

Abstract
The expression of flagella correlates with different aspects of bacterial pathogenicity, ranging from adherence to host cells to activation of inflammatory responses by the innate immune system. In the present study, we investigated the role of flagella in the adherence of an atypical enteropathogenic Escherichia coli (aEPEC) strain (serotype O51:H40) to human enterocytes. Accordingly, isogenic mutants deficient in flagellin (FliC), the flagellar structural subunit; the flagellar cap protein (FliD); or the MotAB proteins, involved in the control of flagellar motion, were generated and tested for binding to differentiated Caco-2 cells. Binding of the aEPEC strain to enterocytes was significantly impaired in strains with the fliC and fliD genes deleted, both of which could not form flagella on the bacterial surface. A nonmotile but flagellated MotAB mutant also showed impaired adhesion to Caco-2 cells. In accordance with these observations, adhesion of aEPEC strain 1711-4 to Caco-2 cells was drastically reduced after the treatment of Caco-2 cells with purified FliD. In addition, incubation of aEPEC bacteria with specific anti-FliD serum impaired binding to Caco-2 cells. Finally, incubation of Caco-2 cells with purified FliD, followed by immunolabeling, showed that the protein was specifically bound to the microvillus tips of differentiated Caco-2 cells. The aEPEC FliD or anti-FliD serum also reduced the adherence of prototype typical enteropathogenic, enterohemorrhagic, and enterotoxigenic E. coli strains to Caco-2 cells. In conclusion, our findings further strengthened the role of flagella in the adherence of aEPEC to human enterocytes and disclosed the relevant structural and functional involvement of FliD in the adhesion process.
INTRODUCTION
Atypical enteropathogenic Escherichia coli (aEPEC) has emerged as an agent of diarrhea in children and adults worldwide (1–3). aEPEC has phenotypic and genotypic properties different from those of typical enteropathogenic E. coli (tEPEC), which causes diarrhea mainly in children <2 years old (4). The frequency of aEPEC strains has increased in both diarrheic and nondiarrheic children to the point that tEPEC strains are often outnumbered in regions where diarrhea due to E. coli is endemic (5, 6). The main difference between aEPEC and tEPEC is the absence of the EPEC adherence factor plasmid (pEAF) in the former group (4, 7). This plasmid encodes the bundle-forming pilus, which is responsible for eukaryotic cell adhesion, as well as adhesion between bacterial cells that culminates in the formation of bacterial clusters on the cell surface. Furthermore, pEAF encodes proteins involved in the regulation of several genes of the locus of enterocyte effacement (LEE), which is a pathogenicity island essential for the dramatic reorganization of the host cell cytoskeleton that leads to the formation of attaching-effacing (A/E) lesion (8). Although aEPEC lacks pEAF, it is also capable of forming A/E lesions. These lesions result from the interaction between the outer membrane adhesin intimin and Tir, the translocated intimin receptor. Tir is translocated to the cytosol by the type 3 secretion system (T3SS) and is inserted into the cell membrane, where it recognizes and interacts with intimin. A number of other LEE-encoded and non-LEE-encoded effector proteins are also translocated by the T3SS, leading to various enterocyte alterations that contribute to the occurrence of tEPEC and aEPEC diarrhea (9, 10).
The first step in the development of diarrheal diseases mediated by bacterial pathogens is gut colonization. In this initial process of interaction between enterocytes and bacterial cells, multiple bacterial surface structures may be involved, including flagella, which contribute to bacterial motility (11, 12). FliC protein (flagellin) is the major structural component of the flagellar filament, whereas the FliD (cap) protein forms a structure exposed at the flagellar tip that is essential for flagellar shaft assembly (13). The flagellar apparatus is associated with the MotA and MotB proteins, which form the stator, a membrane pore channel essential for generation of the proton motive force that is required for flagellar motility (14–16).
Several studies have provided experimental evidence of the contribution of flagella to the pathogenicity of different bacterial species by mediating adhesion and invasion of epithelial cells, as well as contributing to biofilm formation and the activation of innate and adaptive immune responses (11, 16–21). In addition, we have recently shown that flagellum expression by aEPEC strain 1711-4 (serotype O51:H40) is involved in binding to human enterocytes in vitro (22). We hypothesized that the flagellar cap protein (FliD) could mediate the binding of bacteria to microvillus tips, since electron micrographs of Caco-2 cells infected with strain 1711-4 show numerous bacteria adhering to cells by their flagellar tips (22). In the present study, we demonstrated that FliD mediates the adhesion of aEPEC, as well as of other prototype diarrheagenic E. coli strains, to human enterocytes under in vitro conditions and is therefore a relevant virulence-associated factor expressed by these bacteria.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
aEPEC strain 1711-4 (serotype O51:H40) (23) and its isogenic mutants deficient in fliC (17ΔfliC), fliD (17ΔfliD), or motAB (17ΔmotAB), as well as mutants complemented with plasmids pZE21MCS2Kmr (24), pET200/D-TOPO (Invitrogen), and/or pBAD202/D-TOPO (Invitrogen) containing fliC (C1P1-8), fliD [17(pETfliD)] or motAB [17(pBADmotAB)], respectively, were used (Table 1). The 17ΔfliD strain transformed with an empty cloning vector (pET200/D-TOPO) was used as a negative control. The following prototype diarrheagenic E. coli strains were used in inhibition assays with purified recombinant aEPEC 1711-4 FliD and anti-FliD serum: tEPEC E2348/69 (serotype O127:H7), enterohemorrhagic E. coli (EHEC) EDL933 (serotype O157:H7), and enterotoxigenic E. coli (ETEC) H10407 (serotype O78:H11). Strains harboring antibiotic resistance genes were grown in Luria-Bertani (LB) broth containing 50 μg/ml kanamycin (Km), 60 μg/ml zeocin (Zeo), and/or 100 μg/ml apramycin (Apra). All strains were stored in LB medium with 20% glycerol at −80°C.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Genotype and characteristics | Source or reference |
|---|---|---|
| 1711-4 | aEPEC O51:H40, wild type | 23 |
| E2348/69 | Typical EPEC, serotype O127:H7 | 42 |
| EDL933 | EHEC, serotype O157:H7 | 43 |
| H10407 | ETEC, serotype O78:H11 | 44 |
| 17ΔfliD | fliD::ble (Zeor) | This study |
| 17ΔmotAB | motAB::ble (Zeor) | This study |
| 17ΔfliC | fliC::ble (Zeor) | 22 |
| pET200fliD | fliD gene from 1711-4 cloned into pET200/D-TOPO | This study |
| pBADmotAB | motA motB genes from 1711-4 cloned into pET200/D-TOPO | This study |
| pZEKmfliC | fliC gene from 1711-4 cloned into pZE21-MCS2 | 22 |
| pZE21-MCS2 | Plasmid encoding Km resistance (Kmr) | 24 |
| 17(pETfliD) | 17ΔfliD carrying pET200fliD (Zeor Kmr) | This study |
| 17(pBADmotAB) | 17ΔmotAB carrying pBAD202/D-TOPO motAB (Zeor Kmr) | This study |
| C1P1-8 | 17ΔfliC carrying pZEKmfliC (Zeor Kmr) | 22 |
| MC4160-malTΔ224::zeo-(F+) | Source of Zeo cassette (ble gene) | Gift from J. M. Ghigo |
| pKOBEG-Aprar | Plasmid encoding red λ phage operon, Apra resistance (Aprar) | 45 |
Construction of mutant strains deficient in FliD or MotAB.
