

Abstract
Spotted fever group rickettsiae cause potentially life-threatening infections throughout the world. Several members of the Toll-like receptor (TLR) family are involved in host response to rickettsiae, and yet the mechanisms by which these TLRs mediate host immunity remain incompletely understood. In the present study, we found that host susceptibility of MyD88−/− mice to infection with Rickettsia conorii or Rickettsia australis was significantly greater than in wild-type (WT) mice, in association with severely impaired bacterial clearance in vivo. R. australis-infected MyD88−/− mice showed significantly lower expression levels of gamma interferon (IFN-γ), interleukin-6 (IL-6), and IL-1β, accompanied by significantly fewer inflammatory infiltrates of macrophages and neutrophils in infected tissues, than WT mice. The serum levels of IFN-γ, IL-12, IL-6, and granulocyte colony-stimulating factor were significantly reduced, while monocyte chemoattractant protein 1, macrophage inflammatory protein 1α, and RANTES were significantly increased in infected MyD88−/− mice compared to WT mice. Strikingly, R. australis infection was incapable of promoting increased expression of MHC-IIhigh and production of IL-12p40 in MyD88−/− bone marrow-derived dendritic cells (BMDCs) compared to WT BMDCs, although costimulatory molecules were upregulated in both types of BMDCs. Furthermore, the secretion levels of IL-1β by Rickettsia-infected BMDCs and in the sera of infected mice were significantly reduced in MyD88−/− mice compared to WT controls, suggesting that in vitro and in vivo production of IL-1β is MyD88 dependent. Taken together, our results suggest that MyD88 signaling mediates instructive signals in DCs and secretion of IL-1β and type 1 immune cytokines, which may account for the protective inflammatory response during rickettsial infection.
INTRODUCTION
Rickettsiae are obligately intracellular bacteria that cause potentially life-threatening diseases worldwide, with fatality rates as high as 30% if not treated promptly (1, 2). Currently, there is no available U.S. Food and Drug Administration-approved vaccine against rickettsial infections. Our previous studies employing mouse models have identified the critical role of gamma interferon (IFN-γ) and cytotoxic CD8+ T cells in host clearance of rickettsiae in vivo (2). Increased inflammatory responses, such as the serum levels of IFN-γ, tumor necrosis factor alpha (TNF-α), and interleukin-6 (IL-6), have been reported in both human mild and severe rickettsioses (3, 4). However, how rickettsiae initiate these inflammatory immune responses in vivo and the contribution of innate immune elements to host protection still remain elusive.
The initial sensing of infection is mediated by innate pattern recognition receptors (PRRs), which mainly include Toll-like receptors (TLRs) and NOD-like receptors (5). Upon activation by microbes, TLRs transduce signals mostly via an adaptor molecule, MyD88 (6). In addition, activation of TLRs also recruits other adaptor proteins, including TIR domain-containing adaptor-inducing IFN-β (TRIF) and TRIF-related adaptor molecule (TRAM) (7). These MyD88-dependent and -independent pathways activate the transcription factor NF-κB and the induction of proinflammatory cytokines (6–9). MyD88 controls downstream signaling of most TLRs, except for TLR3 and in part TLR4, as well as the IL-1R family (10). Recently, TLR activation by microbial molecules was reported to be essential for upregulation of pro-IL-1β (11). We and others have shown that TLRs are involved in host responses to rickettsial infection. Mice naturally defective in TLR4 signaling (C3H/HeJ) succumb to an inoculum of Rickettsia conorii that is sublethal for TLR4-competent mice (12). TLR2 has been reported to facilitate cell activation in response to Rickettsia akari, the causative agent of rickettsialpox (13). Pretreatment with CpG-B, a TLR9 agonist, protects mice against an ordinarily lethal dose of Rickettsia australis, suggesting that TLR9 is a potential target for vaccine candidates (14). However, the adaptor molecules mediating TLR signaling during rickettsial infection have never been investigated. This problem hampers better understanding of the innate recognition of rickettsiae, which is critical for development of effective therapeutic interventions and vaccine candidates.
Dendritic cells (DCs) are the critical sentinel in antimicrobial immune responses. TLR signaling is one of the most important mediators for DC maturation, which results in priming naive T cells and links the innate and adaptive immune systems (15). Our previous studies have shown that high host susceptibility to R. conorii is associated with an altered DC maturation profile and low levels of IL-12p40 secretion by infected bone marrow-derived DCs (BMDCs), which result in delayed CD4 T cell activation and suppressed Th1 cell polarization (16). Adoptive transfer of rickettsia-stimulated DCs rescues the host from a lethal challenge with R. conorii (17). In the present study, we hypothesized that MyD88 is a critical molecule mediating host-protective inflammation and immunity against fatal murine rickettsioses via signaling TLR activation in innate immune cells such as DCs.
MATERIALS AND METHODS
Rickettsiae and mice.
R. australis (Cutlack strain) and R. conorii (Malish 7 strain) used for animal inoculation were propagated in pathogen-free embryonated chicken eggs (18). For in vitro DC infections, R. australis was cultivated in Vero cells and purified as previously described with modifications (16). Briefly, infected cells were placed on 32, 26, and 20% OptiPrep density gradient medium in a 6× SPG buffer (218 mM sucrose, 3.76 mM KH2PO4, 7.1 mM K2HPO4, 4.9 mM potassium glutamate) bed (Sigma-Aldrich, St. Louis, MO) after sonication. All rickettsial stocks were quantified by plaque assay and stored at −80°C until use. Wild-type (WT) C57BL/6 (B6) mice and TLR2−/− mice were purchased from Jackson Laboratories (Bar Harbor, ME). MyD88−/− mice were kindly provided by Tina Wang at the University of Texas Medical Branch (UTMB) and Shizuo Akira of Osaka University (Japan). For in vivo experiments, mice were inoculated intravenously (i.v.) through the tail vein with R. australis or R. conorii at the doses indicated. After infection, mice were monitored daily for signs of illness until day 20 postinfection (p.i.). To determine the bacterial loads in tissues and immune response in vivo, mice were sacrificed on days 1 and 4 p.i. All the experiments described in this study were performed in a certified biosafety level 3 (BSL3) laboratory at the UTMB. All mice were maintained and manipulated in an animal biosafety level 3 (ABSL3) facility at the UTMB. The UTMB Animal Care and Use Committee approved all experiments and procedures, and experiments in mice were performed according to the guidelines of the Guide for the Care and Use of Laboratory Animals (39).
Quantitative detection of rickettsiae and inflammatory cytokines in infected tissues.
