

Abstract
We demonstrate that mutation of xerC, which reportedly encodes a homologue of an Escherichia coli recombinase, limits biofilm formation in the methicillin-resistant Staphylococcus aureus strain LAC and the methicillin-sensitive strain UAMS-1. This was not due to the decreased production of the polysaccharide intracellular adhesin (PIA) in either strain because the amount of PIA was increased in a UAMS-1 xerC mutant and undetectable in both LAC and its isogenic xerC mutant. Mutation of xerC also resulted in the increased production of extracellular proteases and nucleases in both LAC and UAMS-1, and limiting the production of either class of enzymes increased biofilm formation in the isogenic xerC mutants. More importantly, the limited capacity to form a biofilm was correlated with increased antibiotic susceptibility in both strains in the context of an established biofilm in vivo. Mutation of xerC also attenuated virulence in a murine bacteremia model, as assessed on the basis of the bacterial loads in internal organs and overall lethality. It also resulted in the decreased accumulation of alpha toxin and the increased accumulation of protein A. These findings suggest that xerC may impact the functional status of agr. This was confirmed by demonstrating the reduced accumulation of RNAIII and AgrA in LAC and UAMS-1 xerC mutants. However, this cannot account for the biofilm-deficient phenotype of xerC mutants because mutation of agr did not limit biofilm formation in either strain. These results demonstrate that xerC contributes to biofilm-associated infections and acute bacteremia and that this is likely due to agr-independent and -dependent pathways, respectively.
INTRODUCTION
Staphylococcus aureus is a ubiquitous human pathogen capable of causing a wide variety of infections. These range from acute infections like bacteremia to chronic biofilm-associated infections. It is generally assumed that severe, acute infections are defined by the production of extracellular toxins, but a recent report demonstrated a negative correlation between toxicity for mammalian cells and invasive disease among human isolates (1). Although expression of the accessory gene regulator (agr), which is a primary determinant of toxin production (2), is negatively correlated with biofilm formation (3, 4), no correlation between toxicity and biofilm formation was observed in the current study. In fact, the only correlation was that decreased toxicity was associated with increased fitness, as assessed on the basis of survival in human serum (1).
Nevertheless, it is clear that S. aureus has the capacity to cause diverse forms of infection. In the case of acute infections, the primary therapeutic concern is acquired resistance and the decreasing availability of effective antibiotics (5). In the case of biofilm-associated infections, a critical concern is the biofilm itself, which confers a therapeutically relevant level of intrinsic resistance to both host defenses and conventional antibiotics (6–9). There are multiple reasons for this, including the limited ability to achieve effective concentrations of antibiotic at the site of infection, particularly within the deeper layers of the biofilm, but it has become increasingly evident that biofilms contain subpopulations of bacterial cells that exhibit reduced metabolic activity (e.g., persister cells), thus rendering them less susceptible to conventional antibiotics even if the antibiotic does reach its intended bacterial target (10, 11). It is this intrinsic resistance that makes biofilms a critical virulence factor in the pathogenesis of chronic S. aureus infections; indeed, it is one of the best prognostic indicators of potential therapeutic failure (12). This emphasizes the need for alternative strategies that could be used to limit biofilm formation and thereby increase the therapeutic efficacy of conventional antibiotics.
On the basis of this, much of our research effort has focused on identifying factors that contribute to S. aureus biofilm formation (13–17). To date, the single most promising target that we have identified is the staphylococcal accessory regulator A (sarA), mutation of which limits S. aureus biofilm formation to a degree that can be correlated with increased antibiotic susceptibility and improved therapeutic outcomes in relevant animal models (18–23). We also confirmed that the increased production of extracellular proteases plays an important role in defining the biofilm-deficient phenotype of sarA mutants (14, 16, 17, 23, 24). Moreover, we demonstrated that sarA mutants exhibit reduced virulence in a murine bacteremia model and that this can also be correlated with the increased production of extracellular proteases owing to the decreased accumulation of specific virulence factors, including alpha toxin and phenol-soluble modulins (23, 25). This suggests that inhibitors of sarA-mediated regulation could be used to therapeutic advantage in diverse forms of S. aureus infection, and, in fact, one potential inhibitor has been described (26).
Our recent studies comparing the impact of mutating sarA to that of mutating other regulatory loci demonstrated that mutation of sarA imposes a greater limitation on biofilm formation than mutation of any of the other genes that we examined (17). These studies, as well as additional studies focusing on the genes encoding the proteases themselves (24), also confirmed an inverse correlation between the production of extracellular proteases and biofilm formation in both methicillin-resistant and methicillin-sensitive strains of S. aureus. However, Fey et al. (27) screened the 1,952 transposon insertion mutants in the Nebraska Transposon Mutant Library (NTML) and identified a number of mutants that exhibited increased protease activity but were not included in our earlier experiments. The mutated genes in two of these mutants, xerC (also known as codV) and hslU (also known as clpY), are the first and third genes in a four-gene operon that also includes clpQ and codY (28). ClpQ associates with its ClpY ATPase partner to form a two-component protease, but neither has been evaluated in the context of biofilm formation (28). However, mutation of the genes encoding other Clp ATPases has been shown to impact biofilm formation (29). Mutation of codY has also been shown to impact biofilm formation, but our studies demonstrate that its impact is strain dependent (17). This suggests that multiple genes in the xerC operon, including xerC itself, may be involved in biofilm formation.
Mutation of codY has also been correlated with increased toxin production and increased virulence in animal models of acute S. aureus infection (30, 31). In contrast, Chalker et al. (32) generated a nonpolar mutation in xerC in the S. aureus strain WCUH29 and demonstrated that this mutant exhibited reduced virulence in a murine model of hematogenous pyelonephritis. ClpY/Q play a role in growth at high temperature and the generalized stress response, but mutation of the genes for these proteins did not result in reduced virulence in a murine soft tissue abscess model (28). Nevertheless, these results suggest that genes within this operon, including xerC, may also play a role in acute S. aureus infection. Investigating these issues and doing so in diverse contemporary clinical isolates of S. aureus, including those of the USA300 clonal lineage, were the overall focus of the experiments that we report on here.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Mutant genes generated in the plasmid-cured JE2 derivative of strain LAC (27) were obtained from the Nebraska Transposon Mutant Library (NTML) through BEI Resources (Manassas, VA; http://www.beiresources.org). The xerC mutation was subsequently moved into strains LAC and UAMS-1 and their isogenic sarA mutants via phage-mediated transduction (25). The derivative of LAC that we employed was cured of its plasmid conferring resistance to erythromycin (33), thus allowing erythromycin selection of transductants in both the LAC and UAMS-1 recipients. Confirmation was subsequently obtained by PCR of the targeted genes (data not shown) and, in the case of LAC recipients, by comparison of EcoRI-digested genomic DNA, which confirmed the presence of the small cryptic plasmid and its absence in the JE2 donor (data not shown).
The xerC mutation was also transduced into derivatives of LAC and UAMS-1 with a reduced capacity to produce extracellular proteases and nucleases. The protease-deficient derivative of LAC was unable to produce any extracellular protease other than the proteases encoded by the spl operon; the use of this protease-deficient strain was necessitated by the fact that the spl mutation is defined by resistance to erythromycin (14, 16, 19, 23, 25). However, previous studies confirmed that biofilm formation is comparable in LAC sarA mutants unable to produce any extracellular protease and those that retain the capacity to produce only the spl-encoded proteases (17, 24). The protease-deficient derivative of UAMS-1 was unable to produce aureolysin, SspA, or SspB but retained the capacity to produce the spl-encoded proteases and ScpA both for the reason discussed above and owing to the fact that we were unable to transduce the scpA mutation from the JE2 derivative in NTML into this strain. The nuclease-deficient derivatives of LAC and UAMS-1 were generated by mutation of the nuc-1 gene as previously described (16). These mutations and the identity of the recipient strains were also confirmed by PCR of the targeted gene and additional genes and/or mutations that define each recipient (data not shown).
