

Abstract
Tumor necrosis factor-α (TNF) plays a prominent role in the brain damage and functional deficits that result from ischemic stroke. It was recently reported that the thalidomide analog 3,6′-dithiothalidomide (3,6′-DT) can selectively inhibit the synthesis of TNF in cultured cells. We therefore tested the therapeutic potential of 3,6′-DT in a mouse model of focal ischemic stroke. Administration of 3,6′-DT immediately prior to a stroke or within 3 hr after the stroke reduced infarct volume, neuronal death, and neurological deficits, whereas thalidomide was effective only when administered prior to stroke. Neuroprotection was accompanied by decreased inflammation; 3,6′-DT-treated mice exhibited reduced expression of TNF, interleukin-1β, and inducible nitric oxide synthase; reduced numbers of activated microglia/macrophages, astrocytes, and neutrophils; and reduced expression of intercellular adhesion molecule-1 in the ischemic brain tissue. 3,6′-DT treatment attenuated stroke-induced disruption of the blood–brain barrier by a mechanism that appears to involve suppression of matrix metalloproteinase-9 and preservation of occludin. Treatment with 3,6′-DT did not reduce ischemic brain damage in mice lacking TNF receptors, consistent with a critical role for suppression of TNF production and TNF signaling in the therapeutic action of 3,6′-DT. These findings suggest that anti-inflammatory mechanisms underlie the therapeutic actions of 3,6-DT in an animal model of stroke.
Keywords: ICAM-1, inflammation, ischemia, microglia, neuroprotection, occlusion, stroke, thalidomide, TNF
Stroke remains a major cause of morbidity and mortality worldwide (Roger et al., 2012). Inflammatory responses involving both innate and humoral immune systems contribute to neuronal damage and associated functional deficits in stroke (Wang et al., 2007; Denes et al., 2010). The inflammatory response is characterized by the local expression of proinflammatory cytokines, including tumor necrosis factor-α (TNF) and interleukin-1β (IL-1β) from glial cells (microglia and astrocytes), and by infiltration of circulating lymphocytes and macrophages (Tuttolomondo et al., 2008). The proinflammatory cytokines and microglial activation during the acute phase of ischemic stroke may damage neurons by increasing oxidative stress and promoting excitotoxicity (Buttini et al., 1994; Liu et al., 1994; Mattson, 2003; Offner et al., 2006). Contributing to the brain damage and attendant functional deficits is TNF-mediated expression of inducible nitric oxide synthase (iNOS) in microglia and macrophages (Iaedecola et al., 1995; del Zoppo et al., 2005; Lambertsen et al., 2012).
Inflammation-related molecular and cellular alterations occurring at the blood–brain barrier (BBB) are also believed to contribute to stroke outcome. Cerebral vascular endothelial cells respond to ischemia by producing cell adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1) that bind complementary cell surface adhesion molecules on lymphocytes, allowing migration of the lymphocytes across the BBB and into the brain parenchyma (Stanimirovic and Satoh, 2000). In addition to these proadhesive actions mediated by adhesion molecule such as ICAM-1, TNF plays a role in the induction of ICAM-1 expression and also promotes BBB disruption by inducing the expression of matrix metalloproteinase-9 (MMP-9; Mayhan, 2002; Pan and Kastin, 2007). Previous studies have established the cellular sources of MMP-9 expression in the same stroke model that we used for the present study. During the first 24–48 hr poststroke, MMP-9 is produced in high amounts by activated glial cells and is also produced by neurons and vascular endothelial cells (Romanic et al., 1998; Zhao et al., 2006; Michaluk and Kaczmarek, 2007). MMP-9 increases BBB permeability by degrading tight junction proteins, most notably occludin, leading to BBB leakage and secondary hemorrhagic brain damage (Romanic et al., 1998; Zhao et al., 2006; McColl et al., 2008; Liu et al., 2009). Blocking the innate and humoral immune responses, by deletion of pertinent genes or administration of antibodies against the proteins that they encode, can reduce brain damage and improve functional outcome in mouse stroke models (Arumugam et al., 2007; Tang et al., 2007; Shah et al., 2009).
Given that TNF triggers or facilitates many of the major inflammatory processes involved in the pathogenesis of ischemic brain injury, drugs that suppress TNF production or block its receptors are potential therapeutic agents for stroke patients. Thalidomide (T) is an old drug with anti-inflammatory and anticancer activities that was temporarily “abandoned” because of its teratogenic effects in developing fetuses. However, there has been a resurgence in clinical use of T for the treatment of a range of inflammatory conditions and cancers (Laffitte and Revuz, 2004). T can suppress TNF production in various models of peripheral inflammation, and we have developed a series of synthetic analogs of T exhibiting increased potency in inhibiting TNF production, among which 3,6′-dithiothalidomide (3,6′-DT) is particularly effective (Zhu et al., 2003; Greig et al., 2004; Tweedie et al., 2009). Moreover, 3,6′-DT readily enters the CNS and can reduce TNF levels in the brain in animal models of neuroinflammation (Baratz et al., 2011; Belarbi et al., 2012; Tweedie et al., 2012). Here we show that poststroke treatment with 3,6′-DT is highly effective in suppressing a spectrum of neuroinflammatory processes, preserving BBB integrity and neuronal viability, and improving functional outcome.
MATERIALS AND METHODS
Animals
Male C57BL/6 mice were purchased from the Jackson Laboratories (Bar Harbor, ME). Homozygous TNF-p55/p75 receptor knockout (TNFR-KO) mice on a C57BL/6 × 129 background (Zheng et al., 1995) and wild-type (WT) mice of the same genetic background were maintained in the animal facility of the Biomedical Research Center at the National Institute on Aging Intramural Research Program in Baltimore. All procedures were in accordance with the NIH guidelines for the care and use of animals in research and were performed with approval from the National Institute on Aging Animal Care and Use Committee.