Mouse lung, liver, and spleen samples were collected in RNAlater (Thermo Fisher Scientific, Waltham, MA). Rickettsial loads in the mouse tissues were quantified using quantitative PCR after DNA extraction, as described in our previous study (14, 18). Total RNA was prepared by using a Qiagen RNeasy minikit (Valencia, CA) according to the manufacturer's recommendations. Reverse transcription (RT) was performed using isolated and DNase-treated RNA with an iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). The resulting cDNAs were used as the template for quantitative RT-PCR. The gene expression of individual genes was determined using a SYBR green PCR master mix on a Bio-Rad iCycler IQ. The ΔΔCT method was used as described previously except that we used the β-actin gene as the housekeeping gene (19). Quantification results were expressed as the mRNA relative ratio (2−ΔΔCT) normalized to the amount of β-actin housekeeping gene. Analyzed genes and primers are shown in Table 1 (20–22).
TABLE 1.
Primer sequences used to quantify mRNA of genes of interest
| Gene | Orientationa | Sequence (5′−3′) | Reference(s) |
|---|---|---|---|
| β-Actin | F | GAT TAC TGC TCT GGC TCC TAG C | 20, 21 |
| R | GAC TCA TCG TAC TCC TGC TTG C | ||
| IL-6 | F | CAC AAG TCC GGA GAG GAG AC | 20, 21 |
| R | CAG AAT TGC CAT TGC ACA AC | ||
| IFN-γ | F | GTT ACT GCC ACG GCA CAG TCA TTG | 20 |
| R | ACC ATC CTT TTG CCA GTT CCT CCA G | ||
| IL-10 | F | ACC AAC ATC CTG GTG TCT CC | 22 |
| R | CAT GTC AAA CGT GAG CGA CT | ||
| TNF-α | F | GCA AGC TTC GCT CTT CTG TCT ACT GAA CTT CGG | 20 |
| R | GCT CTA GAA TGA GAT AGC AAA TCG GCT GAC GG | ||
| IL-1β | F | CGC AGC AGC ACA TCA ACA AGA GC | 20, 21 |
| R | TGT CCT CAT CCT GGA AGG TCC ACG |
F, forward; R, reverse.
In vivo systemic cytokine release evaluation.
Serum samples were collected from infected mice on days 1 and 4 p.i. Uninfected mouse sera served as controls. The analysis of 14 cytokines and chemokines was performed on serum samples using a magnetic bead-based multiplex immunoassay (Bio-Plex; Bio-Rad Laboratories, Hercules, CA) according to the manufacturer's instructions. These cytokines and chemokines included IFN-γ, TNF-α, IL-10, IL-4, IL-5, IL-6, IL-12p40, IL-12p70, IL-13, IL-17A, granulocyte colony-stimulating factor (G-CSF), monocyte chemoattractant protein 1 (MCP-1), macrophage inflammatory protein 1α (MIP-1α), and RANTES (regulated on activation, normal T cell expressed and secreted). The plates were read at 450 nm, and the absorbances were transformed to pg/ml using calibration curves prepared with cytokine standards included in the kit.
Histopathological and immunohistochemical analyses.
Formalin-fixed, hematoxylin and eosin (H&E)-stained uninfected and infected tissue sections were evaluated by a pathologist via both low-magnification (×10) and high-magnification (×40) microscopy. Images were taken using an Olympus BX41 photomicroscope (Olympus America, Inc., Center Valley, PA). The frequency and size of the inflammatory infiltrates in the liver were measured by ImageJ as described previously (23). Antigen unmasking was performed by treatment with sodium citrate buffer at pH 6, and tissue sections were stained for neutrophils (Abcam, clone NIMP-R14) and macrophages (Santa Cruz, clone sc-71088). Biotinylated secondary antibodies (Vector Laboratories) were diluted in Dako antibody diluent (Dako, S3022). The tertiary reagents, streptavidin and alkaline phosphatase, were diluted in the same Dako diluent, as were the primary antibodies. 3,3′-Diaminobenzidine (DAB) was used as the chromogen, and hematoxylin was used as the counterstain. All washes were performed with Tris-buffered saline (TBS)–0.1% Tween 20 (Sigma). Sections were dehydrated before the synthetic glass coverslips were mounted with Permount mounting medium. All the sections were photographed with an Olympus DP71 camera (Olympus, Center Valley, PA) attached to an Olympus Ix71 inverted microscope (Olympus, Tokyo, Japan) utilizing a ×20 objective lens. We counted the stained cells using a previously described ImageJ-based method (24).
Generation of BMDCs.
Generation of primary bone marrow-derived dendritic cells (BMDCs) from WT mice and MyD88−/− mice was performed as previously described (16). Briefly, a single-cell suspension from bone marrow was prepared from the mouse femurs and adjusted to 106 cells per 10 ml of complete Iscove modification of Dulbecco modified Eagle medium containing 10% low-endotoxin fetal bovine serum, 1 mM sodium pyruvate, and 50 μM 2-mercaptoethanol. DC culture medium was supplemented with 20 ng/ml recombinant granulocyte-macrophage colony-stimulating factor (GM-CSF; eBioscience, San Diego, CA). At day 3, 6 ml of fresh GM-CSF-containing medium was added, and 10 ml of the culture medium was replaced with fresh GM-CSF-containing medium at day 6. On day 8, cells were analyzed by flow cytometry and used if they contained ≥70% CD11c+ cells.
Flow cytometry.
BMDCs were incubated for 24 h with medium, R. australis, or 500 ng/ml lipopolysaccharide (LPS; Sigma, L4391). Cells were first stained via a Live/Dead fixable staining kit (Life Technologies, Grand Island, NY) for 30 min. Fc receptors were blocked with anti-CD16/32 (Fc Block; BD Biosciences). The cells were then stained with fluorophore-labeled monoclonal antibodies to evaluate the maturation status. Antibodies, including fluorescein isothiocyanate (FITC)-coupled anti-CD80 (clone 16-10A1), and FITC-coupled anti-MHC-II (clone 2G9), were purchased from BD Biosciences (San Jose, CA); the antibodies allophycocyanin (APC)- or peridinin chlorophyll protein (PerCP)-Cy5.5-coupled anti-CD11c (clone N418), APC-conjugated anti-CD40 (clone 1C10), and FITC-coupled anti-CD86 (clone GL1) were purchased from eBioscience (San Diego, CA). The cells were fixed in 1% paraformaldehyde prior to analysis by a FACSCalibur flow cytometer (Becton Dickinson). Data were analyzed using FlowJo software (Tree Star, Inc.).
Cytokine enzyme-linked immunosorbent assay (ELISA).
Supernatants of BMDCs or sera were collected and either filtered or treated with 0.9% sodium azide to eliminate live R. australis. Cytokine concentrations were measured by using an IL-12p40 Quantikine ELISA kit (R&D Systems, Minneapolis, MN) or an IL-1β ELISA kit (eBioscience, San Diego, CA). The absorbance was measured by using a VersaMax microplate reader (Molecular Devices, Sunnyvale, CA). The limits of detection of the ELISA for the cytokine measurements were as follows: IL-1β, 4.0 pg/ml; and IL-12p40, 2.0 pg/ml.
