

Abstract
During epithelial infections, pathogenic bacteria employ an array of strategies to attenuate and evade host immune responses, including the influx of polymorphonuclear leukocytes (PMN; neutrophils). Among the most common bacterial infections in humans are those of the urinary tract, caused chiefly by uropathogenic Escherichia coli (UPEC). During the establishment of bacterial cystitis, UPEC suppresses innate responses via multiple independent strategies. We recently described UPEC attenuation of PMN trafficking to the urinary bladder through pathogen-specific local induction of indoleamine 2,3-dioxygenase (IDO), a tryptophan catabolic enzyme previously shown to have regulatory activity only in adaptive immunity. Here, we investigated the mechanism by which IDO induction attenuates PMN migration. Local tryptophan limitation, by which IDO is known to influence T cell longevity and proliferation, was not involved in its effect on PMN trafficking. Instead, metabolites in the IDO pathway, particularly l-kynurenine, directly suppressed PMN transepithelial migration and induced an attached, spread morphology in PMN both at rest and in the presence of chemotactic stimuli. Finally, kynurenines represent known ligands of the mammalian aryl hydrocarbon receptor (AHR), and UPEC infection of Ahr−/− mice recapitulated the derepressed PMN recruitment observed previously in Ido1−/− mice. UPEC therefore suppresses neutrophil migration early in bacterial cystitis by eliciting an IDO-mediated increase in local production of kynurenines, which act through the AHR to impair neutrophil chemotaxis.
INTRODUCTION
Indoleamine 2,3-dioxygenase (IDO) is a conserved mammalian metabolic enzyme that catalyzes the first step in tryptophan degradation along the kynurenine pathway. In addition to this role in nutrient utilization, IDO has been implicated in a number of biological processes related to regulation of local immunity such as fetal tolerance, autoimmunity, organ transplantation, and tumorigenesis (1). The immunomodulatory capacity of IDO has been best studied in the context of the adaptive immune response, where induction of this enzyme in dendritic cells restricts the proliferation and survival of T effector cells (1–4). This activity is driven by the breakdown of tryptophan, which both reduces the supply of this essential amino acid available to local T cells and generates an array of bioactive downstream metabolites collectively referred to as kynurenines (1, 5, 6).
Recent studies have identified components of the cellular machinery involved in IDO-dependent responses in lymphocytes. IDO-mediated degradation of tryptophan promotes T cell apoptosis and suppresses proliferation, in part because tryptophan starvation activates the GCN2 pathway, which responds to amino acid insufficiency by initiating a stress response that results in general repression of translation (7, 8). The details of how activation of this stress-response pathway alters the T cell activation program are not fully elucidated but likely involve translational regulation of cell cycle progression. In addition to IDO-mediated immune suppression through tryptophan depletion, several kynurenine metabolites act as regulators of T cells, dendritic cells, and microglia and alter the responsiveness of macrophages in vitro (9–12). The kynurenine-responsive aryl hydrocarbon receptor (AHR) has been strongly implicated in these adaptive immune phenotypes. Kynurenine is recognized as an endogenous ligand of AHR (13, 14), a transcription factor best known for its regulation of an adaptive xenobiotic response to exogenous environmental pollutants such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) (15). AHR resides in the cytoplasm of mammalian cells, in a multiprotein complex that includes HSP90 (16) and AHR-interacting protein (AIP) (17). Upon ligation by diverse external and endogenous ligands, AHR is translocated into the nucleus, where it binds the AHR nuclear translocator ARNT and influences gene transcription by binding to motifs termed dioxin-responsive elements (DREs) as well as by nonclassical mechanisms (reviewed in reference 18). The net effects of AHR ligation by an array of activating ligands are variable but largely immunosuppressive (19). While emerging evidence supports the hypothesis of kynurenine-mediated activation of AHR in the context of cancer (14) and autoimmune disease (reviewed in reference 20), the relevance of this mechanism to the immune control of infections is unclear.
Polymorphonuclear leukocytes (PMN; neutrophils) are the first responders to most bacterial infections and therefore represent an important cellular component of the innate immune response. In the case of urinary tract infections (UTIs), PMN recruitment likely contributes to symptomatology and ultimately to bacterial clearance (21, 22), and the presence of PMN in urine (pyuria) is a diagnostic hallmark. Uropathogens have evolved strategies for modulating host inflammatory responses, shifting the balance to favor colonization and disease progression. In addition to directly influencing host cell processes through the secretion of toxins and other exoproteins, bacterial pathogens may activate host anti-inflammatory pathways in order to survive and persist. We recently demonstrated that uropathogenic Escherichia coli (UPEC; the chief cause of community onset and health care-associated UTIs), in contrast to nonpathogenic E. coli, induced local expression of the mammalian IDO1 gene (encoding indoleamine 2,3-dioxygenase), both in vitro and in a murine model of bacterial cystitis. In turn, local activation of IDO was found to inhibit PMN trafficking into infected bladder tissue (23). PMN therefore represent the first identified innate cellular components regulated by IDO activity. In addition, these results illustrate how early induction of IDO by UPEC arriving in the bladder leads to local immune suppression, providing a window of opportunity for UPEC to establish a protected niche by invading superficial epithelial cells of the bladder (24, 25). This niche then provides a haven for rapid bacterial replication while protecting the burgeoning bacterial colony from neutrophils that are subsequently recruited to the bladder in response to multiple chemoattractants.
In order to further define the mechanism by which induction of IDO suppresses PMN recruitment and thereby enhances UPEC survival early in infection, we utilized several models of host-pathogen interaction. We show that, rather than tryptophan limitation, it is local production of kynurenine metabolites that attenuates PMN migration across the infected uroepithelium. These kynurenines exhibited a direct effect on PMN motility, as demonstrated by morphological analysis of adherent neutrophils in vitro. Furthermore, the derepressed PMN influx into the infected bladder that we observed in Ido1−/− mice (23) was here recapitulated in mice lacking expression of Ahr, implicating this transcriptional regulator in control of PMN chemotaxis. In total, our data further elucidate an emerging role for IDO in innate cellular defense and microbial pathogenesis.
MATERIALS AND METHODS
Reagents.
Fibrinogen (Fbg; catalog no. F4883), l-tryptophan (TRP; catalog no. T8491), l-kynurenine (KYN; catalog no. K8625), 3-hydroxy-dl-kynurenine (HK; catalog no. H1771), 3-hydroxyanthranilic acid (HAA; catalog no. 148776), CH-223191 (catalog no. C8124), f-Met-Leu-Phe (fMLF; catalog no. F3506), 1-methyl-d/l-tryptophan (1-MT; catalog no. 860646), dimethyl sulfoxide (DMSO; catalog no. D5879), and fetal bovine serum (FBS; catalog no. F2442) were purchased from Sigma-Aldrich (St. Louis, MO). Recombinant human tumor necrosis factor alpha (TNF-α) was purchased from R&D Systems (Minneapolis, MN).
Bacterial strains and culture.
