

Abstract
Enterotoxigenic Escherichia coli (ETEC) is an important cause of diarrheal disease and death in children <5 years old. ETEC strains that express the heat-stable toxin (ST), with or without the heat-labile toxin, are among the four most important diarrhea-causing pathogens. This makes ST an attractive target for an ETEC vaccine. An ST vaccine should be nontoxic and elicit an immune response that neutralizes native ST without cross-reacting with the human endogenous guanylate cyclase C receptor ligands. To identify variants of ST with no or low toxicity, we screened a library of all 361 possible single-amino-acid mutant forms of ST by using the T84 cell assay. Moreover, we identified mutant variants with intact epitopes by screening for the ability to bind neutralizing anti-ST antibodies. ST mutant forms with no or low toxicity and intact epitopes are termed toxoid candidates, and the top 30 candidates all had mutations of residues A14, N12, and L9. The identification of nontoxic variants of L9 strongly suggests that it is a novel receptor-interacting residue, in addition to the previously identified N12, P13, and A14 residues. The screens also allowed us to map the epitopes of three neutralizing monoclonal antibodies, one of which cross-reacts with the human ligand uroguanylin. The common dominant epitope residue for all non-cross-reacting antibodies was Y19. Our results suggest that it should be possible to rationally design ST toxoids that elicit neutralizing immune responses against ST with minimal risk of immunological cross-reactivity.
INTRODUCTION
Diarrhea caused by enterotoxigenic Escherichia coli (ETEC) contributes to the almost 600,000 annual child deaths due to diarrheal disease in low- and middle-income countries (1). In addition, the 280 to 400 million annual episodes of ETEC diarrhea in children <5 years old (2, 3) contribute to malnutrition and a failure to thrive (4). Thus, the World Health Organization strongly encourages the development of an ETEC vaccine (2). ETEC is also the most common cause of traveler's diarrhea (5).
ETEC causes diarrhea by colonizing the small intestine with subsequent expression of heat-labile (LT) and/or heat-stable toxins (ST; variants STh and STp) that elicit a net efflux of salt and water into the intestinal lumen (6). Both LT and ST are potential vaccine targets, in addition to the many surface-exposed antigens, including the colonization factors. ETEC vaccine development has primarily targeted colonization factors and LT (7, 8), and the most promising vaccine candidate to date is a killed whole-cell vaccine comprising five different ETEC strains that express the most prevalent colonization factors, coadministered with the cholera toxin B subunit, which is a homologue of the LT B subunit (9). This vaccine was found to be efficacious against serious diarrhea among American travelers to Guatemala (10) but not protective when evaluated in Egyptian children (11).
A recently conducted large global multicenter study designed to analyze the etiology of enteric pathogens in children ranked ST-expressing ETEC (with or without LT) among the four most important causes of moderate-to-severe diarrhea (12). This makes ST a highly relevant target for an ETEC vaccine. Moreover, cohort studies in Guinea-Bissau and southern Israel found that ETEC strains with 19-amino-acid STh are more strongly associated with disease than those that express 18-amino-acid STp (13, 14), suggesting that STh should be the primary target of ST vaccine development.
The toxic domain of ST, from the first cysteine to the last (C6 to C18 of STh; see Fig. 1), has been reported to confer the full toxic potential of the peptide (15). The toxic domain is stabilized by three disulfide bridges in a 1-4/2-5/3-6 pattern (Fig. 1), and mutagenesis studies have demonstrated their importance for biological activity (16). Only one residue distinguishes the toxic domains of STh and STp, namely, threonine 16 of STh, which is alanine in STp. STh and STp also have the two tyrosine residues that flank the toxic domain in common (STh: Y4 and Y19). Substitutions in residues N12, P13, and A14 of STh (corresponding to N11, P12, and A13 of STp) have been reported to reduce or eliminate biological activity (17–19). Hence, these residues have been proposed to be directly involved in activation of the receptor through which ST mediates its effects, guanylate cyclase C (GC-C) (20). This is supported by the observation that these three residues are conserved among all reported bacterial GC-C ligands (21).
FIG 1.
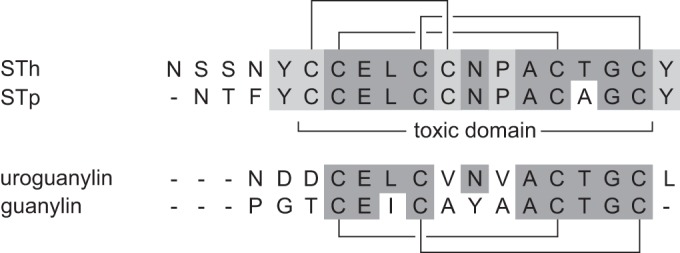
Sequence alignment of STh, STp, uroguanylin, and guanylin. Residues common to at least three of the peptides are shaded dark gray, and those common to only the ST peptides are shaded light gray. The ST disulfide bonding pattern is shown above the alignment, the uroguanylin and guanylin pattern is shown below, and the toxic domain of the ST peptides is indicated in the middle.
STs are similar to the human peptides guanylin and uroguanylin in both sequence (Fig. 1) and structure, and they all activate the GC-C receptor on the epithelial surface of the gut (21). We have recently demonstrated that this similarity may lead to immunological cross-reaction between ST and the human GC-C ligands (22). However, the risk of adverse effects of an ST vaccine may be low, as a cross-reacting anti-STh monoclonal antibody (MAb) had 73-fold less affinity for uroguanylin than for STh and only approximately one-fourth of the antibodies in an anti-STh antiserum cross-reacted with uroguanylin. Furthermore, an anti-STp antiserum and three anti-STp MAbs did not show any cross-reaction to the human GC-C ligands. These data suggest that is should be possible to rationally design a safe ST vaccine with no clinically relevant adverse effects caused by cross-reactivity.
In addition to avoiding immunological cross-reaction, an ST vaccine must be nontoxic and elicit an immune response that neutralizes native ST. Nonimmunogenic by itself, ST can be made immunogenic by polymerization (23) or by coupling to a carrier, either through chemical conjugation (24, 25) or by genetic fusion (26, 27). Frequently, coupling ST to a carrier also reduces toxicity (26, 28), but a safe ST vaccine should additionally be detoxified by mutation (21).
The aim of this study was to identify ST toxoid candidates, which we define as mutant variants with low or no toxicity but with intact epitopes. To this end, we constructed a library of all 361 possible single-amino-acid mutant forms of STh and screened them for effects on toxicity in the T84 cell assay and binding to neutralizing anti-STh antiserum. We also used the mutant library to map the epitopes of three neutralizing MAbs and identified the epitope residues of two neutralizing polyclonal antibodies. One MAb and one antiserum were previously shown to cross-react with uroguanylin (22), and hence, the mapped epitopes and antigenic determinants offer unique insights into the molecular basis of immunological cross-reactivity. Finally, we discuss how the results presented here can be exploited to rationally design a safe and efficacious ST toxoid vaccine.