Statistical analyses.
For the comparison of multiple experimental groups, one-way analysis of variance with Bonferroni's procedure was used. A two-group comparison was conducted using either the Student t test or the Welch t test, depending on whether the variance between two groups was significantly different. To determine whether the difference between survival rates of different mouse groups was significant, data were analyzed by the Gehan-Breslow-Wilcoxon test. All statistical analyses were performed using GraphPad Prism software version 5.01. P values of ≤0.05 were the threshold for statistical significance.
RESULTS
MyD88 mediates host clearance of R. conorii in vivo.
To investigate whether MyD88 is involved in host immunity against rickettsiae, we first compared the survival rates of WT and MyD88−/− mice in response to R. conorii infection. In line with the previous studies, WT mice were resistant to different doses of R. conorii (data not shown) (25). In contrast, 80% of the MyD88−/− mice died on day 1 p.i., and 20% of the MyD88−/− mice died on day 6 p.i. after i.v. inoculation with R. conorii at a dose of 5.7 × 105 PFU (Fig. 1A). These results suggest that MyD88−/− mice died of mouse toxicity resulting from a very high dose of R. conorii (26, 27). In response to a lower dose of R. conorii (3 × 105 PFU), all MyD88−/− mice survived (Fig. 1A), indicating dose-dependent mortality of MyD88−/− mice to rickettsial infection. The concentration of Rickettsia in the liver was dramatically greater in MyD88−/− mice than in WT mice on day 2 p.i. (Fig. 1B). To investigate the involvement of TLR2 in the MyD88-dependent host-protective immune response against R. conorii, we inoculated TLR2−/− and WT mice with the same dose of Rickettsia, and we did not find any significant difference in host survival (data not shown).
FIG 1.
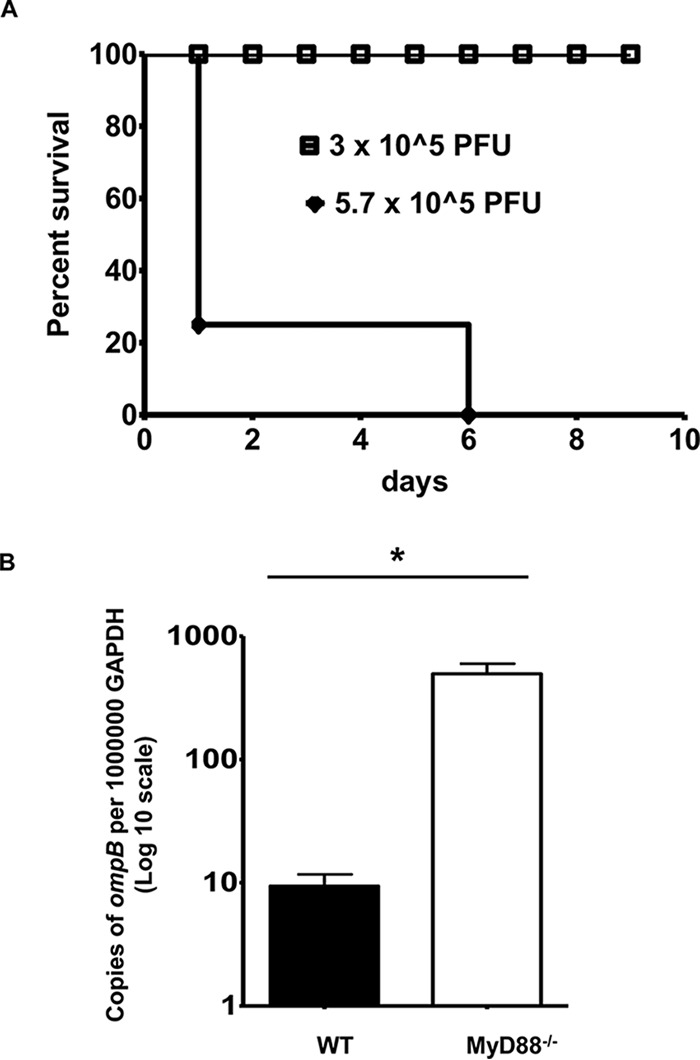
MyD88 mediates host clearance of R. conorii in vivo. (A) MyD88−/− mice were inoculated i.v. with different doses of R. conorii, as indicated. Host survival was monitored daily until day 10 p.i. (B) WT and MyD88−/− mice were injected i.v. with R. conorii at a dose of 3 × 105 PFU/mouse. On day 2 p.i., the mice were sacrificed, and the concentrations of rickettsiae in the liver were quantified by real-time PCR. Data represent means ± the standard deviations (SD) for n = 4 mice for each group. Data represent two independent experiments. *, P < 0.01 for a significant difference between WT and MyD88−/− mice.
MyD88 plays an essential role in host defense against R. australis.
To further determine the contribution of MyD88 to host immunity against rickettsiae, we challenged MyD88−/− mice and WT mice with the same dose of R. australis. R. australis establishes a dose-dependent lethal infection in WT B6 mice (28), which is the murine background on which most of the gene knockout mice have been developed. When challenged with R. australis at a dose of 2.4 × 106 PFU per mouse, all WT mice survived, whereas all MyD88−/− mice died on days 5 and 6 p.i. (Fig. 2A). To determine the growth kinetics of rickettsiae, we inoculated WT and MyD88−/− mice i.v. with R. australis at a dose of 8 × 105 PFU per mouse and measured the bacterial burden in the liver on days 1 and 4 p.i. We observed dramatic and significantly greater rickettsial loads in the livers of MyD88−/− mice than in WT mice on day 4 p.i., but not on day 1 p.i. (Fig. 2B). A significantly greater rickettsial burden was also present in spleen and lung of MyD88−/− mice compared to WT mice on day 4 p.i. (Fig. 2C) (∼3-log higher in each tissue examined). These data illustrate that the decreased host survival of MyD88−/− mice in response to R. australis parallels severely impaired control of bacterial growth in vivo.
FIG 2.
