitable consequence of aging and the predicted rise in dementia as a result of an aging population is not as great as predicted, perhaps because the current definition of old-age dependency is too simplistic[2]. However, the published prevalence of dementia doubles with every 5 years increment in age, according to the World Alzheimer Report 2014 by Alzheimer Disease International[1]. Worldwide prevalence is estimated at 47.5 million with just over half living in middle and low income countries, expected to double by 2030 and treble by 2050 (World Health Organization fact sheet No.362, March 2015). The annual global cost of medical care, social support and informal care was estimated to be US$ 604 billion in 2010, which is only set to increase with the world population of over age 65 years outnumbering the under age 5 years by two-three fold by 2050[3].
On the other hand, delaying the onset of dementia by 5 years would reduce the population prevalence by 50%, greatly reducing its impact in the general population[1]. Currently there is no cure for dementia. Medical and non-medical interventions have had limited success in altering the course of the disease especially as neuropathology is usually extensive by the time the patient has presented with symptoms (Alzheimer’s Disease International 2014 report).
The diagnosis of dementia remains a clinical diagnosis and post-mortem examination of the brain tissue is the only definitive method to establish and confirm the diagnosis. In vivo, various invasive and non-invasive methods are available to support the diagnosis of different sub-types, due to different brain pathology.
Dementia has various causes (Table 1). By far the most important type is Alzheimer’s disease (AD) accounting for 60%-70% of all dementias. Primary dementing conditions have in common abnormal protein or peptide accumulation in the brain: τ and β amyloid in AD; α synuclein in Lewy body dementia (LBD) and τ, Transactive DNA-binding protein (TDP) or Fused in Sarcoma (FUS) in fronto-temporal dementia (FTD). But these conditions can and do often co-exist with other pathologies of aging, most commonly cerebral small vessel disease (CSVD)[4]. Dementia secondary to cerebrovascular disease is the second most common form of dementia.
Table 1.
Causes of dementia and dementia syndromes
| Types of dementia |
| Primary dementias |
| Alzheimer’s disease |
| Late-onset Alzheimer’s disease - most common form 60%-70% of all dementias |
| Early-onset Alzheimer’s disease - under 65 yr of age, chromosome 14 implicated, Down’s syndrome |
| Familial AD - inheritable form present in at least 2 generations within families |
| Dementia with Lewy bodies |
| Frontotemporal dementia |
| Mixed dementia - more than one form of pathology for, e.g., Lewy bodies with Alzheimer’s disease |
| Less common forms |
| Parkinson’s disease |
| Progressive supranuclear palsy |
| Huntington’s disease |
| Secondary dementias |
| Vascular/multi infarct dementia |
| Vascular with Alzheimer’s disease |
| Creutzfeldt-Jakob disease |
| Intracranial mass lesions |
| Normal pressure hydrocephalus |
| Subdural haematomas |
| Trauma |
| Infections - primarily human immunodeficiency virus |
| Alcohol |
| Other documented causes |
| Vitamin deficiencies - vitamins E, B and folic acid are implicated |
| Medications |
| Other causes like depression |
North American, European and United Kingdom National Institute of Health and Care Excellence (NICE) guidelines recommend neuroimaging in all patients at the time of initial diagnosis of dementia[5-8]. Structural and molecular imaging are both useful to support the diagnosis of a dementia-related neuropathology in vivo. Molecular imaging, for example, positron emission tomography (PET) using tracers for amyloid or tau and invasive methods like cerebrospinal fluid (CSF) analysis of amyloid β and τ are also available to support the diagnosis of AD in vivo. However, many of these tools apart from structural neuroimaging remain elusive to regular clinical practice and are confined to specialised centres and to research. Therapeutic interventions in dementia, in particular in AD, have had mixed success, none achieving significant alteration in disease progression. This is largely due to the fact that the process of neuronal damage is quite advanced at the time of clinical presentation. It is widely recognised that early intervention before irreversible neuronal damage occurs is our best hope of delaying the onset and perhaps preventing dementia[9]. Inevitably then it becomes imperative that we learn to identify those individuals who are on the trajectory to develop AD, 15-20 years before clinical dementia. Confusing the picture is the fact that many of these neuronal changes including amyloid deposition occur within the spectrum of normal aging without ever causing dementia. So do we expose these individuals to an intervention that they may never need? Would it be cost effective to do so[10]?
Research has inevitably widened its scope with emphasis now on the pre-clinical stage of the disease so that we could precisely identify those vulnerable individuals with the greatest level of confidence. Individuals affected could potentially be identified for future trials. This has heralded a new era of collaborative global endeavour. Multicentre, collaborative large datasets like the Alzheimer’s Disease Neuroimaging Initiative (ADNI) provide free access to multi-modality data to researchers worldwide, considerably reducing the cost of such research[11]. Molecular imaging and advanced MRI techniques are at the cutting edge of dementia research, primarily in the pre-clinical stage, helping us understand the early life of this devastating condition.
Here, we aim to discuss and provide an overview of imaging in common diseases that cause dementia, both in the clinical setting and within the realm of research. Imaging in dementia has moved away from just ruling out treatable causes of dementia like space occupying lesions or hydrocephalus, to characterising the different types of dementia-related neuropathology with increasing specificity.
AD
A primary neurodegenerative condition, AD is the most common form of late onset dementia (> 65)[12]. Neuropathologically it is characterised by extracellular amyloid plaques and intracellular tau aggregates[13]. Amyloid plaques are aggregates of insoluble fibrillary β-amyloid (Aβ) peptide mostly 40-42 amino acids in length, Aβ42 being the most prevalent[14]. The accumulation of Aβ in turn is thought to trigger a cascade of neurodegenerative events including intracellular aggregation of hyperphosphorylated tau[15] and neuroinflammation[16,17]. Accumulation of Aβ correlates with cognitive decline in some studies, as demonstrated on amyloid PET imaging[18,19]. Lately this is being challenged as there appears to be a certain disconnect between the time of amyloid deposition, which plateaus in late mid-life and progressive cognitive decline. The intracellular tau related neurofibrillary tangles, on the other hand, do correlate with disease severity and cognition at different stages of AD[20,21].
The evolution of AD is a continuum progressing from the asymptomatic pre-clinical stage, decades before the clinical onset of the disease, to the pro-dromal stage where there is onset of cognitive impairment but below the levels of formal dementia diagnosis and eventually to dementia. In the rare autosomal dominant early onset AD, abnormal accumulation of amyloid has been attributed to mutations in the genes regulating amyloid precursor protein (APP) and the presenilins (PSEN 1 and 2)[22]. In sporadic AD, apolipoprotein E gene (APOE4) has been implicated in earlier onset, greater cognitive impairment and more rapid progression[23], but this is not exclusive to AD and is found in other neurodegenerative conditions, such as Parkinson’s disease (PD)[24].
The diagnostic criteria for AD have been recently updated for use in clinical practice as well as research[25]. Endeavours to recognise the disease in the earlier stages have also prompted standardisation of criteria for defining preclinical[26] and pro-dromal [amnestic mild cognitive impairment (MCI)] stages[27] for both clinical and research purposes.
Structural imaging
The evolution of neuropathological changes begins at the entorhinal cortex in the medial temporal lobe which plays an important role in laying down new memory by virtue of its connections to hippocampus. Subsequent hippocampal involvement results in episodic memory loss and, as the disease progresses to involve neocortex, impacts on cognition, language, attention and executive function, affecting the activities of daily life[28]. The typical imaging appearance is that of global brain atrophy with early disproportionate atrophy of medial temporal lobes (MTA), including the hippocampi[29] (Figure 1). MTA can differentiate AD from ageing with a sensitivity and specificity of 80%-85% and is a risk factor for cognitive decline and dementia in normal aging[30] and predicts AD in those with amnestic MCI with a sensitivity and specificity of 73% and 81%[31,32]. Progressive atrophy of posterior temporal and parietal lobes differentiates AD from FTD.
Figure 1.
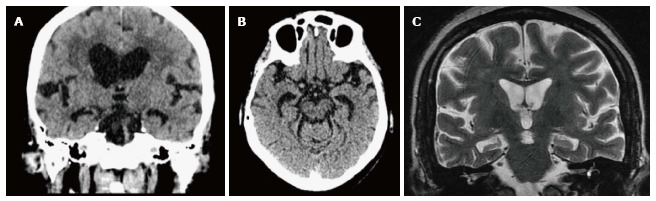
Hippocampal atrophy in an Alzheimer’s disease patient. A: Computed tomography axial; B: Coronal images; C: Medial temporal lobe atrophy on magnetic resonance imaging (not the same patient).
More advanced MRI imaging techniques such as diffusion weighted and diffusion tensor imaging (DWI and DTI), magnetic resonance spectroscopy (MRS) and perfusion imaging are also used in the research context. DWI and DTI techniques measure the integrity of tissue using two different measures, fractional anisotrophy (FA) and mean diffusivity (MD) or apparent diffusion coefficient (ADC). Increased MD/ADC and decreased FA are considered to be markers of neuronal fibre loss and reduced gray matter and white matter integrity (Figure 2)[33]. MRS is a technique to measure the biological metabolites in the target tissue, specifically the metabolites N-acetylaspartate (NAA), a marker of neuronal integrity, which decreases and myo-inositol, a marker of glial proliferation and neuronal damage, which increases. These changes are seen typically in the posterior cingulate gyrus, mesial temporal lobe, parieto-occipital lobes and the fronto-parietal lobes[34]. Cerebral perfusion is imaged using blood flow SPECT, dynamic susceptibility contrast enhanced MRI or arterial spin labelling (ASL) techniques[35,36]. Functional MRI (fMRI) measures brain activity using blood oxygenation level dependent (BOLD) technique demonstrating areas of brain activity by demonstrating the greatest influx of oxygen into the region to compensate increased utilisation. This can be performed in the resting state or during a task[37].