Construction of the xerC complementation plasmid was done by PCR amplification of the xerC open reading frame using genomic DNA from the USA300 strain LAC as the template. For subsequent subcloning, the forward primer contained an NdeI cut site (GGGCATATGTAAATGTATTGAATCATAT, where the NdeI cut site is underlined), and the reverse primer included a BamHI cut site (CCCGGATCCGTAATGTTGTATTACTCAT, where the BamHI cut site is underlined). The amplification product was cloned into the pCR2.1 TOPO vector (Invitrogen, Grand Island, NY) and transformed into Z-Competent E. coli cells (Zymo Research Corp., Irvine, CA). After verification by DNA sequencing (data not shown), the plasmid was digested with NdeI and BamHI and the insert was cloned into the shuttle vector pOS1 (34), such that expression of xerC was under the control of the lipoprotein diacylglycerol transferase promoter (pOS1-plgt) (35). The xerC complementation plasmid was first used to transform the S. aureus strain RN4220 by electroporation. The plasmid was then introduced into the appropriate strains by phage-mediated transduction.
All strains were maintained at −80°C in tryptic soy broth (TSB) containing 25% (vol/vol) glycerol. For each experiment, the strains under study were retrieved from cold storage by plating on tryptic soy agar (TSA) with appropriate antibiotic selection. The following antibiotics were added to the culture media as appropriate at the indicated concentrations: erythromycin, 10 μg ml−1; tetracycline, 5 μg ml−1; kanamycin, 50 μg ml−1; and neomycin, 50 μg ml−1. Kanamycin and neomycin were always used together to avoid the selection of spontaneously resistant strains.
Assessment of biofilm formation.
Biofilm formation was initially assessed in vitro using a 96-well microtiter plate assay (19). Briefly, bacterial cultures were grown at 37°C to stationary phase (16 h) in biofilm medium (BFM) with antibiotics when appropriate. Cultures were standardized to an optical density at 560 nm (OD560) of 0.05 in BFM without antibiotics. The wells of a 96-well microtiter plate that had previously been coated with 20% human plasma were then inoculated with 200 μl of this culture, and the plate was incubated at 37°C for 24 h, at which point the plate was washed three times with 200 μl phosphate-buffered saline (PBS), fixed with 200 μl 100% ethanol (EtOH), stained with 200 μl crystal violet, and washed three times with 200 μl PBS. The stain was then eluted with 150 μl 100% EtOH for 10 min before the eluent was diluted with an equal volume of PBS. The absorbance was measured using a BioTek Synergy 2 microplate reader (BioTek Instruments, Winooski, VT). All assays were performed using at least two biological replicates, each of which contained a minimum of three experimental replicates.
Antibiotic susceptibility assays.
Relative susceptibility to daptomycin and ceftaroline was assessed in vivo using a murine model of catheter-associated biofilm formation (22). All in vivo experiments were approved by the University of Arkansas for Medical Sciences Institutional Animal Care and Use Committee. Specifically, a 1-cm fluorinated ethylene propylene (FEP) catheter segment was placed into a subcutaneous pocket surgically created in each flank of female outbred NIH-Swiss mice (Harlan, Indianapolis, IN). Both to avoid an immune response to human proteins and because the catheter would presumably be coated with the corresponding murine proteins once it was implanted, catheters were not precoated with human plasma. After implantation, 105 CFU of the test strain in a total volume of 100 μl of PBS was introduced directly into the lumen of each catheter. After 24 h, the mice were randomly divided into treatment and nontreatment groups (n = 5). Because each mouse had two catheters implanted and because our previous studies confirmed the absence of cross-contamination between catheters in opposite flanks of the same mouse (data not shown) (22), each catheter was treated as an independent data point (n = 10).
In the untreated groups, 100 μl of sterile PBS was injected into the lumen of each catheter at daily intervals. In the treated groups, 100 μl of sterile PBS containing 5.0 μg/ml daptomycin or 10 μg/ml ceftaroline was injected into the lumen of the catheter at daily intervals for 5 days. These concentrations were chosen on the basis of the results of preliminary in vitro studies employing catheter-associated biofilms (data not shown) and correspond to 5 and 10 times the concentrations defined by the Clinical and Laboratory Standards Institute (CLSI) as the breakpoint MIC for daptomycin and ceftaroline, respectively. Catheters were harvested 24 h after the final treatment, rinsed in PBS to remove nonadherent bacteria, and sonicated in 5.0 ml of PBS to remove adherent bacteria. Aliquots were then plated on TSA, and the number of CFU per catheter was determined after overnight incubation at 37°C.
Bacteremia model.
Groups of 10 5- to 8-week-old female NIH-Swiss mice were infected via tail vein injection with 5 × 107 CFU of LAC or 5 × 108 CFU of UAMS-1. Tissues were harvested from any mouse that died or required compassionate euthanasia; otherwise, tissues were harvested at 6 days postinfection (23). Organs were removed aseptically and homogenized on ice. Serial dilutions of each homogenate were then plated on TSA, and the number of CFU per organ was determined following overnight incubation at 37°C.
Western blotting.
The relative amounts of AgrA and SarA were assessed using whole-cell lysates prepared from stationary-phase cells and rabbit polyclonal anti-SarA or anti-AgrA IgG antibodies (23, 36). Samples were standardized for blotting on the basis of the total protein amounts, as assessed by a Bradford assay. The production of alpha toxin and extracellular protein A (eSpa) was assessed using standardized cell-free supernatants and rabbit polyclonal anti-alpha toxin and anti-protein A IgG antibodies (25, 37). In this case, samples for blotting were standardized by adjusting the optical density of each culture to an equivalent value prior to harvesting of the supernatant; samples were not further standardized on the basis of the Bradford assay because this would potentially mask the impact of extracellular proteases on overall protein production. Secondary antibody was horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (Sigma Chemical Co., St. Louis, MO). Blotting assays were performed using a minimum of three biological replicates. Blots were developed with SuperSignal West Femto chemiluminescent substrate (Thermo Scientific, Rockford, IL) and quantified using a Bio-Rad ChemiDocMP imaging system and Image Lab software (Bio-Rad Laboratories, Inc.).
Assessment of protease and nuclease activity.
Protease activity in standardized samples of the cell-free supernatant from stationary-phase (16 h) cultures grown without antibiotics was assessed using a protease fluorescent detection kit (Sigma Chemical Co., St. Louis, MO) (23). Results are reported as relative fluorescence units and represent those from at least two biological replicates, each of which included four experimental replicates. Nuclease activity was assessed using DNase test agar (Remel, Lenexa, KS) (16).
PIA immunoblot assay.
Production of the polysaccharide intercellular adhesin (PIA) was assessed as previously described with slight modifications (38). Specifically, cultures were grown overnight in BFM with antibiotics as appropriate. After standardization to an OD660 of 8.0, cells were harvested by centrifugation and resuspended in 60 μl 0.5 M EDTA. Cell suspensions were boiled at 105°C for 8 min followed by centrifugation. Forty microliters of supernatant was then incubated for 30 min with 5 μl proteinase K (10 mg per ml) at 37°C to reduce nonspecific background levels. Fifty microliters of Tris-buffered saline (TBS; 20 mM Tris-HCl, 150 mM NaCl [pH 7.4]) was added to the samples. Using a Bio-Dot SF microfiltration apparatus (Bio-Rad Laboratories, Inc., Hercules, CA), 20 μl of this sample was spotted onto a nitrocellulose membrane presoaked with TBS (Roche Diagnostics Corp., Indianapolis, IN). Each well was then rinsed with 200 μl TBS. The membrane was then removed and blocked in 2.5% skim milk overnight at 4°C. PIA production was assessed using anti-PIA antiserum (kindly provided by Kim Jefferson, Virginia Commonwealth University) diluted 1:500 in 2.5% skim milk. Primary antibody was detected using HRP-conjugated goat anti-rabbit IgG secondary antibody (Sigma Chemical Co., St. Louis, MO). The blots were developed and quantified as described above.
RNA isolation and qRT-PCR.