Uropathogenic Escherichia coli (UPEC) isolate UTI89 was isolated from a patient with cystitis (26), while MG1655 is a type 1 piliated K-12 nonpathogenic strain of E. coli (27). For infection of mammalian cells in vitro, bacteria were grown overnight in standing Luria-Bertani (LB) broth at 37°C. Cells were pelleted by centrifugation and resuspended in sterile phosphate-buffered saline (PBS) to an optical density at 600 nm (OD600) of approximately 1.0 and further diluted in RPMI 1640 to achieve a multiplicity of infection (MOI) of approximately 40:1 bladder cell or 10:1 PMN.
Murine cystitis.
C57BL/6, Tnf−/− (The Jackson Laboratory), or Ahr−/− (kind gift of K. Moley) female mice, 8 to 10 weeks of age, were transurethrally inoculated with ∼2 × 107 CFU of UPEC strain UTI89, and bladders were harvested and homogenized at the indicated time points (detailed protocols are published in references 25 and 28). All animal experiments received prior review and approval of the Animal Studies Committee at Washington University School of Medicine. To quantify tissue myeloperoxidase (MPO) activity, a measure of neutrophils present in bladder tissue (29), bladder homogenates stored at −80°C were thawed and cleared by centrifugation, and the lysates were analyzed using a Fluoro MPO kit (Cell Technology, Mountain View, CA) per the manufacturer's instructions.
Cell culture and isolation.
A human bladder epithelial cell line (5637, HTB-9; American Type Culture Collection, Manassas, VA) was cultured in RPMI 1640 medium (Life Technologies, Grand Island, NY) supplemented with 10% FBS. Peripheral blood neutrophils were purified from the venous blood of healthy volunteers by dextran sedimentation and density gradient centrifugation as described previously (29), with informed consent and in accordance with a protocol approved by the Washington University Human Research Protection Office. Independent experiments utilized PMN from different adult donors.
Transwell chemotaxis assay.
A model of PMN transepithelial migration has been described previously (29–31). Briefly, inverted uroepithelial cell layers were grown to confluence on Transwell permeable supports (Costar catalog no. 3472; Corning, New York, NY) (pore size, 3 μm). The cells were washed with RPMI 1640 and mock infected or infected for 1 h with 60 μl of bacterial suspension (E. coli UTI89 or MG1655 at an MOI of approximately 40:1) in RPMI medium with TRP (200 μg/ml) or an appropriate DMSO vehicle control as indicated in Results and in the figure legends. Following infection, the inserts were transferred to a 24-well plate in which the lower reservoir contained 0.6 ml of RPMI 1640, with TRP (200 μg/ml), fMLF (100 nM), KYN (0.125 to 12.5 μM), fMLF (100 nM) plus KYN (0.125 to 12.5 μM), or an appropriate vehicle control as noted. Approximately 106 neutrophils were then added to the upper reservoir in 100 μl RPMI 1640, with TRP (200 μg/ml) or an appropriate DMSO vehicle control as indicated. Samples were incubated at 37°C in 5% CO2 for 1 h to allow transepithelial migration. The top chambers were removed and rinsed, and neutrophils in the lower reservoir were enumerated using a hemacytometer. Neutrophil transepithelial migration is expressed as a percentage of input cells (counted from control wells bearing no Transwell insert). Statistically significant differences were assessed using an unpaired Student t test.
Cytotoxicity assays.
The effect of kynurenine treatment on eukaryotic cell integrity was assessed by the use of a fluorescent assay for lactate dehydrogenase (LDH) release (CytoTox-ONE; Promega, Madison, WI) according to the instructions of the manufacturer. Approximately 106 neutrophils were incubated in a 96-well plate for 1 h at 37°C in 5% CO2 with RPMI 1640 alone or RPMI 1640 containing the indicated concentrations of KYN or vehicle control. Relative fluorescence was measured in a microplate reader (Synergy 2; BioTek, Winooski, VT), and percent cytotoxicity was calculated in comparison to the maximum fluorescence measured with detergent-lysed cells.
Immunofluorescent staining and morphology analysis.
Approximately 1.5 × 105 neutrophils were plated on Fbg-coated glass coverslips (10 μg/ml) and incubated in the presence of fMLF (100 nM), KYN (12.5 μM), fMLF plus KYN, TNF-α (25 ng/ml), TNF-α plus 1-MT (10 μM), or a medium-only control as indicated for 30 min at 37°C in 5% CO2. Cells were fixed, permeabilized, stained for actin with rhodamine phalloidin (1:1,000), and counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (1:10,000) (both stains from Life Technologies) as described previously (31). The coverslips were sealed with an anti-fade reagent (ProLong Gold; Life Technologies) and imaged using a 63× oil-immersion lens on a Zeiss Axioskop 2 MOT Plus fluorescence microscope. Identical exposure times were used for each sample within an individual experiment, and images were acquired with an Axiocam MRm Monochrome camera using Axiovision software. At least 10 random fields of view were imaged after selection with the DAPI channel only, and the morphology of all cells in each image for each condition was assessed (50 to 150 total cells per condition in each of 3 to 4 independent experiments). Images were numbered to blind the analyst to the treatment group. Small, symmetrical cells with no protrusions were categorized as “round.” Elongated cells with asymmetric actin staining and protrusions in a single direction were categorized as “polarized.” Cells that assumed a larger diameter on the surface, contained a ruffled edge or multiple focal adhesions around the cell circumference, and exhibited symmetric actin staining were categorized as “spread.” Statistically significant differences were assessed using an unpaired Student t test.
Gene expression analysis.
The abundance of CYP1A1 transcript has been used as a measure of aryl hydrocarbon receptor activation under various conditions (32). Cultured bladder epithelial cells or human PMN were treated in 24-well plates, and the contents of 4 wells were pooled to isolate sufficient RNA for gene expression analysis. Approximate total numbers of cells per condition were 106 for bladder epithelial cells and 107 for PMN. Conditions included KYN (12.5 μM), KYN (12.5 μM) plus CH-223191 (10 μM), or vehicle-only (DMSO) treatment for 2 h at 37°C in 5% CO2. Cell lysates were prepared and homogenized (Qiashredder; Qiagen, Valencia, CA). Total RNA was isolated by silica membrane binding (RNeasy minikit; Qiagen) according to the instructions of the kit manufacturer. RNA integrity was assessed by spectrophotometry, and first-strand cDNA was synthesized using random primer hexamers (Superscript II; Life Technologies). Real-time PCR was performed in a 10-μl reaction mixture containing approximately 0.1 μg template cDNA, 1× TaqMan assay mix (CYP1A1 assay identification [ID] no. Hs00153120_m1, GAPDH [glyceraldehyde-3-phosphate dehydrogenase] gene assay ID no. Hs99999905_m1; Life Technologies), and TaqMan Fast PCR master mix (Life Technologies) on an Applied Biosystems 7500 Fast instrument (standard cycling conditions: 1 cycle of 2 min at 50°C, 1 cycle of 10 min at 95°C, and 40 cycles of 15 s at 95°C and 1 min at 60°C) using SDS v1.4 software with automatic threshold and baseline corrections. The transcript abundance of the CYP1A1 target gene was normalized to that seen with the endogenous GAPDH control gene, and relative levels of target expression were calculated by the threshold cycle (ΔCT) method (33), where fold change in expression is equal to 2−Δ(ΔCT). Data are presented as fold change in transcript abundance under the experimental condition relative to the mock-treated calibrator condition and represent the means and standard errors of the means (SEM) of results from triplicate assays performed with RNA from three independent experiments. Statistically significant differences were assessed using an unpaired Student's t test.