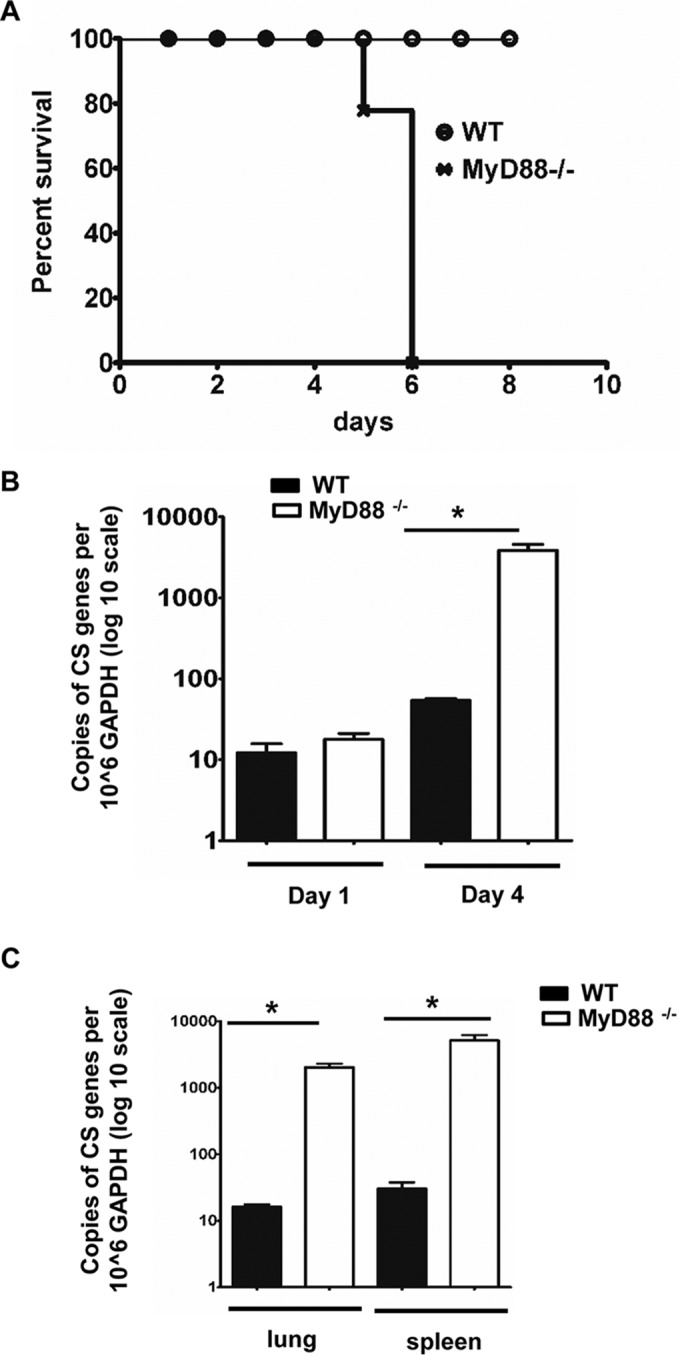
MyD88 is essential for host control of R. australis in vivo. (A) WT and MyD88−/− mice were inoculated i.v. with R. australis at a dose of 2.4 × 106 PFU/mouse. Mice were monitored for survival daily until day 10 p.i. To determine the growth kinetics of R. australis in WT and MyD88−/− mice, the mice were inoculated with 8 × 105 PFU of R. australis i.v. The bacterial burden in liver on days 1 and 4 p.i. (B) and the concentrations of rickettsiae in spleen and lung on day 4 p.i. (C) were determined. CS, citrate synthase; *, P < 0.01 for a significant difference between WT and MyD88−/− mice.
R. australis induces MyD88-dependent and -independent inflammatory cytokines in specific infection sites.
To investigate the cytokines involved in MyD88-mediated host immune protection against rickettsiae, we assayed the proinflammatory and anti-inflammatory cytokines at the transcriptional level by RT-PCR in the specific infection sites during R. australis infection in vivo. On day 1 p.i., the levels of most of the cytokines were not significantly altered except for IFN-γ, IL-6, and IL-1β, which were present at significantly lower levels in the lungs of MyD88−/− mice than in the lungs of WT mice (Fig. 3). These results suggest that i.v. injection of rickettsiae initiated an immediate MyD88-dependent type 1 cytokine response in the lung. On day 4 p.i., the expression levels of IFN-γ (lung, liver, and spleen), TNF-α (lung and spleen), IL-6 (lung and liver), and IL-1β (lung) were all significantly less in MyD88−/− mice than in WT mice, indicating MyD88-dependent production of these inflammatory cytokines in the infection sites. These results suggest that IFN-γ, representing the antimicrobial type 1 immune response, and IL-1β were invoked on day 4 p.i. in a MyD88-dependent manner. Interestingly, the mRNA expression levels of TNF-α and IL-10 in the liver and IL-6 in the spleen were significantly greater in MyD88−/− mice than in WT mice on day 4 p.i. (Fig. 3), suggesting that these cytokines were initiated by a MyD88-independent mechanism or tissue-specific MyD88 signaling. These MyD88-independent cytokines are probably not involved in host protection against rickettsiae. Our previous work has shown the association of IL-10 with the immunosuppression induced by a lethal dose of rickettsial infection (18). IL-6 also mediates the production of T regulatory cells (29). However, it remains unknown whether IL-6 and IL-10 mediate immunosuppression during fatal and severe murine rickettsioses. Taken together, our data suggest that MyD88 mediates host protection against R. australis in vivo mainly through a type 1 immune response.
FIG 3.

In vivo production of MyD88-dependent and -independent inflammatory cytokines upon rickettsial infection. WT and MyD88−/− mice were infected i.v. with R. australis (8 × 105 PFU per mouse). On days 1 and 4 p.i., mice were sacrificed, and tissues were isolated. Total RNA was extracted from these infected tissues. The transcriptional levels of cytokines, including IFN-γ, TNF-α, IL-6, IL-1β, and the anti-inflammatory cytokine IL-10 were determined by RT-PCR as described in Materials and Methods. *, P < 0.05 using an unpaired t test.
R. australis induces circulating MyD88-dependent and -independent cytokines/chemokines.
To further investigate the mechanisms by which MyD88 mediates the protective inflammatory responses upon rickettsial infection in vivo, we measured the production levels of cytokines and chemokines in the sera of WT and MyD88−/− mice infected i.v. with R. australis at a dose of 8 × 105 PFU per mouse. In line with the insignificant changes in bacterial concentration in tissues on day 1 p.i. (Fig. 2B), we did not find any significant difference in serum cytokines/chemokines in WT B6 versus MyD88−/− mice either before infection or on day 1 p.i. (Fig. 4). On day 4 p.i., the levels of IFN-γ, IL-12p40, IL-12p70, IL-6, and G-CSF in serum were significantly lower in MyD88−/− mice than in WT mice (Fig. 4), suggesting that MyD88 signaling mediates a systemic inflammatory response oriented toward a type 1 cytokine profile and that IFN-γ is a critical cytokine mediating the protective immunity against rickettsiae in vivo. Serum levels of TNF-α and IL-10 were not significantly altered in MyD88−/− mice versus WT mice, suggesting that these two cytokines were produced systemically in a MyD88-independent manner. No significant levels of Th2 or Th17 cell cytokines such as IL-4, IL-5, IL-13, and IL-17 were detected in these samples (data not shown), suggesting that a Th1 cell response was dominantly induced. Interestingly, the serum levels of MCP-1, MIP-1α, and RANTES were significantly greater in MyD88−/− mice than in WT mice, suggesting that these chemokines are MyD88 independent or that a deficiency in MyD88 may indirectly promote the production of these chemokines.