S. aureus strains were grown at 37°C to stationary phase (16 h), and RNA was isolated from 1.5 ml of cultures adjusted to an OD660 of 2.0 using RNAzol RT (Molecular Research Center, Inc.) as previously described (39). Quantitative reverse transcription-PCR (qRT-PCR) was performed using primers specific for RNAIII (5′-ATCGACACAGTGAACAAATTCAC-3′ and 5′-CTCTACTAGCAAATGTTACTCAC-3′) and gyrB (5′-TTACAGCAGCGTATTAGAGAGC-3′ and 5′-CCTCATAGTGATAGGAGTCCTC-3′) (40). RNA from 3 independent cultures was analyzed. The level of expression of RNAIII relative to that of the gyrase control was calculated.
Statistical analysis.
Analysis of variance (ANOVA) models with appropriately defined contrasts or unpaired t tests were then used to estimate differences between mutations for each strain and treatment separately. Because many of the data clearly violate the assumptions of these parametric tests, permutation methods were used to calculate P values and when ANOVA models were used to adjust for multiple comparisons. Data involving measures of the numbers of CFU were log10 transformed prior to analysis. Data from experiments assessing protease activity, biofilm formation, the amount of Spa, or the amount of alpha toxin were scaled relative to the mean for the parent strain prior to analysis. Statistical analyses were performed using the statistical programming language R (version 3.2.2; R Development Core Team, Vienna, Austria), SAS software (version 9.4; SAS Institute Inc., Cary, NC), and GraphPad Prism software (version 5.0; GraphPad Software, La Jolla, CA). P values of <0.05 were considered statistically significant.
RESULTS
Impact of xerC on growth, protease production, and biofilm formation.
The NTML was generated in a strain designated JE2, which is a derivative of the USA300 strain LAC cured of both its larger plasmid conferring resistance to erythromycin (p03) and its smaller cryptic plasmid (p01). We obtained seven of the NTML mutants (the brnQ, hslU, ilvE, lacC, mob, xerC, and ydcE mutants) in which the gene mutation is associated with increased protease production (27), and the mutated genes in two of the mutants (hslU and xerC) are contained within the xerC operon. Of these seven mutants, four (the ilvE, lacC, mobB, and xerC mutants) were found to exhibit significantly increased protease activity relative to that of the JE2 parent strain (see Fig. S1 in the supplemental material). All four of these mutants, as well as the hslU mutant, also exhibited a reduced capacity to form a biofilm in vitro (see Fig. S1 in the supplemental material). However, mutation of xerC limited biofilm formation to a much greater extent than mutation of any other gene except sarA.
To facilitate direct comparisons with our earlier studies, the impact of the transposon insertion in xerC was also assessed after transduction of the xerC mutation from JE2 into the same derivative of the USA300 strain LAC used in these experiments. This derivative was cured of the larger plasmid conferring resistance to erythromycin but not the smaller cryptic plasmid (17, 23, 24). Additionally, to assess the extent to which the results that we observed were consistent among diverse clinical isolates, we also transduced the xerC mutation into the USA200 methicillin-sensitive strain UAMS-1 (41). Although a previous report found that mutation of xerC in S. aureus resulted in an increase in the average cell size, as assessed by flow cytometry, but did not impact the overall growth rate (32), we found that it did limit the growth of both LAC and UAMS-1 in rich medium in vitro, but only to a modest extent (<2-fold), and even this difference was not apparent once cultures reached late stationary phase (see Fig. S2 in the supplemental material). Protease production was also increased in both the LAC and UAMS-1 xerC mutants, although in both cases it remained below that observed in the isogenic sarA mutants (Fig. 1). Nevertheless, mutation of xerC did result in a significant decrease in biofilm formation by both LAC and UAMS-1. In LAC, the impact of mutating xerC was comparable to that observed in an isogenic sarA mutant, while in UAMS-1, mutation of sarA had a significantly greater impact than mutation of xerC (Fig. 1).
FIG 1.

Impact of xerC on protease production and biofilm formation. (A) Protease production in LAC (top), UAMS-1 (bottom), their isogenic derivatives with mutations in sarA and/or xerC, and their genetically complemented xerC mutants (xerCC) was assessed using a fluorescence resonance energy transfer-based assay. (B) Biofilm formation in LAC (top), UAMS-1 (bottom), isogenic derivatives with mutations in sarA and/or xerC, and complemented xerC mutants (xerCC) was assessed using a microtiter plate assay. Permutation-based ANOVA models were used to perform these analyses. *, a significant difference relative to the result for the isogenic parent strain; **, a significant difference by comparison to the result for the isogenic sarA mutant. RFU, relative fluorescence units.
The decreased growth rate (see Fig. S2 in the supplemental material) as well as the increased production of extracellular proteases and the decreased capacity to form a biofilm (Fig. 1) were all fully complemented in both LAC and UAMS-1 by introducing an intact copy of xerC alone. This suggests that all of these phenotypes are mediated by the xerC mutation itself rather than a polar effect on downstream genes within the operon. The introduction of codY on a plasmid did restore biofilm formation in a LAC xerC mutant, but this was not the case in a UAMS-1 xerC mutant (see Fig. S3 in the supplemental material). This strain-dependent effect is consistent with our previous observation that mutation of codY limited biofilm formation in LAC but had the opposite effect in UAMS-1 (17). This suggests that the phenotypes observed when we attempted to complement the xerC mutation by introducing codY may reflect the overexpression of codY from a multicopy plasmid. This is consistent with the observation that biofilm formation was increased when the plasmid carrying codY was introduced into LAC but decreased when it was introduced into UAMS-1 (see Fig. S3 in the supplemental material), thereby demonstrating that the impact of introducing codY is independent of the functional status of xerC. Moreover, if the impact of mutating xerC were due to downstream effects on codY, then it would be anticipated that mutation of xerC would have a similar strain-dependent effect on biofilm formation, and this was not the case (Fig. 1).
Mechanistic basis for the biofilm-deficient phenotype of xerC mutants.
To determine if there is a cause-and-effect relationship between the increased protease production and decreased biofilm formation, we introduced the xerC mutation into a derivative of LAC with mutations in the genes encoding aureolysin, SspA, SspB, and ScpA (23) and a derivative of UAMS-1 with mutations in the genes encoding aureolysin, SspA, and SspB. We then compared the relative capacity to form a biofilm in these protease-positive versus protease-deficient xerC mutants. The results confirmed that biofilm formation was almost fully restored in a protease-deficient LAC xerC mutant and partially restored in a protease-deficient UAMS-1 xerC mutant (Fig. 2). The fact that limiting protease production had a greater impact in the LAC xerC mutant than in the UAMS-1 xerC mutant could be interpreted to mean that increased protease production is more important in the former than the latter, but given the difference between these protease-deficient derivatives, it could also suggest an important role for ScpA. Indeed, ScpA has been shown to limit S. aureus biofilm formation under conditions like those employed in these experiments (24, 42). However, we were unable to generate the UAMS-1 xerC aur sspAB scpA mutant required to test this hypothesis. Nevertheless, this does not detract from the general conclusion that the increased production of extracellular proteases plays a role in limiting biofilm formation in both LAC and UAMS-1 xerC mutants.
FIG 2.

Relative biofilm formation in xerC mutants as a function of protease and nuclease production. (A) Biofilm formation in LAC (top), UAMS-1 (bottom), their respective xerC mutants, and their isogenic xerC mutants unable to produce all (LAC) or most (UAMS-1) S. aureus extracellular proteases was assessed using a microtiter plate assay. (B) Biofilm formation in UAMS-1, LAC, their isogenic xerC mutants, and their nuclease-deficient xerC mutants unable to produce the primary S. aureus extracellular nuclease (Nuc1) was assessed. Permutation-based ANOVA models were used to perform these analyses. *, a statistically significant difference between the xerC mutant and its isogenic parent strain with the equivalent capacity to produce extracellular proteases (A) or nucleases (B); **, a statistically significant difference between the protease or nuclease-deficient xerC mutant and the isogenic protease or nuclease-positive xerC strain.