Figure 2.
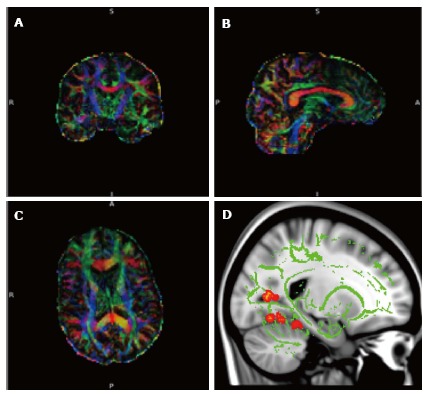
Diffusion tensor imaging. A-C: Diffusion tensor imaging (DTI) data set superimposed on structural image of the brain in 3 orthogonal planes demonstrating colour coded white matter tracts. Blue colour correlate to the tracts in the cranio-caudal direction, red in the transverse direction and green in the antero-posterior direction. (Images kindly prepared by Dr. Gordon D Waiter); D: DTI data of white matter tracts (green) superimposed on T1 image demonstrating statistically significant difference in fractional anisotropy in the fornix (orange areas) compared to the rest of the brain in a subgroup of patients. (Images kindly prepared by Dr. Gordon D Waiter).
A recent review of fMRI studies in dementia demonstrated decreased functional connectivity between precuneus, medial prefrontal cortex, posterior cingulate cortex, anterior cingulate cortex and hippocampus in the resting state, centres which are part of the default mode network (Figure 3) and more than can be accounted for by atrophy. The severity and distribution of decreased functional connectivity at rest is postulated to potentially distinguish MCI patients from AD and AD from other neurodegenerative dementias[38].
Figure 3.
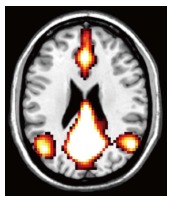
Default mode network, areas active during resting wakeful state. Resting state functional magnetic resonance imaging images using blood oxygenation level dependent technique. Typical areas involved include the medial prefrontal cortices, posterior cingulate, ventral precuneus and parts of parietal lobes (Images kindly prepared by Dr. Michael Stringer).
Molecular imaging
Molecular imaging aims to measure the pathophysiological change within the brain using either tracer that demonstrate normal physiology (non-specific tracers) or that bind to pathological targets (specific tracers). The two main modalities include single photon emission computed tomography (SPECT) and positron emission tomography (PET).
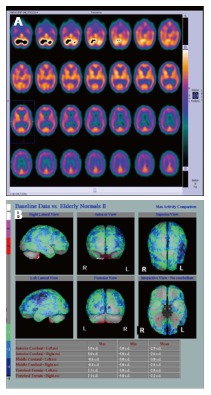
SPECT is used to measure regional cerebral blood flow (rCBF) by intravenously injecting technetium-labelled hexamethylpropylene amine oxime (99Tc-HMPAO). In AD characteristic deficits in posterior temporoparietal, posterior cingulate and inferior frontal regions, reflect underlying neuronal dysfunction and neurodegeneration (Figure 4). Often images demonstrate features secondary to a combination of both Alzheimer’s and vascular pathology (Figure 5).
Figure 4.
Underlying neuronal dysfunction and neurodegeneration. A: Hexamethylpropylene amine oxime (HMPAO) single photon emission computed tomography (SPECT) in normal control subject demonstrating normal almost symmetrical perfusion pattern; B: HMPAO SPECT in Alzheimer’s disease parametric images demonstrate bilateral reduction in perfusion in the temporal lobes especially in the medial temporal regions up to 2 (green) and 3 (blue) standard deviation (Images kindly prepared by Ms Lesley Lovell, Senior technician).
Figure 5.
Hexamethylpropylene amine oxime single photon emission computed tomography in a patient with mixed vascular disease and Alzheimer’s disease. A: Shows reduced perfusion in both the frontal and parietal lobes, especially on the left; B: Parametric images providing an overall view. There was hippocampal atrophy on computed tomography (Images kindly prepared by Dr. Fergus McKiddie).
Like HMPAO SPECT, 18 Fluorodeoxyglucose PET (FDG PET) demonstrates decrease in regional uptake reflecting decreased metabolism in a distribution similar to rCBF. In amnestic MCI, there is bilateral glucose hypometabolism in the limbic system, posterior cingulate cortex, parahippocampal gyri and temporal lobes (inferior temporal gyrus)[39,40], compared to AD patients who had additional profound hypometabolism in precuneus, inferior parietal lobule and middle temporal gyrus along with posterior cingulate cortex[39,41].
Amyloid PET imaging has started new chapters in both clinical and research practice. Amyloid specific ligands such as 11C-Pittsburg compound B (11CPIB), 18F Florbetapir, 18F Flutemetamol, demonstrate amyloid deposition in vivo and show good correlation with autopsy measurements[42]. They show increased uptake in typical locations such as precuneus, posterior cingulate cortex, temporal, parietal and occipital lobes[19,43,44]. A recent review of amyloid imaging studies revealed that even though there was high sensitivity to amyloid across the board with increased uptake in healthy controls, AD, MCI and other dementias like FTD, the sensitivity and specificity to identify AD cases was high and there was a high conversion rate of amyloid positive MCI to AD compared to amyloid negative MCI[3]. Amyloid imaging is now included in the criteria for the diagnosis of AD[25,45]. Both FDA and EMA have approved 18F florbetapir, 18F florbetaben and 18F flutemetamol[46] for clinical use. However, the role of amyloid PET is likely to be greater in early onset AD, than in late onset AD, where neuropathology is more heterogeneous[47]. However, structural MRI and FDG PET are more accurate than amyloid imaging in predicting cognitive status[48].
Ligands targeting the paired helical filament form (PHF) of tau, specific to AD have been developed and are currently close to market[49-51].
Neuroinflammation is also thought to play a role in the neuropathogenesis of AD[52]. PET imaging of neuroinflammatory processes such as microglial activation, reactive astrocytosis and increased phospholipase activity is possible using specific agents[53-56]. Tau imaging and neuro inflammation imaging are out of the realm of clinical practice at present. PET tracers specific for acetylcholinesterase as a proxy measure of acetylcholine synaptic density have been used in a few studies[57-59].
In summary, a multiphase model of neuroimaging corresponding to the stage of evolving neuropathology[60], is most likely with amyloid PET imaging positive during βamyloid accumulation, followed by tau accumulation with reduced rCBF on SPECT and decreased metabolism on FDG PET due to neuronal dysfunction and atrophy on CT and structural MRI following neuronal death.
VASCULAR COGNITIVE IMPAIRMENT AND DEMENTIA
Vascular cognitive impairment (VCI) is the second most common form of late onset dementia and the most common form of secondary dementia. VCI is a heterogenous disease and is due to a number of vascular causes[61] both small and large vessel related. Larger vessel involvement result in cortical infarcts and primary haemorrhages, while small vessel disease manifests as lacunar infarcts, lacunes, white matter hyperintensities (WMH), enlarged perivascular spaces and cerebral microhaemorrhages[62-67] (Figure 6).
Figure 6.
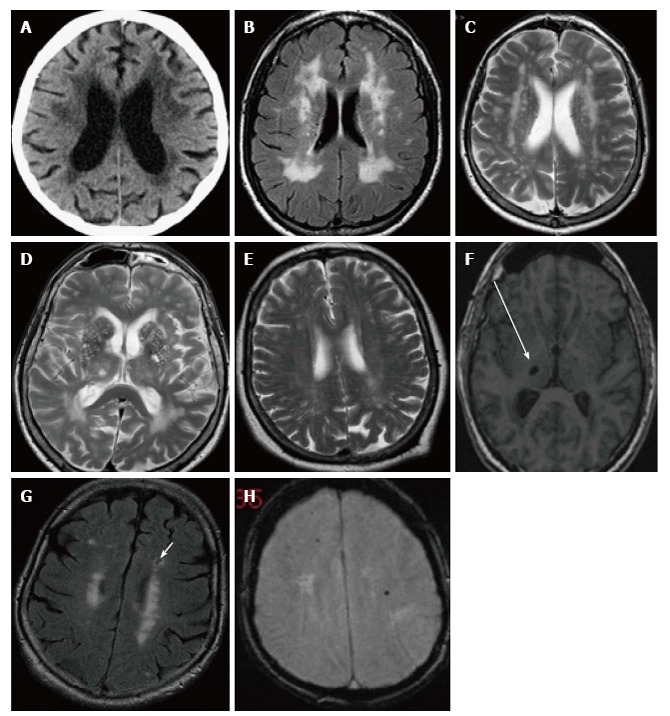
Computed tomography and magnetic resonance imaging images demonstrating structural changes secondary to cerebral small vessel disease. A: Axial image of CT brain demonstrating periventricular white matter low attenuation changes; B and C: The same seen as periventricular white matter high signal areas on FLAIR and T2 MRI; D: Prominent perivascular spaces typically seen in the basal ganglia; E: Centrum semiovale; F: Focal lacune, a cerebrospinal fluid filled space, sequelae of an old lacunar infarct in the right thalamus seen here (arrow) on an axial T1 image; G: Lacune (arrow) in the left frontal lobe on a FLAIR image, usually with a rim of high signal differentiating from a PVS; H: Cerebral microhaemorrhages, seen here as focal rounded black/low signal foci in the white matter of both frontal lobes on T2* gradient echo MRI. MRI: Magnetic resonance imaging; CT: Computed tomography.