FIG 4.

Systemic production of cytokines/chemokines in infected WT and MyD88−/− mice. Mice were infected with R. australis i.v. and then sacrificed on days 1 and 4 p.i. Serum levels of cytokines/chemokines, including IFN-γ, TNF-α, IL-10, IL-4, IL-5, IL-6, IL-12p40, IL-12p70, IL-13, IL-17A, G-CSF, MCP-1, MIP-1α, and RANTES were assessed by Bioplex assay. Cytokines with insignificantly different levels between infected MyD88−/− and WT mice are not shown here, except for IL-10 and TNF-α. The results are means ± the SD of data from 4 to 10 mice per group. *, significant difference in MyD88−/− mice versus WT mice (P < 0.05); n.s., not significant.
MyD88-dependent infiltration of inflammatory cells in infection sites during rickettsial infection in vivo.
We next examined the inflammatory cell recruitment after secretion of inflammatory cytokines/chemokines in lungs and livers of WT and MyD88−/− mice. Histologic analysis of the H&E-stained organ sections showed randomly distributed lobular foci of cellular infiltration consisting of predominantly macrophages, but also lymphocytes and variable numbers of neutrophils in the liver of infected animals (Fig. 5A). The lung sections showed no obviously distinguishing pathological features in infected WT B6 and MyD88−/− mice (see Fig. S1 in the supplemental material). Interestingly, we found a significantly lower frequency and smaller size of inflammatory infiltrations in the livers of MyD88−/− mice than in WT mice on day 4 p.i. with R. australis (Fig. 5A and B).
FIG 5.

MyD88-dependent inflammatory cellular accumulation in vivo. WT and MyD88 −/− mice were infected i.v. with R. australis (8 × 105 PFU per mouse). On day 4 p.i., mice were sacrificed, and tissues were isolated and analyzed. (A) Histology of livers from infected mice under different magnifications (×10 and ×40). Foci of inflammatory infiltration are indicated by arrows. (B) The size and frequency of inflammatory lesions were analyzed using ImageJ (magnification, ×20). (C) Infected tissues were further stained with specific antibodies against F4/80 and NIMP-R14 by immunohistochemistry for the detection of macrophages and neutrophils, respectively. The frequencies of inflammatory cells in different tissues were compared. Fifteen randomly selected microscopic fields from immunohistochemically stained tissues were analyzed for each comparison. These results are representative of two independent experiments (n = 8). *, P < 0.05 using an unpaired t test; n.s., not significant.
To further characterize the inflammatory infiltration mediated by MyD88, we determined the type and frequency of infiltrated inflammatory cells in the liver and lung on day 4 p.i. by immunohistochemical quantification and analysis. Macrophages and neutrophils were stained with individual specific antibodies. In vivo R. australis infection initiated significantly greater infiltration of macrophages in the liver and neutrophils in the lungs of WT B6 mice than in MyD88−/− mice (Fig. 5C). These results suggest that MyD88-dependent expansion or trafficking of macrophages in liver and neutrophils in the lung contributed to the host-protective immune response against R. australis infection.
MyD88 is required for the upregulation of MHC-IIhigh, but not costimulatory molecules, on BMDCs, as well as for IL-12p40 production after rickettsial infection.
To further identify the mechanisms by which MyD88 mediates a protective immune response and the recognition of bacteria by innate PRRs during rickettsial infection, we investigated the maturation of BMDCs of MyD88−/− mice and WT mice upon infection with R. australis. Upon LPS stimulation, the levels of MHC-IIhigh, CD86, CD80, and CD40 in both MyD88−/− and WT mice were greatly enhanced compared to uninfected controls (Fig. 6A). R. australis infection induced full maturation of WT DCs, as evidenced by significantly increased percentages of DCs expressing maturation markers compared to uninfected control cells, including MHC-IIhigh (22.2% versus14.6%), CD86 (19.8% versus 11.9%), CD80 (25.2% versus 18.8%), and CD40 (15.2% versus 8.5%). In contrast, R. australis infection failed to increase the frequency of MyD88−/− DCs expressing MHC-IIhigh (23.9%) compared to uninfected controls (23.6%), although costimulatory molecules in MyD88−/− BMDCs and in WT BMDCs were similarly enhanced upon infection (Fig. 6A). The fold increase in expression levels of MHC-IIhigh on infected WT BMDCs (>1) was significantly higher than those on infected MyD88−/− BMDCs (≤1), suggesting that MHC-IIhigh was only significantly upregulated in WT BMDCs but not MyD88−/− BMDCs upon rickettsial infection (Fig. 6B). No significant difference was observed in the fold increase in expression levels of costimulatory molecules in WT versus MyD88−/− BMDCs (Fig. 6B). These results suggest that R. australis induced the MyD88-independent upregulation of CD86, CD80, and CD40 in BMDCs, while the enhancement of MHC-IIhigh molecules is MyD88 dependent. During maturation, DCs increase their surface expression of MHC-II molecules severalfold (30) and proceed to efficient antigen processing and presentation to T cells. Our results suggest that the full maturation of DCs driven by Rickettsia is MyD88 dependent.
FIG 6.

MyD88-dependent and -independent upregulation of maturation markers on BMDCs after R. australis infection in vitro. BMDCs were isolated from WT and MyD88−/− mice as described in Materials and Methods. The cells were left untreated, were stimulated with LPS (500 ng/ml), or were infected with R. australis at a multiplicity of infection (MOI) of 5 for 24 h. (A) Cells were collected and evaluated for maturation status by expression levels of MHC-II, CD86, CD80, and CD40 on gated CD11c+ cells using flow cytometric analysis. (B) The frequencies of R. australis-infected cells expressing differential maturation markers were calculated for fold increase based on comparison with unstimulated controls. (C) IL-12p40 secretion driven by rickettsial infection was assayed by ELISA and calculated for the fold change based on comparison to uninfected controls. These results are representative of three independent experiments. *, P ≤ 0.05.
IL-12 secreted by DCs is critical and instructive for pathogen-driven Th1 cell polarization (29). Uninfected WT and MyD88−/− BMDCs produced the same levels of IL-12p40 (data not shown). Importantly, no significantly increased secretion of IL-12p40 (fold increase of <1) was detected in MyD88−/− BMDCs upon rickettsial infection, although infected WT BMDCs produced an ∼1.5-fold-greater amount of IL-12p40 than did uninfected controls (Fig. 6C). In addition, the levels of maturation markers on splenic DCs from WT and MyD88−/− mice are consistent with what we have described in vitro (see Fig. S2 in the supplemental material). These results suggest that MyD88 mediates the instructive signals in DCs, which may serve as the key mechanism of host-protective inflammatory response to R. australis.