At the same time, mutation of xerC also appeared to result in increased nuclease activity (see Fig. S4 in the supplemental material), and limiting nuclease production by mutating the nuc-1 gene also restored biofilm formation to a significant extent in both the LAC and UAMS-1 xerC mutants (Fig. 2). These results demonstrate that the increased production of both extracellular proteases and nucleases contributes to the biofilm-deficient phenotype of LAC and UAMS-1 xerC mutants. Additionally, while protease- and nuclease-deficient derivatives of the LAC and UAMS-1 xerC mutants exhibited an increased capacity to form a biofilm by comparison to that of the isogenic xerC mutants, they did not exhibit an increased growth rate (see Fig. S2 in the supplemental material). Thus, the reduced capacity of xerC mutants to form a biofilm cannot be explained by their reduced growth rate.
It has been proposed that biofilm formation in methicillin-sensitive strains is more dependent on the production of the polysaccharide intercellular adhesin (PIA), while methicillin-resistant strains rely on other factors, including extracellular DNA and surface-associated proteins (43). Based on this, we also assessed the impact of mutated xerC on the production of PIA. As was observed in our earlier study (17), we could not detect PIA in LAC or its isogenic xerC mutant, but it was detectable in a LAC codY mutant (Fig. 3). PIA was detectable in UAMS-1, and in this case, its production was increased in both xerC and codY mutants (Fig. 3). This is likely to account for why mutation of xerC had a reduced impact in UAMS-1 in comparison to the impact in LAC (Fig. 1), but to the extent that PIA promotes biofilm formation, it cannot account for the biofilm-deficient phenotype of a UAMS-1 xerC mutant.
FIG 3.
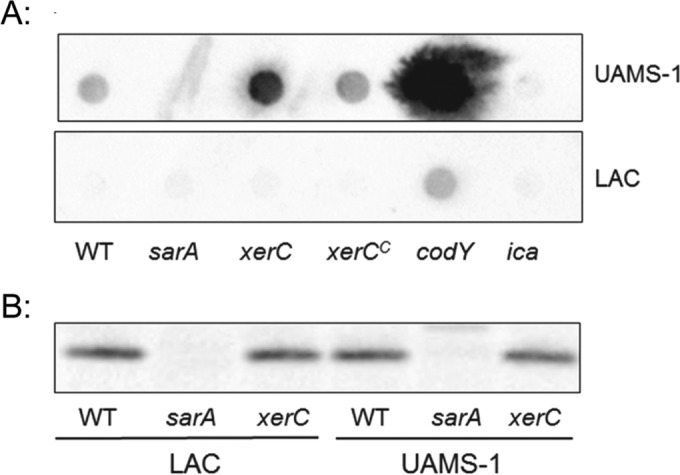
Impact of xerC on PIA and SarA production. (A) PIA production in UAMS-1, LAC, their isogenic sarA, xerC, codY, and ica mutants, and their genetically complemented xerC mutants (xerCC) was assessed by Western dot blotting. The ica and codY mutants were included as negative and positive controls, respectively, on the basis of the fact that ica encodes the enzymes necessary for PIA production, while mutation of codY was previously shown to result in increased PIA production in UAMS-1 (17). (B) The abundance of SarA was assessed by conventional Western blotting. The results shown are representative of those from at least three biological replicates. WT, wild type.
Mutation of sarA also results in decreased biofilm formation and increased production of proteases and nucleases (13, 16, 19). Thus, one explanation for these results is that mutation of xerC limits the production of SarA. However, Western blot analysis confirmed that this was not the case, with the relative levels of SarA being equivalent in LAC, UAMS-1, and their isogenic xerC mutants (Fig. 3). Moreover, mutation of xerC and sarA had opposite effects on the production of PIA (Fig. 3).
Impact of xerC on antibiotic susceptibility in the context of a biofilm.
Many S. aureus regulatory loci have been implicated in biofilm formation (17), but few have been evaluated in the specific context of the clinical problem of reduced antibiotic susceptibility. We previously demonstrated that mutation of sarA results in increased antibiotic susceptibility in vitro and in vivo (21, 22), and the fact that mutation of xerC had an impact comparable to that of mutation of sarA in our microtiter plate biofilm assay (Fig. 1) suggests that mutation of xerC might also limit biofilm formation to an extent that can be correlated with increased antibiotic susceptibility. To test this, we used our in vivo model of catheter-associated biofilm formation (21, 22) to assess the relative susceptibility of LAC, UAMS-1, and their isogenic sarA and xerC mutants to daptomycin and ceftaroline. We chose daptomycin because it is a membrane-active agent that is relatively effective against stationary-phase and nondividing S. aureus cells (44). It was also shown to penetrate staphylococcal biofilms and exhibit a relatively high degree of efficacy in the context of an established biofilm (21, 22, 45–47). Ceftaroline was chosen because it is a beta-lactam antibiotic with activity against methicillin-resistant S. aureus (48).
Although variation was observed between experiments, when the results were assessed by the overall number of CFU per catheter, statistically significant differences were observed between LAC, UAMS-1, and their xerC mutants with respect to the relative capacity to form a biofilm in this model and their relative susceptibility to both daptomycin and ceftaroline (Fig. 4). The potential clinical significance of this is best evidenced by the fact that no catheters colonized with either parent strain were cleared of viable bacteria after 5 days of exposure to either antibiotic (Fig. 5). In contrast, ≥50% of catheters were cleared of viable bacteria, at least within the limit of detection (500 CFU), when the experiments were done with LAC or UAMS-1 xerC mutants, and this was true irrespective of the antibiotic employed. In fact, the results observed with the LAC and UAMS-1 xerC mutants were comparable to those observed with the isogenic sarA mutants, thus suggesting that xerC may also be a viable target for therapeutic intervention in the context of S. aureus biofilm-associated infections.
FIG 4.

Impact of xerC on biofilm formation and antibiotic susceptibility in vivo. Catheters were implanted into the flanks of mice prior to colonization with LAC (A and C), UAMS-1 (B and D), or their isogenic sarA or xerC mutants. After 24 h, the lumen of each catheter was injected with sterile PBS (−) or the antibiotic (+) daptomycin (A and B) or ceftaroline (C and D). This was continued daily for 5 days. At 24 h after the last injection, the catheters were recovered and processed to assess the number of CFU remaining per catheter. The horizontal line within each group indicates the mean for that experimental group. Numbers above each group are permutation-based unpaired t-test P values for that group compared to the result for the parent strain with or without antibiotic exposure as appropriate. NS, not significant.
FIG 5.
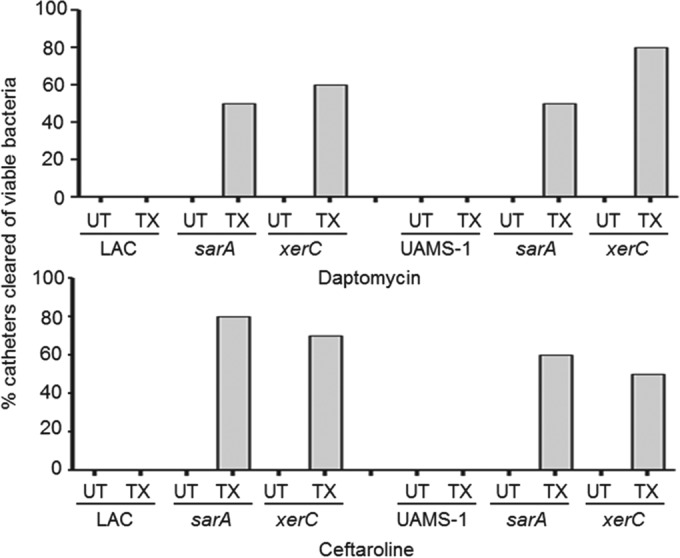
Percentage of catheters cleared by antibiotic exposure. Results illustrate the percentage of catheters (n = 10) colonized with LAC, UAMS-1, or their isogenic xerC and sarA mutants that were cleared of viable bacteria with exposure (treated [TX]) and without exposure (untreated [UT]) to the indicated antibiotic.