The term subcortical ischaemic vascular disease (SIVD) is also used, often synonymous with WMH, the biomarker most significantly correlated with vascular risk factors such as hypertension and impaired glycaemic control[68]. WMH are age related and moderate amounts of WMH is seen up to 30% of normal older population with no significant cognitive dysfunction[69]. WHM in the VCI population on the other hand are significantly associated with not only vascular risk factors, but with cognitive impairment especially executive dysfunction, rapid global functional decline and decline of psychomotor speed and executive control[70,71]. Areas vulnerable to hypoxia, especially in the deep white matter watershed areas when affected are thought to trigger a series of events leading to tissue injury with neuroinflammation, blood-brain barrier (BBB) disruption and axonal damage resulting in white matter loss[72].
Structural imaging
WMH are best seen on structural MRI as bright signal areas on T2 and FLAIR images (Figure 6) in subcortical and periventricular distribution. They are quantified using visual rating scales or automated segmentation methods[73-75]. They are predominantly supratentorial in distribution, although are also common in the pons, and have a predilection for the frontal lobes.
Advanced MRI techniques like DTI, MRS and dynamic contrast enhanced (DCE) MRI demonstrate reduced white matter integrity, evidence of neuronal damage with decrease in NAA and enhancement secondary to BBB breakdown. Techniques to image neuroinflammation demonstrate microglia and macrophages around blood vessels[72]. Abnormal permeability also results in an increase in CSF albumin ratio in patients with vascular dementia[76]. This process repeated over time eventually results in quite significant white matter damage and cognitive impairment.
Diagnosis of VCI is dependent on a combination of the presence of vascular risk factors including hypertension, impaired glycaemic control, renal impairment, WMH on imaging, absence of an AD pattern of atrophy and executive dysfunction on psychometric testing. Memory is less involved[77,78]. Montreal Cognitive Assessment tests executive function and is a more useful tool than MMSE in this group of patients. An attempt is being made to define a set of features that are characteristic of the progressive form of VCI, termed the Binswanger Disease scale score[72].
Molecular imaging
HMPAO SPECT demonstrates decreased perfusion typically distributed in a vascular territory, often bilateral and usually involving the frontal lobes (Figure 7), seen either in combination with AD and in pure vascular dementia.
Figure 7.
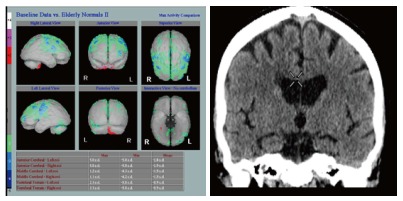
Hexamethylpropylene amine oxime single photon emission computed tomography in a pure cerebral vascular disease patient without Alzheimer’s disease. Note normal hippocampal volumes in the pure cerebral vascular disease patient on computed tomography (Images kindly prepared by Ms Lesley Lovell and Dr Fergus Mckiddie, clinical scientist).
FDG PET and rCBF SPECT demonstrate areas of decreased metabolism and perfusion respectively which may be bilateral, and/or arterial territory in distribution. Rarer causes of vascular dementia include hypercoaguable states (antiphospholipid antibodies), hereditary forms such as congenital autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL), with a temporal lobe distribution of WMH, and leucodystrophies.
In routine clinical practice though, multidetector CT of the brain is the most common, and in most centres the only, imaging performed when a vascular cause is suspected for cognitive impairment.
LEWY BODY DEMENTIA
This is the second most common primary neurodegenerative dementia and accounts for 15% of all dementia in the population and is clinically characterised by cognitive impairment with executive dysfunction, visuospatial impairment, visual, motor parkinsonian features, disordered (rapid eye movement) REM sleep and fluctuation in cognition and arousal[79]. Neuropsychometric tests demonstrate deficits in attention, executive function and visuospatial ability[79].
Pathologically lewy body dementia (LBD) overlaps with PD and is characterised by dopaminergic cell loss and accumulation of α-synuclein particles in presynaptic terminals that aggregate to form intracellular Lewy bodies. Similar to β amyloid pathology, α synuclein can be present as oligomers, fibrils and aggregates, the small oligomers likely being the most neurotoxic. These mainly occur in the cerebral cortex and limbic system, while in PD they exist in the substantia nigra, pars compacta and nigrostriatal projections. Recent work has increased understanding of genetic associations of LBD and PD[80]. Parkinson’s disease dementia (PDD) is pathologically and clinically indistinguishable to LBD, apart from the fact that in PDD, motor symptoms predate cognitive decline by up to 12 mo[79,81]. While the diagnosis of LBD will often be obvious clinically, it may be unclear in a substantial minority of patients, where neuroimaging play a role.
Structural imaging
Structural MRI using Voxel Based Morphometry has demonstrated variable regional brain atrophy in LBD with some studies reporting cortical atrophy in the insula, frontal, inferior parietal, temporal and occipital cortices[82,83] while a larger study has differentiated LBD from AD with more atrophy of hypothalamus, basal forebrain, midbrain, caudate and the putamen with relative preservation of the medial temporal lobe and the hippocampi[84]. The rate of progressive atrophy is increased when compared to normal controls, exaggerated if AD co-exists, but much lower compared to AD. Visual hallucinations and visuo-perceptual deficits, a characteristic feature of LBD do not seem to correlate with occipital lobe involvement[85]. However, correlation with other regions involved in visual processing (visual association areas) and executive functions (inferior frontal lobe) have been reported. If present, hippocampal atrophy is seen in the anterior subfield (CA1)[86], while in AD, CA2 and CA3 are more affected on high resolution MRI.
DTI, ASL and MRS techniques have been used to compare LBD with AD. In general these demonstrate abnormalities in the visual association cortex and posterior putamen in LBD compared with medial temporal lobe and precuneus in AD. The best discrimination will be a result of cumulative data from more than one sequence or imaging modality[86].
Molecular imaging
Increased β amyloid is commonly seen in LBD but not in PD dementia[87]. Amyloid PET imaging demonstrates similar uptake in AD and LBD (apart from occipital lobes which are spared in AD), making it difficult to differentiate between these two conditions. Similarly they are indistinguishable on rCBF SPECT and FDG PET, however involvement of the visual cortex would favour LBD[88-90].
A dopaminergic presynaptic ligand, iodine-123-b-carbo-methoxy-3-b-(4-iodophenyltropane) fluoropropyl (FP-CIT) or ioflupane, is used in SPECT studies. Neuronal loss in the dopaminergic zones are demonstrated by decreased uptake in the posterior putamen and then caudate nuclei when compared to normal controls (Figure 8) and AD patients. Visual image analysis is adequate to make the distinction between normal vs 3 grades of reduced uptake in the striatum, justifying routine use in clinical practice[91] as recommended by both NICE in United Kingdom and European Federation of Neurological Sciences in Europe. Quantitative analysis of FP-CIT images using shape analysis is as accurate as expert observer assessment and more reproducible[92]. Low dopamine transporter uptake in basal ganglia demonstrated by SPECT or PET imaging is the only imaging feature in the diagnostic criteria for LBD[79]. However, FP-CIT SPECT is not indicated to distinguish between different parkinsonian syndromes[93]. FP-CIT SPECT scan has a sensitivity of 78% and a specificity of 90% with an overall accuracy of 80% to distinguish between normal (or AD) and a parkinsonian syndrome (LBD)[94].
Figure 8.
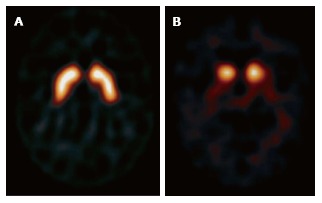
Iodine-123-b-carbo-methoxy-3-b-(4-iodophenyltropane) fluropropyl. A: Normal example symmetrical uptake in the caudate heads and putamen bilaterally; B: Absent uptake in the putamen in a patient with Lewy body dementia.
Cholinergic neuronal loss and reduced presynaptic choline acetyltransferace activity is seen in both LBD and AD. There is however differential uptake with reductions in medical occipital cortex in LBD and temporal lobe in AD[95]. Cardiac sympathetic denervation in LBD and PD predates neuronal loss can be measured using 123I MIGB, an analogue of noradrenaline in myocardial scintigraphy. Yoshita et al[96] demonstrated that the cut-off value of heart-to-mediastinum ratio of 1.68 yielded a sensitivity of 100% and a specificity of 100% for differentiating LBD from AD.