MyD88-dependent IL-1β production in vitro and in vivo.
IL-1β is a well-documented cytokine mediating the inflammatory response. Recent studies have demonstrated the critical role of inflammasomes in the production of IL-1β, as well as the involvement of MyD88 in the induction of pro-IL-1β (11). To further investigate whether IL-1β is one of the downstream effectors mediated by MyD88 during rickettsial infection, we measured the in vitro and in vivo production of IL-1β in WT and MyD88−/− mice. We did not find a significant level of IL-1β produced by uninfected BMDCs (data not shown). Upon R. australis infection, WT BMDCs produced a significantly higher level of IL-1β than that in MyD88−/− mice (Fig. 7A), suggesting that MyD88 is required for secretion of IL-1β in these infected cells. In line with the in vitro findings, the levels of IL-1β in the sera of infected MyD88−/− mice were significantly lower than in WT mice (Fig. 7B), suggesting that IL-1β production is dependent on MyD88 in vivo. Taken together, our results suggest that inflammasome-derived IL-1β is the critical component of the MyD88-mediated inflammatory response to rickettsial infection, as demonstrated both in vitro and in vivo.
FIG 7.
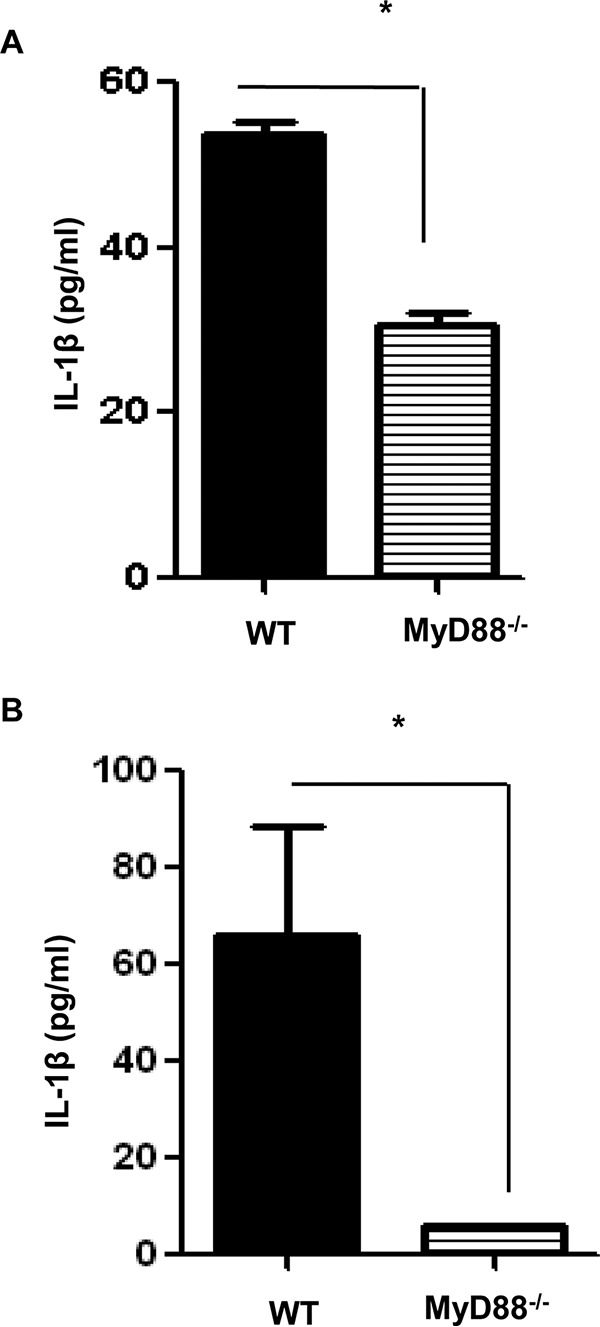
MyD88-dependent secretion of IL-1β during rickettsial infection in vitro and in vivo. (A) BMDCs isolated from WT and MyD88−/− mice were infected with R. australis (MOI = 5) for 24 h. IL-1β secretion by these infected cells was assayed by ELISA. (B) WT and MyD88−/− mice were infected i.v. with R. australis (8 × 105 PFU per mouse). On day 4 p.i., the mice were sacrificed, and the levels of IL-1β in sera were evaluated by ELISA. These results are representative of two independent experiments. *, P < 0.05.
DISCUSSION
Although the available evidence suggests that multiple TLRs, including TLR2, TLR4 and TLR9 (12–14), are involved in host responses to Rickettsia, it is not clear how signals from different TLRs are orchestrated in generating the immune response and how they contribute to host resistance against rickettsiae in vivo. In this study, we demonstrated that MyD88 was essential for host resistance to R. australis as evidenced by significantly greater control of rickettsial replication and enhanced host survival in infected WT mice compared to MyD88−/− mice. Our results also suggest that MyD88, which likely serves as the essential adaptor of key TLRs and priming signal for biologically functional IL-1β secretion, was required for sensing rickettsiae and inducing a protective inflammatory cytokine response, as well as cellular infiltrations, during infection in vivo with this endothelium-targeting intracellular bacterium.
MyD88 signaling plays distinct roles in host responses against several intracellular pathogens closely related to rickettsiae. MyD88-deficient mice infected with Ehrlichia muris exhibit reduced serum and bone marrow concentrations of IFN-γ, which is required for controlling infection, compared to WT mice (31, 32). Interestingly, although the in vivo clearance of Anaplasma phagocytophilum is completely independent of MyD88 (33), MyD88−/− mice have less severe histopathological changes than infected controls (34). In contrast, our results illustrated that MyD88 not only mediated host clearance of rickettsiae in vivo but also contributed to inflammatory cellular infiltrations in vivo during R. australis infection.