Ido1 transcript abundance was measured in bladder homogenates of infected C57BL/6 and Tnf−/− mice in analogous fashion (Ido1 assay ID no. Mm00492586_m1, GAPDH gene assay ID no. Mm99999915_g1; Life Technologies), with RNA prepared as previously described (23). Aggregate data from three independent experiments are presented as fold change in transcript abundance in infected mice relative to mock-infected mice of the same strain and represent the means and SEM of results of triplicate assays performed with RNA from at least 8 mice total per group. Statistically significant differences were assessed using the Mann-Whitney U test.
Statistical analysis.
Statistical tests for significance were performed using Prism software version 6 for Windows (GraphPad, San Diego, CA). P values of <0.05 were considered significant.
RESULTS
UPEC suppression of neutrophil migration is not mediated by tryptophan limitation.
We reported previously that epithelial infection with UPEC (but not with nonpathogenic E. coli) induces local expression of the tryptophan catabolic enzyme IDO1, resulting in suppression of PMN migration in both in vitro and in vivo models of the early inflammatory response (23). To determine if this immunomodulatory mechanism was dependent on local alteration in tryptophan availability (as reported for T lymphocytes [7]), we measured the effect of tryptophan supplementation on PMN transepithelial migration in vitro. We observed that excess tryptophan did not restore PMN transepithelial migration across UPEC-infected epithelial layers (and similarly had no effect on migration in response to nonpathogenic E. coli; Fig. 1).
FIG 1.
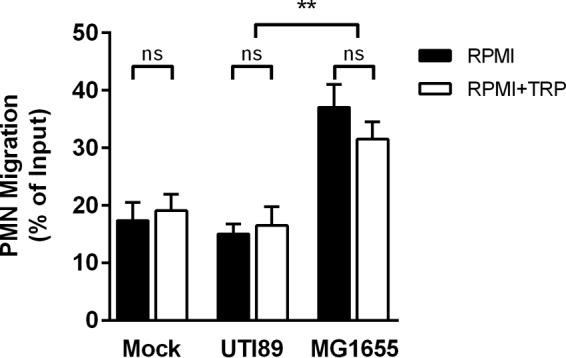
Tryptophan depletion is not required for UPEC suppression of neutrophil transepithelial migration. Neutrophils (approximately 106) were added to the upper chamber of a 3-μm-pore-size Transwell insert seeded with an inverted layer of bladder epithelial cells that were first infected for 1 h with uropathogenic E. coli (UTI89) or with nonpathogenic E. coli (MG1655) or were mock infected with PBS. The experiment was carried out in either unmodified RPMI 1640 (which contains minimal tryptophan) (RPMI) or RPMI 1640 supplemented with 200 μg/ml l-tryptophan (RPMI+TRP). The number of PMN migrating to the bottom chamber after 1 h is shown as a percentage of input. Data represent the means and SEM of sample results from 4 experiments with different PMN donors, each with 2 to 3 assays per condition. No significant difference (ns) was observed between samples from cells infected with the same strain in the presence or absence of excess tryptophan. Consistent with our previous data, the expected difference between samples infected with UTI89 and those infected with MG1655 was significant under both media conditions (**, P < 0.01 [by unpaired Student's t test]).
Kynurenine metabolites inhibit transepithelial neutrophil migration.
While suppression of neutrophil migration was not due to tryptophan limitation, the possibility remained that an increased local concentration of the IDO1 reaction product (namely, l-kynurenine) or of one or more of its downstream metabolites was important for this effect. To test this hypothesis, we measured neutrophil transepithelial migration upon the addition of l-kynurenine or one of several metabolites downstream in the kynurenine pathway of tryptophan degradation (Fig. 2). Starting concentrations of kynurenine metabolites were based on supernatant concentrations after UPEC infection of cultured bladder epithelial cells (10 to 15 μM; data not shown) and on observations with multiple other cell types (see, e.g., references 34, 35, and 36). The addition of l-kynurenine (KYN) or 3-hydroxykynurenine (HK) resulted in a significant decrease in PMN transepithelial migration toward the chemoattractant fMLF compared to the vehicle control results (Fig. 3A). Neutrophil migration upon addition of an equivalent concentration of 3-hydroxyanthranilic acid (HAA) trended similarly but did not reach statistical significance (Fig. 3A). In addition, the suppressive effect of l-kynurenine was dose dependent (Fig. 3B). Of note, the measured effect of l-kynurenine on neutrophil migration in this in vitro model was not due to neutrophil cytotoxicity, as determined by a standard assay for membrane integrity (Fig. 3C).
FIG 2.

The kynurenine pathway of tryptophan catabolism. Indoleamine 2,3-dioxygenase (IDO) catalyzes the rate-limiting step of the pathway. Major metabolites present along the pathway are indicated, and the metabolites assayed as described for Fig. 3A are shown here in bold.
FIG 3.