MATERIALS AND METHODS
Construction and expression of STh mutant library.
The sta3 gene from ETEC strain H10407, encoding the STh prepropeptide, was amplified by PCR with primers STA2-left-PagI (CGTGTTTCGGAGGTATCATGAAAAAATC) and STA3-right (TTAATAGCACCCGGTACAAGCAG). The PCR product was cloned into the pBAD-TOPO TA vector (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Positive colonies were identified by PCR initially and then verified by sequencing. To remove the leading peptide of the vector, the plasmid was linearized by partial digestion with PagI in the presence of ethidium bromide (30 μg/ml). The full-length linearized plasmid was separated from smaller fragments by 1% agarose gel electrophoresis, purified with a QIAquick gel extraction kit according to the manufacturer's instructions (Qiagen), and subsequently digested with NcoI. The full-length plasmid was subjected to another round of gel separation and purification as described above, circularized by ligation with T4 DNA ligase, and then transformed into E. coli One Shot TOP10 cells (Invitrogen, Carlsbad, CA), resulting in pBAD-TOPO-STh. STh was expressed from the pBAD-TOPO-STh plasmid in TOP10 cells grown in LB medium supplemented with ampicillin (100 μg/ml). The araBAD promoter of the pBAD-TOPO-STh plasmid allows tuneable expression by induction with 0.00002 to 0.2% arabinose. When induced at arabinose levels above 0.002%, mature STh was released into the culture medium at levels detectable in the T84 cell assay. The maximum level of STh in the culture medium was obtained with 0.2% arabinose, which was used in all subsequent studies. A library of all 361 possible single-amino-acid mutant STh variants was generated by gene site saturation mutagenesis (29) (GenScript). The pBAD-TOPO-STh plasmid was used as the basis of a plasmid library, and the mutant forms of STh were expressed in TOP10 cells as described above, in groups of 19 mutant forms of STh expressed together with three native STh cultures as controls. To ensure consistency and comparability within and between groups, the same expression conditions were used for both controls and mutant forms of STh. The STh (mutant) culture supernatants were isolated by centrifugation at 4,000 × g for 10 min at 4°C, filtered through a 0.2-μm cellulose acetate filter (Whatman), and stored at −20°C.
T84 cell assay.
The T84 cell assay was performed essentially as described previously (22). Briefly, T84 cells (ATCC) were used to seed 24-well plates and grown to confluence in Dulbecco's modified Eagle medium (DMEM:F12; Lonza, Walkersville, MD). DMEM:F12 was supplemented with 10% fetal bovine serum and 0.2% gentamicin. Cells were washed three times with 500 μl of DMEM:F12 and preincubated with 200 μl of DMEM containing 1 mM 3-isobutyl-1-methylxanthine for 45 to 60 min at 37°C. A 200-μl sample volume (filtered culture supernatant) was added to each well and incubated for 60 min at 37°C. All samples were tested in duplicate wells. Following incubation, the reaction medium was aspirated and the cells were lysed with 0.1 M HCl at 20°C for 20 min. The lysates were centrifuged at 16,000 × g for 10 min, and supernatants were collected for analysis. Cyclic GMP (cGMP) levels were measured with a cGMP enzyme immunoassay kit according to the manufacturer's instructions (Enzo Life Sciences Inc., Farmingdale, NY).
Polyclonal ST ELISAs.
Competitive ST enzyme-linked immunosorbent assays (ELISAs) with polyclonal anti-STh and anti-STp antibodies were performed essentially as described previously (22, 30). Both antibodies were raised against bovine serum albumin (BSA) glutaraldehyde conjugates in rabbits (Bethyl Laboratories) and protein A purified. ELISA coating was prepared by glutaraldehyde conjugation of STh and STp, respectively, to ovalbumin as described previously (22). Briefly, microtiter plates (76 341 05; Thermo Fisher Scientific, Waltham, MA) were coated overnight at 37°C with STh- or STp-ovalbumin conjugate in 150 μl of ELISA buffer (128 mM NaCl, 2.7 mM KCl, 1.5 mM KH2PO4, 8.1 mM Na2HPO4, pH 7.2). We used 0.11 μg of conjugate per well for the STh ELISA and 0.16 μg of conjugate per well for the STp ELISA. Wells were then washed and blocked with 1% ovalbumin in ELISA buffer for 1 h at 37°C. Subsequently, 75 μl of sample was mixed with 75 μl of diluted primary antibody and incubated for 2 h at 37°C (anti-STh antibody, 1:2,000 final dilution; anti-STp antibody, 1:10,000 final dilution). After washing, plates were incubated with 100 μl of an anti-rabbit secondary antibody (alkaline phosphatase conjugate, A8025; Sigma-Aldrich, St. Louis, MO) at a dilution of 1:400 for 1 h at room temperature. Plates were developed for approximately 30 min with 100 μl of freshly prepared developing buffer (1 mg/ml para-nitrophenylphosphate in 9.7% [vol/vol] 0.5 mM MgCl2, pH 9.8), and stopped by the addition of 50 μl of 2 M NaOH. Optical density was measured at 405 nm.
Monoclonal ST ELISAs.
The competitive ELISAs with MAbs differed only in coating and antibody concentrations. All three MAbs were raised in mice. The anti-STh MAb ST:G8, kindly provided by Sandhya S. Visweswariah, Indian Institute of Science, was raised against a mutant variant of STh (SThY19F) conjugated to BSA with glutaraldehyde. The two anti-STp MAbs (Fitzgerald Industries International, North Acton, MA), C29 (10-1013) and C30 (10-1014), were raised against a conjugate where synthetic STp with native disulfide bridges (Bachem H-6248) was conjugated to keyhole limpet hemocyanin. The conjugation was performed to selectively target carboxyl groups. Briefly, ELISA plates (Nunc Immobilizer Amino) were coated with 100 μl of STh-ovalbumin conjugate in phosphate-buffered saline (PBS; 146 mM NaCl, 4 mM Na2HPO4, 1.1 mM KH2PO4, pH 7.2) and incubated overnight at 4°C. For the ST:G8 ELISA, 0.020 μg of conjugate per well was used, and for the C29 and C30 ELISAs, plates were coated with 0.005 μg of conjugate per well. Plates were subsequently blocked with 1% ovalbumin in PBS-T (PBS with 0.05% [vol/vol] Tween 20) for 1 h at 20°C with shaking. Plates were then washed with PBS-T, and 60 μl of sample and 60 μl of primary antibody were added to wells and incubated for 90 min at 20°C with shaking. The final antibody dilutions were as follows: ST:G8, 1:6,500; C29, 1:16,000; C30, 1:30,000. The plates were then washed with PBS-T and incubated at room temperature for 1 h with 100 μl of anti-mouse secondary antibody (alkaline phosphatase conjugate A4312; Sigma-Aldrich) diluted in PBS-T. The final dilutions were as follows: ST:G8, 1:400; C29 and 30, 1:2,000. After a final washing, the plates were developed with 100 μl of developing buffer (described above) for 15 to 20 min, the reaction was stopped by adding 50 μl of 2 M NaOH, and optical density was measured at 405 nm.