Focal Ischemic Stroke Model and Evaluation of Functional Outcome and Brain Damage
The middle cerebral artery occlusion/reperfusion (MCAO/R) stroke model was performed via the intraluminal suture technique with 1 hr of MCAO as reported previously (Canugovi et al., 2012) and described in the Supporting Information. Cerebral blood flow was monitored by laser Doppler flowmetry, and only mice in which the blood flow was reduced by at least 85% below the baseline level were included in the study. The functional consequences of MCAO injury were evaluated using a five-point neurological deficit score (0, no deficit; 1, failure to extend left paw; 2, circling to the left; 3, falling to the left; 4, unable to the walk spontaneously) and were assessed in a blinded fashion. Immediately after behavioral evaluation, mice were euthanized, and 2-mm-thick coronal brain sections were prepared and stained with 2% 2,3,5-triphenyltetrazolium chloride (TTC) and evaluated for infarct size using standard methodology (Swanson and Sharp, 1994), as described in the Supporting Information. To quantify neuronal death, brain tissue sections were stained by using the TUNEL method and caspase-3 immunoreactivity, and positive (apoptotic) cells were counted (see Supporting Information for details).
Drug Treatment
3,6′-DT and T were prepared as a suspension in 1% carboxymethyl cellulose (CMC) and were administered by intraperitoneal (i.p.) injection. For pretreatment studies, mice were injected with 3,6′-DT (14, 28, 56, or 112 mg/kg) or T (50 mg/kg) or vehicle (1% CMC) 1 hr before MCAO/R. For poststroke treatments, 3,6′-DT (56 mg/kg) or T (50 mg/kg) was administered 1 or 3 hr after MCAO/R. TNFR-KO mice and corresponding WT mice were injected with 3,6′-DT (56 mg/kg) or vehicle (1% CMC) 3 hr after MCAO/R. The 56 mg/kg concentration of 3,6′-DT is equimolar to 50 mg/kg T.
Measurements of Cytokine Concentrations
Mice were euthanized, and brains were quickly removed. Both the ipsilateral (stroke side) and the contralateral hemispheres were then dissected. Each hemisphere was homogenized in PBS, and the homogenates were centrifuged at 4°C and 14,000 rpm for 15 min. Supernatants were assayed in duplicate by using TNF and IL-1β assay kits (eBioscience, San Diego, CA) according to the manufacturer’s guidelines. The cytokine contents in the brain tissue samples were expressed as picograms per milligram protein.
Histochemistry
The ischemic penumbra was defined by examination of sections stained with hematoxylin and eosin as described previously (Borlongan et al., 2000). The ischemic core was defined as the region where there was no detectable survival of neurons or glial cells. The ischemic penumbra was defined as the region immediately adjacent to the core that contained viable neurons and abundant glial cells. Glial fibrillary acidic protein (GFAP), Iba-1 (specific for microglia and macrophage), ICAM-1, iNOS, and terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labeling (TUNEL) staining were performed on brain sections from mice euthanized 72 hr after the stroke. Myeloperoxidase (MPO) and MMP-9 stainings were performed on brain sections from mice euthanized 24 and 48 hr after the stroke, respectively. Digital images of brain sections were acquired and analyzed as described in the Supporting Information.
Assessment of BBB Integrity
BBB integrity was evaluated by Evans blue extravasation. Evans blue (4% in saline, 4 ml/kg; Sigma, St. Louis, MO) was intravenously administered via tail vein at 48 hr poststroke. One hour later, the mice were anesthetized and perfused transcardially with saline to remove the intravascular dye. While still under anesthesia, the mice were decapitated, brains were quickly removed, images of the entire dorsal surface of the brain were acquired, and then each cerebral hemisphere was homogenized in 1 ml 50% trichloroacetic acid. Samples were then centrifuged at 1,000g for 30 min, and relative levels of Evans blue in the supernatants were determined by spectrophotometry (absorbance at 620 nm λ). The changes in BBB integrity after cerebral ischemia were expressed as the ratio of absorbance in the ipsilateral hemisphere to that in the contralateral hemisphere (Evans blue index).
Immunoblot Analysis
For immunoblotting, brain tissue samples were homogenized in a lysis buffer, and proteins were separated by polyacrylamide gel electrophoresis and transferred to a nitrocellulose sheet, which was then immunoreacted with antibodies against iNOS, ICAM-1, MMP-9, occludin, β-actin, and GAPDH as described in the Supporting Information.
Statistical Analysis
All data are presented as mean ± SEM. Statistical analysis was performed by one-way ANOVA, followed by Bonferroni post hoc test for pairwise comparisons. P < 0.05 was considered statistically significant.
RESULTS
3,6′-DT Treatment Reduces Infarct Size and Neuronal Death and Improves Functional Outcome in Experimental Ischemic Stroke
We first evaluated the potential abilities of 3,6′-DT to protect the brain against focal ischemic stroke by administering increasing concentrations of 3,6′-DT (0, 14, 28, 56, or 112 mg/kg) to mice 1 hr prior to the onset of ischemia. In addition, a group of mice was given 50 mg/kg T. There was a progressive reduction in brain infarct volume with doses of 14, 28, and 56 mg/kg 3,6′-DT, although the highest dose of 3,6′-DT was not protective; the magnitude of the reduction in brain damage with the 56 mg/kg dose of 3,6′-DT was similar to that achieved with T (Fig. 1A, Supp. Info. Fig. 1A). Ischemic brain damage was significantly reduced and functional outcome improved when treatment with 3,6′-DT was delayed for 1 or 3 hr after the experimental stroke (Fig. 1C,D). In contrast, T did not afford significant protection when treatment was delayed for 3 hr. Measurements of cerebral blood flow by laser Doppler flowmetry established that 3,6′-DT had no significant effect on blood flow during the ischemia or after reperfusion (data not shown).
Fig. 1.