DCs mediate pathogen-driven T cell polarization through antigen-specific signal presented by MHC class II molecules, costimulatory molecules, and polarizing cytokines (35). MyD88-dependent or -independent TLRs in DCs play a critical role in sensing microbial pathogens resulting in DC maturation and subsequent T cell priming. Maturation of DCs stimulated by both Brucella abortus and Mycobacterium tuberculosis requires MyD88, as demonstrated by abrogated upregulation of CD80, CD86, and MHC class II, as well as IL-12 production, in MyD88-deficient DCs upon infection (9, 36). MyD88-deficient BMDCs infected with respiratory syncytial virus upregulate costimulatory molecules but do not upregulate class II as efficiently as do WT BMDCs (37), which is very similar to our findings with rickettsial infection. It is known that MyD88-deficient BMDCs do not mature in response to bacterial DNA, the ligand for TLR9, but undergo full maturation upon intracytoplasmic TLR4 activation by LPS (15). As shown in Fig. 6, our results suggest that R. australis activates TLRs other than TLR9, possibly TLR4, to promote MyD88-independent upregulation of costimulatory molecules in DCs. However, R. australis activates TLRs other than intracytoplasmic TLR4 to promote MyD88-dependent upregulation of MHC-IIhigh and IL-12 production in DCs, which are both essential for induction of antigen-specific Th1 cell responses. Although Jordan et al. demonstrated the critical role of TLR4 in mediating host protection against R. conorii in vivo, the maturation profile of DCs from mice carrying the TLR4 point mutation is not known (12). It is an attractive hypothesis that rickettsial antigen, which accounts for MyD88-dependent enhancement of MHC-IIhigh on DCs, can be used as the most potent or essential component of a vaccine candidate for controlling of rickettsial infection via type 1 immune responses. Although future investigations are required to reveal whether MyD88 also functions as an adaptor protein downstream of the IL-1R family during rickettsial infection, our current data at least suggest the contribution of MyD88-dependent TLR signaling to innate recognition of rickettsiae in vivo.
Our study clearly demonstrated the immune mechanisms mediated by MyD88 during in vivo infection with rickettsiae. However, it is worth noting that the production levels of CC chemokines (MCP-1, RANTES, and MIP-1α) in circulation were MyD88 independent. Rickettsia has been shown to induce these CC chemokines in endothelial cells in vitro (38). Given the severity of rickettsioses in MyD88−/− mice as indicated by dramatically greater concentrations of rickettsiae in infected tissues, these CC chemokines were likely produced by infected endothelial cells and may play a role in the late stage of pathogenesis of rickettsial diseases. We propose that significantly increased serum levels of MCP-1, RANTES, and MIP-1α could potentially be used as biomarkers of human rickettsioses. Of note, on day 4 p.i., although the expression of cytokine transcripts, including TNF-α and IL-10 in the liver and IL-6 in the spleen, was significantly upregulated in MyD88−/− mice (Fig. 3), the serum levels of TNF-α, IL-10, and IL-6 either were insignificantly altered or were significantly decreased in MyD88−/− mice (Fig. 4). These results suggest that the in vivo induction of these proinflammatory (TNF-α and IL-6) and anti-inflammatory (IL-10) cytokines by rickettsial infection may involve MyD88-independent or tissue-specific MyD88 signaling.
Overall, our studies revealed that MyD88 is a key molecule in the immune surveillance system mediating the innate recognition of rickettsiae in vivo. These findings suggest that MyD88-mediated signaling pathways could serve as potential targets in vaccine design and therapeutic interventions during this intracellular bacterial infection.
Supplementary Material
ACKNOWLEDGMENTS
We thank Kerry Graves, UTMB, for professional assistance with the immunohistochemical staining.
This study was supported by grants AI021242 and AI101413 from the National Institute of Allergy and Infectious Diseases. J.B. is supported at UTMB by Biodefense Training Grant PT32-AI060549.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01361-15.
REFERENCES
- 1.Edwards MS, Feigin RD. 2004. Rickettsial diseases, p 2497–2515. In Feigin RD, Cherry JD, Demmler GJ, Kaplan SL (ed), Textbook of pediatric infectious diseases, 5th ed WB Saunders Co, New York, NY. [Google Scholar]
- 2.Walker DH. 2007. Rickettsiae and rickettsial infections: the current state of knowledge. Clin Infect Dis 45(Suppl 1):S39–S44. doi: 10.1086/518145. [DOI] [PubMed] [Google Scholar]
- 3.Cillari E, Milano S, D'Agostino P, Arcoleo F, Stassi G, Galluzzo A, Richiusa P, Giordano C, Quartararo P, Colletti P, Gambino G, Mocciaro C, Spinelli A, Vitale G, Mansueto S. 1996. Depression of CD4 T cell subsets and alteration in cytokine profile in boutonneuse fever. J Infect Dis 174:1051–1057. doi: 10.1093/infdis/174.5.1051. [DOI] [PubMed] [Google Scholar]
- 4.Jensenius M, Ueland T, Fournier PE, Brosstad F, Stylianou E, Vene S, Myrvang B, Raoult D, Aukrust P. 2003. Systemic inflammatory responses in African tick-bite fever. J Infect Dis 187:1332–1336. doi: 10.1086/368415. [DOI] [PubMed] [Google Scholar]
- 5.Takeuchi O, Akira S. 2010. Pattern recognition receptors and inflammation. Cell 140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 6.Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, Janeway CA Jr. 1998. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol Cell 2:253–258. doi: 10.1016/S1097-2765(00)80136-7. [DOI] [PubMed] [Google Scholar]
- 7.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. 2003. Role of adaptor TRIF in the MyD88-independent Toll-like receptor signaling pathway. Science 301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 8.Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. 1999. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity 11:115–122. doi: 10.1016/S1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- 9.Schnare M, Barton GM, Holt AC, Takeda K, Akira S, Medzhitov R. 2001. Toll-like receptors control activation of adaptive immune responses. Nat Immunol 2:947–950. doi: 10.1038/ni712. [DOI] [PubMed] [Google Scholar]