Deletion of fliD or motAB was carried out by exchange of the target gene with a gene encoding Zeo resistance (ble) (25). Recombination events were facilitated by proteins encoded by the λ red phage genes contained in the pKOBEG plasmid (26), which was inserted into the wild-type strain by electroporation. The ble gene was amplified by PCR with primers described in Table 2. The purified PCR products were inserted by electroporation into the wild-type strain previously transformed with pKOBEG. The preparations were incubated at 30°C for 45 min and then plated on LB agar containing Zeo; the plates were incubated at 30°C in ambient air for 24 h. Subsequently, transformants were cultivated at 37°C for 18 to 24 h to obtain pKOBEG-cured clones. Zeo-resistant clones that were unable to grow in Apra-containing medium were selected and stored at −80°C. Homologous recombination events were confirmed by PCR with primers flanking the fliD gene (fliD.verifF-5′/fliD.verifR-3′) (Table 2) (22).
TABLE 2.
Primers used for PCR amplification or sequencing
| Primer | Nucleotide sequence | Amplicon size (bp) |
|---|---|---|
| motAB-FW | 5′ GCTTATATCCATGCTCACGCTG 3′ | 1,800 |
| motAB-RV | 5′ GGTCAACAGTGGAAGGATGAT 3′ | |
| motAB.zeo.RV | 5′ CTGAACATCCTGTCATGGTCAACAGTGGAAGGATGATGTCGTCATCGCTTGCATTAGAAAGG 3′ | 642 |
| motAB.zeo.FW | 5′ AAATGTCTGATAAAAATCGCTTATATCCATGCTCACGCTGGAATGATGCAGAGATGTAAG 3′ | |
| pBAD-202-motAB-FW | 5′ CACCTGAGTGCTTATCTTATTAGGTTAC 3′ | 1,953 |
| pBAD-202-motAB-RV-1953 | 5′ AGCCCGAAAGATGGCATTCA 3′ | |
| fliCH40.zeo-5 | 5′ GCCCAATAACATCAAGTTGTAATTGATAAGGAAAAGATCATGGTCATCGCTTGCATTAGAAAG 3′ | 642 |
| fliCH40.zeo-3 | 5′ AACCCCGCCGGTGGCGGGGTTTGAGCGATAAGTGTAAATTAGAATGATGCAGAGATGTAAG 3′ | |
| PrfliCH40F | 5′ AAATTTCTCGAGACCTAATTCCTTTTGATTGCAAAC 3′ | 1,833 |
| fliCH40R | 5′ AAATTTTCTAGACAGGGGTTATTTGGGGGTTA 3′ | |
| Pet fliD FW | 5′ CACCATGGCAAGTATTTCATGC 3′ | 1,450 |
| fliD-ext-3L | 5′ TTACTTGGAATTACTGTTGTTTTCG 3′ | |
| fliD.zeo.5 | 5′ TATTCGTTTTACGTGTCGAAAGATAAAAGGAAATCGCATGGTCATCGCTTGCATTAGAAAGG 3′ | 642 |
| fliD.zeo.3 | 5′ TTGCCGCGTACATGACCTGTCTCCCGATGAATATTGATTAGAATGATGCAGAGATGTAAG 3′ | |
| fliD-FW-pET-Topo | 5′ CACCATGGCAAGTATTTCATCG 3′ | 1,600a |
| fliD-RV-pET-Topo | 5′ TCGCTTGGAATTACTGTTGTTTT 3′ | 200b |
| fliD.verifF-5′ | 5′ CGTCAACCCTGTTATCGTCTG 3′ | 1,765 |
| fliD.verifR-3′ | 5′ AAACAGGCTCGCTCTAACCA 3′ | |
| pBADverf FW | 5′ ATGCCAATAGCATTTTTATCC 3′ | 685b |
| pBADverf RV | 5′ TCTGATTTAATCTGTATCAGG 3′ | 2,496a |
| TrxFus-FWc | 5′ TTCCTCGACGCTAACCTG 3′ | 238b |
| pBAD-RVc | 5′ GATTTAATCTGTATCAGG 3′ | 2,049a |
With insert.
Without insert.
Sequencing primer.
Complementation of mutants deficient in FliD or MotAB.
Primers designed according to the pBAD Directional TOPO kit (Invitrogen) manufacturer's instructions were employed to amplify the motAB genes (Table 2). These genes were amplified by using the pBAD-202-motAB-FW and pBAD-202-motAB-RV-1953 primers (Table 2) with genomic DNA from strain 1711-4 and the High Fidelity Phusion Taq DNA polymerase (Finnzymes, Finland). The conditions used were 30 s at 98°C; 35 cycles of 98°C for 10 s, 57°C for 15 s, and 72°C for 30 s; and a final extension at 72°C for 10 min. After amplification, the PCR product was purified and ligated into pBAD202/D-TOPO at a 2:1 insert-to-vector ratio. Afterwards, the ligation products were used to transform competent TOP10 cells by electroporation (E. coli Pulser Apparatus; Bio-Rad). Clones transfected with the MotAB-encoding plasmid were selected after plating bacterial cells on LB agar plates containing 50 μg/ml Km, followed by incubation for 18 h at 37°C. Recombinant clones were screened by PCR with three primer pairs, pBADverf FW/pBADverf RV, pBADverf FW/pBAD-202-motAB-RV-1953, and TrxFus-FW/pBAD-RV (Table 2). Taq MasterMix (Promega) was used under the following conditions: 5 min at 94°C; 30 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 2 min; and a final extension at 72°C for 7 min. The same procedure was used to purify the plasmid containing the motAB genes with the QIAprep Spin kit (Qiagen). Electrocompetent cells mutated in the motAB genes were prepared and subsequently transfected with the recombinant plasmid. The suspension was plated on LB agar containing 50 μg/ml Km and 60 μg/ml Zeo and incubated for 18 h at 37°C. The colonies were selected and stored at 80°C.
For complementation of fliD mutants, pET200/D-TOPO from the pET Directional TOPO kit (Invitrogen) was used. The fliD-FW-pET-Topo and fliD-RV-pET-Topo primers were used to amplify the fliD gene (Table 2). The correct orientation of the insert was confirmed by sequencing the PCR product with primers flanking the cloning sites, provided in the pET Directional TOPO kit.
Motility test.
The mutants and wild-type strains were assayed for motility as previously described by Sampaio et al. (22). Motility was demonstrated by bacterial spreading around the initial inoculum, as evidenced by increased turbidity.
Recombinant FliD protein production.
The PCR product was cloned and expressed by the E. coli pET200/D-TOPO expression system (Invitrogen) according to standard protocols. Purification was performed with an ÄKTA Avant chromatography system with a HIS-Trap HP column (GE Healthcare Life Science). The column was washed with Ni-nitrilotriacetic acid (NTA) buffer (20 mM Tris, 100 mM NaCl, and 10 mM imidazole adjusted to pH 8.0 with NaOH), and the protein was eluted with Ni-NTA buffer containing 250 mM imidazole. The concentrated recombinant FliD protein was dialyzed into a buffer solution (20 mM Tris [pH 8.0], 100 mM NaCl, 10% glycerol). Protein concentration was determined with the Bradford protein assay (Bio-Rad Laboratories) and measurement of absorbance at 280 nm, assuming an extinction coefficient of 25,900 cm−1 · M−1.
FliD polyclonal antiserum preparation.