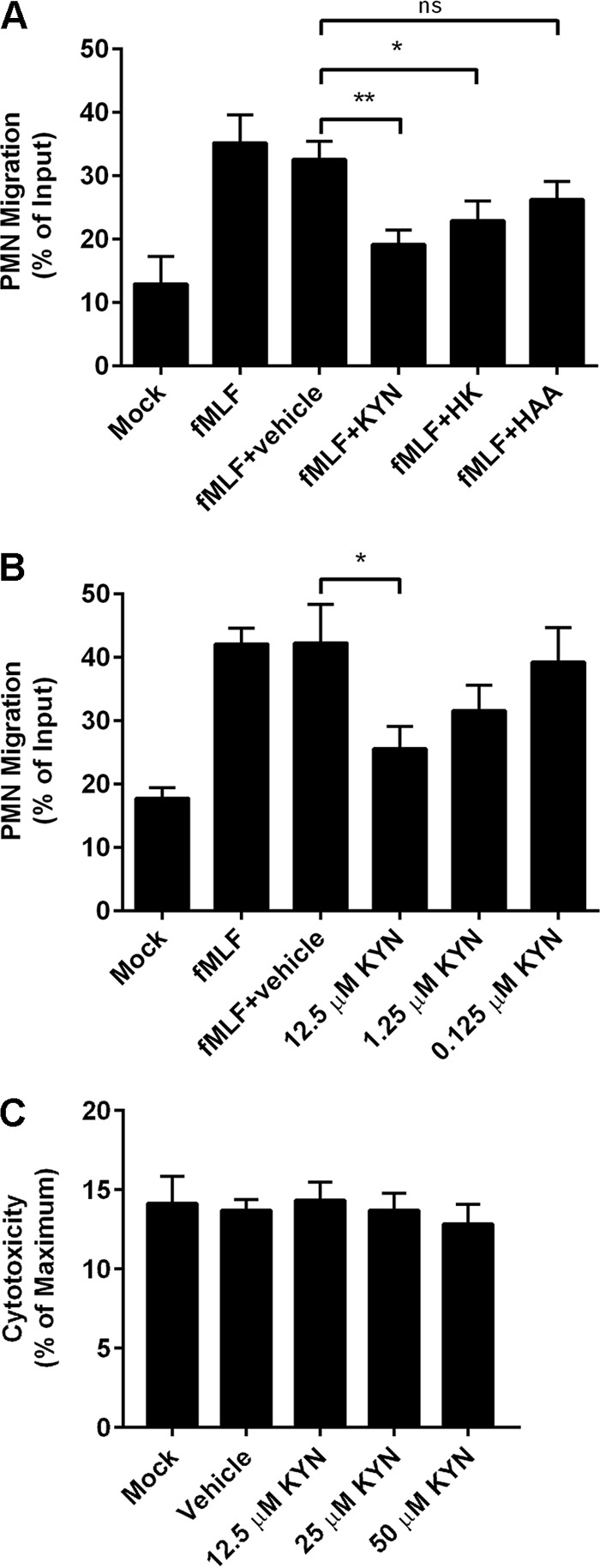
Kynurenines inhibit neutrophil transepithelial migration. (A and B) Approximately 106 neutrophils were added to the upper chamber of a 3-μm-pore-size Transwell insert seeded with an inverted layer of bladder epithelial cells. The lower chamber contained unmodified RPMI 1640 (Mock), RPMI 1640 plus fMLF (100 nM), or RPMI 1640 with fMLF plus the indicated kynurenine metabolite (12.5 μM) or vehicle control (A) or unmodified RPMI 1640 (Mock), RPMI 1640 with fMLF (100 nM), or RPMI 1640 with fMLF plus the indicated concentration of l-kynurenine (KYN) or vehicle control (B). The number of PMN migrating to the bottom chamber after 1 h is shown as a percentage of input. Data represent the means and SEM of sample results from 3 experiments with different PMN donors, each performed with 2 to 3 assays per condition. Statistical significance compared to the vehicle control is indicated (ns, not significant; *, P < 0.05; **, P < 0.01 [by unpaired Student's t test]). (C) As assessed by LDH release assay, no significant cytotoxicity to PMN was observed in the presence of the indicated concentrations of l-kynurenine.
Kynurenine exposure alters neutrophil morphology.
Because neutrophil polarization and rearrangement of the actin cytoskeleton are critical to the process of neutrophil chemotaxis, we characterized neutrophil morphology on a fibrinogen-coated surface in response to l-kynurenine in the presence or absence of the chemoattractant fMLF. The majority of unstimulated PMN were small and round in appearance, with minimal, evenly distributed f-actin staining (Fig. 4; Untreated). Upon exposure to fMLF, neutrophils have been shown to exhibit a polarized morphology featuring localization of f-actin filaments to the leading edge or pseudopod, correlating with chemotactic activity (31, 37). Our fMLF treatment also yielded this expected cellular morphology (Fig. 4; fMLF). In contrast to fMLF, treatment with l-kynurenine alone resulted in a significant percentage of cells adopting an attached, spread morphology (Fig. 4; KYN). Furthermore, kynurenine treatment in the presence of fMLF resulted in a significant decrease in the percentage of polarized cells and a significant increase in the percentage of spread cells compared to fMLF treatment alone (Fig. 4; fMLF+KYN). These data suggest that kynurenine directly alters neutrophil morphology and impairs chemotaxis.
FIG 4.
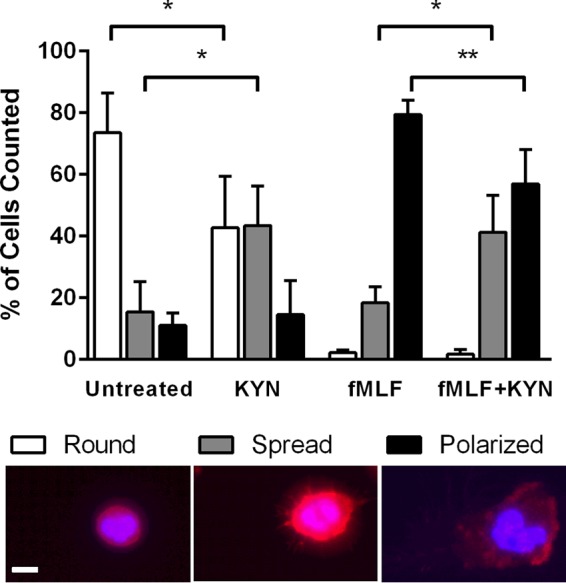
Kynurenine exposure alters neutrophil morphology. Neutrophils were allowed to adhere on fibrinogen-coated slides in unmodified RPMI 1640 (Untreated) or RPMI 1640 with l-kynurenine (KYN; 12.5 μM), fMLF (fMLF; 100 nM), or both (fMLF+KYN) for 30 min at 37°C and 5% CO2. Samples were processed as described in Materials and Methods and stained for actin with rhodamine phalloidin (red). Nuclei were labeled with DAPI (blue). The morphology of all cells in 10 random fields of view was categorized as round, spread, or polarized (as described in Materials and Methods), and the number of cells in each category is shown as a percentage of total cells counted. Data represent the means and SEM of sample results from 4 experiments with PMN from different donors, each with 50 to 150 cells counted per condition. Significant differences (Untreated versus KYN and fMLF versus fMLF+KYN) are indicated (*, P < 0.05; **, P < 0.01 [by unpaired Student's t test]). A representative example of each morphological type is shown below the graph; scale bar, 10 μm.
In addition, the spread morphology of kynurenine-treated PMN was reminiscent of that observed upon neutrophil treatment with TNF-α, previously proposed to provide a “stop signal” that counters PMN chemotaxis (38). As TNF-α has been demonstrated to induce IDO activity in various mammalian cell types (reviewed recently in reference 39), we wondered whether TNF-α served as an intermediary in the UPEC-dependent IDO induction that we previously observed in the murine bladder during bacterial cystitis (23). Indeed, IDO induction was sharply reduced in TNF-α-deficient mice within the UPEC-infected bladder 6 h postinfection (hpi; Fig. 5A). Of note, IDO induction was evident by 24 hpi in these Tnf−/− mice (Fig. 5A), complementing our earlier studies showing that other IDO inducers, specifically interferons, are important at this later interval (23). However, treatment with the IDO inhibitor 1-MT did not block TNF-α effects on PMN morphology (Fig. 5B), suggesting that TNF-α influences PMN chemotaxis via an alternative mechanism.
FIG 5.