FRONTOTEMPORAL DEMENTIA
Frontotemporal dementia is a heterogenous group of diseases that account for approximately 5% of late onset dementia but is the second commonest cause of early onset dementia after AD[97]. Clinical presentation is often in the 5th and 6th decade, at least 10 years younger than AD and patients have a family history in about 50% of the cases[98].
The two main clinical syndromes of frontotemporal dementia (FTD) are behavioural variant FTD (bvFTD) characterised by deterioration in social function and personality and primary progressive aphasia (PPA) where there is an insidious decline in language skills. There are various subtypes of PPA such as semantic dementia (svPPA), progressive non-fluent aphasia (nfvPPA), logopenic aphasia (LPA - an AD variant) and progressive apraxia of speech, based on speech pattern involved[99]. Pathologically, based on the protein involved, they are divided into the following three categories: (1) FTLD-tau: Including tauopathies such as progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), multisystem tauopathy with dementia and Pick’s disease; (2) FTLD-TDP43: Transactive DNA-binding protein (TDP) 43 related abnormalities, a subgroup may also have motor neuron disease (MND)[100]; and (3) FTLD-FUS: Fused in sarcoma (FUS) protein[101].
As above FTD may be associated with overlap syndromes of MND or PSP, if so indicating likely molecular pathologies of TDP43 or tau respectively.
Structural imaging
Varying patterns of regional brain atrophy is the hallmark of these conditions depending on the clinical phenotype and the reporting radiologist may be the first to suggest FTD as the diagnosis in these patients.
bvFTD: Bilateral mesial frontal, orbitofrontal, anterior insular cortices and anterior cingulate cortex atrophy with more involvement on the right[102,103]. The frontal-insula-anterior cingulate are suggested to be part of a structurally and functionally connected neural network (a salience network) which demonstrates decreased functional connectivity during resting state fMRI[102,103] (Figure 9).
Figure 9.
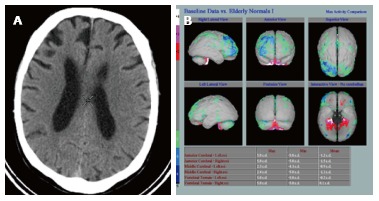
Computed tomography showing atrophy. A: Asymmetric right frontal lobe atrophy in fronto-temporal dementia; B: Hexamethylpropylene amine oxime single photon emission computed tomography in the same patient (Images kindly prepared by Ms Lewley Lovell, and Dr. Fergus Mckiddie).
svPPA: Bilateral, typically highly asymmetrical, usually left sided, atrophy of the anterior temporal lobes. As disease progresses the atrophy extends inferiorly to involve the posterior temporal lobes and superiorly to involve the inferior frontal lobes.
nfvPPA: Anterior perisylvian especially the dominant hemisphere, in particular the left frontal operculum - Broca’s areas 44, 45 and 47.
Quantification of regional atrophy rates on MRI could potentially be a useful biomarker of progression in FTD[49]. DTI has shown decreased white matter integrity in the respective regions affected depending on the clinical phenotype[104]. On fMRI FTD can be differentiated from AD by reduced connectivity in the salience network and increased connectivity in the DMN, opposite to that of AD[105,106].
Molecular imaging
FDG PET demonstrates frontal and anterior temporal lobe hypometabolism, which is useful in differentiating FTD from AD especially in the heterogenous group of progressive aphasias and in CBD[107]. However, PET imaging is not usually required as the diagnosis of FTD as frontal atrophy is usually obvious on structural imaging.
IMAGING IN OTHER DEMENTIAS
There are numerous less common causes of dementia. All these types of dementias can occur in people younger than 65 years but more often have a genetic cause and those affected generally tend to have accelerated progression. Dementias in people younger than 35 years are rare and more unusual causes such as infection or autoimmune encephalopathies need to be considered[108]. Imaging in this group and two other unusual causes of dementia will be discussed here.
AUTOIMMUNE DEMENTIAS
Previously termed as “limbic encephalitis”, these are a heterogeneous group of disorders that include various encephalopathies with specific clinical, electroencephalographic or CSF features[109]. They may present with cognitive impairment, seizures and are responsive to steroids. Imaging features are variable, MRI may show high signal intensity on T2 weighted and FLAIR images in the areas involved, typically in the limbic system. About 50% of autoimmune dementia patients, who have neuron-specific CSF autoantibodies, will have a paraneoplastic syndrome and whole body FDG-PET CT is appropriate to identify an underlying tumor[110].
PRION PROTEIN DISEASES
Accumulation of abnormal prion proteins can occur sporadically [sporadic Creutzfeld Jakob disease (CJD)], due to exposure to food (variant CJD) or infected tissues (iatrogenic CJD) due to genetic variation in the prion protein gene (PrnP), fatal familial insomnia. sCJD and vCJD typically present as rapidly progressive dementia with an earlier age at onset in vCJD. Other features at presentation could be hemiparesis, myoclonus in sCJD and painful sensory symptoms in vCJD supplemented by typical abnormal complexes on EEG. On MRI typical T2 and FLAIR hyperintensity is seen in the pulvinar of the thalami in vCJD, which is virtualy pathognomonic, and in the caudate heads and cortices (“cortical ribboning”) in sCJD which can be asymmetrical[111]. These abnormalities are best seen on DWI where they demonstrate diffusion restriction.
HUMAN IMMUNODEFICIENCY VIRUS ASSOCIATED NEUROCOGNITIVE DISORDER
HIV associated dementia is the most severe HIV associated neurocognitive disorder and presents as impairment in executive function, motor activities and memory. On structural MRI global cortical atrophy is seen with predilection for the anterior cingulate, lateral temporal, primary motor and sensory cortices. White matter hyperintensities too are seen, some presenting as progressive multifocal leukoencephalopathy characterised by focal white matter lesions typically in subcortical regions[112,113]. DTI studies demonstrate reduced white matter integrity in the cortical white matter, corona radiata and the corpus callosum are associated with cognitive impairment[114-116]. Other imaging modalities include MRS, fMRI, FDG PET and dopamine transporter imaging and demonstrate evidence of neuronal loss, impaired functional connectivity, hypometabolism and decreased uptake in the putamina and ventral striatum respectively.
Some studies suggest these imaging abnormalities are reversible following retroviral therapies, however additional research is needed[104].
CONCLUSION
Imaging in neurodegenerative disorders that cause dementia has evolved from the days of ruling out other pathologies to diagnosis of specific likely underlying neuropathologies. MRI studies, without doubt, are far superior to MDCT in providing information on the structural and functional changes corresponding to the pathological evolution of the disease. Newer techniques in MRI and PET are readily embraced by researchers in the quest for earlier detection of the disease before irreversible neuronal damage occurs, now believed to be the best current approach the global community can adopt to tackle these devastating conditions. Current and future interventions need to target individuals who are most at risk before the manifestation of dementia. Large multicentre datasets like ADNI, which are freely available, are invaluable for providing new research opportunities are important for future progress.
Future PET tracers for specific proteinopathies (tau, TDP-43, α synuclein) would provide more information and offer more challenges. Development of specific imaging correlates of different proteinopathies is a research goal that will offer an opportunity to observe the disease processes in their earliest of stages and do not wait for clinical manifestation. The clinical challenge will be to identify those at risk at the earliest opportunity.
Large longitudinal cohort studies are a necessity to explore the influence of cognitive reserve and early life factors, which are increasingly gaining importance and attention.
Table 2 summarises the pathophysiology and the imaging features of all the dementias discussed (Figure 10). Demonstrates the regional atrophy in FTD and AD.
Table 2.