Mouse anti-FliD sera were generated by subcutaneous immunization of mice with 10 μg of purified recombinant FliD protein emulsified in Freund's complete adjuvant. The animals were given booster injections two times with the same protein concentration in incomplete Freund's adjuvant at intervals of 2 weeks. Nonspecific antibodies were removed from the anti-FliD mouse polyclonal serum by negative affinity purification with a whole-cell lysate of E. coli BL21 (Invitrogen). Briefly, bacteria cultivated overnight in LB broth (100 ml) were centrifuged, washed with phosphate-buffered saline (PBS), and lysed by sonication, and the protein concentration was adjusted to 10 mg/ml of protein solution. Proteins were cross-linked with formaldehyde (37%) and washed with PBS. The anti-FliD polyclonal serum was added to the cross-linked protein pellet and incubated at 4°C for 3 h with constant mixing. The supernatant was transferred to a new tube after a final centrifugation step at 8,000 × g for 10 min.
Reactivity of the antisera was assessed and titers were determined with native FliD protein of E. coli 1711-4 by immunoblotting and enzyme-linked immunosorbent assay.
Immunodetection of FliD in purified native flagellin and mutants strains.
Purified recombinant FliD, native flagellin from aEPEC 1711-4, and extracts of mutants strains (Δeae, ΔfliC, and ΔfliD) were separated by SDS-PAGE (12.5%), transferred to a nitrocellulose membrane, and processed for immunoblot assays that were developed with the SuperSignal detection kit (Pierce) after reaction with diluted anti-FliD specific polyclonal serum samples and a horseradish peroxidase-conjugated rabbit anti-mouse IgG antibody (Sigma) as the secondary detection reagent.
Eukaryotic cell lines and culture conditions.
Human colorectal adenocarcinoma epithelial cells (Caco-2; ATCC HTB37) were cultivated in Dulbecco's minimum Eagle medium (DMEM; GIBCO-BRL) supplemented with 10% inactivated fetal bovine serum (FBS; Gibco-BRL), 1% nonessential amino acids (Gibco-BRL), and 1% penicillin-streptomycin. After 7 days of cultivation, cells were exposed to a trypsin solution (0.25% in EDTA). Caco-2 cells were counted and distributed in six-well plates (Corning Life Sciences) at approximately 105 per well.
Caco-2 monolayer infection experiments.
Postconfluent polarized and differentiated Caco-2 cells were prepared and used as reported earlier (22). Briefly, cells were cultivated in six-well plates for 10 days. Each well was inoculated with 40 μl of a suspension of bacteria grown in LB broth for 18 h at 37°C and diluted 1:100 in PBS (∼107 CFU/ml). Infected cells were incubated for 3 h at 36 ± 1°C in an atmosphere of 5% CO2. Monolayers were washed with PBS to remove nonadherent bacteria and used in quantitative and qualitative assays (determination of the number of cell-adherent bacteria). For the quantitative assays, infected cells were lysed with 1% Triton X-100, plated on LB agar, and incubated at 37°C for 18 h. To further assess the adherence of the MotAB-deficient mutant to Caco-2 cells, a group of plates was submitted to centrifugation for 5 min at 730 × g immediately after inoculation. For the qualitative assays, a group of infected monolayers was fixed with methanol, stained with May-Grünwald-Giemsa, and then mounted on glass coverslips for examination with a light microscope.
Negative staining.
Bacterial suspensions (100 μl) were immediately centrifuged for 3 min at 500 × g, and pellets were washed with 400 μl of PBS, centrifuged again, and resuspended in a 2.0% (vol/vol) formaldehyde–2.5% (vol/vol) glutaraldehyde buffered solution. A drop of each preparation was placed under a Formvar/carbon-coated nickel grid and allowed to dry for 5 min. The preparation was washed with water and stained with saturated uranyl acetate solution. The material was examined at 80 kV with an electron microscope (JEOL 1200 EX II; JEOL, Japan) equipped with a GATAN 791 MultiScan camera (GATAN, Pleasanton, CA) for image acquisition.
Transmission electron microscopy (TEM) of infected Caco-2 cells.
After infected Caco-2 cell monolayers were fixed with a 2.0% (vol/vol) formaldehyde–2.5% (vol/vol) glutaraldehyde buffered solution for 24 h at room temperature, they were rinsed with 0.1 M cacodylate buffer, pH 7.4, and postfixed in 1% (wt/vol) osmium tetroxide. Specimens were then exposed to a graded ethanol series and to propylene oxide. After being embedded in epoxy resin and polymerized at 60°C for 48 h, ultrathin sections were placed on Formvar-coated copper grids and stained with saturated aqueous uranyl acetate and lead citrate. Samples were examined with an electron microscope as described above.
Pre-embedding immunogold labeling for FliD protein for scanning electron microscopy (SEM) and TEM.
Caco-2 cell monolayers were grown on glass coverslips for SEM immunogold labeling (Thermanox 174950; Thermo Scientific) or Aclar film disks for TEM immunogold labeling (Ted Pella 10501-25). For FliD localization on Caco-2 cells, pre-embedding immunogold labeling with 10-nm nanogold conjugate (goat anti-rabbit 10-nm conjugate EMS 25108) was performed. Caco-2 cells were washed with PBS and placed in fresh medium with 0.25 μg/ml of purified FliD protein. The preparations were incubated for 30 min at 36 ± 1°C in an atmosphere of 5% CO2. The cell monolayers were fixed with 4.0% formaldehyde. A blocking buffer containing 1% bovine serum albumin (BSA) was added before incubation at room temperature for 1 h. For some preparations, the primary murine anti-FliD antibody was incubated (1:100 in 0.1 M PBS [pH 7.4], 0.1% BSA) for 30 min at room temperature and then for 18 h at 4°C. Cells were then washed with PBS and incubated with goat anti-rabbit 10-nm conjugate (1:10 in 0.1 M PBS [pH 7.4], 0.1% BSA). Cells were washed with PBS, postfixed with 1% glutaraldehyde, and washed four times with double-distilled water at 5-min intervals. Samples were washed with 0.1 M cacodylate buffer.
For SEM immunogold labeling, samples were washed with 0.1 M PBS, treated for 1 h with 1% (wt/vol) osmium tetroxide in 0.1 M cacodylate buffer, and then washed with 0.1 M cacodylate buffer. Cells were subsequently incubated with 1% aqueous tannic acid for 30 min, washed in distilled water, treated with 1% osmium tetroxide in 0.1 M cacodylate buffer for 30 min, and washed with distilled water. Samples were then dehydrated with a graded ethanol series and then critical point dried with CO2 (Balzers CPD 030; Balzers, Germany). Specimens were mounted on stubs and sputter coated with 25-nm gold particles (Leica EM SCD 500; Leica, Germany). Images were obtained with a Quanta FEG 250 scanning electron microscope (FEI, Austria). For TEM immunogold labeling, preparations were rinsed with 0.1 M cacodylate buffer, pH 7.4, and postfixed in 1% osmium tetroxide. Specimens were then exposed to a graded ethanol series and to propylene oxide for polymerization in epoxy resin. Ultrathin sections were placed on Formvar-coated copper grids and stained with saturated aqueous uranyl acetate and lead citrate. Samples were examined with a 1200EX II transmission electron microscope (JEOL, Japan) at 80 kV.
Inhibition of bacterial adherence to differentiated Caco-2 cells by antibodies or recombinant FliD protein.
Rabbit anti-H40 antiserum (Centers for Disease Control and Prevention) and preimmune rabbit serum were diluted 1:10, 1:20, and 1:50. Mouse polyclonal anti-FliD antibody (this study) or preimmune mouse serum was serially diluted from 1:103 to 1:107. The independently tested concentrations of recombinant FliD or BSA were 0.03, 0.06, 0.12, 0.25, and 0.5 μg/ml. All dilutions were prepared in DMEM with 2% FBS. For assays with mouse or rabbit serum, a total of 40 μl of bacterial culture incubated for 18 h at 37°C (∼107 CFU/ml) in 1 ml of each serum dilution tested was added separately to wells containing Caco-2 cell monolayers.