Impact of xerC in acute hematogenous infection.
We also evaluated the relative ability of xerC mutants to cause acute infection following direct introduction into the bloodstream. The results confirmed that mutation of xerC in LAC limits virulence, as assessed by the colony counts in the spleen and heart but not those in the kidney (Fig. 6). In contrast, mutation of sarA resulted in a significant reduction in virulence, as assessed in all three of these organs. Overall virulence assessed on the basis of lethality was also reduced to a significant extent in both LAC xerC and sarA mutants (Fig. 6). These same relationships were observed with UAMS-1 xerC mutants, with colony counts being significantly reduced in the spleen and heart but not the kidney and with overall virulence assessed on the basis of lethality being significantly reduced in the mutants in comparison to that in the isogenic parent strain (Fig. 7).
FIG 6.

Virulence of LAC xerC mutants in hematogenous infection. Mice were infected by tail vein injection with the indicated strains. Tissues were harvested either upon compassionate euthanasia or after 6 days. The results shown are the colony counts obtained from each tissue in individual mice, with the horizontal line within each group indicating the mean within that experimental group (A to C). Permutation-based ANOVA models were used to analyze these data. The numbers above each group are P values for that group compared to the result for the isogenic parent strain. NS, not significant. (D) Kaplan-Meier survival curves for mice hematogenously infected with LAC and its isogenic xerC or sarA mutant. Log rank tests were used to compare the results for the mutant and parent strains. *, statistically significant difference by comparison to the result for the parent strain.
FIG 7.

Virulence of UAMS-1 xerC mutants in hematogenous infection. Mice were infected by tail vein injection with UAMS-1 (U1) or its isogenic xerC mutant. Tissues were harvested after compassionate euthanasia or at 6 days after infection. (A) The results shown are the colony counts obtained from each tissue in individual mice, with the horizontal line within each group indicating the mean within that experimental group. Permutation-based unpaired t tests were used to compare the result for the isogenic xerC mutant to that for the parent strain. The numbers above each xerC mutant are P values for that group compared to the result for the parent strain. NS, not significant. (B) Kaplan-Meier survival curves for mice hematogenously infected with UAMS-1 and its isogenic xerC mutant. *, a statistically significant difference by the log-rank test when the result for the isogenic xerC mutant is compared to that for the parent strain.
LAC and UAMS-1 xerC mutants were also found to produce increased amounts of protein A (Fig. 8). In contrast, the amount of alpha toxin was reduced in a LAC xerC mutant (UAMS-1 does not produce alpha toxin). In the isogenic sarA mutants, both protein A and alpha toxin were essentially absent. Moreover, as would be expected on the basis of the findings of our previous study (23), limiting protease production restored the accumulation of both protein A and alpha toxin in a LAC sarA mutant, but it had no impact on the abundance of either protein in a LAC xerC mutant (Fig. 8). This suggests that xerC plays a more direct regulatory role in the production of these proteins. Increased production of protein A and decreased production of alpha toxin are characteristic of S. aureus agr mutants. Based on this, we also assessed the impact of mutating xerC on the accumulation of AgrA and RNAIII, and the results confirmed that the relative amounts of both were reduced in xerC mutants (Fig. 9).
FIG 8.

Abundance of protein A and alpha toxin in xerC mutants. (A) The relative abundance of protein A (Spa), as assessed by measuring the amount of extracellular Spa (eSpa) in clarified supernatants, was assessed in LAC, UAMS-1, and the indicated mutants by Western blotting. (B) The relative amounts of alpha toxin in LAC and the indicated mutants were assessed by Western blotting. An alpha toxin mutant (hla) and purified alpha toxin were included as controls. UAMS-1 does not produce alpha toxin and thus was not included in this experiment. *, a statistically significant difference by comparison to the results for the isogenic parent strain; **, a statistically significant difference by comparison to the results for the isogenic sarA mutant. (C and D) The relative amounts of eSpa (C) and alpha toxin (D) in LAC, its sarA and xerC mutants, and protease-deficient derivatives of each of these strains were assessed by Western blotting. *, a statistically significant difference between the results for the protease-deficient derivative and the isogenic protease-positive strain. Permutation-based ANOVA models were used to perform these analyses.
FIG 9.

Impact of xerC on agr. (A) The abundance of AgrA in LAC, UAMS-1, their isogenic sarA, xerC and agr mutants, and their genetically complemented xerC mutants (xerCC) was assessed by Western blotting. (B) The abundance of RNAIII in the indicated strains was assessed by qRT-PCR. *, a statistically significant decrease relative to the amount for the isogenic parent strain. Permutation-based ANOVA models were used to perform these analyses.
These results suggest that the reduced virulence of xerC mutants observed in the context of bacteremia is likely due to reduced agr function. However, this cannot account for the decreased capacity of xerC mutants to form a biofilm because mutation of agr did not decrease biofilm formation in either strain (Fig. 10). A UAMS-1 agr xerC double mutant exhibited a reduced capacity to form a biofilm, thereby demonstrating that mutation of xerC limits biofilm formation irrespective of the functional status of agr, while the increased capacity to form a biofilm observed in a LAC agr mutant was also evident in the isogenic agr xerC mutant.
FIG 10.
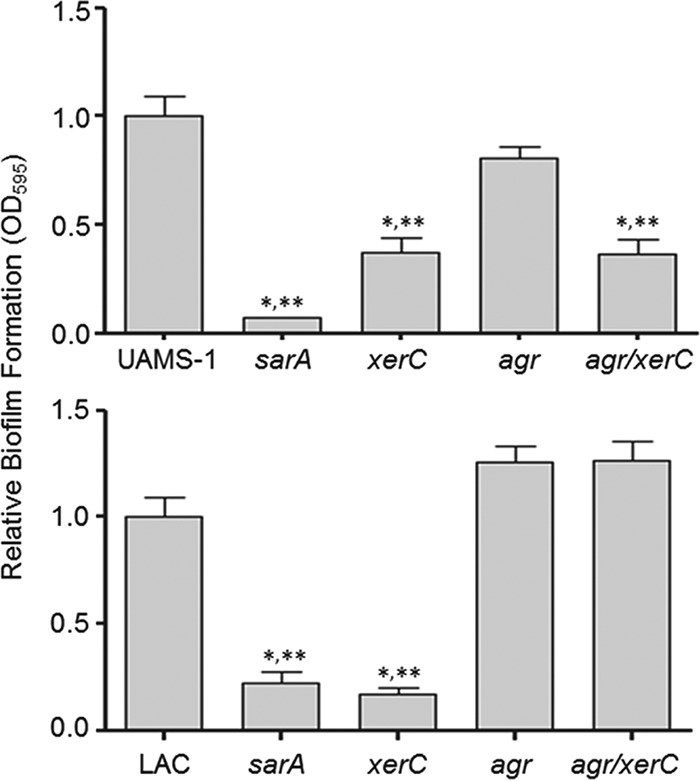
Relative impact of agr and xerC on biofilm formation. Biofilm formation was assessed in UAMS-1 (top), LAC (bottom), and their isogenic sarA, agr, xerC, and agr/xerC mutants. *, a statistically significant difference relative to the result for the isogenic parent strain; **, a statistically significant difference by comparison to the result for the isogenic agr mutant. Permutation-based ANOVA models were used to perform these analyses.
In LAC, one possible explanation for these results is that biofilm formation is mediated by surface proteins that are produced in increased amounts in an isogenic agr mutant and that the relatively modest increase in protease production observed in a xerC mutant does not reverse this as it does in a sarA mutant (23). However, given the comparable levels of protease production in the two xerC mutants, this would presumably also be true in a UAMS-1 agr xerC mutant. Also, the difference cannot be a function of the impact of mutating xerC on PIA production in UAMS-1 (Fig. 3), which would be expected to increase rather than decrease biofilm formation. Thus, at present, we do not have an explanation for this strain-dependent difference. However, in the context of whether the impact of mutating xerC on biofilm formation is mediated via an agr-dependent mechanism, this difference is irrelevant, in that mutation of agr did not limit biofilm formation in UAMS-1 or LAC, while mutation of xerC did (Fig. 10).