STh mutant library screen.
The T84 cell assay and all five ELISAs were used to screen the STh mutant library and were carried out as follows. All 19 STh variants harboring mutations of the same residue of native STh were analyzed together and related to one of the three native STh control samples that had been coexpressed with that group of mutant forms of STh. A serial dilution of the native STh control supernatant was used to create a standard curve, and the measured optical densities of mutant forms of STh were used to calculate activity relative to that of STh based on the standard curve. The standard curve was plotted on a log scale, and only the linear area was used for curve fitting and calculations of relative activities. In the T84 assay, samples were analyzed undiluted in duplicate wells. In the ELISAs, all samples were analyzed in triplicate. Generally, samples were also analyzed undiluted in the ELISAs, but in some positions of STh where several mutant forms had activity at the native STh level, it was necessary to dilute samples by a factor of 1:3 or 1:4. The dilution was corrected for when calculating relative STh activity. Six mutant forms lacked T84 cell assay data, i.e., C11A, C11D, C11E, C11F, C11G, and C11H, and only the noncysteine residue mutant forms were screened with the MAb ELISAs.
Data and statistical analysis.
The six screen data sets all have missing values, as the mutant forms with the most pronounced effects had undetectable antigenicities or toxicities. To include as many of these mutations as possible in subsequent data analysis, they were conservatively set to the detection limit of the assay according to the following rules: for the ELISA screens, if the measured relative toxicity was greater than or equal to the detection limit; for the T84 assay screen, if at least one ELISA had an estimated relative antigenicity assigned (the detection limit of the T84 assay is lower than those of all of the ELISAs). The polyclonal anti-STp ELISA screen of the Y5 mutant forms had a poorer detection limit than that of the others, and hence, missing Y5 values were not assigned values. For the number of measured and assigned values and the assay thresholds used to assign missing values, see Table S1 in the supplemental material. The R statistical computing environment was used for correlation analysis and principal-component analysis (PCA) (http://www.r-project.org/). Pearson correlation analysis was performed to assess pairwise correlations between the screen data sets with the corr.test function of the R package psych (https://cran.r-project.org/web/packages/psych/). PCA was performed with the R prcomp function. PCA plots were made with the R factoextra package (https://github.com/kassambara/factoextra). Other plots were made with the R ggplot2 system (http://ggplot2.org/).
RESULTS
Saturation mutagenesis and expression of mutant forms of STh.
To construct a comprehensive library of mutant forms of STh, all 19 codons of the STh peptide-encoding region of the sta3 gene were subjected to saturation mutagenesis, resulting in 361 single-amino-acid mutant forms of STh. To promote proper folding and disulfide bridge formation, we expressed STh and the mutant forms thereof as pre-pro-ST, thereby mimicking native ST secretion by ETEC. Groups of 19 mutant forms were expressed with three native STh controls. The culture supernatants were filtered and used directly for further analyses. Competitive anti-STh ELISAs of the controls showed that STh levels, on average, varied 1.7-fold between culture filtrates within a group (the maximum observed difference was 3.3-fold).
STh mutations affect culture filtrate toxicity differently.
To identify detoxifying STh mutations, the effects of the mutant forms of STh on culture filtrate toxicity were assessed with the T84 assay (Fig. 2A). A total of 247 mutant forms of STh had measurable activities in the T84 assay, with toxicities relative to that of native STh ranging from 0.0014 to 3.4. The amino acid residues of STh can be classified into three groups on the basis of the effects of their mutations on toxicity. The first group comprises STh residues where the majority of the mutant forms of STh had toxicities close to that of native STh (relative toxicity, >0.2) and includes the five N-terminal residues plus E8 and Y19. The second group includes residues where the majority of mutant forms of STh had undetectable toxicity: C7, C10, A14, C15, and C18. The final group consists of residues where mutant forms of STh had variable effects on toxicity and includes C6, L9, C11, N12, P13, T16, and G17. Frequently, mutations to amino acids with similar physicochemical properties gave comparable results (Fig. 2A).
FIG 2.

Toxicity and αSTh antigenicity of culture filtrates harboring mutant forms of STh relative to those of native STh. (A, B) Toxicity was measured with the T84 assay (A), and αSTh antigenicity was measured with an anti-STh polyclonal antibody (αSTh) in a competitive ELISA (B). The relative toxicities and antigenicities are presented in 15 graphs, each representing an amino acid position of STh. The data points are marked with single-letter amino acid codes to highlight individual mutations. The region from 0.3- to 3.3-fold relative toxicity or antigenicity is shaded gray and represents changes that may be attributed to variations in expression levels, as observed in native STh controls. The amino acids are ordered by physicochemical properties (acidic, D and E; basic, R and K; polar, H, S, T, Q, and N; nonpolar, C, M, P, G, A, I, L, and V; aromatic, F, Y, and W). Mutant forms of STh that did not display detectable culture filtrate toxicity or antigenicity are plotted below the dotted lines, which represent the detection limits of the assays, which were 0.0014 for toxicity and 0.0034 for antigenicity. A total of 273 mutant forms of STh are represented in panels A and B. None of the mutant forms of STh in positions C7, C10, C15, and C18 had detectable toxicities or antigenicities, with the exception of C18Y, which had a relative antigenicity of 0.2. Graphs are not shown for these positions.
An anti-STh antiserum binds differently to STh mutant culture filtrates.
To be useful in a vaccine, detoxifying mutations must not compromise the ability to induce an immune response that can neutralize native ST. This property of the mutations was indirectly assessed in a competitive ELISA with a neutralizing rabbit antiserum raised against native STh (αSTh) (22). The assumption is that mutant forms of STh that bind to αSTh with an affinity similar to that of native STh would be able to induce an immune response capable of neutralizing native ST. Figure 2B shows the antigenicity of the culture filtrates in the αSTh ELISA (αSTh antigenicity).
A total of 219 mutant forms had measurable αSTh antigenicities, ranging from 0.0034 to 2 relative to that of native STh. Classification of the amino acid residues of STh, based on the effects of mutations on αSTh antigenicity, resulted in groups similar to those for toxicity. Notable exceptions were residues N1, A14, and Y19. Mutation of these residues seemed to affect toxicity and αSTh antigenicity differently. The effect of mutation of residues N1 and Y19 on antigenicity was generally more pronounced than the effect on toxicity, whereas most mutations of residue A14 affected toxicity more than antigenicity.