For assays with recombinant FliD, differentiated Caco-2 cells were treated with different FliD or BSA concentrations and incubated for 30 min at 37°C in an atmosphere of 5% CO2. The preparations were washed with PBS before 1 ml of fresh DMEM with 2% FBS containing 40 μl of an 18-h-old broth culture of strain 1711-4 (∼107 CFU/ml) was added. For all experiments, after 3 h of incubation at 37°C in 5% CO2, the preparations were washed and lysed with 1% Triton X-100. Suspensions were plated on MacConkey agar for determination of the total CFU count in each well. Assays were independently performed three times in triplicate.
To evaluate the ability of FliD to inhibit the adherence of different E. coli pathotypes, a single concentration of FliD (0.25 μg/ml) or BSA (0.25 μg/ml) was tested in Caco-2 cells before incubation with the prototype tEPEC, EHEC, and ETEC strains. In experiments performed with serum samples, bacterial cells were incubated with a single dilution of anti-FliD or preimmune mouse serum (1:1,000) and subsequently transferred to wells containing Caco-2 cells. Assays were independently performed three times in duplicate.
Immunofluorescence assays.
Caco-2 cell monolayers were treated with recombinant FliD or murine anti-FliD serum, fixed with 4% paraformaldehyde for 30 min, and then treated with permeabilization buffer (saponin solution; Sigma). Monolayers were next washed and treated with an anti-mouse secondary antibody conjugated to fluorescein isothiocyanate (FITC, green) before a final washing step. Actin and nuclear DNA were labeled with 4′,6-diamino-2-phenylindole and tetramethyl rhodamine isocyanate (TRITC)-phalloidin, respectively. The slides were examined with a fluorescence microscope.
Statistical analyses.
Values were obtained from the average of three independent experiments performed in triplicate, and statistical analyses were performed with Prism version 5.03 (GraphPad Software Inc.). Analysis of variance and post hoc Bonferroni analysis were performed to evaluate all results.
RESULTS
Lack of the flagellar structure or motility reduces the adherence of aEPEC strain 1711-4 to Caco-2 cells.
The presence and function of flagella in wild-type aEPEC strain 1711-4 and its isogenic mutants deficient in the fliC (17ΔfliC), fliD (17ΔfliD), and motAB (17ΔmotAB) genes, as well as the corresponding complemented mutants, C1P1-8, 17(pETfliD), and 17(pBADmotAB), respectively, were evaluated by negative staining and motility assays in semisolid agar (0.3%). Negative-staining analysis revealed that the 1711-4 and 17ΔmotAB mutant strains produced flagella, while 17ΔfliC and 17ΔfliD did not (Fig. 1A).The 1711-4 wild-type strain and the three complemented mutants were motile, as evidenced by growth on the surface of agar plates (Fig. 1B), whereas the isogenic fliC, fliD, and motAB mutants were nonmotile (Fig. 1C to E). As expected, the 17ΔfliD strain transformed with pET200/D-TOPO, used as a negative control, was nonmotile (Fig. 1F).
FIG 1.
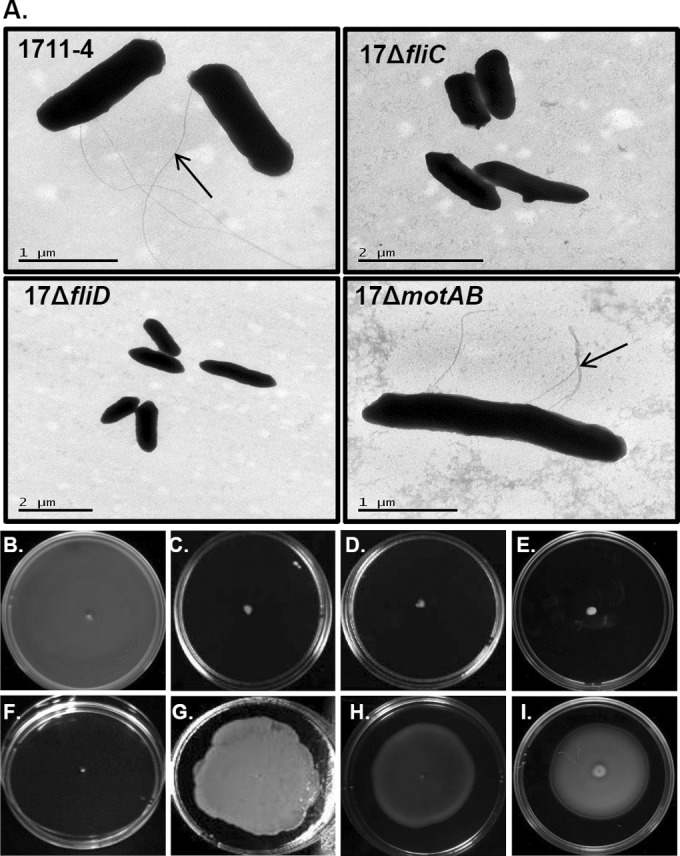
TEM and negative staining of aEPEC 1711-4 cells and isogenic derivatives mutated in the fliC (17ΔfliC), fliD (17ΔfliD), and motAB (17ΔmotAB) genes. The flagellar structure is indicated by black arrows. Note the absence of flagella in mutants deficient in fliC and fliD (A). Also shown are motility tests of the wild-type strain (B); isogenic mutants deficient in fliC (C), fliD (D), or motAB (E); complemented mutants C1P1-8 (G), 17(pETfliD) (H), and 17(pBADmotAB) (I); and the 17ΔfliD mutant transformed with the empty cloning vector (pET200/D-TOPO) (F).
The strain 1711-4 derivatives mutated in fliC, fliD, or motAB and the corresponding complemented strains were tested for the ability to adhere to Caco-2 cells. Examination by light microscopy showed that fewer bacteria adhered to Caco-2 cells infected with the 17ΔfliD, 17ΔfliC, or 17ΔmotAB mutant (Fig. 2A). Complementation of the mutants (for FliD, FliC, or MotAB) restored the ability to adhere to Caco-2 cells (Fig. 2A). Three hours after infection, the expected loose bacterial clusters were observed on monolayers infected with strain 1711-4 or complemented mutants, but a significant decrease in the number of adherent bacteria of all three mutants was observed (Fig. 2A). Indeed, a significant quantitative decrease (P < 0.005) (∼40-fold) in Caco-2 cell adherence was detected for all three deficient mutants (17ΔfliD, 17ΔfliC, and 17ΔmotAB strains) compared to that of the 1711-4 parental strain (Fig. 2B). Of note, centrifugation did not enhance the adherence of the 17ΔfliD, 17ΔfliC, and 17ΔmotAB strains to Caco-2 cells (Fig. 2B).
FIG 2.