Summary
| Dementia | Pathological feature | Structural imaging CT/MRI | Molecular imaging (non-specific) | Molecular imaging (specific) | Research |
| Alzheimer’s disease | Primary neurodegenerative, extracellular amyloid plaques (Aβ42), intracellular tau aggregates[13], Autosomal dominant early onset inherited form - presenelins are also implicated[22] | Hippocampal-medial temporal lobe (CA2 and CA3 hippocampal subregions are more affected), posterior cingulate gyrus and postero-medial parietal lobe atrophy on MRI and CT | SPECT1- ↓perfusion FDG PET2- ↓glucose uptake in medial temporal lobe and hippocampi[39-41] | 11C PIB, Florbetapir3 uptake in amyloid plaques[42] | Tau specific ligands -PET, MRI-BOLD, fMRI-↓connectivity in DMN, MR perfusion[38], MR spectroscopy, DTI -↓medial temporal lobe and precuneus[34], VBM |
| LBD | Intracellular Lewy bodies-aggregates of α-synuclein particles in pre-synaptic terminals Overlaps with Parkinson’s disease | Atrophy in inferior frontal lobe, visual cortex, insula, hypothalamus, midbrain, caudate, putamen and anterior hippocampi (CA1 subregion)[86] | SPECT -↓in putamen and caudate, visual cortex[88,89] FDG PET -↓in visual cortices[88-90] | FP-CIT-↓uptake in putamen and caudate[79] Cholinergic PET/SPECT- ↓in medial occipital lobe[95] 123I MIBG-↓cardiac uptake[96] | Diffusion weighted MR-DTI, ↓ in visual association cortex and posterior putamen MRS, fMRI ASL-MR |
| FTD | Various proteins including tauopathies, TDP43, FUS- clinically can overlap with PSP, MSA, MND[100,101] | Variable-predominantly anterior frontal, temporal and insular atrophy[102,103] | FDG PET and SPECT-↓anterior, frontal and temporal uptake[107] | - | fMRI, DTI-↓ in WM of affected regions[104] fMRI-↓"salient" network’ but ↑DMN connectivity on resting fMRI- unlike AD[105,106] |
| Vascular dementia | Small and large vessel disease - vascular risk factors like HT, smoking and DM implicated[61] CADASIL- hereditary form | CT-cortical infarct, macrohaemorrhage, frontal subcortical and periventricular WMH, lacunes[62-67] MRI-CT features as above and PVS, CMB | FDG PET and rCBF SPECT-↓ frontal and periventricular regions | - | - |
| CJD sCJD vCJD | Prion protein - sources include food, tissues, genetic variation | MRI-↑signal on T2W and DWI in the caudate and cortex ("cortical ribboning") MRI-↑ on T2W and DWI in the pulvinar of thalami | |||
| Autoimmune encephalitis related dementia | Previously limbic encephalitis -neuron specific CSF autoantibodies Paraneoplastic syndrome | MRI-↑ signal on T2W and FLAIR in the mesial temporal lobe | FDG PET -↑ uptake in the medial temporal lobe Whole body PET to identify underlying primary malignancy[110] |
SPECT-radiotracer is 99mTc hexamethylpropylene amine oxime;
FDG PET-radiotracer is 18F-FDG;
Recently approved by FDA for clinical use in specific cases, primarily to exclude Alzheimer’s disease.↑: Increased; ↓: Decreased; Aβ42: Beta amyloid protein with 42 amino acids; CA1, CA2, CA3: Subfields of hippocampus; ASL- MR: Arterial spin labelling MR; BOLD: Blood oxygenation level dependent; CADASIL: Congenital autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy; CMB: Cerebral microbleeds; CSF: Cerebrospinal fluid; DM: Diabetes mellitus; DTI: Diffusion tensor imaging; FDG: Fludeoxyglucose; FLAIR: Fluid-attenuated inversion–recovery; fMRI: Functional MRI; DMN: Default mode network; FP-CIT: Dopaminergic presynaptic ligand iodine-123-b-carbo-methoxy-3 b-(4-iodophenyltropane) fluoropropyl; FUS: Fused in sarcoma protein; HT: Hypertension; LBD: Lewy body dementia; MSA: Multisystem atrophy; MND: Motor neuron disease; MRS: MR spectroscopy; PET: Positron emission tomography; PIB: Pittsburgh compound B; PSP: Progressive supranuclear palsy; PVS: Perivascular spaces; rCBF SPECT: Regional cerebral blood flow SPECT; sCJD: Sporadic form of Creutzfeldt–Jacob disease; vCJD: Variant form of Creutzfeldt–Jacob disease; SPECT: Single photon emission computed tomography; T2W: T2 weighted; TDP43: Transactive DNA-binding protein; VBM: Voxel-based morphometry; WMH: White matter hyperintensities.
Figure 10.
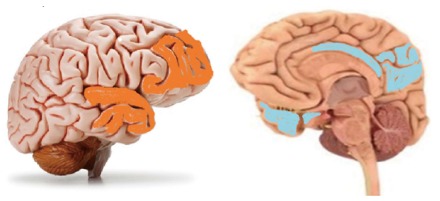
Regions of atrophy in fronto-temporal dementia (shaded orange) and Alzheimer’s disease (shaded light blue).
Footnotes
Conflict-of-interest statement: No conflicts of interest or financial support.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: August 26, 2015
First decision: October 27, 2015
Article in press: January 7, 2016
P- Reviewer: Altamura C S- Editor: Qi Y L- Editor: A E- Editor: Lu YJ
References
- 1. Available from: https://www.alz.co.uk/research/WorldAlzheimerReport2014.pdf.
- 2.Spijker J, MacInnes J. Population ageing: the timebomb that isn’t? BMJ. 2013;347:f6598. doi: 10.1136/bmj.f6598. [DOI] [PubMed] [Google Scholar]
- 3.Haub C. World population aging: clocks illustrate growth in population under age5 and over age 65. Population Bulletin. Available from: http: //www.prb.org/Articles/2011/agingpopulationclocks.aspx.
- 4.Qiu C, Xu W, Fratiglioni L. Vascular and psychosocial factors in Alzheimer’s disease: epidemiological evidence toward intervention. J Alzheimers Dis. 2010;20:689–697. doi: 10.3233/JAD-2010-091663. [DOI] [PubMed] [Google Scholar]
- 5.Dubois B, Feldman HH, Jacova C, Dekosky ST, Barberger-Gateau P, Cummings J, Delacourte A, Galasko D, Gauthier S, Jicha G, et al. Research criteria for the diagnosis of Alzheimer’s disease: revising the NINCDS-ADRDA criteria. Lancet Neurol. 2007;6:734–746. doi: 10.1016/S1474-4422(07)70178-3. [DOI] [PubMed] [Google Scholar]
- 6.Hort J, O’Brien JT, Gainotti G, Pirttila T, Popescu BO, Rektorova I, Sorbi S, Scheltens P. EFNS guidelines for the diagnosis and management of Alzheimer’s disease. Eur J Neurol. 2010;17:1236–1248. doi: 10.1111/j.1468-1331.2010.03040.x. [DOI] [PubMed] [Google Scholar]
- 7.Jack CR, Albert MS, Knopman DS, McKhann GM, Sperling RA, Carrillo MC, Thies B, Phelps CH. Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:257–262. doi: 10.1016/j.jalz.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.NCC for Mental Health. Dementia: The NICE-SCIE Guideline on supporting people with dementia and their carers in health and social care (National Clinical Practice Guideline) USA: British Psychological RCPsych publications; 2007. [PubMed] [Google Scholar]
- 9.Sperling R, Mormino E, Johnson K. The evolution of preclinical Alzheimer’s disease: implications for prevention trials. Neuron. 2014;84:608–622. doi: 10.1016/j.neuron.2014.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bermingham SL. The appropriate use of neuroimaging in the diagnostic work-up of dementia: an economic literature review and cost-effectiveness analysis. Ont Health Technol Assess Ser. 2014;14:1–67. [PMC free article] [PubMed] [Google Scholar]
- 11.Toga AW. The clinical value of large neuroimaging data sets in Alzheimer’s disease. Neuroimaging Clin N Am. 2012;22:107–118, ix. doi: 10.1016/j.nic.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gauthier S, Reisberg B, Zaudig M, Petersen RC, Ritchie K, Broich K, Belleville S, Brodaty H, Bennett D, Chertkow H, et al. Mild cognitive impairment. Lancet. 2006;367:1262–1270. doi: 10.1016/S0140-6736(06)68542-5. [DOI] [PubMed] [Google Scholar]
- 13.Masters CL, Selkoe DJ. Biochemistry of amyloid beta-protein and amyloidz deposits in AD. Cold Spring Harb Perspect Med. 2012;2:a006262. doi: 10.1101/cshperspect.a006262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in AD and Down syndrome. Proc Natl Acad Sci USA. 1985;82:4245–4259. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jack CR, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, Shaw LM, Vemuri P, Wiste HJ, Weigand SD, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207–216. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eikelenboom P, Stam FC. Immunoglobulins and complement factors in senile plaques. An immunoperoxidase study. Acta Neuropathol. 1982;57:239–242. doi: 10.1007/BF00685397. [DOI] [PubMed] [Google Scholar]
- 17.Eikelenboom P, Hack CE, Rozemuller JM, Stam FC. Complement activation in amyloid plaques in Alzheimer’s dementia. Virchows Arch B Cell Pathol Incl Mol Pathol. 1989;56:259–262. doi: 10.1007/BF02890024. [DOI] [PubMed] [Google Scholar]