DISCUSSION
The xerC gene encodes a homologue of an Escherichia coli recombinase previously shown to resolve multimers of DNA replicons for efficient partitioning into daughter cells (49). The overwhelming majority of studies investigating xerC have focused on this function (50–52), with very few considering its potential role in pathogenesis. One exception is the report by Chalker et al. (32), who found that mutation of xerC in S. aureus limited virulence in a murine model of hematogenous pyelonephritis.
We were unable to demonstrate a significant decrease in bacterial burdens in the kidney, but we did confirm that mutation of xerC results in significant attenuation, as assessed by the bacterial burdens of the spleen and heart and as assessed on the basis of overall lethality. Moreover, this was true in both the USA300 methicillin-resistant S. aureus strain LAC and the USA200 methicillin-sensitive S. aureus strain UAMS-1. Our results also demonstrate that mutation of xerC limits biofilm formation to a degree that can be correlated with increased antibiotic susceptibility in vivo in both of these strains. Thus, the results of the experiments that we report are the first to suggest that xerC is potentially a viable therapeutic target in diverse forms of infection and in genetically and phenotypically diverse strains of S. aureus. Our results also demonstrate that xerC impacts the ability to cause biofilm-associated infections and acute hematogenous infections via agr-independent and agr-dependent pathways, respectively. Mutation of sarA also limits virulence in both of these clinical contexts, and in this case, the mechanistic basis for this attenuation is largely due to the increased production of extracellular proteases and the consequent decreased accumulation of both surface-associated and extracellular virulence factors (23, 25). In the case of xerC, this also appears to be true in biofilm-associated infections but not in acute hematogenous infections. This suggests that the impact of mutating xerC could be at least partially mediated through sarA, which is also known to function via both agr-dependent and agr-independent pathways (3, 53). However, mutation of xerC did not result in the decreased accumulation of SarA. According to the Staphylococcus aureus transcriptome meta-database (SATMD), mutation of sarA also has no impact on the transcription of xerC (54).
Thus, the full mechanistic basis for the impact of xerC remains unknown. At the same time, it has been suggested that SarA functions as an architectural protein capable of promoting DNA recombination (55), and as noted above, it has been demonstrated that xerC also encodes a recombinase (49). This suggests that SarA and XerC may function through a common mechanism in which the reduced capacity to control the DNA architecture has a global impact on gene transcription. Alternatively, like sarA (56), xerC could impact the production of S. aureus virulence factors via a posttranscriptional effect on mRNA stability. Both of these possibilities are under investigation.
Irrespective of the mechanism(s) involved, a primary motivation behind the experiments that we report on here was to determine whether mutation of xerC limits S. aureus biofilm formation to a therapeutically relevant degree. The presence of a biofilm in S. aureus infections confers a therapeutically relevant degree of intrinsic resistance to both host defenses and conventional antibiotics, thus necessitating an interdisciplinary clinical approach that often includes surgical debridement of bone infections and infected tissues and definite removal of colonized medical devices (7, 57, 58). The therapeutic recalcitrance of these infections to conventional antibiotic therapy, the fact that their incidence is predicted to increase dramatically in coming years, particularly in the context of orthopedics (59, 60), and the growing impact of acquired antibiotic resistance in S. aureus (61) all emphasize the need for the development of novel therapeutic strategies to combat S. aureus infections.
Our previous studies, when viewed collectively, strongly suggest that sarA is a viable target in this regard and that, at least in the context of biofilm-associated infection, may in fact be the best target owing to the fact that mutation of sarA limits biofilm formation to a greater degree than mutation of any other regulatory locus that we have examined (16, 17, 19, 24, 25). Additionally, in those few cases in which a given mutation enhanced biofilm formation (e.g., mutation of agr, fur, or mgrA), concomitant mutation of sarA reversed this effect (17), thereby suggesting that therapeutic strategies targeting sarA may be less subject to the development of resistance owing to mutations in other aspects of S. aureus regulatory circuits.
The fact that all of the in vivo phenotypes that we observed with LAC and UAMS-1 xerC mutants were comparable to those observed with the isogenic sarA mutants suggests that xerC, which was not included in our previous comparisons (17), may also be a viable therapeutic target worthy of additional exploration. One observation that argues against that is the increased production of PIA in a UAMS-1 xerC mutant, although the impact of this was not evident in any of our in vivo experiments. A second relevant observation is that biofilm formation was not reduced in a LAC agr xerC mutant. This is important because agr dysfunction is common among clinical isolates and has been associated with both increased mortality and decreased antibiotic susceptibility (1, 62, 63). Thus, mutations leading to agr dysfunction would presumably compromise therapeutic strategies targeting xerC in at least some strains of S. aureus, at least in the context of biofilm-associated infections, while such mutations would not be expected to compromise therapeutic strategies targeting sarA.
Supplementary Material
ACKNOWLEDGMENTS
We thank Cody J. Story, Carrie A. Binyon, Weston B. Mills, and William B. Lynn for technical assistance.
Additional support was provided by core facilities supported by the Translational Research Institute (UL1TR000039).
The content is solely the responsibility of the authors and does not represent the views of the NIH.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01462-15.
REFERENCES
- 1.Laabei M, Uhlemann AC, Lowy FD, Austin ED, Yokoyama M, Ouadi K, Feil E, Thorpe HA, Williams B, Perkins M, Peacock SJ, Clarke SR, Dordel J, Holden M, Votintseva AA, Bowden R, Crook DW, Young BC, Wilson DJ, Recker M, Massey RC. 2015. Evolutionary trade-offs underlie the multi-faceted virulence of Staphylococcus aureus. PLoS Biol 13:e1002229. doi: 10.1371/journal.pbio.1002229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lowy FD. 1998. Staphylococcus aureus infections. N Engl J Med 339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 3.Beenken KE, Mrak LN, Griffin LM, Zielinska AK, Shaw LN, Rice KC, Horswill AR, Bayles KW, Smeltzer MS. 2010. Epistatic relationships between sarA and agr in Staphylococcus aureus biofilm formation. PLoS One 5:e10790. doi: 10.1371/journal.pone.0010790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boles BR, Horswill AR. 2008. Agr-mediated dispersal of Staphylococcus aureus biofilms. PLoS Pathog 4:e1000052. doi: 10.1371/journal.ppat.1000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bassetti M, Merelli M, Temperoni C, Astilean A. 2013. New antibiotics for bad bugs: where are we? Ann Clin Microbiol Antimicrob 12:22. doi: 10.1186/1476-0711-12-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bjarnsholt T, Ciofu O, Molin S, Givskov M, Hoiby N. 2013. Applying insights from biofilm biology to drug development—can a new approach be developed? Nat Rev Drug Discov 12:791–808. doi: 10.1038/nrd4000. [DOI] [PubMed] [Google Scholar]