Adherence of aEPEC 1711-4 bacteria and the corresponding deleted and complemented derivatives to Caco-2 cells. (A) Bacterial adherence to Caco-2 cells was analyzed by optical microscopy 3 h after infection. Adherence tests were performed with differentiated Caco-2 cells and bacteria with or without a centrifugation step. Bacterial clusters attached to Caco-2 cells (white arrows) were observed in assays performed with wild-type (WT) strain 1711-4 and the complemented C1P1-8, 17(pETfliD), and/or 17(pBADmotAB) derivatives. Few adherent bacteria and no clusters were observed on Caco-2 cells incubated with the 17ΔfliC, 17ΔfliD, and 17motAB mutants. Uninfected Caco-2 cells were used as a control. (B) Error bars indicate standard deviation (SD), and an asterisk indicates a statistically significant difference between aEPEC 1711-4 and each deficient mutant tested (17ΔfliD, 17ΔfliC, or 17ΔmotAB). Note the decreased adherence of the mutant derivatives with regard to the wild-type strain (∼40-fold) both in the presence and in the absence of a centrifugation step. For the complemented mutants, the number of bacteria recovered was higher (∼30-fold) than that of the wild-type strain (P < 0.005). Results are representative of three independent experiments performed in triplicate.
Recombinant FliD inhibits adherence of aEPEC strain 1711-4 to Caco-2 cells.
To further investigate the role of FliD in bacterial adherence to human enterocytes, we tested if purified recombinant FliD would interfere with the adherence of strain 1711-4 to Caco-2 cells by preincubating the cells with different concentrations of purified recombinant FliD. BSA was used as a negative control. Pretreatment of Caco-2 cells with FliD, but not with BSA, decreased bacterial adherence in a dose-dependent manner, with saturation observed at 0.25 μg/ml of purified FliD (Fig. 3A). The number of bacteria recovered in tests performed without previous treatment with FliD (2.5 × 107 CFU) was approximately 100-fold higher than in tests performed with Caco-2 cells preincubated with 0.25 μg/ml of purified FliD (3.5 × 105 CFU) (Fig. 3A). Similar results were observed in stained samples examined by light microscopy (Fig. 3B). These results indicated that purified FliD interfered with the adherence of aEPEC strain 1711-4 to Caco-2 cells.
FIG 3.

Recombinant FliD inhibits adherence of aEPEC strain 1711-4 to differentiated Caco-2 cells. (A) Quantitative plate assays were performed with different FliD or BSA concentrations. Note the binding saturation obtained with FliD at 0.25 μg/ml. The number of CFU detected in cell monolayers treated with 0.5 μg/ml (4.1 × 105 CFU) was similar to that observed with 0.25 μg/ml (3.5 × 105 CFU). The presence of BSA did not interfere with bacterial cell adherence. Error bars indicate standard deviation (SD). Values were obtained from three independent experiments performed in triplicate. (B) Light microscopy of Caco-2 monolayers pretreated with recombinant FliD or left untreated. Note the significant reduction in the adherence of bacteria to Caco-2 cells pretreated with FliD at 0.25 μg/ml. Packed bacterial clusters adhering to the cells were observed on cell monolayers not treated with FliD, in contrast to the few bacteria present on monolayers pretreated with FliD and infected with 1711-4. WT, wild type.
Mouse anti-FliD and rabbit anti-H40 antibodies inhibit adherence of strain 1711-4 to differentiated Caco-2 cells.
To further investigate the role of FliD in the adherence of strain 1711-4 to human enterocytes, we first generated a monospecific anti-FliD antibody by immunizing mice with purified FliD and subsequently removing contaminating nonspecific antibodies reacting with other proteins expressed by the E. coli strain. The specificity of the anti-FliD antibody was evaluated by immunoblotting with whole-cell lysates and culture supernatants of wild-type strain 1711-4 and its isogenic mutants deficient in the eae, fliC, or fliD gene. The anti-FliD polyclonal antibody was highly specific for FliD, and no reaction with the 17ΔfliD strain was detected (Fig. 4A). The FliD protein was detected mainly in whole-cell preparations of both strain 1711-4 and the eae-negative isogenic strain. Only a residual reaction was detected in whole-cell extracts of the 17ΔfliC strain (Fig. 4A). Next, we treated strain 1711-4 with the anti-FliD antibody, as well as with anti-H40 serum prepared with whole flagella before infection of Caco-2 monolayers. The experiments were also performed with preimmune mouse or rabbit serum as a control. Preincubation of bacteria with the antibodies, but not with preimmune serum, resulted in a dose-dependent reduction in bacterial adherence to differentiated Caco-2 cells (Fig. 4B and C). The decrease in the number of bacteria adherent to Caco-2 cells ranged from 400-fold with the anti-FliD antibody at a final dilution of 1/1,000 (from 3.5 × 107 to 8.0 × 104 CFU) (Fig. 4B) to 20-fold with the anti-FliC-H40 antibodies at a final dilution of 1/10 (from 1.2 × 107 to 6.0 × 105 CFU) (Fig. 4C). To confirm the binding of anti-H40 antibodies to bacterial flagella, cells of strain 1711-4 were treated with anti-H40 serum, labeled with protein A-colloidal gold particles, and examined by TEM. As shown in Fig. 4D, flagella of strain 1711-4 were labeled by the anti-H40 antibodies.
FIG 4.

Specificity of anti-FliD antibody and inhibition of Caco-2 cell binding by anti-FliD and anti-H40 antibodies. (A) The specific recognition of recombinant FliD protein by anti-FliD antibody was confirmed by immunoblotting. Note the intense reaction obtained with recombinant FliD protein and purified aEPEC 1711-4 native flagellin (PP), which is absent from the FliD-deficient mutant and drastically reduced in the FliC mutant. (B) Inhibition of adherence of strain 1711-4 to Caco-2 cells after incubation with anti-FliD or preimmune serum. (C) Inhibition of adherence of strain 1711-4 to Caco-2 cells by rabbit anti-FliC-H40 serum. Values were obtained from three independent experiments performed in triplicate. (D) The specificity of the anti-FliC-H40 serum was evaluated by immunostaining. Note the localization of colloidal gold nanoparticles on the flagellar structure (arrow). WT, wild type; SD, standard deviation.
Recombinant FliD binds to microvillus tips of human enterocytes.
In order to determine if FliD mediates aEPEC interaction with microvilli or other specific structures on the Caco-2 cell surface, the recombinant protein was added to Caco-2 cell monolayers and subsequently observed by immunofluorescence. As shown in Fig. 5A to C, cells treated at 0.25 μg/ml with purified FliD, the anti-FliD antibody, and FITC-conjugated anti-mouse IgG were specifically labeled on the cell surface in patches located at the microvillus tips. In the reaction controls, cells incubated only with FliD or the anti-FliD antibody were not labeled. The same experiment was performed with cells treated with FliD, the anti-FliD antibody, and protein A conjugated with gold nanoparticles. As shown by SEM, only cells incubated with FliD and the anti-FliD antibody were labeled with gold nanoparticles (Fig. 5). In addition, the labeling was clearly detected at the microvillus tips (Fig. 5D). Again, cells treated only with FliD or anti-FliD polyclonal antibody were not labeled (Fig. 5E and F). A more detailed analysis was carried out with samples analyzed by TEM. As shown in Fig. 6, gold particles were specifically bound to the microvillus tips of differentiated Caco-2 cells (Fig. 6A). These results indicated that the aEPEC FliD protein specifically recognized a receptor located at microvillus tips of human enterocytes.
FIG 5.

FliD protein adheres to the Caco-2 cell surface. Cell monolayers were treated with the recombinant FliD protein, anti-FliD antibodies, and/or FITC-conjugated anti-mouse IgG (green). Actin and nuclear DNA were labeled with TRITC-phalloidin (red) and 6-diamino-2-phenylindole (blue), respectively. Cells not treated with anti-FliD antibodies (A) or FliD protein (C) showed no labeling. Note the presence of FliD in clusters (green) on the Caco-2 cell surface (arrows) (B). SEM of Caco-2 cells treated with purified FliD, anti-FliD serum, and protein A labeled with gold particles (10 nm). Note the aggregation of gold particles at the tips of microvilli (white arrows) only after the cells were treated with FliD and anti-FliD serum (D). Note the absence of labeling in cell monolayers treated only with FliD (E) or anti-FliD serum (F). DAPI, 4′,6-diamidino-2-phenylindole.