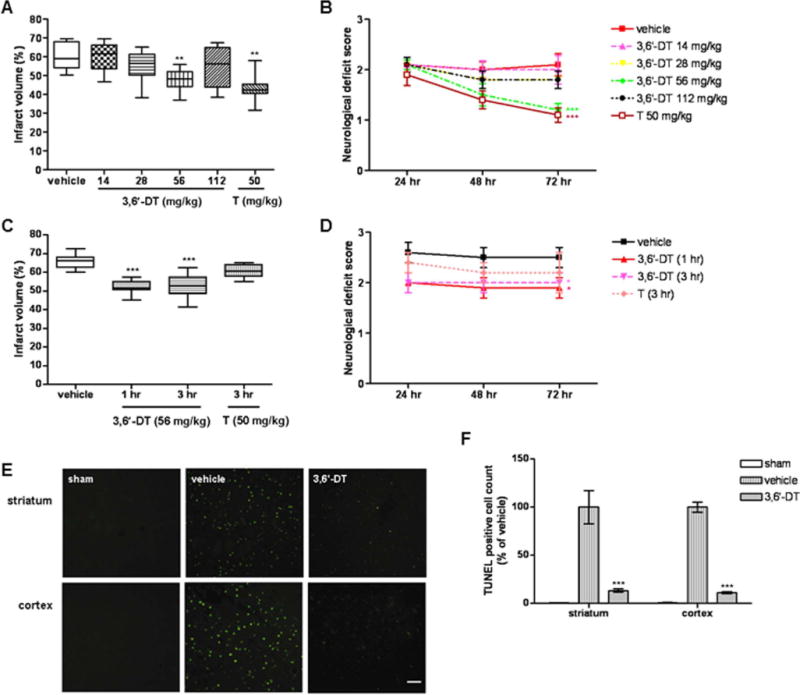
3,6′-DT reduces brain damage and improves functional outcome in a mouse stroke model. A: Ischemic infarct volumes 72 hr after MCAO/R in mice treated with vehicle (10 mice) or the indicated concentrations of 3,6-dithiothalidomide (3,6′-DT; 10 mice per group) or thalidomide (T; eight mice per group). B: Neurological deficit scores were recorded at the indicated poststroke time points. C: Infarct volumes in mice treated with vehicle (3 hr poststroke; n = 8) or with 3,6′-DT (n = 8) or T (n = 6) at the indicated poststroke time points. D: Neurological deficit scores for the mice in the experiment described for C. E: Images showing TUNEL-stained cells (n = 6 per group) in the cerebral cortex of mice in the indicated groups. F: Results of quantification of numbers of TUNEL-stained cells in the striatum and cerebral cortex of mice in the indicated groups. **P < 0.01, ***P < 0.001 compared with the value for vehicle-treated mice. Scale bar = 50 μm. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
To determine whether 3,6′-DT treatment did, in fact, preserve neurons, we performed TUNEL staining of brain tissue sections from mice treated with either 56 mg/kg 3,6′-DT or vehicle 3 hr poststroke as well as sham-operated control mice. At 72 hr after MCAO/R, numerous TUNEL-positive cells were present in the striatum and cerebral cortex of vehicle-treated mice, whereas mice treated with 3,6′-DT exhibited significantly fewer TUNEL-positive cells in the striatum and cortex (Fig. 1E,F).
3,6′-DT Treatment Attenuates Stroke-Induced Elevation of Proinflammatory Cytokines and iNOS
Previous studies have shown that inflammation contributes to neuronal dysfunction after ischemic stroke and that elevated circulating and brain TNF levels are associated with the severity of ischemic stroke injury (Wang et al., 2007; Denes et al., 2010). To elucidate the mechanism by which 3,6′-DT protects neurons against ischemic injury, the involvement of inflammation in ischemic injury was examined. We first measured levels of proinflammatory cytokines (TNF and IL-1β) in both ipsilateral and contralateral hemispheres. Levels of TNF in both hemispheres of vehicle-treated mice were markedly elevated at 2 hr and remained elevated through 72 hr after MCAO/R (Fig. 2A,B). Treatment of mice with 3,6′-DT resulted in a significant attenuation of stroke-induced elevation of TNF and IL-1β levels in both cerebral hemispheres (Fig. 2A,B).
Fig. 2.
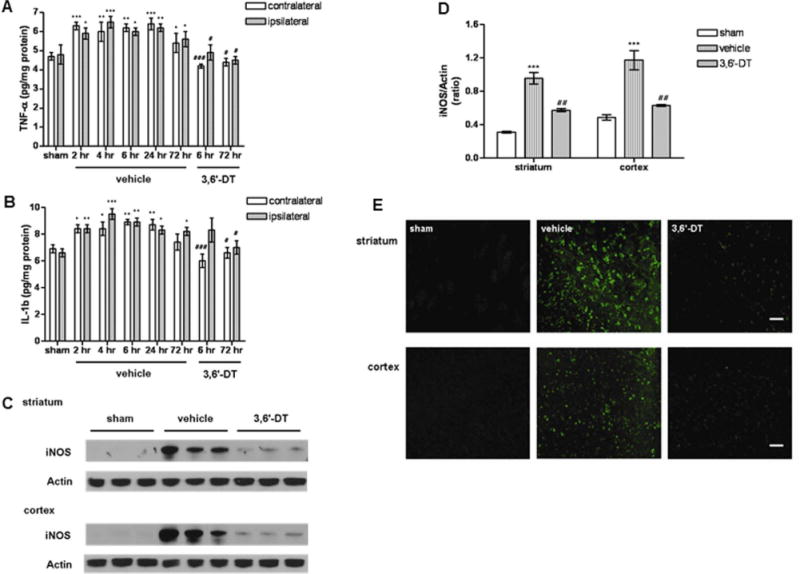
3,6′-DT treatment suppresses stroke-induced production of proinflammatory cytokines (TNFα and IL-1β) and iNOS. Levels of TNF (A) and IL-1β (B) levels were measured in the ipsilateral and contralateral hemispheres at the indicated poststroke time points and were compared with their levels in sham-operated control mice. The mice (n = 6 for each time point) were treated with either vehicle or 56 mg/kg 3,6′-DT 1 hr prior to MCAO/R. *P < 0.05, **P < 0.01, ***P < 0.001 compared with the value for sham mice. #P < 0.05, ###P < 0.001 compared with the value for vehicle-treated mice at the same time point. C: Immunoblot showing examples of relative levels of iNOS in tissue samples from striatum and cortex in vehicle-treated or 3,6′-DT-treated mice at 72 hr after ischemic stroke. D: Results of densitometric analysis of iNOS immunoblots. ***P < 0.001 compared with the value for sham mice. ##P < 0.01 compared with the value for vehicle-treated mice. E: Representative image of iNOS immunoreactivity in the indicated groups of mice. Scale bars = 100 μm. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Previous work has demonstrated that TNF induces upregulation of iNOS in various inflammatory cells (Iaedecola et al., 1995; del Zoppo et al., 2000; Lambertsen et al., 2012). We therefore measured iNOS protein levels at 72 hr after MCAO/R. Immunoblot analysis showed a significant increase in iNOS in the ischemic striatum and cortex of vehicle-treated mice compared with sham control mice. In 3,6′-DT-treated mice, the level of iNOS protein was significantly lower in the ischemic striatum and cortex compared with vehicle-treated mice (Fig. 2C,D). Consistent with the immunoblot results, MCAO/R resulted in a large increase of iNOS-immunoreactive cells in the striatum and cortex of vehicle-treated mice, and 3,6′-DT treatment suppressed the accumulation of the iNOS-positive cells (Fig. 2E).