- 18.Morris JC, Roe CM, Grant EA, Head D, Storandt M, Goate AM, Fagan AM, Holtzman DM, Mintun MA. Pittsburgh compound B imaging and prediction of progression from cognitive normality to symptomatic Alzheimer disease. Arch Neurol. 2009;66:1469–1475. doi: 10.1001/archneurol.2009.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jack CR, Lowe VJ, Weigand SD, Wiste HJ, Senjem ML, Knopman DS, Shiung MM, Gunter JL, Boeve BF, Kemp BJ, et al. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer’s disease: implications for sequence of pathological events in Alzheimer’s disease. Brain. 2009;132:1355–1365. doi: 10.1093/brain/awp062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Delacourte A, David JP, Sergeant N, Buée L, Wattez A, Vermersch P, Ghozali F, Fallet-Bianco C, Pasquier F, Lebert F, et al. The biochemical pathway of neurofibrillary degeneration in aging and Alzheimer’s disease. Neurology. 1999;52:1158–1165. doi: 10.1212/wnl.52.6.1158. [DOI] [PubMed] [Google Scholar]
- 21.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 22.Bertram L, Tanzi RE. Thirty years of Alzheimer’s disease genetics: the implications of systematic meta-analyses. Nat Rev Neurosci. 2008;9:768–778. doi: 10.1038/nrn2494. [DOI] [PubMed] [Google Scholar]
- 23.Chang YL, Fennema-Notestine C, Holland D, McEvoy LK, Stricker NH, Salmon DP, Dale AM, Bondi MW. APOE interacts with age to modify rate of decline in cognitive and brain changes in Alzheimer’s disease. Alzheimers Dement. 2014;10:336–348. doi: 10.1016/j.jalz.2013.05.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li YJ, Hauser MA, Scott WK, Martin ER, Booze MW, Qin XJ, Walter JW, Nance MA, Hubble JP, Koller WC, et al. Apolipoprotein E controls the risk and age at onset of Parkinson disease. Neurology. 2004;62:2005–2009. doi: 10.1212/01.wnl.0000128089.53030.ac. [DOI] [PubMed] [Google Scholar]
- 25.McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263–269. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR, Kaye J, Montine TJ, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, Gamst A, Holtzman DM, Jagust WJ, Petersen RC, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:270–279. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 29.Harper L, Barkhof F, Scheltens P, Schott JM, Fox NC. An algorithmic approach to structural imaging in dementia. J Neurol Neurosurg Psychiatry. 2014;85:692–698. doi: 10.1136/jnnp-2013-306285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ikram MA, Vrooman HA, Vernooij MW, den Heijer T, Hofman A, Niessen WJ, van der Lugt A, Koudstaal PJ, Breteler MM. Brain tissue volumes in relation to cognitive function and risk of dementia. Neurobiol Aging. 2010;31:378–386. doi: 10.1016/j.neurobiolaging.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 31.Duara R, Loewenstein DA, Potter E, Appel J, Greig MT, Urs R, Shen Q, Raj A, Small B, Barker W, et al. Medial temporal lobe atrophy on MRI scans and the diagnosis of Alzheimer disease. Neurology. 2008;71:1986–1992. doi: 10.1212/01.wnl.0000336925.79704.9f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frisoni GB, Fox NC, Jack CR, Scheltens P, Thompson PM. The clinical use of structural MRI in Alzheimer disease. Nat Rev Neurol. 2010;6:67–77. doi: 10.1038/nrneurol.2009.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jack CR Jr, Bernstein MA, Borowski BJ, Gunter JL, Fox NC, Thompson PM, Schuff N, Krueger G, Killiany RJ, Decarli CS, et al. Update on the magnetic resonance imaging core of the Alzheimer’s disease neuroimaging initiative. Alzheimers Dement. 2010;6:212–220. doi: 10.1016/j.jalz.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao F, Barker PB. Various MRS application tools for Alzheimer disease and mild cognitive impairment. AJNR Am J Neuroradiol. 2014;35:S4–11. doi: 10.3174/ajnr.A3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alsop DC, Dai W, Grossman M, Detre JA. Arterial spin labeling blood flow MRI: its role in the early characterization of Alzheimer’s disease. J Alzheimers Dis. 2010;20:871–880. doi: 10.3233/JAD-2010-091699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Saykin AJ, Pfeuffer J, Lin C, Mosier KM, Shen L, Kim S, Hutchins GD. Regional reproducibility of pulsed arterial spin labeling perfusion imaging at 3T. Neuroimage. 2011;54:1188–1195. doi: 10.1016/j.neuroimage.2010.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ogawa S, Tank DW, Menon R, Ellermann JM, Kim SG, Merkle H, Ugurbil K. Intrinsic signal changes accompanying sensory stimulation: functional brain mapping with magnetic resonance imaging. Proc Natl Acad Sci USA. 1992;89:5951–5955. doi: 10.1073/pnas.89.13.5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hafkemeijer A, van der Grond J, Rombouts SA. Imaging the default mode network in aging and dementia. Biochim Biophys Acta. 2012;1822:431–441. doi: 10.1016/j.bbadis.2011.07.008. [DOI] [PubMed] [Google Scholar]
- 39.Shivamurthy VK, Tahari AK, Marcus C, Subramaniam RM. Brain FDG PET and the diagnosis of dementia. AJR Am J Roentgenol. 2015;204:W76–W85. doi: 10.2214/AJR.13.12363. [DOI] [PubMed] [Google Scholar]
- 40.Sanabria-Diaz G, Martínez-Montes E, Melie-Garcia L. Glucose metabolism during resting state reveals abnormal brain networks organization in the Alzheimer’s disease and mild cognitive impairment. PLoS One. 2013;8:e68860. doi: 10.1371/journal.pone.0068860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Del Sole A, Clerici F, Chiti A, Lecchi M, Mariani C, Maggiore L, Mosconi L, Lucignani G. Individual cerebral metabolic deficits in Alzheimer’s disease and amnestic mild cognitive impairment: an FDG PET study. Eur J Nucl Med Mol Imaging. 2008;35:1357–1366. doi: 10.1007/s00259-008-0773-6. [DOI] [PubMed] [Google Scholar]
- 42.Clark CM, Schneider JA, Bedell BJ, Beach TG, Bilker WB, Mintun MA, Pontecorvo MJ, Hefti F, Carpenter AP, Flitter ML, et al. Use of florbetapir-PET for imaging beta-amyloid pathology. JAMA. 2011;305:275–283. doi: 10.1001/jama.2010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jack CR, Lowe VJ, Senjem ML, Weigand SD, Kemp BJ, Shiung MM, Knopman DS, Boeve BF, Klunk WE, Mathis CA, et al. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer’s disease and amnestic mild cognitive impairment. Brain. 2008;131:665–680. doi: 10.1093/brain/awm336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergström M, Savitcheva I, Huang GF, Estrada S, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 45.Dubois B, Feldman HH, Jacova C, Hampel H, Molinuevo JL, Blennow K, DeKosky ST, Gauthier S, Selkoe D, Bateman R, et al. Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol. 2014;13:614–629. doi: 10.1016/S1474-4422(14)70090-0. [DOI] [PubMed] [Google Scholar]
- 46.Zwan MD, Okamura N, Fodero-Tavoletti MT, Furumoto S, Masters CL, Rowe CC, Villemagne VL. Voyage au bout de la nuit: Aβ and tau imaging in dementias. Q J Nucl Med Mol Imaging. 2014;58:398–412. [PubMed] [Google Scholar]
- 47.Ossenkoppele R, Jansen WJ, Rabinovici GD, Knol DL, van der Flier WM, van Berckel BN, Scheltens P, Visser PJ, Verfaillie SC, Zwan MD, et al. Prevalence of amyloid PET positivity in dementia syndromes: a meta-analysis. JAMA. 2015;313:1939–1949. doi: 10.1001/jama.2015.4669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kadir A, Almkvist O, Forsberg A, Wall A, Engler H, Långström B, Nordberg A. Dynamic changes in PET amyloid and FDG imaging at different stages of Alzheimer’s disease. Neurobiol Aging. 2012;33:198.e1–198.14. doi: 10.1016/j.neurobiolaging.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 49.Gordon E, Rohrer JD, Kim LG, Omar R, Rossor MN, Fox NC, Warren JD. Measuring disease progression in frontotemporal lobar degeneration: a clinical and MRI study. Neurology. 2010;74:666–673. doi: 10.1212/WNL.0b013e3181d1a879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Steinacker P, Hendrich C, Sperfeld AD, Jesse S, von Arnim CA, Lehnert S, Pabst A, Uttner I, Tumani H, Lee VM, et al. TDP-43 in cerebrospinal fluid of patients with frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Arch Neurol. 2008;65:1481–1487. doi: 10.1001/archneur.65.11.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kasai T, Tokuda T, Ishigami N, Sasayama H, Foulds P, Mitchell DJ, Mann DM, Allsop D, Nakagawa M. Increased TDP-43 protein in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta Neuropathol. 2009;117:55–62. doi: 10.1007/s00401-008-0456-1. [DOI] [PubMed] [Google Scholar]
- 52.Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ahmed RM, Paterson RW, Warren JD, Zetterberg H, O’Brien JT, Fox NC, Halliday GM, Schott JM. Biomarkers in dementia: clinical utility and new directions. J Neurol Neurosurg Psychiatry. 2014;85:1426–1434. doi: 10.1136/jnnp-2014-307662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Royal College of Radiologists. Evidence based indications for the use of PET-CT in the UK 2013. Available from: https://www.rcr.ac.uk/sites/default/files/publication/2013_PETCT_RCP_RCR.pdf.