FIG 6.

TEM immunostaining analyses of Caco-2 cells incubated with purified FliD. (A) In the bottom right corner is a higher magnification of a microvillus tip with bound gold particles (arrows). (B) Nanoparticles were not observed in cell monolayers not treated with FliD protein.
Pretreatment with FliD or anti-FliD mouse serum significantly reduces the adhesion of different E. coli pathotypes to Caco-2 cells.
Since FliD is conserved among different E. coli strains, we determined the interference of purified FliD or anti-FliD mouse serum on the adherence of prototype tEPEC, EHEC, and ETEC strains to Caco-2 cells. As shown in Fig. 7, statistically significant differences (P < 0.001) in the adherence of the different E. coli strains tested to Caco-2 cells were observed in assays carried out with cells previously incubated with purified FliD or bacterial cells treated with anti-FliD serum. In experiments performed with Caco-2 cells incubated with purified FliD, the levels of adherence of the E. coli strains tested were 56-fold (aEPEC 1711-4), 15-fold (tEPEC E2348/69), 154-fold (EHEC EDL933), and 90-fold (ETEC H10407) less than the values recorded in experiments carried out with Caco-2 cells previously incubated with BSA. Similarly, in experiments performed with bacterial cells incubated with anti-FliD serum, the reductions in adherence to Caco-2 cells were 269-fold (aEPEC 1711-4), 86-fold (tEPEC E2348/69), 70-fold (EHEC EDL933), and 27-fold (ETEC H10407) less than the values recorded in experiments performed with preimmune mouse serum (Fig. 7).
FIG 7.

FliD-mediated adhesion of different E. coli pathotypes. Values are mean ± standard deviation (SD). Note the significant decrease in the adherence of all of the E. coli strains tested after incubation with Caco-2 cells with purified FliD or after treatment of bacterial cells with anti-FliD serum, compared with that in assays performed with Caco-2 cells pretreated with BSA or preimmune mouse serum (P < 0.001). Values were obtained in three independent experiments performed in duplicate.
DISCUSSION
Flagella belong to the virulence armamentarium of many bacteria. Besides motility, bacterial flagella also contribute to the adhesion and invasion of epithelial cells, biofilm formation, and induction of inflammatory responses (17, 20, 22, 27–31). In this study, we demonstrated for the first time that the FliD protein of O51:H40 aEPEC strain 1711-4 contributes significantly to bacterial adhesion to enterocytes in vitro and behaves as a true adhesin. Our results are in accordance with and extend previous findings reported by our group demonstrating the interaction between the tips of flagella expressed by aEPEC and Caco-2 cell microvillus tips (22). In addition, our results indicate that the FliD-mediated adherence to Caco-2 cells is also present in different E. coli pathotypes, which discloses an important physiological role of flagella that may impact the behavior of these bacterial strains under in vivo conditions.
Our results demonstrate that the adherence of aEPEC to Caco-2 cells also requires functional flagellar machinery, since a mutant defective in the expression of MotAB showed impaired adherence to Caco-2 cells. This impairment was not overcome when infected Caco-2 cell monolayers were submitted to a centrifugation step. The MotA and MotB proteins form the flagella stator channel are located in the bacterial membrane (14). The MotAB-deficient mutant assembles intact but nonfunctional flagella at the bacterial surface. Considering that the external portion of the flagellar filament is composed of FliC and FliD, our findings also indicate that flagellar movement is important for the interaction of FliC or FliD with putative cognate receptors on Caco-2 cells. Flagellar movement probably increases the probability that flagellated bacteria will find and interact with specific receptors on the microvillus tips, but functional active flagella are required for efficient adherence to intestinal cell microvilli. Future experiments will address additional features of FliD-associated adherence to Caco-2 cells.
The involvement of FliD in the adherence of an aEPEC strain to human enterocytes was demonstrated experimentally for the first time in this study. We confirmed that FliD-mediated adherence occurs through the interaction with receptors located at the microvillus tips of Caco-2 cells. Taken together, our results indicate that FliD is the relevant adhesin for aEPEC strains expressing the H40 flagellar type.
Previous studies have addressed the role of flagella in the adherence of pathogenic E. coli to eukaryotic cells but produced conflicting evidence. Girón and colleagues reported that a typical EPEC mutant strain deficient in MotB did not show any significantly impaired adherence to HeLa cells compared to that of the parental strain (19). Nonetheless, HeLa cells, derived from cervical cancer cells infected with papillomavirus, are known not to form microvilli (32), which may affect flagellin-mediated adherence. On the other hand, bacterial flagella have been associated with the process of adherence of several pathogenic bacteria to eukaryotic cells (31, 33–35).
It is noteworthy that the structural organization of the flagellar shaft is similar to that found in different fimbrial adhesins, such as type I fimbriae of uropathogenic E. coli strains, which comprise a main stem and a tip-associated adhesin (FimH) that recognizes numerous host receptors, including uroplakins (36). Like FimH, FliD is located at the tip of the flagellar structure and could, in a similar way, recognize a receptor on enterocytes. Similarly, the CFA/I fimbriae expressed by ETEC strains are composed of a polymeric protein (CfaB), forming the fimbriae shaft, and a tip-localized minor subunit (CfaE) with adhesive properties (37).
Aiming to elucidate the role of FliD as an adhesin, we evaluated the adherence of aEPEC strain 1711-4 to Caco-2 cell monolayers pretreated with different concentrations of purified recombinant FliD protein. We found that recombinant FliD inhibited aEPEC 1711-4 adherence to Caco-2 cells in a dose-dependent manner. Kim et al. demonstrated that purified FliD protein was essential for flagellum assembly, host colonization, and motility in Helicobacter pylori in a mouse infection model (38). Other groups showed that purified FliD protein is essential for adhesion to mucus in an axenic cecal mouse model and participates in the initial process of Pseudomonas aeruginosa adhesion to human tracheobronchial mucins (39). Tasteyre and colleagues showed that radiolabeled cultured cells bound with high affinity to the FliD protein of Clostridium difficile and weakly to intact flagella (28). The same authors demonstrated in a mouse model that the adherence of a nonflagellated C. difficile strain to cecal tissue was 10-fold lower than that of a flagellated strain of the same serogroup (40). The present results suggest that recombinant FliD specifically interacts with receptors expressed on microvillus tips of Caco-2 cells and, similarly to other bacterial species, ascribe an active role to flagella in the adherence of aEPEC strain 1711-4 to human intestinal cells.
Adherence of strain 1711-4 to host cells was also reduced after the incubation of bacterial cells with monospecific anti-FliD serum in a dose-dependent manner. The same result was obtained, but at a much lower dilution, with anti-H40 antiserum. Mahajan and colleagues evaluated the role of FliC as an adhesin in the interaction of EHEC (O157:H7) with bovine intestinal epithelium by using a commercial polyclonal antiflagellin serum (33). Commercial polyclonal antibodies and the polyclonal anti-FliC antibody used in this work were produced with crude flagellin extracts. These extracts contain smaller amounts of FliD in association with FliC, and consequently, the finding that antiflagellin serum inhibited bacterial adherence may therefore be associated with the presence of anti-FliD antibodies.