3,6′-DT Treatment Suppresses Glial Activation, Neutrophil Infiltration, and ICAM-1 Expression
Activated microglia and astrocytes and infiltrating blood-borne immune cells contribute to ischemic damage by releasing proinflammatory cytokines and reactive oxygen species (ROS). We found that the stroke-induced accumulation of activated microglia (Iba-1 immunoreactive cells) and astrocytes (GFAP-positive cells) in the ischemic striatum and cortex was attenuated significantly in mice treated with 3,6′-DT compared with vehicle-treated mice (Fig. 3A–C, Supp. Info. Fig. 2). We also immunostained brain sections with an antibody against myeloperoxidase (MPO), an inflammatory enzyme secreted by neutrophils and microglia/macrophages (Breckwoldt et al., 2008). Stroke induced a dramatic increase in numbers of MPO-positive cells in the striatum and cortex, and 3,6′-DT treatment significantly reduced numbers of MPO-positive cells (Fig. 3A,D).
Fig. 3.

The thalidomide analog 3,6′-DT treatment decreases glial activation, cell adhesion molecule expression, and neutrophil infiltration after ischemic stroke. A: Examples of immunoreactivity for markers of activated microglia (Iba-1), astrocytes (GFAP), neutrophils (MPO), and the vascular endothelial cell adhesion molecule ICAM-1 in the cerebral cortex of mice from the indicated treatment groups. B–D: Results of quantification of numbers of cells immunoreactive with Iba-1 (B), GFAP (C), and MPO (D) in the ipsilateral cerebral cortex at 72 hr poststroke. E: Immunoblot showing examples of relative levels of ICAM-1 in tissue samples from cortex in vehicle-treated or 3,6′-DT-treated mice at 72 hr after ischemic stroke. F: Results of densitometry analysis of ICAM-1 immunoblots (n = 6 mice per group). The mice were treated with either vehicle or 56 mg/kg 3,6′-DT 1 hr prior to MCAO/R. *P < 0.05, **P < 0.01, ***P < 0.001 compared with the value for sham mice. #P < 0.05, ##P < 0.01 compared with the value for vehicle-treated mice. Scale bar = 50 μm. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The ischemia-induced entry of circulating leukocytes and macrophages into the brain parenchyma is mediated, in part, by binding to ICAM-1 on the surface of vascular endothelial cells. As expected from previous studies (Yilmaz and Granger, 2010), levels of ICAM-1, evaluated by immunostaining and immunoblot analysis, were increased in the ischemic cortex (Fig. 3A,E,F). The ischemia-induced increase in ICAM-1 levels was attenuated significantly in mice treated with 3,6′-DT, consistent with an anti-inflammatory action of 3,6′-DT on vascular endothelial cells. Because ICAM-1 mediates, in part, the infiltration of immune cells through the BBB after a stroke, we next determined whether 3,6′-DT modifies stroke-induced disruption of the BBB.
3,6′-DT Treatment Attenuates BBB Disruption by a Mechanism Involving Inhibition of MMP-9 Expression and Preservation of Occludin
When infused into the bloodstream, the dye Evans blue will not cross an intact BBB but will pass through a BBB compromised as the result of a stroke. The Evans blue concentration in both the ipsilateral and the contralateral hemispheres of mice 48 hr after MCAO/R was measured to determine an Evans blue index (EBI; ischemic/nonischemic hemisphere Evans blue concentrations). The EBI in vehicle-treated mice subjected to MCAO/R was significantly higher than that in sham mice, whereas 3,6′-DT-treated MCAO/R mice had a significantly lower EBI compared with vehicle-treated MCAO/R mice (Fig. 4A,B). The mechanism of BBB disruption in stroke involves increased expression of MMP-9 and degradation of the endothelial cell tight junction protein occludin (Fernández-López et al., 2012). We found that 3,6′-DT treatment resulted in a significant attenuation of the stroke-induced upregulation of MMP-9 (Fig. 4C–F). Levels of occludin were significantly reduced after MCAO/R, and occludin was completely preserved in mice treated with 3,6′-DT 1 hr prior to MCAO/R (Fig. 4G,H). Collectively, the data to this point demonstrated that delayed treatment with 3,6′-DT can reduce brain damage and improve functional outcome in the MCAO/R mouse stroke model by a mechanism involving suppression of multiple inflammatory processes, including the production of proinflammatory cytokines, disruption of the BBB, and infiltration of leukocytes.
Fig. 4.