- 7.Brady RA, Leid JG, Calhoun JH, Costerton JW, Shirtliff ME. 2008. Osteomyelitis and the role of biofilms in chronic infection. FEMS Immunol Med Microbiol 52:13–22. doi: 10.1111/j.1574-695X.2007.00357.x. [DOI] [PubMed] [Google Scholar]
- 8.Lewis K. 2008. Multidrug tolerance of biofilms and persister cells. Curr Top Microbiol Immunol 322:107–131. [DOI] [PubMed] [Google Scholar]
- 9.Trotonda MP, Tamber S, Memmi G, Cheung AL. 2008. MgrA represses biofilm formation in Staphylococcus aureus. Infect Immun 76:5645–5654. doi: 10.1128/IAI.00735-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Donlan RM. 2001. Biofilm formation: a clinically relevant microbiological process. Clin Infect Dis 33:1387–1392. doi: 10.1086/322972. [DOI] [PubMed] [Google Scholar]
- 11.Conlon BP, Rowe SE, Lewis K. 2015. Persister cells in biofilm associated infections. Adv Exp Med Biol 831:1–9. doi: 10.1007/978-3-319-09782-4_1. [DOI] [PubMed] [Google Scholar]
- 12.Costerton JW, Stewart PS, Greenberg EP. 1999. Bacterial biofilms: a common cause of persistent infections. Science 284:1318–1322. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- 13.Beenken KE, Spencer H, Griffin LM, Smeltzer MS. 2012. Impact of extracellular nuclease production on the biofilm phenotype of Staphylococcus aureus under in vitro and in vivo conditions. Infect Immun 80:1634–1638. doi: 10.1128/IAI.06134-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beenken KE, Mrak LN, Zielinska AK, Atwood DN, Loughran AJ, Griffin LM, Matthews KA, Anthony AM, Spencer HJ, Skinner RA, Post GR, Lee CY, Smeltzer MS. 2014. Impact of the functional status of saeRS on in vivo phenotypes of Staphylococcus aureus sarA mutants. Mol Microbiol 92:1299–1312. doi: 10.1111/mmi.12629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cassat JE, Hammer ND, Campbell JP, Benson MA, Perrien DS, Mrak LN, Smeltzer MS, Torres VJ, Skaar EP. 2013. A secreted bacterial protease tailors the Staphylococcus aureus virulence repertoire to modulate bone remodeling during osteomyelitis. Cell Host Microbe 13:759–772. doi: 10.1016/j.chom.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsang LH, Cassat JE, Shaw LN, Beenken KE, Smeltzer MS. 2008. Factors contributing to the biofilm-deficient phenotype of Staphylococcus aureus sarA mutants. PLoS One 3:e3361. doi: 10.1371/journal.pone.0003361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Atwood DN, Loughran AJ, Courtney AP, Anthony AC, Meeker DG, Spencer HJ, Gupta RK, Lee CY, Beenken KE, Smeltzer MS. 2015. Comparative impact of diverse regulatory loci on Staphylococcus aureus biofilm formation. Microbiologyopen 4:436–451. doi: 10.1002/mbo3.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abdelhady W, Bayer AS, Seidl K, Moormeier DE, Bayles KW, Cheung A, Yeaman MR, Xiong YQ. 2014. Impact of vancomycin on sarA-mediated biofilm formation: role in persistent endovascular infections due to methicillin-resistant Staphylococcus aureus. J Infect Dis 209:1231–1240. doi: 10.1093/infdis/jiu007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beenken KE, Blevins JS, Smeltzer MS. 2003. Mutation of sarA in Staphylococcus aureus limits biofilm formation. Infect Immun 71:4206–4211. doi: 10.1128/IAI.71.7.4206-4211.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Valle J, Toledo-Arana A, Berasain C, Ghigo JM, Amorena B, Penades JR, Lasa I. 2003. SarA and not sigmaB is essential for biofilm development by Staphylococcus aureus. Mol Microbiol 48:1075–1087. doi: 10.1046/j.1365-2958.2003.03493.x. [DOI] [PubMed] [Google Scholar]
- 21.Weiss EC, Spencer HJ, Daily SJ, Weiss BD, Smeltzer MS. 2009. Impact of sarA on antibiotic susceptibility of Staphylococcus aureus in a catheter-associated in vitro model of biofilm formation. Antimicrob Agents Chemother 53:2475–2482. doi: 10.1128/AAC.01432-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weiss EC, Zielinska A, Beenken KE, Spencer HJ, Daily SJ, Smeltzer MS. 2009. Impact of sarA on daptomycin susceptibility of Staphylococcus aureus biofilms in vivo. Antimicrob Agents Chemother 53:4096–4102. doi: 10.1128/AAC.00484-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zielinska AK, Beenken KE, Mrak LN, Spencer HJ, Post GR, Skinner RA, Tackett AJ, Horswill AR, Smeltzer MS. 2012. sarA-mediated repression of protease production plays a key role in the pathogenesis of Staphylococcus aureus USA300 isolates. Mol Microbiol 86:1183–1196. doi: 10.1111/mmi.12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loughran AJ, Atwood DN, Anthony AC, Harik NS, Spencer HJ, Beenken KE, Smeltzer MS. 2014. Impact of individual extracellular proteases on Staphylococcus aureus biofilm formation in diverse clinical isolates and their isogenic sarA mutants. Microbiologyopen 3:897–909. doi: 10.1002/mbo3.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zielinska AK, Beenken KE, Joo HS, Mrak LN, Griffin LM, Luong TT, Lee CY, Otto M, Shaw LN, Smeltzer MS. 2011. Defining the strain-dependent impact of the staphylococcal accessory regulator (sarA) on the alpha-toxin phenotype of Staphylococcus aureus. J Bacteriol 193:2948–2958. doi: 10.1128/JB.01517-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arya R, Ravikumar R, Santhosh RS, Princy SA. 2015. SarA based novel therapeutic candidate against Staphylococcus aureus associated with vascular graft infections. Front Microbiol 6:416. doi: 10.3389/fmicb.2015.00416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fey PD, Endres JL, Yajjala VK, Widhelm TJ, Boissy RJ, Bose JL, Bayles KW. 2013. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. mBio 4:e00537-12. doi: 10.1128/mBio.00537-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frees D, Thomsen LE, Ingmer H. 2005. Staphylococcus aureus ClpYQ plays a minor role in stress survival. Arch Microbiol 183:286–291. doi: 10.1007/s00203-005-0773-x. [DOI] [PubMed] [Google Scholar]
- 29.Frees D, Chastanet A, Qazi S, Sorensen K, Hill P, Msadek T, Ingmer H. 2004. Clp ATPases are required for stress tolerance, intracellular replication and biofilm formation in Staphylococcus aureus. Mol Microbiol 54:1445–1462. doi: 10.1111/j.1365-2958.2004.04368.x. [DOI] [PubMed] [Google Scholar]
- 30.Rivera FE, Miller HK, Kolar SL, Stevens SM Jr, Shaw LN. 2012. The impact of CodY on virulence determinant production in community-associated methicillin-resistant Staphylococcus aureus. Proteomics 12:263–268. doi: 10.1002/pmic.201100298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Montgomery CP, Boyle-Vavra S, Roux A, Ebine K, Sonenshein AL, Daum RS. 2012. CodY deletion enhances in vivo virulence of community-associated methicillin-resistant Staphylococcus aureus clone USA300. Infect Immun 80:2382–2389. doi: 10.1128/IAI.06172-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chalker AF, Lupas A, Ingraham K, So CY, Lunsford RD, Li T, Bryant A, Holmes DJ, Marra A, Pearson SC, Ray J, Burnham MK, Palmer LM, Biswas S, Zalacain M. 2000. Genetic characterization of gram-positive homologs of the XerCD site-specific recombinases. J Mol Microbiol Biotechnol 2:225–233. [PubMed] [Google Scholar]