Since FliD is conserved among flagellated E. coli strains, we evaluated the role of FliD expressed by different E. coli pathotypes in adherence to Caco-2 cells. Experiments performed with Caco-2 cells previously incubated with purified FliD or bacterial cells treated with anti-FliD serum clearly showed that FliD-associated adherence is also present in the E. coli pathotypes tested, including tEPEC, EHEC, and ETEC. Roy et al. (41) demonstrated that the EtpA protein mediates the binding of ETEC flagella to enterocytes and that FliD partially inhibits the interaction of EtpA with FliC, but no similar gene sequence (etpA) could be amplified from the genome of aEPEC 1711-4 (data not shown). The present results suggest that FliD can interact with a receptor(s) present on the microvilli of enterocytes in the absence of EtpA, as demonstrated with aEPEC strain 1711-4. On the other hand, FliD-associated adherence reinforces the concept of bacterial adhesion as a complex and multifactorial event.
ACKNOWLEDGMENTS
This study was supported by grants from the São Paulo Research Foundation (FAPESP 2011/12664-5) to Tânia A. T. Gomes. Suely C. F. Sampaio received felowships from FAPESP (2009/50399-1) and the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq; 150833/2012-1).
We thank André Aguillera and Márcia Tanakai (Centro de Microscopia Eletrônica Universidade Federal de São Paulo, Escola Paulista de Medicina UNIFESP) for assistance with microscopy preparations. We are also grateful to Jean-Marc Ghigo (Unité de Génétique des Biofilms, Department Microbiologie, Institut Pasteur) for providing the KOBEG-Apra plasmid. A. Leyva provided English editing of the manuscript.
Funding Statement
This study was supported by grants from São Paulo Research Foundation (FAPESP 2011/12664-5) to Tânia A. T. Gomes. Suely Sampaio received fellowships from FAPESP (2009/50399-1) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq; 150833/2012-1).
REFERENCES
- 1.Scotland SM, Willshaw GA, Smith HR, Said B, Stokes N, Rowe B. 1993. Virulence properties of Escherichia coli strains belonging to serogroups O26, O55, O111 and O128 isolated in the United Kingdom in 1991 from patients with diarrhoea. Epidemiol Infect 111:429–438. doi: 10.1017/S0950268800057150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scotland SM, Smith HR, Cheasty T, Said B, Willshaw GA, Stokes N, Rowe B. 1996. Use of gene probes and adhesion tests to characterise Escherichia coli belonging to enteropathogenic serogroups isolated in the United Kingdom. J Med Microbiol 44:438–443. doi: 10.1099/00222615-44-6-438. [DOI] [PubMed] [Google Scholar]
- 3.Bokete TN, Whittam TS, Wilson RA, Clausen CR, O'Callahan CM, Moseley SL, Fritsche TR, Tarr PI. 1997. Genetic and phenotypic analysis of Escherichia coli with enteropathogenic characteristics isolated from Seattle children. J Infect Dis 175:1382–1389. doi: 10.1086/516470. [DOI] [PubMed] [Google Scholar]
- 4.Kaper JB. 1996. Defining EPEC. Rev Microbiol 7:130–133. [Google Scholar]
- 5.Hernandes RT, Elias WP, Vieira MA, Gomes TA. 2009. An overview of atypical enteropathogenic Escherichia coli. FEMS Microbiol Lett 297:137–149. doi: 10.1111/j.1574-6968.2009.01664.x. [DOI] [PubMed] [Google Scholar]
- 6.Ochoa TJ, Barletta F, Contreras C, Mercado E. 2008. New insights into the epidemiology of enteropathogenic Escherichia coli infection. Trans R Soc Trop Med Hyg 102:852–856. doi: 10.1016/j.trstmh.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trabulsi LR, Keller R, Gomes TAT. 2002. Typical and atypical enteropathogenic Escherichia coli. Emerg Infect Dis 8:508–513. doi: 10.3201/eid0805.010385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Girón JA, Ho AS, Schoolnik GK. 1991. An inducible bundle-forming pilus of enteropathogenic Escherichia coli. Science 254:710–713. doi: 10.1126/science.1683004. [DOI] [PubMed] [Google Scholar]
- 9.Kaper JB, Nataro JP, Mobley HL. 2004. Pathogenic Escherichia coli. Nat Rev Microbiol 2:123–140. doi: 10.1038/nrmicro818. [DOI] [PubMed] [Google Scholar]
- 10.Wong AR, Pearson JS, Bright MD, Munera D, Robinson KS, Lee SF, Frankel G, Hartland EL. 2011. Enteropathogenic and enterohaemorrhagic Escherichia coli: even more subversive elements. Mol Microbiol 80:1420–1438. doi: 10.1111/j.1365-2958.2011.07661.x. [DOI] [PubMed] [Google Scholar]
- 11.Haiko J, Westerlund-Wikström B. 2013. The role of the bacterial flagellum in adhesion and virulence. Biology (Basel) 2:1242–1267. doi: 10.3390/biology2041242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nougayrède JP, Fernandes PJ, Donnenberg MS. 2003. Adhesion of enteropathogenic Escherichia coli to host cells. Cell Microbiol 5:359–372. doi: 10.1046/j.1462-5822.2003.00281.x. [DOI] [PubMed] [Google Scholar]
- 13.Yonekura K, Maki-Yonekura S, Namba K. 2002. Growth mechanism of the bacterial flagellar filament. Res Microbiol 153:191–197. doi: 10.1016/S0923-2508(02)01308-6. [DOI] [PubMed] [Google Scholar]
- 14.Berg HC. 2003. The rotary motor of bacterial flagella. Annu Rev Biochem 72:19–54. doi: 10.1146/annurev.biochem.72.121801.161737. [DOI] [PubMed] [Google Scholar]
- 15.Chevance FF, Hughes KT. 2008. Coordinating assembly of a bacterial macromolecular machine. Nat Rev Microbiol 6:455–465. doi: 10.1038/nrmicro1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yonekura K, Maki S, Morgan DG, DeRosier DJ, Vonderviszt F, Imada K, Namba K. 2000. The bacterial flagellar cap as the rotary promoter of flagellin self-assembly. Science 290:2148–2152. doi: 10.1126/science.290.5499.2148. [DOI] [PubMed] [Google Scholar]
- 17.O'Toole GA, Kolter R. 1998. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol Microbiol 30:295–304. doi: 10.1046/j.1365-2958.1998.01062.x. [DOI] [PubMed] [Google Scholar]
- 18.Allen-Vercoe E, Woodward MJ. 1999. The role of flagella, but not fimbriae, in the adherence of Salmonella enterica serotype Enteritidis to chick gut explant. J Med Microbiol 48:771–780. doi: 10.1099/00222615-48-8-771. [DOI] [PubMed] [Google Scholar]
- 19.Girón JA, Torres AG, Freer E, Kaper JB. 2002. The flagella of enteropathogenic Escherichia coli mediate adherence to epithelial cells. Mol Microbiol 44:361–379. doi: 10.1046/j.1365-2958.2002.02899.x. [DOI] [PubMed] [Google Scholar]