Evidence that 3,6′-DT treatment inhibits stroke-induced disruption of the blood–brain barrier. A: Images of stroke-induced extravasation of Evans blue dye into the cerebral parenchyma (evaluated at 48 hr poststroke) in vehicle-treated and 3,6′-DT-treated (56 mg/kg; 1 hr prior to MCAO/R) mice. B: Evans blue index in mice treated with vehicle or 3,6′-DT 1 hr prior to MCAO/R in comparison with sham-operated mice (eight or nine mice per group). C: MMP-9 immunoreactivity in sections of striatum and cerebral cortex from mice in the indicated treatment groups. D: Results of quantification of numbers of MMP-9-immunoreactive cells (n = 6 mice per group). E: Immunoblot showing relative levels of MMP-9 in the striatum and cortex of mice in the indicated treatment groups (48 hr poststroke). F: Results of densitometric analysis of MMP-9 immunoblots. G: Immunoblot showing relative levels of occludin in the striatum and cortex of mice in the indicated treatment groups (48 hr poststroke). H: Results of densitometric analysis of occludin immunoblots. *P < 0.05, **P < 0.01, ***P < 0.001 compared with the value for sham mice. #P < 0.05, ##P < 0.01, ###P < 0.001 compared with the value for vehicle-treated mice. Scale bar = 50 μm. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Neuroprotection by 3,6′-DT Requires TNF Signaling
Because TNF expression is increased rapidly in response to MCAO/R and because TNF can induce or promote each of the inflammatory processes evaluated in the present study, we designed an experiment to determine whether suppression of TNF signaling is sufficient to account for the neuroprotective actions of 3,6′-DT in this stroke model. Mice lacking both TNF receptors (TNFR KO) and wild-type (WT) control mice were treated with either 3,6′-DT or vehicle 1 hr prior to MCAO/R, and brain infarct size and functional deficits were measured at 72 hr poststroke. Whereas 3,6′-DT treatment reduced brain damage and improved functional outcome in WT mice, 3,6′-DT had no significant effect on brain damage or functional deficits in TNFR KO mice (Fig. 5). Thus, genetic disruption of TNF signaling abolished the neuroprotective efficacy of 3,6′-DT treatment in a mouse model of focal ischemic stroke.
Fig. 5.
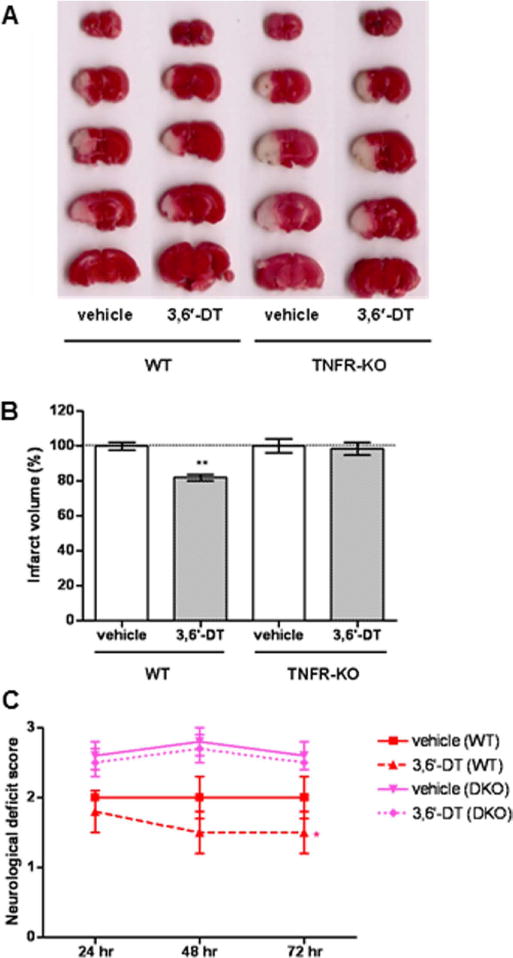
The therapeutic effect of 3,6′-DT treatment is abolished in mice lacking TNF receptors. Wild-type mice (WT) and TNF receptor-deficient mice (TNFR KO) were treated with either vehicle or 56 mg/kg 3,6′-DT 1 hr prior to MCAO/R (five or six mice per group). A: Examples of TTC-stained serial brain sections from mice at 72 hr after ischemic stroke. Infarct volumes (B) and neurological deficit scores (C) are shown. **P < 0.01 compared with the value for vehicle-treated mice. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
DISCUSSION
Our results show that 3,6′-DT reduces neuronal loss and improves functional outcome in a mouse model of focal ischemic stroke. Treatment with 3,6′-DT inhibited the expression of TNF, IL-1β, and iNOS and reduced apoptotic cell death in the ischemic hemisphere. TNF production occurs early (within several hours after a stroke) and is sustained at high levels for several days as a result of glial cell activation and infiltration of circulating inflammatory leukocytes and macrophages (Liu et al., 1994; Wang et al., 2007). Moreover, previous studies have shown that TNF signaling mediates multiple inflammatory processes that occur in the brain parenchyma and cerebral vasculature in response to a stroke, including activation of microglia/macrophages (and their production of other proinflammatory cytokines and nitric oxide), upregulation of ICAM-1, disruption of the BBB, and leukocyte infiltration (Mayhan, 2002; Pan and Kastin, 2007). The inhibition of poststroke TNF production by 3,6′-DT may therefore be responsible for its ability to suppress microglia/macrophage activation, BBB disruption, and leukocyte inflammation, thereby limiting brain damage and improving functional outcome.
In support of inhibition of TNF production as the primary mechanism for the neuroprotective action of 3,6′-DT, we found that the ability of 3,6′-DT to reduce ischemic brain damage was abolished in mice lacking TNF receptors. Functional deficits were also not lessened in TNFR-KO mice treated with 3,6′-DT. There are two different TNF receptors, TNFR1 and TNFR2. In our previous study (Bruce et al., 1996), we found that mice lacking both TNFR1 and TNFR2 exhibit increased vulnerability to seizure- and ischemia-induced neuronal damage. Data from subsequent studies suggest that activation of TNFR1 in microglia and in neurons (Fontaine et al., 2002; He et al., 2012) contributes to ischemic neuronal injury, whereas activation of TNFR2 in neurons protects them against ischemia via a nuclear factor-κB-mediated pathway (Barger et al., 1995; Fontaine et al., 2002). In response to ischemic stroke, microglia are a major source of endogenous TNF, and suppression of this TNF production by 3,6′DT likely protects neurons by reducing production of microglia-derived toxins and/or by preventing activation of TNFR1 in neurons. Selective inhibition of TNFR1 might therefore be expected to prevent the detrimental actions of TNF while preserving the neuroprotective effects of TNFR2-mediated signaling (Chadwick et al., 2008). Previous studies have shown that both T and 3,6′-DT can inhibit TNF production in cultured immune cells (Tweedie et al., 2009). Interestingly, we found that both T and 3,6′-DT reduced brain damage and improved functional outcome when administered prior to MCAO/R, but only 3,6′-DT was effective when treatment was delayed for 3 hr after the stroke. This differential efficacy of T and 3,6′-DT with delayed treatment may be the result of the different abilities to enter and be retained within the brain, in line with the ability of 3,6′-DT but not T to lower brain TNF levels and improve cognition in an animal model of Alzheimer’s disease (Gabbita et al., 2012).