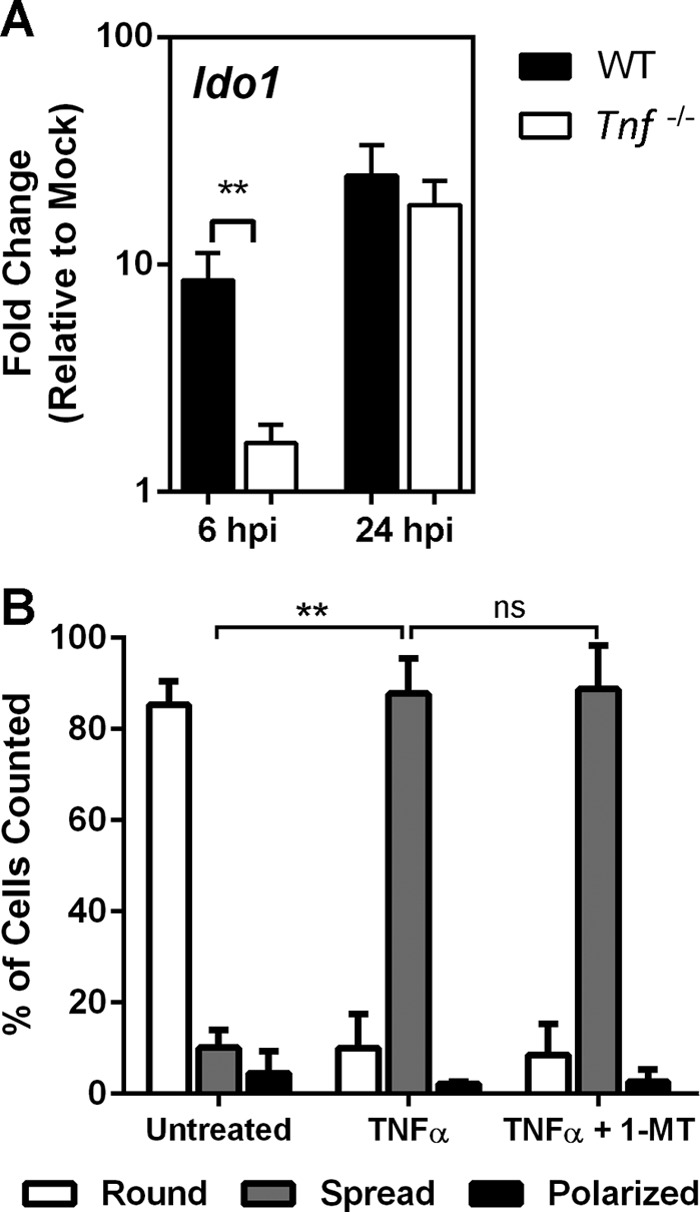
TNF-α mediates IDO induction in the infected bladder but impairs chemotaxis independently of IDO. (A) C57BL/6 (WT) or TNF-α-deficient mice were inoculated transurethrally with 107 CFU of E. coli strain UTI89. Bladder homogenates prepared at the indicated times (hpi [hours postinfection]) were subjected to RNA isolation and quantitative PCR for Ido1 transcript analysis. Data are represented as fold change compared to mock-infected mice. (B) Human PMN were allowed to adhere on fibrinogen-coated slides in unmodified RPMI 1640 (Untreated) or in RPMI 1640 with TNF-α (25 ng/ml) or TNF-α plus 1-MT (10 μM), with processing and morphological classification as round, spread, or polarized performed as described for Fig. 4. Data represent the means and SEM of the results of 3 experiments performed with different donors. TNF-α treatment induced a predominantly spread morphology (**, P < 0.01 [versus Untreated]), which was unaffected by 1-MT treatment (ns, not significant).
The kynurenine-responsive aryl hydrocarbon receptor participates in IDO modulation of neutrophil function.
It has been previously shown in other cell types (e.g., T cells and brain tumor cells) that l-kynurenine is an endogenous ligand for the aryl hydrocarbon receptor (AHR) (13, 14). AHR-ligand interaction is complicated, in that ligands can behave as agonists, antagonists, or both depending on context (40); furthermore, receptor stimulation and downstream effects are dependent on cell type and participation of other components in a multimeric transcription factor complex (18). We sought first to demonstrate a relationship between l-kynurenine treatment and cellular expression of the canonical AHR-responsive CYP1A1 gene (32), which encodes a cytochrome P450-associated hydroxylase acting on aryl hydrocarbons. In both human bladder epithelial cells and primary neutrophils, the normalized abundance of CYP1A1 transcript was upregulated upon l-kynurenine treatment; this response was prevented by the addition of a specific inhibitor of AHR (CH-223191; Fig. 6A). In order to demonstrate a role for AHR in modulation of neutrophil function, we introduced UPEC into the bladders of AHR-deficient mice and measured bladder tissue MPO levels, a surrogate for neutrophil influx (23, 29), at 6 hpi. As we observed previously in Ido1−/− mice (23), PMN influx (as reflected by tissue MPO content) was significantly higher in Ahr−/− mice than in wild-type BL/6 mice (Fig. 6B). Meanwhile, there was no decrement in bladder bacterial load in Ahr−/− mice 6 hpi (data not shown), matching what was seen in Ido1−/− mice at this time point (23).
FIG 6.
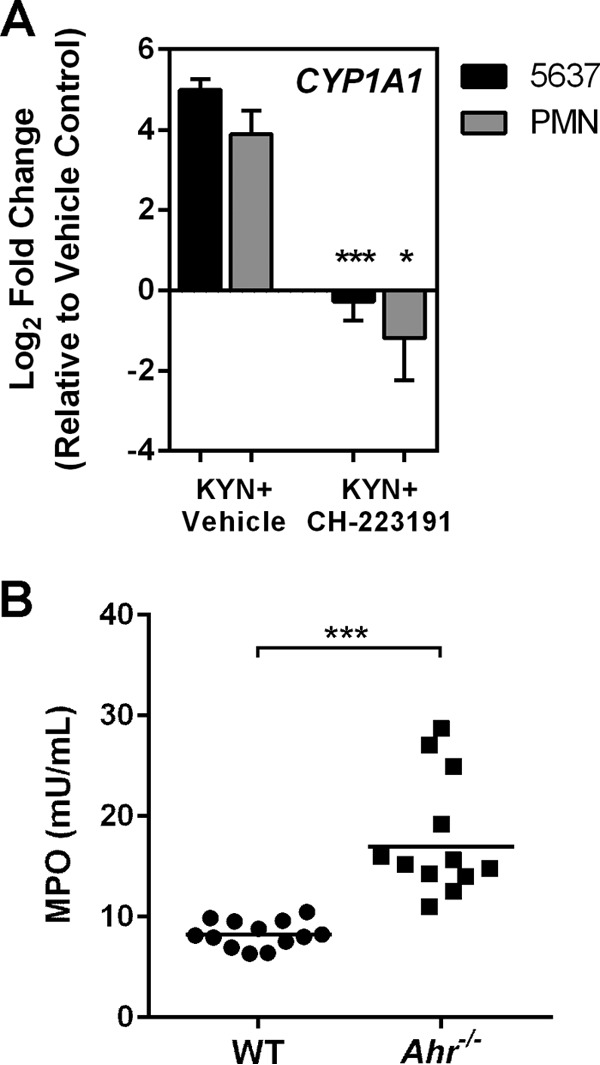
Kynurenine activates the aryl hydrocarbon receptor (AHR), suppressing neutrophil migration. (A) Neutrophils or cultured bladder epithelial cells were treated with l-kynurenine (KYN; 12.5 μM) plus either an AHR antagonist (CH-223191; 10 μM) or vehicle control for 2 h. Transcript abundance of the AHR-responsive CYP1A1 gene normalized to that of the GAPDH gene is shown relative to cells treated with DMSO vehicle only. Data represent the means and SEM of sample results from 3 experiments performed with different PMN donors, analyzed in triplicate. Kynurenine treatment of either bladder epithelial cells or PMN increased CYP1A1 transcription 4-fold to 5-fold compared with vehicle control results. Addition of CH-223191 resulted in statistically significant decreases in CYP1A1 transcript abundance (*, P < 0.05; ***, P < 0.001 [by unpaired Student's t test]). (B) Wild-type C57BL/6 mice or Ahr−/− mice were infected transurethrally with UPEC strain UTI89; at 6 hpi, bladders were homogenized and analyzed by fluorescent myeloperoxidase assay. Aggregate data from three independent experiments are shown. Bladder MPO content, shown in relative fluorescence units (RFU), was significantly higher in Ahr−/− mice (***, P < 0.001).