Identification of STh toxoid candidate mutations and STh residues important for toxicity.
To identify STh toxoid candidates, the effects of individual mutations on toxicity and αSTh antigenicity were compared directly (Fig. 3A). Pairwise correlation analysis of the two data sets revealed that mutational effects on toxicity overall correlated weakly but significantly with those on αSTh antigenicity (r = 0.36, P < 0.001; Table 1). This suggests that a significant number of mutations affect toxicity and αSTh antigenicity in a similar manner but, more importantly, that there are mutations that selectively affect either toxicity or antigenicity. The measured activities (toxicity or antigenicity) of the mutant STh-harboring culture filtrates are the product of intrinsic peptide activity and peptide concentration. But since the same samples were used for both toxicity and antigenicity measurements, the αSTh antigenicity-toxicity fold changes (diagonal dimension in Fig. 3A) do not depend on concentrations and reflect the intrinsic peptide properties. The mutant forms of STh can be ranked according to their αSTh antigenicity-toxicity fold change, and the best toxoid candidates, with the greatest fold changes, are listed in Table 2.
FIG 3.

T84 toxicity compared to αSTh antigenicity. (A) The graphs represent STh amino acid positions and show the relative T84 toxicity values (horizontal axis) of each mutant form of STh plotted against the relative αSTh antigenicity values (vertical axis). Mutant forms of STh with unmeasurable relative toxicity (<0.0014) but measurable antigenicity were plotted outside the toxicity scale to the left. Similarly, mutant forms of STh with unmeasurable relative antigenicity (<0.0034) but measurable toxicity were plotted outside the antigenicity scale at the bottom. The diagonal lines represent the antigenicity-toxicity ratio (fold change): the full line represents no change, the dotted lines to the left represent 10- and 100-fold increases, and those to the right represent 10- and 100-fold reductions. Differences in antigenicity-toxicity fold changes are illustrated with a two-color gradient, where no change is white, positive fold changes are red, and negative ones are blue. (B) The bubble chart shows the median relative T84 toxicity values (horizontal axis) for amino acid positions of STh plotted against the median relative αSTh antigenicity values (vertical axis). The bubble sizes reflect the numbers of mutant forms of STh for each position for which T84 toxicity, αSTh antigenicity, or both values were successfully determined. For the calculation of median values, mutant forms of STh that had measurable activity in only one assay had the missing value set to the detection limit of the cognate assay (toxicity, 0.0014; antigenicity, 0.0034). Median antigenicity-toxicity ratios (fold changes) are represented as in panel A. A total of 273 mutant forms are represented in panels A and B. (C) Structural model of STh (22), with two views of the structure shown, rotated 180° along the x axis in relation to each other. Amino acids are labeled and colored according to the median antigenicity-toxicity fold changes of each amino acid position as in panel B. Graphs are not shown for positions C7, C10, C15, and C18 (see the legend to Fig. 2).
TABLE 1.
Pairwise correlations of the six screen data sets
| Data setc | Correlation with: |
|||||
|---|---|---|---|---|---|---|
| αSTh | ST:G8 | αSTp | C29 | C30 | T84 | |
| αSTh | ||||||
| ST:G8 | 0.44a | |||||
| αSTp | 0.54a | 0.40a | ||||
| C29 | 0.71a | 0.59a | 0.61a | |||
| C30 | 0.68a | 0.64a | 0.60a | 0.95a | ||
| T84 | 0.36a | 0.30a | 0.12b | 0.30a | 0.24a | |
P < 0.001.
P > 0.05.
Abbreviations: αSTh, anti-STh serum; ST:G8, anti-STh MAb; αSTp, anti-STp serum; C29, anti-STp MAb; C30, anti-STp MAb; T84, T84 cell assay.
TABLE 2.
Best STh toxoid candidates
| STh mutation | Anti-STh antigenicity | Toxicity | Anti-STh antigenicity-toxicity fold change |
|---|---|---|---|
| A14H | 0.69 | 0.0014a | ≥494 |
| A14Q | 0.56 | 0.0014a | ≥401 |
| A14R | 0.56 | 0.0014a | ≥397 |
| A14T | 0.42 | 0.0014a | ≥298 |
| A14E | 0.27 | 0.0014a | ≥193 |
| N12K | 0.17 | 0.0014a | ≥122 |
| A14I | 0.16 | 0.0014a | ≥118 |
| A14L | 0.13 | 0.0014a | ≥93 |
| A14K | 0.13 | 0.0014a | ≥92 |
| N12V | 1.10 | 0.0123 | 90 |
| N12T | 2.00 | 0.0250 | 80 |
| A14W | 0.11 | 0.0014a | ≥79 |
| A14N | 0.09 | 0.0014a | ≥64 |
| A14 M | 0.08 | 0.0014a | ≥61 |
| N12E | 0.15 | 0.0026 | 59 |
| A14D | 0.08 | 0.0014a | ≥57 |
| N12R | 0.08 | 0.0014a | ≥57 |
| A14F | 0.07 | 0.0014a | ≥51 |
| A14V | 0.07 | 0.0014a | ≥49 |
| N12S | 0.83 | 0.0187 | 44 |
| N12G | 0.06 | 0.0014a | ≥40 |
| A14Y | 0.05 | 0.0014a | ≥33 |
| L9G | 0.04 | 0.0014a | ≥29 |
| N12A | 0.33 | 0.0115 | 29 |
| L9S | 0.04 | 0.0014a | ≥28 |
| L9A | 0.04 | 0.0014a | ≥28 |
| L9E | 0.04 | 0.0014a | ≥27 |
| L9P | 0.04 | 0.0014a | ≥25 |
| N12W | 0.11 | 0.0042 | 25 |
| N12Q | 0.36 | 0.0142 | 25 |
Toxicity values were not detectable in the T84 assay and were set to the detection limit of the assay (0.0014).
As evident from Fig. 3A, the individual STh amino acid residues were differently affected by mutations. Further, individual mutations of a particular residue tended to cluster. This implies that it is meaningful to consider the average mutational effect on individual STh residues as a measure of their toxic and antigenic role. Figure 3B shows the median αSTh antigenicity-toxicity fold changes. Interestingly, the five residues that have median αSTh antigenicity-toxicity changes of >1.5-fold, which implies that toxicity is affected more than antigenicity, form a structural cluster (shown in shades of red in Fig. 3C). This group of STh residues includes the reported receptor-interacting residues (N12, P13, and A14), as may have been expected, but also L9.
Identification of epitope residues and determinants of cross-reactivity.