TNF is known to induce the expression of ICAM-1 in vascular endothelial cells and to increase MMP-9 levels at the BBB (Siesjo and Siesjo, 1996; Rosenberg, 1999; Gidday et al., 2006; Rosell et al., 2008). An early and major pathological action of MMP-9 in cerebral ischemia is to degrade the tight junction protein occludin, thereby increasing the permeability of the BBB to circulating factors and cells in the blood. We found that the disruption of the BBB normally observed after MCAO/R was largely prevented by 3,6′-DT treatment. Because TNF is known to induce the expression of both ICAM-1 and MMP-9 (Dobbie et al., 1999; Takata et al., 2011), suppression of TNF production is sufficient to account for the BBB-preserving actions of 3,6′-DT. In this way, 3,6′-DT may limit the entry of proinflammatory monocytes, lymphocytes, and macrophages into the ischemic brain parenchyma. The increase in MMP-9 protein levels and the decrease in occludin levels (a well-established substrate for MMP-9) support the conclusion that MMP-9 activity is increased in response to ischemic stroke. Conversely, the suppression of both the increase in MMP-9 levels and the decrease of occludin levels by 3,6′-DT suggest a reduction of MMP-9 activity by this T analog.
There has been considerable interest in the development of drugs that block pathological actions of TNF in a range of inflammatory conditions. TNF antibodies are marketed for the treatment of arthritis, inflammatory bowel disorders, and sepsis (Qiu et al., 2011; Aaltonen et al., 2012; Ordás et al., 2012). T is known to inhibit TNF production and has exhibited therapeutic efficacy in clinical trials in patients with a range of TNF-mediated disorders, including cancer cachexia and arthritic disorders (Ossandon et al., 2002; Gordon et al., 2005). Recent preclinical studies have also provided evidence that T can protect neurons and improve function outcome in animal models of spinal cord injury (Esposito and Cuzzocrea, 2011) and excitotoxic injury to striatal neurons (Ryu et al., 2009). We previously developed a series of thiothalidomide analogs of T and found that 3,6′-DT was more potent than T in inhibiting TNF synthesis (Zhu et al., 2003). The mechanism by which 3,6′-DT inhibits TNF synthesis appears to be by an interaction with the 3′UTR of the TNF mRNA (Zhu et al., 2003). Although this has yet to be tested in humans, the greater potency and brain penetrability of 3,6′-DT compared with T, taken together with its superior therapeutic window in the present stroke model, suggest a potential use for 3,6′-DT in stroke patients.
Acknowledgments
Contract grant sponsor: National Institute on Aging Intramural Research Program.
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- Aaltonen KJ, Virkki LM, Malmivaara A, Konttinen YT, Nordström DC, Blom M. Systematic review and meta-analysis of the efficacy and safety of existing TNF blocking agents in treatment of rheumatoid arthritis. PLoS One. 2012;7:e30275. doi: 10.1371/journal.pone.0030275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arumugam TV, Tang SC, Lathia JD, et al. Intravenous immunoglobulin (IVIG) protects the brain against experimental stroke by preventing complement-mediated neuronal cell death. Proc Natl Acad Sci U S A. 2007;104:14104–14109. doi: 10.1073/pnas.0700506104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baratz R, Tweedie D, Rubovitch V, et al. Tumor necrosis factor-α synthesis inhibitor, 3,6′-dithiothalidomide, reverses behavioral impairments induced by minimal traumatic brain injury in mice. J Neurochem. 2011;118:1032–1042. doi: 10.1111/j.1471-4159.2011.07377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barger SW, Hörster D, Furukawa K, Goodman Y, Krieglstein J, Mattson MP. Tumor necrosis factors alpha and beta protect neurons against amyloid beta-peptide toxicity: evidence for involvement of a kappa B-binding factor and attenuation of peroxide and Ca2+ accumulation. Proc Natl Acad Sci U S A. 1995;92:9328–9332. doi: 10.1073/pnas.92.20.9328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belarbi K, Jopson T, Tweedie D, et al. TNF-α protein synthesis inhibitor restores neuronal function and reverse cognitive deficits induced chronic neuroinflammation. J Neuroinflamm. 2012;9:23. doi: 10.1186/1742-2094-9-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borlongan CV, Yamamoto M, Takei N, Kumazaki M, Ungsuparkorn C, Hida H, Sanberg PR, Nishino H. Glial cell survival is enhanced during melatonin-induced neuroprotection against cerebral ischemia. FASEB J. 2000;14:1307–1317. doi: 10.1096/fj.14.10.1307. [DOI] [PubMed] [Google Scholar]
- Breckwoldt MO, Chen JW, Stangenberg L, Aikawa E, Rodriguez E, Qiu S, Moskowitz MA, Weissleder R. Tracking the inflammatory response in stroke in vivo by sensing the enzyme myeloperoxidase. Proc Natl Acad Sci U S A. 2008;105:18584–18589. doi: 10.1073/pnas.0803945105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce AJ, Boling W, Kindy MS, Peschon J, Kraemer PJ, Carpenter MK, Holtsberg FW, Mattson MP. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat Med. 1996;2:788–794. doi: 10.1038/nm0796-788. [DOI] [PubMed] [Google Scholar]
- Buttini M, Sauter A, Boddeke HW. Induction of interleukin-1 beta mRNA after focal cerebral ischemia in the rat. Brain Res Mol Brain Res. 1994;23:126–134. doi: 10.1016/0169-328x(94)90218-6. [DOI] [PubMed] [Google Scholar]