DISCUSSION
Alteration of local immune function within the host is a common theme in bacterial pathogenesis; though bacterial effectors and mechanistic strategies vary widely, many pathogens have evolved specific capacity to modulate host immunity in a variety of niches. Within the mammalian bladder, UPEC employs an array of distinct strategies to attenuate the initial recruitment of PMN, providing an opportunity for the pathogen to establish residence within uroepithelial cells (41). The PMN that ultimately respond to infection specifically locate and surround these infected epithelial cells, but the replicating UPEC within are sheltered (42). We previously reported one such strategy, namely, UPEC-specific induction of local IDO1 activity (23). In the present study, we delineate the mechanism by which IDO1 activity regulates PMN chemotaxis in response to epithelial bacterial infection. A large body of work has examined the role of IDO in adaptive immune regulation; this topic is not exhaustively reviewed here, but in brief, IDO activity can attenuate T effector cell proliferation by limiting the available tryptophan pool (5) and also generates broadly bioactive kynurenine metabolites (18). Here, we demonstrate that in the regulation of innate cellular responses, PMN transepithelial migration is inhibited via the action of kynurenines, rather than by tryptophan limitation. Of note, one recent study showed diminished PMN recruitment to infected cecum in Ido1-deficient mice (43). In that paper, l-kynurenine was found to promote apoptosis of murine bone marrow-derived neutrophils, though only in tryptophan-deficient media. Our in vitro experiments revealed no cytotoxicity to human PMNs upon exposure to physiologically relevant concentrations of kynurenine metabolites. In total, our current findings suggest that the influence of IDO and l-kynurenine in PMNs relates to chemotaxis more than to apoptosis.
Kynurenines are known endogenous ligands for AHR (13, 14), and this interaction acts to modulate inflammatory responses by promoting the generation of regulatory T cells (44, 45). Here, we found that expression of the canonical AHR-regulated CYP1A1 target gene increased in the presence of kynurenine and that infections of Ahr−/− mice recapitulated the augmented PMN influx previously observed in Ido1−/− mice. Our findings indicate that kynurenine ligation of AHR leads to impaired PMN chemotaxis, though it is notable that data in other systems are not uniformly congruent with this concept. For example, pretreatment of mice with TCDD suppressed neutrophil recruitment to the murine lung following intranasal inoculation with Streptococcus pneumoniae (46). However, in potential contrast, TCDD activation of AHR-dependent signaling in respiratory epithelial cells appeared to increase neutrophil recruitment to the lungs of influenza virus-infected mice, while neutrophil numbers were not increased systemically or in hematopoietic tissue (7, 47). These apparently incompatible results may arise from effects of dosage, host and microbial contexts, relative contributions of multiple cell types to the phenotypic results, and the presence of additional AHR ligands in the respective model systems. Our findings add to the rapidly expanding body of literature on AHR function in health, pregnancy, cancer, and immunology that has begun to reveal a highly complex set of ligand-AHR and AHR-target interactions—including the integration of endogenous and exogenous ligands (48), potential allosteric effects, and multiple noncanonical means by which AHR may regulate gene expression (18). An intriguing future direction will be to define how AHR signaling in human PMN might intersect with that triggered by ligation of formyl peptide receptors (which recognize fMLF and influence multiple PMN functions [reviewed recently in reference 49]) to exert a net effect on chemotaxis. Finally, our data do not discount the possibility that suppression of PMN migration by kynurenines is also mediated in parallel via mechanisms distinct from AHR.
The observed alteration in PMN morphology upon kynurenine treatment recapitulates a PMN phenotype reported previously with TNF-α treatment. Specifically, TNF-α was shown to initiate a p38 mitogen-activated protein kinase (MAPK)-dependent “stop signal” that induces neutrophil adhesion and not polarization, resulting in diminished PMN motility toward chemoattractants (38). Existing data indicate that UPEC induces local TNF-α secretion during cystitis (21, 50), and we show that in the bladder as a whole, TNF-α influences early IDO induction in response to UPEC (Fig. 5A). However, our present data (Fig. 5B) indicate that TNF-α effects on neutrophil chemotaxis are not attributable to IDO induction, as chemical inhibition of IDO did not abrogate the TNF-α “stop signal” (Fig. 7). Another piece of this model that remains to be specified is the UPEC-specific effector or attribute that promotes IDO activation; of note, the recently identified PMN migration suppressor YbcL (51, 52) does not rely on IDO for its action (unpublished data).
FIG 7.
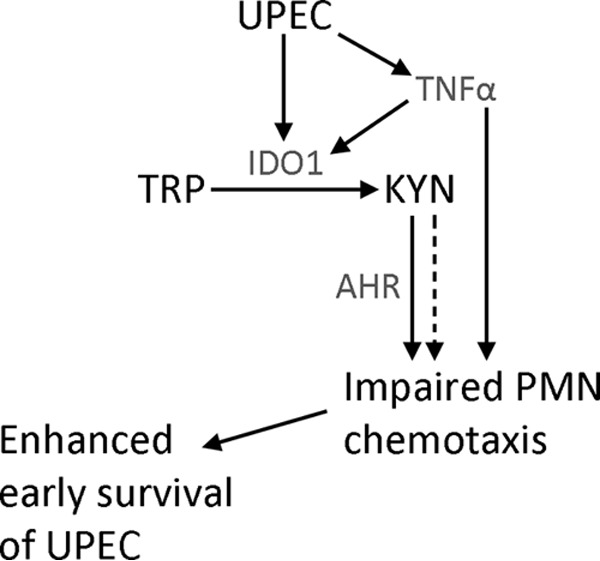
Model for UPEC suppression of PMN chemotaxis. In the acute phase of bladder infection, UPEC induces the expression of IDO1, first via TNF-α and later via interferons. Enzymatic activity of IDO1 catabolizes tryptophan (TRP) to l-kynurenine (KYN), which can be further metabolized to other downstream bioactive compounds. TNF-α dampens PMN chemotaxis in an IDO-independent way, as chemical inhibition of IDO did not reverse the PMN morphological change induced by TNF-α. l-Kynurenine, via ligation of AHR (but potentially also through alternative mechanisms), impairs early PMN chemotaxis, which offers UPEC sufficient opportunity to establish a replicative niche (the intracellular bacterial community) within uroepithelial cells that is protected from subsequently arriving PMN.