STh residues whose mutation affects antigenicity more than toxicity are likely to be epitope residues. Hence, N1, C6, E8, T16, G17, and Y19 are potential αSTh epitope residues (Fig. 3B). Y19 seems to be the most prominent epitope residue, as it displayed the lowest αSTh antigenicity-toxicity fold change.
We recently characterized the immunological cross-reactivity of a panel of anti-ST antibodies against the human GC-C ligands guanylin and uroguanylin (22). This panel included αSTh and four other anti-ST antibodies with neutralizing ability: an anti-STh MAb (ST:G8), an anti-STp polyclonal antibody (αSTp), and two anti-STp MAbs (C29 and C30). To identify epitope residues and determinants of cross-reactivity, the STh mutant library was analyzed with these four additional antibodies. The αSTp serum was used to screen the full library, whereas only the noncysteine residue mutant forms were analyzed with the MAbs. The results are summarized, together with the T84 toxicity and αSTh antigenicity data for comparison, in Fig. 4A. Pairwise correlation analysis of all six screen data sets suggests that the ELISAs, in general, correlated better with each other than with the T84 toxicity assay (Table 1). This was also reflected by a PCA of the data sets, where the first principal component mainly captured the variance in the ELISAs, and the second principal component mainly reflected the variance in the T84 assay (Fig. 4B). Interestingly, the anti-STh ELISA vectors (αSTh and ST:G8) are almost identical and slightly different from those of the highly similar anti-STp ELISA vectors (αSTp, C29, and C30).
FIG 4.

Summary of T84 toxicity and antigenicities of culture filtrates harboring mutant forms of STh relative to those of native STh. (A) The graphs shown represent STh amino acid positions, and each one contains box plots of data from six different assays. The T84 assay data (T84) and the anti-STh polyclonal ELISA data (αSTh) are the same as in Fig. 2 and 3. In addition, the data are from an anti-STp polyclonal ELISA (αSTp), an anti-STh MAb ELISA (ST:G8), and two anti-STp MAb ELISAs (C29 and C30). Activity (toxicity or antigenicity) relative to that of native STh is shown on the vertical axis. The median relative activities of the mutant forms of STh are displayed as horizontal black lines; the bottom and top of the boxes represent the first and third quartiles, respectively; the upper whiskers represent the highest value that is within 1.5 times the interquartile range (IQR) of the third quartile; and the lower whisker represents the lowest value within 1.5 times the IQR of the first quartile. Outliers are shown as black points. The number of mutant forms that had detectable activity in each assay is reported for each amino acid position at the top of each graph. Values for mutant forms of STh with unmeasurable relative toxicity in the T84 assay but with detectable antigenicities were set to the assay detection limit (0.0014; dotted lines). Mutant forms of STh with changes at the cysteine positions were not analyzed in the MAb ELISAs. Graphs are not shown for positions C7, C10, C15, and C18 (see the legend to Fig. 2). (B) PCA correlation circle map of the six screen data sets. The first (PC1) and second (PC2) principal components are plotted (percent variance is in parentheses), showing a positive correlation between the ELISA data sets and a weak correlation between the ELISA data sets and the T84 assay data set. Prior to the PCA, missing values were set to the detection limit of the assays according to the rules described in Materials and Methods.
The five N-terminal residues displayed similar results for all antibodies, including the marked reduction in antigenicity observed for many N1 mutant forms with αSTh (Fig. 4A). Most other positions showed differing antigenicities for the various antibodies. For example, E8 seems to be an epitope residue for αSTh, L9 for ST:G8, T16 for αSTh, and G17 and Y19 for all but ST:G8. Also worth noting is that the results presented in Fig. 4A suggest that the majority of the mutant forms are expressed at a near-native level, except for the cysteine residue mutant forms and the P13 mutant forms, which seem to be expressed at a lower level.
C29 and C30 recognize the same epitope, which is distinct from that of ST:G8.
To map the epitopes of the MAbs, we performed pairwise comparisons of their mutational screen data. Pairwise comparisons of data obtained from the same culture filtrates eliminate the effect of peptide concentration, and mutant forms with antigenicity-antigenicity changes much larger or smaller than 1-fold are indications of antigenic determinants that are unique to one of the MAbs. Figure 4A suggests that the C29 and C30 MAbs may have similar epitopes, as only minor differences were observed. This was also reflected by the nearly perfect correlation between the two data sets (r = 0.95; Table 1). The largest difference between the two MAbs was observed at position Y19 (see Fig. S1 in the supplemental material), suggesting that the two epitopes are similar but not identical.
The ST:G8 MAb showed pronounced differences from the C29 and C30 MAbs in the binding of mutant forms (Fig. 4A), which was reflected by the lower correlation coefficients (Table 1). A direct pairwise comparison of the C29 and ST:G8 antigenicities of individual mutant forms highlights these differences (Fig. 5A). The differences are visualized in the STh structure model (Fig. 5B and C), where potential C29 and ST:G8 epitope residues are in shades of green and blue, respectively, according to their median antigenicity-antigenicity fold changes. The results suggest that the ST:G8 epitope is distinct from that of C29/C30, that the dominant epitope residue is L9, and that E8, N12, and P13 are additional epitope residues. The dominant epitope residue of C29/C30 seems to be Y19, with G17 and P13 as additional epitope residues.
FIG 5.

Epitope mapping. (A) The graphs represent STh amino acid positions and show the relative C29 antigenicity values (horizontal axis) of each mutant form of STh plotted against the relative ST:G8 antigenicity values (vertical axis). C29 antigenicity and ST:G8 antigenicity were measured with the anti-STp MAb (C29) and the anti-STh MAb (ST:G8) ELISAs, respectively. Values for mutant forms of STh with unmeasurable antigenicity for one MAb were plotted outside the scale to the left (C29; <0.013) or bottom (ST:G8; <0.003). The diagonal lines represent the ST:G8 antigenicity/C29 antigenicity ratio (fold change) as follows: the full line represents no change, the dotted lines to the left represent 10- and 100-fold increases, and those to the right represent 10- and 100-fold reductions. A total of 229 mutant forms of STh are represented. Mutant forms of STh with changes at the cysteine residue positions were not screened. (B) Structural model of STh (22) with two views of the structure shown, rotated 180° along the x axis in relation to each other. Amino acids are colored according to the median ST:G8 antigenicity-C29 antigenicity changes of each amino acid position. For the C29 MAb, increases are in shades of green, and for the ST:G8 MAb, reductions are in shades of blue, reflecting their values.