- Canugovi C, Yoon JS, Feldman N, et al. Endonuclease 8-like 1 promotes short-term spatial memory retention and protects from ischemic stroke induced death and brain dysfunction in mice. Proc Natl Acad Sci U S A. 2012 doi: 10.1073/pnas.1204156109. [E-pub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadwick W, Magnus T, Martin B, Keselman A, Mattson MP, Maudsley S. Targeting TNF-alpha receptors for neurotherapeutics. Trends Neurosci. 2008;31:504–511. doi: 10.1016/j.tins.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Zoppo G, Ginis I, Hallenbeck JM, Iadecola C, Wang X, Feuerstein GZ. Inflammation and stroke: putative role for cytokines, adhesion molecules and iNOS in brain response to ischemia. Brain Pathol. 2000;10:95–112. doi: 10.1111/j.1750-3639.2000.tb00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denes A, Thornton P, Rothwell NJ, et al. Inflammation and brain injury: acute cerebral ischemia, peripheral and central inflammation. Brain Behav Immun. 2010;24:708–723. doi: 10.1016/j.bbi.2009.09.010. [DOI] [PubMed] [Google Scholar]
- Dobbie MS, Hurst RD, Klein NJ, Surtees RA. Upregulation of intercellular adhesion molecule-1 expression on human endothelial cells by tumour necrosis factor-alpha in an in vitro model of the blood–brain barrier. Brain Res. 1999;830:330–336. doi: 10.1016/s0006-8993(99)01436-5. [DOI] [PubMed] [Google Scholar]
- Esposito E, Cuzzocrea S. Anti-TNF therapy in the injured spinal cord. Trends Pharmacol Sci. 2011;32:107–115. doi: 10.1016/j.tips.2010.11.009. [DOI] [PubMed] [Google Scholar]
- Fernández-López D, Faustino J, Daneman R, Zhou L, Lee SY, Derugin N, Wendland MF, Vexler ZS. Blood–brain barrier permeability is increased after acute adult stroke but not neonatal stroke in the rat. J Neurosci. 2012;32:9588–9600. doi: 10.1523/JNEUROSCI.5977-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine V, Mohand-Said S, Hanoteau N, Fuchs C, Pfizenmaier K, Eisel U. Neurodegenerative and neuroprotective effects of tumor necrosis factor (TNF) in retinal ischemia: opposite roles of TNF receptor 1 and TNF receptor 2. J Neurosci. 2002;22:RC216. doi: 10.1523/JNEUROSCI.22-07-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbita SP, Srivastava MK, Eslami P, et al. Early intervention with a small molecule inhibitor for tumor necrosis factor-α prevents cognitive deficits in a triple transgenic mouse model of Alzheimer’s disease. J Neuroinflamm. 2012;9:99. doi: 10.1186/1742-2094-9-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gidday JM, Gasche YG, Copin JC, et al. Leukocyte-derived matrix metalloproteinsase-9 mediates blood–brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am J Physiol Heart Circ Physiol. 2005;289:H558–H568. doi: 10.1152/ajpheart.01275.2004. [DOI] [PubMed] [Google Scholar]
- Gordon JN, Trebble TM, Ellis RD, Duncan HD, Johns T, Goggin PM. Thalidomide in the treatment of cancer cachexia: a randomised placebo controlled trial. Gut. 2005;54:540–545. doi: 10.1136/gut.2004.047563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greig NH, Giordano T, Zhu X, Yu QS, Perry TA, Holloway HW, Brossi A, Rogers JT, Sambamurti K, Lahiri DK. Thalidomide-based TNF-alpha inhibitors for neurodegenerative diseases. Acta Neurobiol Exp. 2004;64:1–9. doi: 10.55782/ane-2004-1486. [DOI] [PubMed] [Google Scholar]
- He P, Liu Q, Wu J, Shen Y. Genetic deletion of TNF receptor suppresses excitatory synaptic transmission via reducing AMPA receptor synaptic localization in cortical neurons. FASEB J. 2012;26:334–345. doi: 10.1096/fj.11-192716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Zhang F, Xu S, et al. Inducible nitric oxide synthase gene expression in brain following cerebral ischemia. J Cereb Blood Flow Metab. 1995;15:378–384. doi: 10.1038/jcbfm.1995.47. [DOI] [PubMed] [Google Scholar]
- Laffitte E, Revuz J. Thalidomide: an old drug with new clinical applications. Expert Opin Drug Saf. 2004;3:47–56. doi: 10.1517/14740338.3.1.47. [DOI] [PubMed] [Google Scholar]
- Lambertsen KL, Biber K, Finsen B. Inflammatory cytokines in experimental and human stroke. J Cereb Blood Flow Metab. 2012 doi: 10.1038/jcbfm.2012.88. [E-pub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Clark RK, McDonnell PC, et al. Tumor necrosis factor-alpha expression in ischemic neurons. Stroke. 1994;25:1481–1488. doi: 10.1161/01.str.25.7.1481. [DOI] [PubMed] [Google Scholar]
- Liu W, Hendren J, Qin XJ, et al. Normobaric hyperoxia attenuates early blood–brain barrier disruption by inhibiting MMP-9-mediated occludin degradation in focal cerebral ischemia. J Neurochem. 2009;108:811–820. doi: 10.1111/j.1471-4159.2008.05821.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Excitotoxic and excitoprotective mechanisms: abundant targets for the prevention and treatment of neurodegenerative disorders. Neuromol Med. 2003;3:65–94. doi: 10.1385/NMM:3:2:65. [DOI] [PubMed] [Google Scholar]
- Mayhan WG. Cellular mechanisms by which tumor necrosis factor-alpha produces disruption of the blood–brain barrier. Brain Res. 2002;927:144–152. doi: 10.1016/s0006-8993(01)03348-0. [DOI] [PubMed] [Google Scholar]
- McColl BW, Rothwell NJ, Allan SM. Systemic inflammation alters the kinetics of cerebrovascular tight junction disruption after experimental stroke in mice. Neurobiol Dis. 2008;28:9451–9462. doi: 10.1523/JNEUROSCI.2674-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaluk P, Kaczmarek L. Matrix metalloproteinase-9 in glutamate-dependent adult brain function and dysfunction. Cell Death Differ. 2007;14:1255–1258. doi: 10.1038/sj.cdd.4402141. [DOI] [PubMed] [Google Scholar]