- 10.Akira S, Takeda K. 2004. Toll-like receptor signaling. Nat Rev Immunol 4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 11.Franchi L, Muñoz-Planillo R, Núñez G. 2012. Sensing and reacting to microbes through the inflammasomes. Nat Immunol 13:325–332. doi: 10.1038/ni.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jordan JM, Woods ME, Olano J, Walker DH. 2008. The absence of Toll-like receptor 4 signaling in C3H/HeJ mice predisposes them to overwhelming rickettsial infection and decreased protective Th1 responses. Infect Immun 76:3717–3724. doi: 10.1128/IAI.00311-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quevedo-Diaz MA, Song C, Xiong Y, Chen H, Wahl LM, Radulovic S, Medvedev AE. 2010. Involvement of TLR2 and TLR4 in cell responses to Rickettsia akari. J Leukoc Biol 88:675–685. doi: 10.1189/jlb.1009674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xin L, Shelite TR, Gong B, Mendell NL, Soong L, Fang R, Walker DH. 2012. Systemic treatment with CpG-B after sublethal rickettsial infection induces mouse death through indoleamine 2,3-dioxygenase (IDO). PLoS One 7:e34062. doi: 10.1371/journal.pone.0034062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaisho T, Takeuchi O, Kawai T, Hoshino K, Akira S. 2001. Endotoxin-induced maturation of MyD88-deficient dendritic cells. J Immunol 166:5688–5694. doi: 10.4049/jimmunol.166.9.5688. [DOI] [PubMed] [Google Scholar]
- 16.Fang R, Ismail N, Soong L, Popov VL, Whitworth T, Bouyer DH, Walker DH. 2007. Differential interaction of dendritic cells with Rickettsia conorii: impact on host susceptibility to murine spotted fever rickettsiosis. Infect Immun 75:3112–3123. doi: 10.1128/IAI.00007-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jordan JM, Woods ME, Feng HM, Soong L, Walker DH. 2007. Rickettsiae-stimulated dendritic cells mediate protection against lethal rickettsial challenge in an animal model of spotted fever rickettsiosis. J Infect Dis 196:629–638. doi: 10.1086/519686. [DOI] [PubMed] [Google Scholar]
- 18.Fang R, Ismail N, Shelite T, Walker DH. 2009. CD4+ CD25+ Foxp3− T-regulatory cells produce both gamma interferon and interleukin-10 during acute severe murine spotted fever rickettsiosis. Infect Immun 77:3838–3849. doi: 10.1128/IAI.00349-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 20.Wu M, McClellan SA, Barrett RP, Hazlett LD. 2009. Beta-defensin-2 promotes resistance against infection with P. aeruginosa. J Immunol 182:1609–1616. doi: 10.4049/jimmunol.182.3.1609. [DOI] [PubMed] [Google Scholar]
- 21.Deng Q, Sun M, Yang K, Zhu M, Chen K, Yuan J, Wu M, Huang X. 2013. MRP8/14 enhances corneal susceptibility to Pseudomonas aeruginosa infection by amplifying inflammatory responses. Invest Ophthalmol Vis Sci 54:1227–1234. doi: 10.1167/iovs.12-10172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jung YO, Cho ML, Lee SY, Oh HJ, Park JS, Park MK, Park MJ, Ju JH, Kim SI, Park SH, Kim HY, Min JK. 2009. Synergism of Toll-like receptor 2 (TLR2), TLR4, and TLR6 ligation on the production of tumor necrosis factor (TNF)-alpha in a spontaneous arthritis animal model of interleukin (IL)-1 receptor antagonist-deficient mice. Immunol Lett 123:138–143. doi: 10.1016/j.imlet.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 23.Tang XN, Berman AE, Swanson RA, Yenari MA. 2010. Digitally quantifying cerebral hemorrhage using Photoshop and ImageJ. J Neurosci Methods 190:240–243. doi: 10.1016/j.jneumeth.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tuominen VJ, Ruotoistenmäki S, Viitanen A, Jumppanen M, Isola J. 2010. ImmunoRatio: a publicly available web application for quantitative image analysis of estrogen receptor (ER), progesterone receptor (PR), and Ki-67. Breast Cancer Res 12:R56. doi: 10.1186/bcr2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fang R, Ismail N, Walker DH. 2012. Contribution of NK cells to the innate phase of host protection against an intracellular bacterium targeting systemic endothelium. Am J Pathol 181:185–195. doi: 10.1016/j.ajpath.2012.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bell EJ, Pickens EG. 1953. A toxic substance associated with the rickettsias of the spotted fever group. J Immunol 70:461–472. [PubMed] [Google Scholar]
- 27.Feng HM, Walker DH. 2003. Cross-protection between distantly related spotted fever group rickettsiae. Vaccine 21:3901–3905. doi: 10.1016/S0264-410X(03)00301-3. [DOI] [PubMed] [Google Scholar]
- 28.Feng HM, Wen J, Walker DH. 1993. Rickettsia australis infection: a murine model of a highly invasive vasculopathic rickettsiosis. Am J Pathol 142:1471–1482. [PMC free article] [PubMed] [Google Scholar]
- 29.Pasare C, Medzhitov R. 2003. Toll pathway-dependent blockade of CD4+ CD25+ T cell-mediated suppression by dendritic cells. Science 299:1033–1036. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 30.Villadangos JA, Schnorrer P, Wilson NS. 2005. Control of MHC class II antigen presentation in dendritic cells: a balance between creative and destructive forces. Immunol Rev 207:191–205. doi: 10.1111/j.0105-2896.2005.00317.x. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Y, Jones M, McCabe A, Winslow GM, Avram D, MacNamara KC. 2013. MyD88 signaling in CD4 T cells promotes IFN-γ production and hematopoietic progenitor cell expansion in response to intracellular bacterial infection. J Immunol 190:4725–4735. doi: 10.4049/jimmunol.1203024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koh YS, Koo JE, Biswas A, Kobayashi KS. 2010. MyD88-dependent signaling contributes to host defense against ehrlichial infection. PLoS One 5:e11758. doi: 10.1371/journal.pone.0011758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.von Loewenich FD, Scorpio DG, Reischl U, Dumler JS, Bogdan C. 2004. Frontline: control of Anaplasma phagocytophilum, an obligate intracellular pathogen, in the absence of inducible nitric oxide synthase, phagocyte NADPH oxidase, tumor necrosis factor, Toll-like receptor (TLR)2 and TLR4, or the TLR adaptor molecule MyD88. Eur J Immunol 34:1789–1797. doi: 10.1002/eji.200425029. [DOI] [PubMed] [Google Scholar]
- 34.Scorpio DG, von Loewenich FD, Göbel H, Bogdan C, Dumler JS. 2006. Innate immune response to Anaplasma phagocytophilum contributes to hepatic injury. Clin Vaccine Immunol 13:806–809. doi: 10.1128/CVI.00092-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kapsenberg ML. 2003. Dendritic-cell control of pathogen-driven T-cell polarization. Nat Rev Immunol 3:984–993. doi: 10.1038/nri1246. [DOI] [PubMed] [Google Scholar]
- 36.Macedo GC, Magnani DM, Carvalho NB, Bruna-Romero O, Gazzinelli RT, Oliveira SC. 2008. Central role of MyD88-dependent dendritic cell maturation and proinflammatory cytokine production to control Brucella abortus infection. J Immunol 180:1080–1087. doi: 10.4049/jimmunol.180.2.1080. [DOI] [PubMed] [Google Scholar]
- 37.Rudd BD, Schaller MA, Smit JJ, Kunkel SL, Neupane R, Kelley L, Berlin AA, Lukacs NW. 2007. MyD88-mediated instructive signals in dendritic cells regulate pulmonary immune responses during respiratory virus infection. J Immunol 178:5820–5827. doi: 10.4049/jimmunol.178.9.5820. [DOI] [PubMed] [Google Scholar]
- 38.Clifton DR, Rydkina E, Huyck H, Pryhuber G, Freeman RS, Silverman DJ, Sahni SK. 2005. Expression and secretion of chemotactic cytokines IL-8 and MCP-1 by human endothelial cells after Rickettsia rickettsii infection: regulation by nuclear transcription factor NF-κB. Int J Med Microbiol 295:267–278. doi: 10.1016/j.ijmm.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 39.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.