- 20.Zhou X, Girón JA, Torres AG, Crawford JA, Negrete E, Vogel SN, Kaper JB. 2003. Flagellin of enteropathogenic Escherichia coli stimulates interleukin-8 production in T84 cells. Infect Immun 71:2120–2129. doi: 10.1128/IAI.71.4.2120-2129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luck SN, Badea L, Bennett-Wood V, Robins-Browne R, Hartland EL. 2006. Contribution of FliC to epithelial cell invasion by enterohemorrhagic Escherichia coli O113:H21. Infect Immun 74:6999–7004. doi: 10.1128/IAI.00435-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sampaio SC, Gomes TA, Pichon C, du Merle L, Guadagnini S, Abe CM, Sampaio JL, Le Bouguenec C. 2009. The flagella of an atypical enteropathogenic Escherichia coli strain are required for efficient interaction with and stimulation of interleukin-8 production by enterocytes in vitro. Infect Immun 77:4406–4413. doi: 10.1128/IAI.00177-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gomes TAT, Irino K, Girao DM, Girao VB, Guth BE, Vaz TM, Moreira FC, Chinarelli SH, Vieira MA. 2004. Emerging enteropathogenic Escherichia coli strains? Emerg Infect Dis 10:1851–1855. doi: 10.3201/eid1010.031093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lutz R, Bujard H. 1997. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res 25:1203–1210. doi: 10.1093/nar/25.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Drocourt D, Calmels T, Reynes JP, Baron M, Tiraby G. 1990. Cassettes of the Streptoalloteichus hindustanus ble gene for transformation of lower and higher eukaryotes to phleomycin resistance. Nucleic Acids Res 18:4009. doi: 10.1093/nar/18.13.4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Link AJ, Phillips D, Church GM. 1997. Methods for generating precise deletions and insertions in the genome of wild-type Escherichia coli: application to open reading frame characterization. J Bacteriol 179:6228–6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steiner TS, Nataro JP, Poteet-Smith CE, Smith JA, Guerrant RL. 2000. Enteroaggregative Escherichia coli expresses a novel flagellin that causes IL-8 release from intestinal epithelial cells. J Clin Invest 105:1769–1777. doi: 10.1172/JCI8892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tasteyre A, Barc MC, Collignon A, Boureau H, Karjalainen T. 2001. Role of FliC and FliD flagellar proteins of Clostridium difficile in adherence and gut colonization. Infect Immun 69:7937–7940. doi: 10.1128/IAI.69.12.7937-7940.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kirov SM, Castrisios M, Shaw JG. 2004. Aeromonas flagella (polar and lateral) are enterocyte adhesins that contribute to biofilm formation on surfaces. Infect Immun 72:1939–1945. doi: 10.1128/IAI.72.4.1939-1945.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pichon C, Hechard C, du Merle L, Chaudray C, Bonne I, Guadagnini S, Vandewalle A, Le Bouguenec C. 2009. Uropathogenic Escherichia coli AL511 requires flagellum to enter renal collecting duct cells. Cell Microbiol 11:616–628. doi: 10.1111/j.1462-5822.2008.01278.x. [DOI] [PubMed] [Google Scholar]
- 31.Rogers TJ, Thorpe CM, Paton AW, Paton JC. 2012. Role of lipid rafts and flagellin in invasion of colonic epithelial cells by Shiga-toxigenic Escherichia coli O113:H21. Infect Immun 80:2858–2867. doi: 10.1128/IAI.00336-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adey A, Burton JN, Kitzman JO, Hiatt JB, Lewis AP, Martin BK, Qiu R, Lee C, Shendure J. 2013. The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line. Nature 500:207–211. doi: 10.1038/nature12064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mahajan A, Currie CG, Mackie S, Tree J, McAteer S, McKendrick I, McNeilly TN, Roe A, La Ragione RM, Woodward MJ, Gally DL, Smith DG. 2009. An investigation of the expression and adhesin function of H7 flagella in the interaction of Escherichia coli O157:H7 with bovine intestinal epithelium. Cell Microbiol 11:121–137. doi: 10.1111/j.1462-5822.2008.01244.x. [DOI] [PubMed] [Google Scholar]
- 34.Sampaio SC, Andrade JR, Sampaio JL, Carneiro CR, Freymuller E, Gomes TA. 2011. Distinct interaction of two atypical enteropathogenic Escherichia coli strains with enterocytes in vitro. Open Microbiol J 5:65–71. doi: 10.2174/1874285801105010065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Turroni F, Serafini F, Foroni E, Duranti S, O'Connell Motherway M, Taverniti V, Mangifesta M, Milani C, Viappiani A, Roversi T, Sanchez B, Santoni A, Gioiosa L, Ferrarini A, Delledonne M, Margolles A, Piazza L, Palanza P, Bolchi A, Guglielmetti S, van Sinderen D, Ventura M. 2013. Role of sortase-dependent pili of Bifidobacterium bifidum PRL2010 in modulating bacterium-host interactions. Proc Natl Acad Sci U S A 110:11151–11156. doi: 10.1073/pnas.1303897110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thumbikat P, Berry RE, Zhou G, Billips BK, Yaggie RE, Zaichuk T, Sun TT, Schaeffer AJ, Klumpp DJ. 2009. Bacteria-induced uroplakin signaling mediates bladder response to infection. PLoS Pathog 5:e1000415. doi: 10.1371/journal.ppat.1000415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li YF, Poole S, Nishio K, Jang K, Rasulova F, McVeigh A, Savarino SJ, Xia D, Bullitt E. 2009. Structure of CFA/I fimbriae from enterotoxigenic Escherichia coli. Proc Natl Acad Sci U S A 106:10793–10798. doi: 10.1073/pnas.0812843106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim JS, Chang JH, Chung SI, Yum JS. 1999. Molecular cloning and characterization of the Helicobacter pylori fliD gene, an essential factor in flagellar structure and motility. J Bacteriol 181:6969–6976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arora SK, Ritchings BW, Almira EC, Lory S, Ramphal R. 1998. The Pseudomonas aeruginosa flagellar cap protein, FliD, is responsible for mucin adhesion. Infect Immun 66:1000–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tasteyre A, Karjalainen T, Avesani V, Delmee M, Collignon A, Bourlioux P, Barc MC. 2001. Molecular characterization of fliD gene encoding flagellar cap and its expression among Clostridium difficile isolates from different serogroups. J Clin Microbiol 39:1178–1183. doi: 10.1128/JCM.39.3.1178-1183.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roy K, Hilliard GM, Hamilton DJ, Luo J, Ostmann MM, Fleckenstein JM. 2009. Enterotoxigenic Escherichia coli EtpA mediates adhesion between flagella and host cells. Nature 457:594–598. doi: 10.1038/nature07568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Levine MM, Bergquist EJ, Nalin DR, Waterman DH, Hornick RB, Young CR, Sotman S. 1978. Escherichia coli strains that cause diarrhoea but do not produce heat-labile or heat-stable enterotoxins and are non-invasive. Lancet i:1119–1122. [DOI] [PubMed] [Google Scholar]
- 43.Riley LW, Remis RS, Helgerson SD, McGee HB, Wells JG, Davis BR, Hebert RJ, Olcott ES, Johnson LM, Hargrett NT, Blake PA, Cohen ML. 1983. Hemorrhagic colitis associated with a rare Escherichia coli serotype. N Engl J Med 308:681–685. doi: 10.1056/NEJM198303243081203. [DOI] [PubMed] [Google Scholar]
- 44.Evans DG, Silver RP, Evans DJ Jr, Chase DG, Gorbach SL. 1975. Plasmid-controlled colonization factor associated with virulence in Escherichia coli enterotoxigenic for humans. Infect Immun 12:656–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chaveroche MK, Ghigo JM, d'Enfert C. 2000. A rapid method for efficient gene replacement in the filamentous fungus Aspergillus nidulans. Nucleic Acids Res 28:E97. doi: 10.1093/nar/28.22.e97. [DOI] [PMC free article] [PubMed] [Google Scholar]