- Offner H, Subrammanian S, Parker SM, et al. Experimental stroke induces massive, rapid activation of peripheral immune system. J Cereb Blood Flow Metab. 2006;26:654–665. doi: 10.1038/sj.jcbfm.9600217. [DOI] [PubMed] [Google Scholar]
- Ordás I, Mould DR, Feagan BG, Sandborn WJ. Anti-TNF monoclonal antibodies in inflammatory bowel disease: pharmacokinetics-based dosing paradigms. Clin Pharmacol Ther. 2012;91:635–646. doi: 10.1038/clpt.2011.328. [DOI] [PubMed] [Google Scholar]
- Ossandon A, Cassarà EA, Priori R, Valesini G. Thalidomide: focus on its employment in rheumatologic diseases. Clin Exp Rheumatol. 2002;20:709–718. [PubMed] [Google Scholar]
- Pan W, Kastin A. Tumor necrosis factor and stroke: role of the blood–brain barrier. Prog Neurobiol. 2007;83:363–374. doi: 10.1016/j.pneurobio.2007.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu P, Cui X, Barochia A, Li Y, Natanson C, Eichacker PQ. The evolving experience with therapeutic TNF inhibition in sepsis: considering the potential influence of risk of death. Expert Opin Invest Drugs. 2011;20:1555–1564. doi: 10.1517/13543784.2011.623125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roger VL, Go AS, Lloyd-Jones DM, et al. Heart disease and stroke statistics—2012 update: a report from the American Heart Association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanic AM, White RF, Arleth AJ, et al. Matrix metalloproteinases expression increases after cerebral focal ischemia in rats: inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke. 1998;29:1020–2030. doi: 10.1161/01.str.29.5.1020. [DOI] [PubMed] [Google Scholar]
- Rosell A, Cuadrado E, Ortega-Aznar A, et al. MMP-9-positive neutrophil infiltration is associated to blood–brain barrier breakdown and basal lamina type IV collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke. 2008;39:1121–1126. doi: 10.1161/STROKEAHA.107.500868. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA. Ischemic brain edema. Prog Cardiovasc Dis. 1999;42:209–216. doi: 10.1016/s0033-0620(99)70003-4. [DOI] [PubMed] [Google Scholar]
- Ryu JK, Jantaratnotai N, McLarnon JG. Thalidomide inhibition of vascular remodeling and inflammatory reactivity in the quinolinic acid-injected rat striatum. Neuroscience. 2009;163:601–608. doi: 10.1016/j.neuroscience.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Shah IM, Macrae IM, Napoli DI. Neuroinflammation and neuroprotective strategies in acute ischaemic stroke—from bench to bedside. Curr Mol Med. 2009;9:336–354. doi: 10.2174/156652409787847236. [DOI] [PubMed] [Google Scholar]
- Siesjo BK, Siesjo P. Mechanisms of secondary brain injury. Eur J Anaesthesiol. 1996;13:247–268. [PubMed] [Google Scholar]
- Stanimirovic D, Satoh K. Inflammatory mediators of cerebral endothelium: a role in ischemic brain inflammation. Brain Pathol. 2000;10:113–126. doi: 10.1111/j.1750-3639.2000.tb00248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson RA, Sharp FR. Infarct measurement methodology. J Cereb Blood Flow Metab. 1994;14:697–698. doi: 10.1038/jcbfm.1994.88. [DOI] [PubMed] [Google Scholar]
- Takata F, Dohgu S, Matsumoto J, Takahashi H, Machida T, Wakigawa T, Harada E, Miyaji H, Koga M, Nishioku T, Yamauchi A, Kataoka Y. Brain pericytes among cells constituting the blood–brain barrier are highly sensitive to tumor necrosis factor-α, releasing matrix metalloproteinase-9 and migrating in vitro. J Neuroinflamm. 2011;8:106. doi: 10.1186/1742-2094-8-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SC, Arumugam TV, Xu X, et al. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci U S A. 2007;104:13798–13803. doi: 10.1073/pnas.0702553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuttolomondo A, Raimondo DD, Sciacca RD, et al. Inflammatory cytokines in acute ischemic stroke. Curr Pharm Des. 2008;14:3574–3589. doi: 10.2174/138161208786848739. [DOI] [PubMed] [Google Scholar]
- Tweedie D, Luo W, Short RG, Brossi A, Holloway HW, Li Y, Yu QS, Greig NH. A cellular model of inflammation for identifying TNF-alpha synthesis inhibitors. J Neurosci Methods. 2009;183:182–187. doi: 10.1016/j.jneumeth.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tweedie D, Ferguson RA, Fishman K, et al. Tumor necrosis factor-α synthesis inhibitor 3,6-′-dithiothalidomide attenuates markers of inflammation, Alzheimer pathology and behavioral deficits in animal models of neuroinflammation and Alzheimer’s disease. J Neuroinflamm. 2012;9:106. doi: 10.1186/1742-2094-9-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Tang XN, Yenari MA. The inflammatory response in stroke. J Neuroimmunol. 2007;184:53–68. doi: 10.1016/j.jneuroim.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz G, Granger DN. Leukocyte recruitment and ischemic brain injury. Neuromol Med. 2010;12:193–204. doi: 10.1007/s12017-009-8074-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao BQ, Wang A, Kim HY, et al. Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat Med. 2006;4:441–445. doi: 10.1038/nm1387. [DOI] [PubMed] [Google Scholar]
- Zheng L, Fisher G, Miller RE, Peschon J, Lynch DH, Lenardo MJ. Induction of apoptosis in mature T cells by tumour necrosis factor. Nature. 1995;377:348–351. doi: 10.1038/377348a0. [DOI] [PubMed] [Google Scholar]
- Zhu X, Giordano T, Yu Q, et al. Thiothalidomides: novel isosteric analogues of thalidomide with enhanced TNF-alpha inhibitory activity. J Med Chem. 2003;46:5222–5229. doi: 10.1021/jm030152f. [DOI] [PubMed] [Google Scholar]