DISCUSSION
To rationally design a safe vaccine that elicits antibodies that neutralize ST, a comprehensive understanding of ST's structure and function is required. We have generated a library that encompasses all 361 single-amino-acid mutant forms of the STh peptide and screened them for effects on the ability to activate the GC-C receptor (biological activity/toxicity) and the ability to bind to a panel of antibodies with toxin-neutralizing ability (antigenicity). The toxic domain of ST, comprising the residues from the first cysteine to the last (C6 to C18), has been reported to retain full biological activity (15). Other reports suggest that residues outside the toxic domain are also required for full potency (31). Our results show that all ST amino acid residues had sets of mutational variants that displayed detrimental effects on toxicity, antigenicity, or both (Fig. 4A). As expected, the most pronounced effects on toxicity were observed in the residues that constitute the toxic domain. Mutations of cysteines 7, 10, 15, and 18 showed no detectable toxicity or antigenicity, except for C18Y, which was detected in the T84 assay. These four cysteines form the two disulfide bridges (C7-C15, C10-C18) that are shared with guanylin and uroguanylin (32). In contrast, the ST-specific disulfide bridge cysteines, C6 and C11, were surprisingly tolerant to mutations. Both C6 mutant forms and C11 mutant forms had reduced but measurable activities in the T84 assay and the polyclonal anti-STh ELISA. These findings suggest that the C6-C11 bridge is not essential for function but that it is necessary for full toxic potency. This is in line with the hypothesis that the C6-C11 disulfide bridge locks ST in a conformation that resembles the active conformer of the endogenous ligands guanylin and uroguanylin (33).
Previous mutagenesis studies have identified N12, P13, and A14 as particularly important for biological activity (17–19), and they are frequently referred to as the receptor-interacting residues. Our results confirm the functional importance of these residues (Fig. 4A). A14 is the residue with the most prominent effects from mutations: all but two A14 mutant forms were inactive in the T84 assay (Fig. 2A). Most of the N12 and P13 mutant forms had reduced toxicities, some had unmeasurable toxicities, and some had near-native activities (Fig. 2A).
Mutations in the remaining toxic domain residues, E8, L9, T16, and G17, also showed effects on biological activity (Fig. 4A). The T16 and G17 residues are both shared with guanylin and uroguanylin (Fig. 1), and their mutant forms have median toxicities that are at least an order of magnitude lower than that of native ST. Most E8 mutant forms had near-native activity in the T84 assay, but mutations that reversed the negative charge, namely, E8K and E8R, led to a 500-fold reduction in biological activity (Fig. 2A). This suggests that E8, which is shared with guanylin and uroguanylin (Fig. 1), is not directly involved in receptor interaction. Furthermore, it seems that the net negative charge of ST is not necessary for full biological activity but that an ST molecule with a net positive charge interacts poorly with the receptor. L9 mutant forms had variable biological activities, from undetectable to near native, and the median effect on toxicity is 2 orders of magnitude greater than the effect on antigenicity in the C29 and C30 ELISAs (Fig. 4A). This suggests that L9, which is shared with uroguanylin and replaced with isoleucine in guanylin (Fig. 1), is a receptor-interacting residue, in addition to the previously reported N12, P13, and A14 residues.
Mutational variants of the N1 residue displayed the most pronounced effects observed outside the toxic domain, with a negative impact on toxicity but an even greater negative impact on antigenicity (Fig. 4A). This is reflected by the larger range of effects on antibody binding than on toxicity and the fact that 18 mutant forms had detectable biological activity, and only 7 to 12 had detectable antigenicity in the different ELISAs. As three of the five antibodies were generated against conjugates where the ST peptide was attached to a carrier via the N terminus, it is unlikely that N1 is part of an epitope that is recognized by these antibodies. A possible explanation for the effects of N1 mutant forms is that the flexible N terminus folds back and interacts with the toxic domain in native ST and that some mutant residues interfere with this interaction in a manner that disturbs binding to toxic domain epitopes.
All of the antibodies used in this study have neutralizing ability, but only the αSTh serum and the ST:G8 MAb displayed cross-reaction with uroguanylin (22). Pairwise comparison of the relative antigenicities of mutant forms of STh obtained with the C29 and ST:G8 ELISAs allowed us to map epitope residues that form two distinct epitopes (Fig. 5). The C29/C30 epitope is located at the C terminus and comprises residues P13, G17, and Y19. Y19 and P13 are both unique to the ST peptides (Fig. 1), and this may explain why the C29 and C30 MAbs do not cross-react with (uro)guanylin. The ST:G8 epitope is located centrally and comprises residues E8, L9, N12, and P13. Three of the residues are shared with uroguanylin, and this may explain the observed cross-reaction of the ST:G8 MAb. It is unlikely, however, that the 73-fold lower affinity of the ST:G8 MAb for uroguanylin (22) is due solely to P13 being changed to valine in uroguanylin. One additional difference that may contribute to this low affinity is the ST-specific C6-C11 disulfide bridge that connects the ST:G8 epitope residues identified (Fig. 5B).
The αSTh and αSTp antisera both seem to have Y19 as the dominant epitope residue and G17 as an additional one (Fig. 4A), suggesting that they contain antibody specificities that are similar to those of the C29 and C30 MAbs and that ST may have a limited repertoire of epitopes. This is corroborated by an epitope mapping study where two anti-STh MAbs also had Y19 as the dominant epitope residue, a third recognized L9 (the dominant epitope residue of ST:G8), and the last one recognized N4/Y5 (23). However, the αSTh serum seems to contain additional antibody specificities that recognize both E8 and T16 (Fig. 3 and 4A). In contrast to Y19, the latter two residues are shared with the (uro)guanylin peptides (Fig. 1) and thus may contribute to the partial cross-reaction between αSTh and uroguanylin (22).
An ST vaccine should be nontoxic and elicit an immune response that neutralizes native ST but does not cross-react with the human GC-C receptor ligands (22). Although coupling of ST to a carrier may reduce or abolish toxicity, detoxifying mutations should be introduced for safety. In this respect, it is encouraging that ST mutant variants genetically fused in triplicate to LT elicited neutralizing antibody responses in mice (34). Because of the small size of ST, single-amino-acid mutations may affect protective epitopes and undesired cross-reacting epitopes, in addition to having a detoxifying effect. Hence, it is important to carefully select suitable mutations. The top 30 toxoid candidates (Table 2) consist of 15 A14 mutant forms, 10 N12 mutant forms, and 5 L9 mutant forms. Interestingly, N12 and A14 are shared with both guanylin and uroguanylin and L9 is shared with uroguanylin. Hence, mutations in these residues also have the potential to reduce the risk of immunological cross-reactivity.
The A14 residue seems to be the most attractive residue to mutate for detoxification, as all but two A14 mutant forms had undetectable toxicities. Moreover, most of the A14 mutant forms had near-native antigenicities in the polyclonal αSTh and αSTp ELISAs, and the mapping of the neutralizing MAb's epitopes suggests that A14 is not part of those epitopes. This also implies that A14 mutations are unlikely to reduce the risk of cross-reactivity with guanylin and uroguanylin.