In summary, our findings support the idea of an expanded role for IDO in innate cellular responses through the AHR-mediated effects of kynurenine metabolites on neutrophil function, in addition to the previously identified roles in adaptive immune regulation. Beyond this, our data illuminate a mechanism by which a prototypic bacterial pathogen leverages these innate host pathways to facilitate the establishment of epithelial infection.
ACKNOWLEDGMENTS
We thank the neutrophil donors, and we acknowledge K. Moley for the Ahr−/− mice and M. Dinauer for critical review of the manuscript.
D.A.H. serves on the Board of Directors of BioVersys AG, Basel, Switzerland. The other authors have no potential conflicts to disclose.
REFERENCES
- 1.Mellor AL, Munn DH. 2004. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol 4:762–774. doi: 10.1038/nri1457. [DOI] [PubMed] [Google Scholar]
- 2.Hwu P. 2000. Indoleamine 2,3-dioxygenase production by human dendritic cells results in the inhibition of T cell proliferation. J Immunol 164:3596–3599. doi: 10.4049/jimmunol.164.7.3596. [DOI] [PubMed] [Google Scholar]
- 3.Mellor AL. 2003. Cutting edge: induced indoleamine 2,3 dioxygenase expression in dendritic cell subsets suppresses T cell clonal expansion. J Immunol 171:1652–1655. doi: 10.4049/jimmunol.171.4.1652. [DOI] [PubMed] [Google Scholar]
- 4.Munn DH. 2002. Potential regulatory function of human dendritic cells expressing indoleamine 2,3-dioxygenase. Science 297:1867–1870. doi: 10.1126/science.1073514. [DOI] [PubMed] [Google Scholar]
- 5.Fallarino F, Grohmann U, Vacca C, Bianchi R, Orabona C, Spreca A, Fioretti MC, Puccetti P. 2002. T cell apoptosis by tryptophan catabolism. Cell Death Differ 9:1069–1077. doi: 10.1038/sj.cdd.4401073. [DOI] [PubMed] [Google Scholar]
- 6.Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. 1999. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med 189:1363–1372. doi: 10.1084/jem.189.9.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Teske S, Bohn AA, Hogaboam JP, Lawrence BP. 2008. Aryl hydrocarbon receptor targets pathways extrinsic to bone marrow cells to enhance neutrophil recruitment during influenza virus infection. Toxicol Sci 102:89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang P. 2002. The GCN2 eIF2α kinase is required for adaptation to amino acid deprivation in mice. Mol Cell Biol 22:6681–6688. doi: 10.1128/MCB.22.19.6681-6688.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grohmann U, Fallarino F, Puccetti P. 2003. Tolerance, DCs and tryptophan: much ado about IDO. Trends Immunol 24:242–248. doi: 10.1016/S1471-4906(03)00072-3. [DOI] [PubMed] [Google Scholar]
- 10.Stone TW, Darlington LG. 2002. Endogenous kynurenines as targets for drug discovery and development. Nat Rev Drug Discov 1:609–620. doi: 10.1038/nrd870. [DOI] [PubMed] [Google Scholar]
- 11.Moffett JR, Namboodiri MA. 2003. Tryptophan and the immune response. Immunol Cell Biol 81:247–265. doi: 10.1046/j.1440-1711.2003.t01-1-01177.x. [DOI] [PubMed] [Google Scholar]
- 12.Barth MC, Ahluwalia N, Anderson TJ, Hardy GJ, Sinha S, Alvarez-Cardona JA, Pruitt IE, Rhee EP, Colvin RA, Gerszten RE. 2009. Kynurenic acid triggers firm arrest of leukocytes to vascular endothelium under flow conditions. J Biol Chem 284:19189–19195. doi: 10.1074/jbc.M109.024042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Veldhoen M, Hirota K, Christensen J, O'Garra A, Stockinger B. 2009. Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. J Exp Med 206:43–49. doi: 10.1084/jem.20081438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, Schumacher T, Jestaedt L, Schrenk D, Weller M, Jugold M, Guillemin GJ, Miller CL, Lutz C, Radlwimmer B, Lehmann I, von Deimling A, Wick W, Platten M. 2011. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478:197–203. doi: 10.1038/nature10491. [DOI] [PubMed] [Google Scholar]
- 15.Denison MS, Nagy SR. 2003. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol 43:309–334. doi: 10.1146/annurev.pharmtox.43.100901.135828. [DOI] [PubMed] [Google Scholar]
- 16.Perdew GH. 1988. Association of the Ah receptor with the 90-kDa heat shock protein. J Biol Chem 263:13802–13805. [PubMed] [Google Scholar]
- 17.Ma Q, Whitlock JP Jr. 1997. A novel cytoplasmic protein that interacts with the Ah receptor, contains tetratricopeptide repeat motifs, and augments the transcriptional response to 2,3,7,8-tetrachlorodibenzo-p-dioxin. J Biol Chem 272:8878–8884. doi: 10.1074/jbc.272.14.8878. [DOI] [PubMed] [Google Scholar]
- 18.Stockinger B, Di Meglio P, Gialitakis M, Duarte JH. 2014. The aryl hydrocarbon receptor: multitasking in the immune system. Annu Rev Immunol 32:403–432. doi: 10.1146/annurev-immunol-032713-120245. [DOI] [PubMed] [Google Scholar]
- 19.Hao N, Whitelaw ML. 2013. The emerging roles of AhR in physiology and immunity. Biochem Pharmacol 86:561–570. doi: 10.1016/j.bcp.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 20.Nguyen NT, Nakahama T, Le DH, Van Son L, Chu HH, Kishimoto T. 2014. Aryl hydrocarbon receptor and kynurenine: recent advances in autoimmune disease research. Front Immunol 5:551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ingersoll MA, Kline KA, Nielsen HV, Hultgren SJ. 2008. G-CSF induction early in uropathogenic Escherichia coli infection of the urinary tract modulates host immunity. Cell Microbiol 10:2568–2578. doi: 10.1111/j.1462-5822.2008.01230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hagberg L, Hull R, Hull S, McGhee JR, Michalek SM, Svanborg Eden C. 1984. Difference in susceptibility to Gram-negative urinary tract infection between C3H/HeJ and C3H/HeN mice. Infect Immun 46:839–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Loughman JA, Hunstad DA. 2012. Induction of indoleamine 2,3-dioxygenase by uropathogenic bacteria attenuates innate responses to epithelial infection. J Infect Dis 205:1830–1839. doi: 10.1093/infdis/jis280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anderson GG, Palermo JJ, Schilling JD, Roth R, Heuser J, Hultgren SJ. 2003. Intracellular bacterial biofilm-like pods in urinary tract infections. Science 301:105–107. doi: 10.1126/science.1084550. [DOI] [PubMed] [Google Scholar]