- 33.Boles BR, Thoendel M, Roth AJ, Horswill AR. 2010. Identification of genes involved in polysaccharide-independent Staphylococcus aureus biofilm formation. PLoS One 5:e10146. doi: 10.1371/journal.pone.0010146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bubeck Wardenburg J, Williams WA, Missiakas D. 2006. Host defenses against Staphylococcus aureus infection require recognition of bacterial lipoproteins. Proc Natl Acad Sci U S A 103:13831–13836. doi: 10.1073/pnas.0603072103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Torres VJ, Attia AS, Mason WJ, Hood MI, Corbin BD, Beasley FC, Anderson KL, Stauff DL, McDonald WH, Zimmerman LJ, Friedman DB, Heinrichs DE, Dunman PM, Skaar EP. 2010. Staphylococcus aureus fur regulates the expression of virulence factors that contribute to the pathogenesis of pneumonia. Infect Immun 78:1618–1628. doi: 10.1128/IAI.01423-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Graham JW, Lei MG, Lee CY. 2013. Trapping and identification of cellular substrates of the Staphylococcus aureus ClpC chaperone. J Bacteriol 195:4506–4516. doi: 10.1128/JB.00758-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mrak LN, Zielinska AK, Beenken KE, Mrak IN, Atwood DN, Griffin LM, Lee CY, Smeltzer MS. 2012. saeRS and sarA act synergistically to repress protease production and promote biofilm formation in Staphylococcus aureus. PLoS One 7:e38453. doi: 10.1371/journal.pone.0038453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beenken KE, Dunman PM, McAleese F, Macapagal D, Murphy E, Projan SJ, Blevins JS, Smeltzer MS. 2004. Global gene expression in Staphylococcus aureus biofilms. J Bacteriol 186:4665–4684. doi: 10.1128/JB.186.14.4665-4684.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lei MG, Cue D, Roux CM, Dunman PM, Lee CY. 2011. Rsp inhibits attachment and biofilm formation by repressing fnbA in Staphylococcus aureus MW2. J Bacteriol 193:5231–5241. doi: 10.1128/JB.05454-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luong TT, Dunman PM, Murphy E, Projan SJ, Lee CY. 2006. Transcription profiling of the mgrA regulon in Staphylococcus aureus. J Bacteriol 188:1899–1910. doi: 10.1128/JB.188.5.1899-1910.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gillaspy AF, Hickmon SG, Skinner RA, Thomas JR, Nelson CL, Smeltzer MS. 1995. Role of the accessory gene regulator (agr) in pathogenesis of staphylococcal osteomyelitis. Infect Immun 63:3373–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mootz JM, Malone CL, Shaw LN, Horswill AR. 2013. Staphopains modulate Staphylococcus aureus biofilm integrity. Infect Immun 81:3227–3238. doi: 10.1128/IAI.00377-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McCarthy H, Rudkin JK, Black NS, Gallagher L, O'Neill E, O'Gara JP. 2015. Methicillin resistance and the biofilm phenotype in Staphylococcus aureus. Front Cell Infect Microbiol 5:1. doi: 10.3389/fcimb.2015.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mascio CT, Alder JD, Silverman JA. 2007. Bactericidal action of daptomycin against stationary-phase and nondividing Staphylococcus aureus cells. Antimicrob Agents Chemother 51:4255–4260. doi: 10.1128/AAC.00824-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.LaPlante KL, Woodmansee S. 2009. Activities of daptomycin and vancomycin alone and in combination with rifampin and gentamicin against biofilm-forming methicillin-resistant Staphylococcus aureus isolates in an experimental model of endocarditis. Antimicrob Agents Chemother 53:3880–3886. doi: 10.1128/AAC.00134-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.LaPlante KL, Mermel LA. 2007. In vitro activity of daptomycin and vancomycin lock solutions on staphylococcal biofilms in a central venous catheter model. Nephrol Dial Transplant 22:2239–2246. doi: 10.1093/ndt/gfm141. [DOI] [PubMed] [Google Scholar]
- 47.Stewart PS, Davison WM, Steenbergen JN. 2009. Daptomycin rapidly penetrates a Staphylococcus epidermidis biofilm. Antimicrob Agents Chemother 53:3505–3507. doi: 10.1128/AAC.01728-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Holmes NE, Howden BP. 2014. What's new in the treatment of serious MRSA infection? Curr Opin Infect Dis 27:471–478. doi: 10.1097/QCO.0000000000000101. [DOI] [PubMed] [Google Scholar]
- 49.Blakely G, Colloms S, May G, Burke M, Sherratt D. 1991. Escherichia coli XerC recombinase is required for chromosomal segregation at cell division. New Biol 3:789–798. [PubMed] [Google Scholar]
- 50.Barre FX, Soballe B, Michel B, Aroyo M, Robertson M, Sherratt D. 2001. Circles: the replication-recombination-chromosome segregation connection. Proc Natl Acad Sci U S A 98:8189–8195. doi: 10.1073/pnas.111008998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lesterlin C, Barre FX, Cornet F. 2004. Genetic recombination and the cell cycle: what we have learned from chromosome dimers. Mol Microbiol 54:1151–1160. doi: 10.1111/j.1365-2958.2004.04356.x. [DOI] [PubMed] [Google Scholar]
- 52.Segall AM, Craig NL. 2005. New wrinkles and folds in site-specific recombination. Mol Cell 19:433–435. doi: 10.1016/j.molcel.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 53.Cheung AL, Projan SJ. 1994. Cloning and sequencing of sarA of Staphylococcus aureus, a gene required for the expression of agr. J Bacteriol 176:4168–4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nagarajan V, Elasri MO. 2007. SAMMD: Staphylococcus aureus microarray meta-database. BMC Genomics 8:351. doi: 10.1186/1471-2164-8-351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fujimoto DF, Higginbotham RH, Sterba KM, Maleki SJ, Segall AM, Smeltzer MS, Hurlburt BK. 2009. Staphylococcus aureus SarA is a regulatory protein responsive to redox and pH that can support bacteriophage lambda integrase-mediated excision/recombination. Mol Microbiol 74:1445–1458. doi: 10.1111/j.1365-2958.2009.06942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morrison JM, Anderson KL, Beenken KE, Smeltzer MS, Dunman PM. 2012. The staphylococcal accessory regulator, SarA, is an RNA-binding protein that modulates the mRNA turnover properties of late-exponential and stationary phase Staphylococcus aureus cells. Front Cell Infect Microbiol 2:26. doi: 10.3389/fcimb.2012.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fazli M, Bjarnsholt T, Kirketerp-Moller K, Jorgensen B, Andersen AS, Krogfelt KA, Givskov M, Tolker-Nielsen T. 2009. Nonrandom distribution of Pseudomonas aeruginosa and Staphylococcus aureus in chronic wounds. J Clin Microbiol 47:4084–4089. doi: 10.1128/JCM.01395-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fowler VG Jr, McIntyre LM, Yeaman MR, Peterson GE, Barth Reller L, Corey GR, Wray D, Bayer AS. 2000. In vitro resistance to thrombin-induced platelet microbicidal protein in isolates of Staphylococcus aureus from endocarditis patients correlates with an intravascular device source. J Infect Dis 182:1251–1254. doi: 10.1086/315812. [DOI] [PubMed] [Google Scholar]
- 59.Kurtz SM, Lau E, Schmier J, Ong KL, Zhao K, Parvizi J. 2008. Infection burden for hip and knee arthroplasty in the United States. J Arthroplasty 23:984–991. doi: 10.1016/j.arth.2007.10.017. [DOI] [PubMed] [Google Scholar]
- 60.Parvizi J, Pawasarat IM, Azzam KA, Joshi A, Hansen EN, Bozic KJ. 2010. Periprosthetic joint infection: the economic impact of methicillin-resistant infections. J Arthroplasty 25:103–107. doi: 10.1016/j.arth.2010.04.011. [DOI] [PubMed] [Google Scholar]
- 61.Pasberg-Gauhl C. 2014. A need for new generation antibiotics against MRSA resistant bacteria. Drug Discov Today Technol 11:109–116. doi: 10.1016/j.ddtec.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 62.Fowler VG Jr, Sakoulas G, McIntyre LM, Meka VG, Arbeit RD, Cabell CH, Stryjewski ME, Eliopoulos GM, Reller LB, Corey GR, Jones T, Lucindo N, Yeaman MR, Bayer AS. 2004. Persistent bacteremia due to methicillin-resistant Staphylococcus aureus infection is associated with agr dysfunction and low-level in vitro resistance to thrombin-induced platelet microbicidal protein. J Infect Dis 190:1140–1149. doi: 10.1086/423145. [DOI] [PubMed] [Google Scholar]
- 63.Viedma E, Sanz F, Orellana MA, San Juan R, Aguado JM, Otero JR, Chaves F. 2014. Relationship between agr dysfunction and reduced vancomycin susceptibility in methicillin-susceptible Staphylococcus aureus causing bacteraemia. J Antimicrob Chemother 69:51–58. doi: 10.1093/jac/dkt337. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.