The L9 residue is the dominant epitope residue of the cross-reacting ST:G8 epitope, with N12 as an additional one. Hence, mutations in these residues may disrupt epitopes that are likely to elicit a cross-reacting immune response. According to the toxoid candidate ranking, the N12 residue seems to be the second most attractive residue to mutate, despite the fact that only two of the N12 mutant forms in Table 2 had undetectable toxicities. Although several L9 mutant forms had undetectable toxicities, they rank poorly as toxoid candidates, as αSTh antigenicity is also affected by the mutations. However, it is possible that the αSTh antibody clones that are affected by the L9 mutations are the same that cross-react with the human peptides, implying that L9 is a more attractive residue to mutate than suggested by the αSTh-based ranking.
The antigenic determinant common to all of the non-cross-reacting antibodies is Y19. This is not surprising, as Y19 is specific to ST and not shared with the human GC-C receptor peptides. The fact that all of these antibodies also have neutralizing abilities suggests that optimizing the presentation of the Y19-dominated epitope may provide a path toward an ST vaccine that is safe from immunological cross-reaction. Such a strategy is also compatible with the introduction of detoxifying mutations in the conserved L9, N12, and A14 residues. Elimination of both the toxic and the cross-reactive potentials of an ST vaccine candidate may ultimately require the combination of two or more mutations.
An ST vaccine has the potential to confer protection against a wide range of ETEC strains. The present study is the first analysis of a complete STh mutant library that includes all 361 single-amino-acid mutant forms of STh. By screening the STh mutant library, we have identified L9 as a receptor-interacting residue, in addition to N12, P13, and A14. Furthermore, we have ranked the best 30 single-amino-acid mutant ST toxoid candidates that have reduced or undetectable toxicity but retain antigenicity. Finally, we have mapped the epitopes of three neutralizing MAbs, one of which cross-reacts with uroguanylin. These results provide information that can feed into rational design approaches to ST vaccine development. Our findings suggest that it is indeed possible to construct ST toxoids that elicit neutralizing immune responses against ST, with a minimal risk of immunological cross-reactivity. Further studies will advance our candidate toxoids by ascertaining their ability to protect animals and humans from ETEC diarrhea.
Supplementary Material
ACKNOWLEDGMENTS
We thank Sandhya S. Visweswariah for providing the ST:G8 MAb and Thomas A. Aloysius and Elisabeth Silden for their skilled technical assistance.
The research leading to these results was funded by The Research Council of Norway (GLOBVAC program, grants 185872 to EntVac and 234364 to ETECvac) and PATH (grant 102290-002), with additional support from the European Union's Seventh Framework Programme for research, technological development, and demonstration under grant agreement 261472 (to STOPENTERICS). A.M.T. was supported by the University of Bergen.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01225-15.
REFERENCES
- 1.Liu L, Oza S, Hogan D, Perin J, Rudan I, Lawn JE, Cousens S, Mathers C, Black RE. 2015. Global, regional, and national causes of child mortality in 2000–13, with projections to inform post-2015 priorities: an updated systematic analysis. Lancet 385:430–440. doi: 10.1016/S0140-6736(14)61698-6. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization. 2006. Future directions for research on enterotoxigenic Escherichia coli vaccines for developing countries. Wkly Epidemiol Rec 81:97–104. [PubMed] [Google Scholar]
- 3.Wennerås C, Erling V. 2004. Prevalence of enterotoxigenic Escherichia coli-associated diarrhoea and carrier state in the developing world. J Health Popul Nutr 22:370–382. [PubMed] [Google Scholar]
- 4.Guerrant RL, DeBoer MD, Moore SR, Scharf RJ, Lima AA. 2013. The impoverished gut—a triple burden of diarrhoea, stunting and chronic disease. Nat Rev Gastroenterol Hepatol 10:220–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hill DR, Beeching NJ. 2010. Travelers' diarrhea. Curr Opin Infect Dis 23:481–487. doi: 10.1097/QCO.0b013e32833dfca5. [DOI] [PubMed] [Google Scholar]
- 6.Sears CL, Kaper JB. 1996. Enteric bacterial toxins: mechanisms of action and linkage to intestinal secretion. Microbiol Rev 60:167–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Darsley MJ, Chakraborty S, DeNearing B, Sack DA, Feller A, Buchwaldt C, Bourgeois AL, Walker R, Harro CD. 2012. The oral, live attenuated enterotoxigenic Escherichia coli vaccine ACE527 reduces the incidence and severity of diarrhea in a human challenge model of diarrheal disease. Clin Vaccine Immunol 19:1921–1931. doi: 10.1128/CVI.00364-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tobias J, Svennerholm AM. 2012. Strategies to overexpress enterotoxigenic Escherichia coli (ETEC) colonization factors for the construction of oral whole-cell inactivated ETEC vaccine candidates. Appl Microbiol Biotechnol 93:2291–2300. doi: 10.1007/s00253-012-3930-6. [DOI] [PubMed] [Google Scholar]
- 9.Svennerholm AM, Lundgren A. 2012. Recent progress toward an enterotoxigenic Escherichia coli vaccine. Expert Rev Vaccines 11:495–507. doi: 10.1586/erv.12.12. [DOI] [PubMed] [Google Scholar]
- 10.Sack DA, Shimko J, Torres O, Bourgeois AL, Francia DS, Gustafsson B, Karnell A, Nyquist I, Svennerholm AM. 2007. Randomised, double-blind, safety and efficacy of a killed oral vaccine for enterotoxigenic E. coli diarrhoea of travellers to Guatemala and Mexico. Vaccine 25:4392–4400. doi: 10.1016/j.vaccine.2007.03.034. [DOI] [PubMed] [Google Scholar]
- 11.Walker RI, Steele D, Aguado T. 2007. Analysis of strategies to successfully vaccinate infants in developing countries against enterotoxigenic E. coli (ETEC) disease. Vaccine 25:2545–2566. doi: 10.1016/j.vaccine.2006.12.028. [DOI] [PubMed] [Google Scholar]
- 12.Kotloff KL, Nataro JP, Blackwelder WC, Nasrin D, Farag TH, Panchalingam S, Wu Y, Sow SO, Sur D, Breiman RF, Faruque AS, Zaidi AK, Saha D, Alonso PL, Tamboura B, Sanogo D, Onwuchekwa U, Manna B, Ramamurthy T, Kanungo S, Ochieng JB, Omore R, Oundo JO, Hossain A, Das SK, Ahmed S, Qureshi S, Quadri F, Adegbola RA, Antonio M, Hossain MJ, Akinsola A, Mandomando I, Nhampossa T, Acacio S, Biswas K, O'Reilly CE, Mintz ED, Berkeley LY, Muhsen K, Sommerfelt H, Robins-Browne RM, Levine MM. 2013. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet 382:209–222. doi: 10.1016/S0140-6736(13)60844-2. [DOI] [PubMed] [Google Scholar]