- 55.Seppälä TT, Nerg O, Koivisto AM, Rummukainen J, Puli L, Zetterberg H, Pyykkö OT, Helisalmi S, Alafuzoff I, Hiltunen M, et al. CSF biomarkers for Alzheimer disease correlate with cortical brain biopsy findings. Neurology. 2012;78:1568–1575. doi: 10.1212/WNL.0b013e3182563bd0. [DOI] [PubMed] [Google Scholar]
- 56.Moghekar A, Li S, Lu Y, Li M, Wang MC, Albert M, O’Brien R. CSF biomarker changes precede symptom onset of mild cognitive impairment. Neurology. 2013;81:1753–1758. doi: 10.1212/01.wnl.0000435558.98447.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rinne JO, Kaasinen V, Järvenpää T, Någren K, Roivainen A, Yu M, Oikonen V, Kurki T. Brain acetylcholinesterase activity in mild cognitive impairment and early Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2003;74:113–115. doi: 10.1136/jnnp.74.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Herholz K, Weisenbach S, Kalbe E, Diederich NJ, Heiss WD. Cerebral acetylcholine esterase activity in mild cognitive impairment. Neuroreport. 2005;16:1431–1434. doi: 10.1097/01.wnr.0000177007.21732.7b. [DOI] [PubMed] [Google Scholar]
- 59.Sabri O, Kendziorra K, Wolf H, Gertz HJ, Brust P. Acetylcholine receptors in dementia and mild cognitive impairment. Eur J Nucl Med Mol Imaging. 2008;35 Suppl 1:S30–S45. doi: 10.1007/s00259-007-0701-1. [DOI] [PubMed] [Google Scholar]
- 60.Jack CR, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9:119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bowler JV. Modern concept of vascular cognitive impairment. Br Med Bull. 2007;83:291–305. doi: 10.1093/bmb/ldm021. [DOI] [PubMed] [Google Scholar]
- 62.Kearney-Schwartz A, Rossignol P, Bracard S, Felblinger J, Fay R, Boivin JM, Lecompte T, Lacolley P, Benetos A, Zannad F. Vascular structure and function is correlated to cognitive performance and white matter hyperintensities in older hypertensive patients with subjective memory complaints. Stroke. 2009;40:1229–1236. doi: 10.1161/STROKEAHA.108.532853. [DOI] [PubMed] [Google Scholar]
- 63.Raz N, Rodrigue KM, Kennedy KM, Acker JD. Vascular health and longitudinal changes in brain and cognition in middle-aged and older adults. Neuropsychology. 2007;21:149–157. doi: 10.1037/0894-4105.21.2.149. [DOI] [PubMed] [Google Scholar]
- 64.Fischer P, Krampla W, Mostafaie N, Zehetmayer S, Rainer M, Jungwirth S, Huber K, Bauer K, Hruby W, Riederer P, et al. VITA study: white matter hyperintensities of vascular and degenerative origin in the elderly. J Neural Transm Suppl. 2007;(72):181–188. doi: 10.1007/978-3-211-73574-9_23. [DOI] [PubMed] [Google Scholar]
- 65.Jagust WJ, Zheng L, Harvey DJ, Mack WJ, Vinters HV, Weiner MW, Ellis WG, Zarow C, Mungas D, Reed BR, et al. Neuropathological basis of magnetic resonance images in aging and dementia. Ann Neurol. 2008;63:72–80. doi: 10.1002/ana.21296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Doubal FN, MacLullich AM, Ferguson KJ, Dennis MS, Wardlaw JM. Enlarged perivascular spaces on MRI are a feature of cerebral small vessel disease. Stroke. 2010;41:450–454. doi: 10.1161/STROKEAHA.109.564914. [DOI] [PubMed] [Google Scholar]
- 67.Vernooij MW, de Groot M, van der Lugt A, Ikram MA, Krestin GP, Hofman A, Niessen WJ, Breteler MM. White matter atrophy and lesion formation explain the loss of structural integrity of white matter in aging. Neuroimage. 2008;43:470–477. doi: 10.1016/j.neuroimage.2008.07.052. [DOI] [PubMed] [Google Scholar]
- 68.Murray AD, Staff RT, Shenkin SD, Deary IJ, Starr JM, Whalley LJ. Brain white matter hyperintensities: relative importance of vascular risk factors in nondemented elderly people. Radiology. 2005;237:251–257. doi: 10.1148/radiol.2371041496. [DOI] [PubMed] [Google Scholar]
- 69.Hunt AL, Orrison WW, Yeo RA, Haaland KY, Rhyne RL, Garry PJ, Rosenberg GA. Clinical significance of MRI white matter lesions in the elderly. Neurology. 1989;39:1470–1474. doi: 10.1212/wnl.39.11.1470. [DOI] [PubMed] [Google Scholar]
- 70.Inzitari D, Pracucci G, Poggesi A, Carlucci G, Barkhof F, Chabriat H, Erkinjuntti T, Fazekas F, Ferro JM, Hennerici M, et al. Changes in white matter as determinant of global functional decline in older independent outpatients: three year follow-up of LADIS (leukoaraiosis and disability) study cohort. BMJ. 2009;339:b2477. doi: 10.1136/bmj.b2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jokinen H, Kalska H, Ylikoski R, Madureira S, Verdelho A, van der Flier WM, Scheltens P, Barkhof F, Visser MC, Fazekas F, et al. Longitudinal cognitive decline in subcortical ischemic vascular disease--the LADIS Study. Cerebrovasc Dis. 2009;27:384–391. doi: 10.1159/000207442. [DOI] [PubMed] [Google Scholar]
- 72.Rosenberg GA, Bjerke M, Wallin A. Multimodal markers of inflammation in the subcortical ischemic vascular disease type of vascular cognitive impairment. Stroke. 2014;45:1531–1538. doi: 10.1161/STROKEAHA.113.004534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fazekas F, Chawluk JB, Alavi A, Hurtig HI, Zimmerman RA. MR signal abnormalities at 1.5 T in Alzheimer’s dementia and normal aging. AJR Am J Roentgenol. 1987;149:351–356. doi: 10.2214/ajr.149.2.351. [DOI] [PubMed] [Google Scholar]
- 74.Scheltens P, Barkhof F, Leys D, Pruvo JP, Nauta JJP, Vermersch P, Steinling M, Valk J. A semiquantitative rating scale for the assessment of signal hyperintensities on magnetic resonance imaging. J Neurol Sci. 1993;114:7–12. doi: 10.1016/0022-510x(93)90041-v. [DOI] [PubMed] [Google Scholar]
- 75.de Boer R, Vrooman HA, van der Lijn F, Vernooij MW, Ikram MA, van der Lugt A, Breteler MM, Niessen WJ. White matter lesion extension to automatic brain tissue segmentation on MRI. Neuroimage. 2009;45:1151–1161. doi: 10.1016/j.neuroimage.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 76.Wallin A, Blennow K, Fredman P, Gottfries CG, Karlsson I, Svennerholm L. Blood brain barrier function in vascular dementia. Acta Neurol Scand. 1990;81:318–322. doi: 10.1111/j.1600-0404.1990.tb01562.x. [DOI] [PubMed] [Google Scholar]
- 77.Hachinski V, Iadecola C, Petersen RC, Breteler MM, Nyenhuis DL, Black SE, Powers WJ, DeCarli C, Merino JG, Kalaria RN, et al. National Institute of Neurological Disorders and Stroke-Canadian Stroke Network vascular cognitive impairment harmonization standards. Stroke. 2006;37:2220–2241. doi: 10.1161/01.STR.0000237236.88823.47. [DOI] [PubMed] [Google Scholar]
- 78.Roman GC, Erkinjuntti T, Wallin A, Pantoni L, Chui HC. Subcortical ischaemic vascular dementia. Lancet Neurol. 2002;1:426–436. doi: 10.1016/s1474-4422(02)00190-4. [DOI] [PubMed] [Google Scholar]
- 79.McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, Cummings J, Duda JE, Lippa C, Perry EK, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65:1863–1872. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- 80.Bekris LM, Mata IF, Zabetian CP. The genetics of Parkinson disease. J Geriatr Psychiatry Neurol. 2010;23:228–242. doi: 10.1177/0891988710383572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Leverenz JB, Quinn JF, Zabetian C, Zhang J, Montine KS, Montine TJ. Cognitive impairment and dementia in patients with Parkinson disease. Curr Top Med Chem. 2009;9:903–912. [PMC free article] [PubMed] [Google Scholar]
- 82.Burton EJ, Karas G, Paling SM, Barber R, Williams ED, Ballard CG, McKeith IG, Scheltens P, Barkhof F, O’Brien JT. Patterns of cerebral atrophy in dementia with Lewy bodies using voxel-based morphometry. Neuroimage. 2002;17:618–630. [PubMed] [Google Scholar]
- 83.Burton EJ, McKeith IG, Burn DJ, Williams ED, O’Brien JT. Cerebral atrophy in Parkinson’s disease with and without dementia: a comparison with Alzheimer’s disease, dementia with Lewy bodies and controls. Brain. 2004;127:791–800. doi: 10.1093/brain/awh088. [DOI] [PubMed] [Google Scholar]
- 84.Whitwell JL, Weigand SD, Shiung MM, Boeve BF, Ferman TJ, Smith GE, Knopman DS, Petersen RC, Benarroch EE, Josephs KA, et al. Focal atrophy in dementia with Lewy bodies on MRI: a distinct pattern from Alzheimer’s disease. Brain. 2007;130:708–719. doi: 10.1093/brain/awl388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Firbank MJ, Blamire AM, Teodorczuk A, Teper E, Burton EJ, Mitra D, O’Brien JT. High resolution imaging of the medial temporal lobe in Alzheimer’s disease and dementia with Lewy bodies. J Alzheimers Dis. 2010;21:1129–1140. doi: 10.3233/jad-2010-100138. [DOI] [PubMed] [Google Scholar]