- 25.Mulvey MA, Lopez-Boado YS, Wilson CL, Roth R, Parks WC, Heuser J, Hultgren SJ. 1998. Induction and evasion of host defenses by type 1-piliated uropathogenic Escherichia coli. Science 282:1494–1497. doi: 10.1126/science.282.5393.1494. [DOI] [PubMed] [Google Scholar]
- 26.Hultgren SJ, Schwan WR, Schaeffer AJ, Duncan JL. 1986. Regulation of production of type 1 pili among urinary tract isolates of Escherichia coli. Infect Immun 54:613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blattner FR, Plunkett G III, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453–1474. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 28.Hung CS, Dodson KW, Hultgren SJ. 2009. A murine model of urinary tract infection. Nat Protoc 4:1230–1243. doi: 10.1038/nprot.2009.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loughman JA, Hunstad DA. 2011. Attenuation of human neutrophil migration and function by uropathogenic bacteria. Microbes Infect 13:555–565. doi: 10.1016/j.micinf.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lau ME, Hunstad DA. 2013. Quantitative assessment of human neutrophil migration across a cultured bladder epithelium. J Vis Exp 2013:e50919. doi: 10.3791/50919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lokuta MA, Nuzzi PA, Huttenlocher A. 2007. Analysis of neutrophil polarization and chemotaxis. Methods Mol Biol 412:211–229. doi: 10.1007/978-1-59745-467-4_14. [DOI] [PubMed] [Google Scholar]
- 32.Whitlock JP., Jr 1999. Induction of cytochrome P4501A1. Annu Rev Pharmacol Toxicol 39:103–125. doi: 10.1146/annurev.pharmtox.39.1.103. [DOI] [PubMed] [Google Scholar]
- 33.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 34.De Ravin SS, Zarember KA, Long-Priel D, Chan KC, Fox SD, Gallin JI, Kuhns DB, Malech HL. 2010. Tryptophan/kynurenine metabolism in human leukocytes is independent of superoxide and is fully maintained in chronic granulomatous disease. Blood 116:1755–1760. doi: 10.1182/blood-2009-07-233734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guillemin GJ, Kerr SJ, Smythe GA, Smith DG, Kapoor V, Armati PJ, Croitoru J, Brew BJ. 2001. Kynurenine pathway metabolism in human astrocytes: a paradox for neuronal protection. J Neurochem 78:842–853. doi: 10.1046/j.1471-4159.2001.00498.x. [DOI] [PubMed] [Google Scholar]
- 36.Ibana JA, Belland RJ, Zea AH, Schust DJ, Nagamatsu T, AbdelRahman YM, Tate DJ, Beatty WL, Aiyar AA, Quayle AJ. 2011. Inhibition of indoleamine 2,3-dioxygenase activity by levo-1-methyl tryptophan blocks gamma interferon-induced Chlamydia trachomatis persistence in human epithelial cells. Infect Immun 79:4425–4437. doi: 10.1128/IAI.05659-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dekker LV, Segal AW. 2000. Perspectives: signal transduction. Signals to move cells. Science 287:982–983, 985. [DOI] [PubMed] [Google Scholar]
- 38.Lokuta MA, Huttenlocher A. 2005. TNF-α promotes a stop signal that inhibits neutrophil polarization and migration via a p38 MAPK pathway. J Leukoc Biol 78:210–219. doi: 10.1189/jlb.0205067. [DOI] [PubMed] [Google Scholar]
- 39.Munn DH, Mellor AL. 2013. Indoleamine 2,3-dioxygenase and metabolic control of immune responses. Trends Immunol 34:137–143. doi: 10.1016/j.it.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barouki R, Aggerbeck M, Aggerbeck L, Coumoul X. 2012. The aryl hydrocarbon receptor system. Drug Metabol Drug Interact 27:3–8. [DOI] [PubMed] [Google Scholar]
- 41.Hunstad DA, Justice SS. 2010. Intracellular lifestyles and immune evasion strategies of uropathogenic Escherichia coli. Annu Rev Microbiol 64:203–221. doi: 10.1146/annurev.micro.112408.134258. [DOI] [PubMed] [Google Scholar]
- 42.Olson PD, Hunstad DA. 2016. Subversion of host innate immunity by uropathogenic Escherichia coli. Pathogens 5:pii:E2. doi: 10.3390/pathogens5010002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.El-Zaatari M, Chang YM, Zhang M, Franz M, Shreiner A, McDermott AJ, van der Sluijs KF, Lutter R, Grasberger H, Kamada N, Young VB, Huffnagle GB, Kao JY. 2014. Tryptophan catabolism restricts IFNγ-expressing neutrophils and Clostridium difficile immunopathology. J Immunol 193:807–816. doi: 10.4049/jimmunol.1302913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. 2010. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol 185:3190–3198. doi: 10.4049/jimmunol.0903670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nguyen NT, Kimura A, Nakahama T, Chinen I, Masuda K, Nohara K, Fujii-Kuriyama Y, Kishimoto T. 2010. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc Natl Acad Sci U S A 107:19961–19966. doi: 10.1073/pnas.1014465107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang T, Wyrick KL, Pecka MR, Wills TB, Vorderstrasse BA. 2012. Mechanistic exploration of AhR-mediated host protection against Streptococcus pneumoniae infection. Int Immunopharmacol 13:490–498. doi: 10.1016/j.intimp.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wheeler JL, Martin KC, Lawrence BP. 2013. Novel cellular targets of AhR underlie alterations in neutrophilic inflammation and inducible nitric oxide synthase expression during influenza virus infection. J Immunol 190:659–668. doi: 10.4049/jimmunol.1201341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Julliard W, Fechner JH, Mezrich JD. 2014. The aryl hydrocarbon receptor meets immunology: friend or foe? A little of both. Front Immunol 5:458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dorward DA, Lucas CD, Chapman GB, Haslett C, Dhaliwal K, Rossi AG. 2015. The role of formylated peptides and formyl peptide receptor 1 in governing neutrophil function during acute inflammation. Am J Pathol 185:1172–1184. doi: 10.1016/j.ajpath.2015.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Olson PD, Hruska KA, Hunstad DA. 8 October 2015. Androgens enhance male urinary tract infection severity in a new model. J Am Soc Nephrol doi: 10.1681/ASN.2015030327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lau ME, Danka ES, Tiemann KM, Hunstad DA. 2014. Bacterial lysis liberates the neutrophil migration suppressor YbcL from the periplasm of uropathogenic Escherichia coli. Infect Immun 82:4921–4930. doi: 10.1128/IAI.01838-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lau ME, Loughman JA, Hunstad DA. 2012. YbcL of uropathogenic Escherichia coli suppresses transepithelial neutrophil migration. Infect Immun 80:4123–4132. doi: 10.1128/IAI.00801-12. [DOI] [PMC free article] [PubMed] [Google Scholar]