- 13.Porat N, Levy A, Fraser D, Deckelbaum RJ, Dagan R. 1998. Prevalence of intestinal infections caused by diarrheagenic Escherichia coli in Bedouin infants and young children in southern Israel. Pediatr Infect Dis J 17:482–488. doi: 10.1097/00006454-199806000-00010. [DOI] [PubMed] [Google Scholar]
- 14.Steinsland H, Valentiner-Branth P, Perch M, Dias F, Fischer TK, Aaby P, Molbak K, Sommerfelt H. 2002. Enterotoxigenic Escherichia coli infections and diarrhea in a cohort of young children in Guinea-Bissau. J Infect Dis 186:1740–1747. doi: 10.1086/345817. [DOI] [PubMed] [Google Scholar]
- 15.Yoshimura S, Ikemura H, Watanabe H, Aimoto S, Shimonishi Y, Hara S, Takeda T, Miwatani T, Takeda Y. 1985. Essential structure for full enterotoxigenic activity of heat-stable enterotoxin produced by enterotoxigenic Escherichia coli. FEBS Lett 181:138–142. doi: 10.1016/0014-5793(85)81129-7. [DOI] [PubMed] [Google Scholar]
- 16.Okamoto K, Okamoto K, Yukitake J, Kawamoto Y, Miyama A. 1987. Substitutions of cysteine residues of Escherichia coli heat-stable enterotoxin by oligonucleotide-directed mutagenesis. Infect Immun 55:2121–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirayama T. 1995. Heat-stable enterotoxin of Escherichia coli, p 281–296. In Moss J, Iglewski B, Vaughan M, Tu AT (ed), Handbook of natural toxins. Marcel Dekker, Inc., New York, NY. [Google Scholar]
- 18.Okamoto K, Okamoto K, Yukitake J, Miyama A. 1988. Reduction of enterotoxic activity of Escherichia coli heat-stable enterotoxin by substitution for an asparagine residue. Infect Immun 56:2144–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamasaki S, Sato T, Hidaka Y, Ozaki H, Ito H, Hirayama T, Takeda Y, Sugimura T, Tai A, Shimonishi Y. 1990. Structure-activity relationship of Escherichia coli heat-stable enterotoxin: role of Ala residue at position 14 in toxin-receptor interaction. Bull Chem Soc Jpn 63:2063–2070. doi: 10.1246/bcsj.63.2063. [DOI] [Google Scholar]
- 20.Ozaki H, Sato T, Kubota H, Hata Y, Katsube Y, Shimonishi Y. 1991. Molecular structure of the toxin domain of heat-stable enterotoxin produced by a pathogenic strain of Escherichia coli. A putative binding site for a binding protein on rat intestinal epithelial cell membranes. J Biol Chem 266:5934–5941. [PubMed] [Google Scholar]
- 21.Taxt A, Aasland R, Sommerfelt H, Nataro J, Puntervoll P. 2010. Heat-stable enterotoxin of enterotoxigenic Escherichia coli as a vaccine target. Infect Immun 78:1824–1831. doi: 10.1128/IAI.01397-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taxt AM, Diaz Y, Bacle A, Grauffel C, Reuter N, Aasland R, Sommerfelt H, Puntervoll P. 2014. Characterization of immunological cross-reactivity between enterotoxigenic Escherichia coli heat-stable toxin and human guanylin and uroguanylin. Infect Immun 82:2913–2922. doi: 10.1128/IAI.01749-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takeda T, Nair GB, Suzuki K, Zhe HX, Yokoo Y, De Mol P, Hemelhof W, Butzler JP, Takeda Y, Shimonishi Y. 1993. Epitope mapping and characterization of antigenic determinants of heat-stable enterotoxin (STh) of enterotoxigenic Escherichia coli by using monoclonal antibodies. Infect Immun 61:289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brandwein H, Deutsch A, Thompson M, Giannella R. 1985. Production of neutralizing monoclonal antibodies to Escherichia coli heat-stable enterotoxin. Infect Immun 47:242–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takeda T, Takeda Y, Aimoto S, Takao T, Ikemura H, Shimonishi Y, Miwatani T. 1983. Neutralization of activity of two different heat-stable enterotoxins (STh and STp) of enterotoxigenic Escherichia coli by homologous and heterologous antisera. FEMS Microbiol Lett 20:357–359. doi: 10.1111/j.1574-6968.1983.tb00147.x. [DOI] [Google Scholar]
- 26.Clements JD. 1990. Construction of a nontoxic fusion peptide for immunization against Escherichia coli strains that produce heat-labile and heat-stable enterotoxins. Infect Immun 58:1159–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Löwenadler B, Lake M, Elmblad A, Holmgren E, Holmgren J, Karlstrom A, Svennerholm AM. 1991. A recombinant Escherichia coli heat-stable enterotoxin (STa) fusion protein eliciting anti-STa neutralizing antibodies. FEMS Microbiol Lett 66:271–277. [DOI] [PubMed] [Google Scholar]
- 28.Klipstein FA, Engert RF, Clements JD, Houghten RA. 1983. Protection against human and porcine enterotoxigenic strains of Escherichia coli in rats immunized with a cross-linked toxoid vaccine. Infect Immun 40:924–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robertson DE, Steer BA. 2004. Recent progress in biocatalyst discovery and optimization. Curr Opin Chem Biol 8:141–149. doi: 10.1016/j.cbpa.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 30.Lockwood DE, Robertson DC. 1984. Development of a competitive enzyme-linked immunosorbent assay (ELISA) for Escherichia coli heat-stable enterotoxin (STa). J Immunol Methods 75:295–307. doi: 10.1016/0022-1759(84)90113-3. [DOI] [PubMed] [Google Scholar]
- 31.Waldman SA, O'Hanley P. 1989. Influence of a glycine or proline substitution on the functional properties of a 14-amino-acid analog of Escherichia coli heat-stable enterotoxin. Infect Immun 57:2420–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Forte LR., Jr 2004. Uroguanylin and guanylin peptides: pharmacology and experimental therapeutics. Pharmacol Ther 104:137–162. doi: 10.1016/j.pharmthera.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 33.Marx UC, Klodt J, Meyer M, Gerlach H, Rosch P, Forssmann WG, Adermann K. 1998. One peptide, two topologies: structure and interconversion dynamics of human uroguanylin isomers. J Pept Res 52:229–240. [DOI] [PubMed] [Google Scholar]
- 34.Ruan X, Robertson DC, Nataro JP, Clements JD, Zhang W. 2014. Characterization of heat-stable (STa) toxoids of enterotoxigenic Escherichia coli fused to double mutant heat-labile toxin peptide in inducing neutralizing anti-STa antibodies. Infect Immun 82:1823–1832. doi: 10.1128/IAI.01394-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.