- 86.Kantarci K, Lowe VJ, Boeve BF, Weigand SD, Senjem ML, Przybelski SA, Dickson DW, Parisi JE, Knopman DS, Smith GE, et al. Multimodality imaging characteristics of dementia with Lewy bodies. Neurobiol Aging. 2012;33:2091–2105. doi: 10.1016/j.neurobiolaging.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rowe CC, Ng S, Ackermann U, Gong SJ, Pike K, Savage G, Cowie TF, Dickinson KL, Maruff P, Darby D, et al. Imaging beta-amyloid burden in aging and dementia. Neurology. 2007;68:1718–1725. doi: 10.1212/01.wnl.0000261919.22630.ea. [DOI] [PubMed] [Google Scholar]
- 88.Ishii K, Imamura T, Sasaki M, Yamaji S, Sakamoto S, Kitagaki H, Hashimoto M, Hirono N, Shimomura T, Mori E. Regional cerebral glucose metabolism in dementia with Lewy bodies and Alzheimer’s disease. Neurology. 1998;51:125–130. doi: 10.1212/wnl.51.1.125. [DOI] [PubMed] [Google Scholar]
- 89.Teune LK, Bartels AL, de Jong BM, Willemsen AT, Eshuis SA, de Vries JJ, van Oostrom JC, Leenders KL. Typical cerebral metabolic patterns in neurodegenerative brain diseases. Mov Disord. 2010;25:2395–2404. doi: 10.1002/mds.23291. [DOI] [PubMed] [Google Scholar]
- 90.Ishii K, Soma T, Kono AK, Sofue K, Miyamoto N, Yoshikawa T, Mori E, Murase K. Comparison of regional brain volume and glucose metabolism between patients with mild dementia with lewy bodies and those with mild Alzheimer’s disease. J Nucl Med. 2007;48:704–711. doi: 10.2967/jnumed.106.035691. [DOI] [PubMed] [Google Scholar]
- 91.Booij J, Speelman JD, Horstink MWIM, Wolters EC. The clinical benefit of imaging striatal dopamine transporters with FP-CIT SPECT in differentiating patients with presynaptic parkinsonism from those with other forms of parkinsonism. Eur J Nucl Med. 2001;28:266–272. doi: 10.1007/s002590000460. [DOI] [PubMed] [Google Scholar]
- 92.Staff RT, Ahearn TS, Wilson K, Counsell CE, Taylor K, Caslake R, Davidson JE, Gemmell HG, Murray AD. Shape analysis of 123I-N-omega-fluoropropyl-2-beta-carbomethoxy-3beta-(4-iodophenyl) nortropane single-photon emission computed tomography images in the assessment of patients with parkinsonian syndromes. Nucl Med Commun. 2009;30:194–201. doi: 10.1097/MNM.0b013e328314b863. [DOI] [PubMed] [Google Scholar]
- 93.Tatsch K, Poepperl G. Nigrostriatal dopamine terminal imaging with dopamine transporter SPECT: an update. J Nucl Med. 2013;54:1331–1338. doi: 10.2967/jnumed.112.105379. [DOI] [PubMed] [Google Scholar]
- 94.O’Brien JT. Role of imaging techniques in the diagnosis of dementia. Br J Radiol. 2007;80 Spec No 2:S71–S77. doi: 10.1259/bjr/33117326. [DOI] [PubMed] [Google Scholar]
- 95.Shimada H, Hirano S, Shinotoh H, Aotsuka A, Sato K, Tanaka N, Ota T, Asahina M, Fukushi K, Kuwabara S, et al. Mapping of brain acetylcholinesterase alterations in Lewy body disease by PET. Neurology. 2009;73:273–278. doi: 10.1212/WNL.0b013e3181ab2b58. [DOI] [PubMed] [Google Scholar]
- 96.Yoshita M, Taki J, Yokoyama K, Noguchi-Shinohara M, Matsumoto Y, Nakajima K, Yamada M. Value of 123I-MIBG radioactivity in the differential diagnosis of DLB from AD. Neurology. 2006;66:1850–1854. doi: 10.1212/01.wnl.0000219640.59984.a7. [DOI] [PubMed] [Google Scholar]
- 97.Ratnavalli E, Brayne C, Dawson K, Hodges JR. The prevalence of frontotemporal dementia. Neurology. 2002;58:1615–1621. doi: 10.1212/wnl.58.11.1615. [DOI] [PubMed] [Google Scholar]
- 98.Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–1554. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- 99.Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, Ogar JM, Rohrer JD, Black S, Boeve BF, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76:1006–1014. doi: 10.1212/WNL.0b013e31821103e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hu WT, Seelaar H, Josephs KA, Knopman DS, Boeve BF, Sorenson EJ, McCluskey L, Elman L, Schelhaas HJ, Parisi JE, et al. Survival profiles of patients with frontotemporal dementia and motor neuron disease. Arch Neurol. 2009;66:1359–1364. doi: 10.1001/archneurol.2009.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rohrer JD. Structural brain imaging in frontotemporal dementia. Biochim Biophys Acta. 2012;1822:325–332. doi: 10.1016/j.bbadis.2011.07.014. [DOI] [PubMed] [Google Scholar]
- 102.Rosen HJ, Gorno-Tempini ML, Goldman WP, Perry RJ, Schuff N, Weiner M, Feiwell R, Kramer JH, Miller BL. Patterns of brain atrophy in frontotemporal dementia and semantic dementia. Neurology. 2002;58:198–208. doi: 10.1212/wnl.58.2.198. [DOI] [PubMed] [Google Scholar]
- 103.Jason DW, Jonathan DR. Frontotemporal dementia. BMJ. 2013;6:347. [Google Scholar]
- 104.Risacher SL, Saykin AJ. Neuroimaging biomarkers of neurodegenerative diseases and dementia. Semin Neurol. 2013;33:386–416. doi: 10.1055/s-0033-1359312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pievani M, de Haan W, Wu T, Seeley WW, Frisoni GB. Functional network disruption in the degenerative dementias. Lancet Neurol. 2011;10:829–843. doi: 10.1016/S1474-4422(11)70158-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhou J, Greicius MD, Gennatas ED, Growdon ME, Jang JY, Rabinovici GD, Kramer JH, Weiner M, Miller BL, Seeley WW. Divergent network connectivity changes in behavioural variant frontotemporal dementia and Alzheimer’s disease. Brain. 2010;133(Pt 5):1352–1367. doi: 10.1093/brain/awq075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rohrer JD, Schott JM. Primary progressive aphasia: defining genetic and pathological subtypes. Curr Alzheimer Res. 2011;8:266–272. doi: 10.2174/156720511795563728. [DOI] [PubMed] [Google Scholar]
- 108.Rossor MN, Fox NC, Mummery CJ, Schott JM, Warren JD. The diagnosis of young-onset dementia. Lancet Neurol. 2010;9:793–806. doi: 10.1016/S1474-4422(10)70159-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Flanagan EP, McKeon A, Lennon VA, Boeve BF, Trenerry MR, Tan KM, Drubach DA, Josephs KA, Britton JW, Mandrekar JN, et al. Autoimmune dementia: clinical course and predictors of immunotherapy response. Mayo Clin Proc. 2010;85:881–897. doi: 10.4065/mcp.2010.0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Titulaer MJ, Soffietti R, Dalmau J, Gilhus NE, Giometto B, Graus F, Grisold W, Honnorat J, Sillevis Smitt PA, Tanasescu R, et al. Screening for tumours in paraneoplastic syndromes: report of an EFNS task force. Eur J Neurol. 2011;18:19–e3. doi: 10.1111/j.1468-1331.2010.03220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wu HM, Lu CS, Huang CC, Lyu RK, Chen RS, Wu YR, Chen YC, Lai SC, Hung J, Tsai YT, et al. Asymmetric involvement in sporadic creutzfeldt-jakob disease: clinical, brain imaging, and electroencephalographic studies. Eur Neurol. 2010;64:74–79. doi: 10.1159/000315148. [DOI] [PubMed] [Google Scholar]
- 112.Chang L, Ernst T, Tornatore C, Aronow H, Melchor R, Walot I, Singer E, Cornford M. Metabolite abnormalities in progressive multifocal leukoencephalopathy by proton magnetic resonance spectroscopy. Neurology. 1997;48:836–845. doi: 10.1212/wnl.48.4.836. [DOI] [PubMed] [Google Scholar]
- 113.Mark AS, Atlas SW. Progressive multifocal leukoencephalopathy in patients with AIDS: appearance on MR images. Radiology. 1989;173:517–520. doi: 10.1148/radiology.173.2.2798883. [DOI] [PubMed] [Google Scholar]
- 114.Ragin AB, Storey P, Cohen BA, Epstein LG, Edelman RR. Whole brain diffusion tensor imaging in HIV-associated cognitive impairment. AJNR Am J Neuroradiol. 2004;25:195–200. [PMC free article] [PubMed] [Google Scholar]
- 115.Tate DF, Conley J, Paul RH, Coop K, Zhang S, Zhou W, Laidlaw DH, Taylor LE, Flanigan T, Navia B, et al. Quantitative diffusion tensor imaging tractography metrics are associated with cognitive performance among HIV-infected patients. Brain Imaging Behav. 2010;4:68–79. doi: 10.1007/s11682-009-9086-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Leite SC, Corrêa DG, Doring TM, Kubo TT, Netto TM, Ferracini R, Ventura N, Bahia PR, Gasparetto EL. Diffusion tensor MRI evaluation of the corona radiata, cingulate gyri, and corpus callosum in HIV patients. J Magn Reson Imaging. 2013;38:1488–1493. doi: 10.1002/jmri.24129. [DOI] [PubMed] [Google Scholar]