

Abstract
Human enteric viruses are among the main causative agents of shellfish-associated outbreaks. In this study, the kinetics of viral bioaccumulation in live oysters and the heat stabilities of the predominant enteric viruses were determined both in tissue culture and in oyster tissues. A human norovirus (HuNoV) GII.4 strain, HuNoV surrogates (murine norovirus [MNV-1], Tulane virus [TV]), hepatitis A virus (HAV), and human rotavirus (RV) bioaccumulated to high titers within oyster tissues, with different patterns of bioaccumulation for the different viruses. We tested the thermal stability of each virus at 62, 72, and 80°C in culture medium. The viruses can be ranked from the most heat resistant to the least stable as follows: HAV, RV, TV, MNV-1. In addition, we found that oyster tissues provided protection to the viruses during heat treatment. To decipher the mechanism underlying viral inactivation by heat, purified TV was treated at 80°C for increasing time intervals. It was found that the integrity of the viral capsid was disrupted, whereas viral genomic RNA remained intact. Interestingly, heat treatment leading to complete loss of TV infectivity was not sufficient to completely disrupt the receptor binding activity of TV, as determined by the porcine gastric mucin–magnetic bead binding assay. Similarly, HuNoV virus-like particles (VLPs) and a HuNoV GII.4 strain retained some receptor binding ability following heat treatment. Although foodborne viruses have variable heat stability, 80°C for >6 min was sufficient to completely inactivate enteric viruses in oysters, with the exception of HAV.
INTRODUCTION
Approximately 7.6 million to 14.5 million illnesses in the United States are attributed to the consumption of contaminated seafood each year, and enteric viruses are responsible for more than 50% of these cases (1). In a review of the available epidemiological evidence, human norovirus (HuNoV) and hepatitis A virus (HAV) were the leading viruses associated with shellfish, accounting for 83.7% and 12.8% of outbreaks, respectively (2). The type of shellfish most frequently associated with viral outbreaks was oysters, which were the vehicle in 58.4% of outbreaks (2). In some regions, human enteric viruses are practically ubiquitous in harvested shellfish. Keller et al. (3) showed that 100% of shellfish samples collected from Vitória Bay, Espírito Santo, Brazil, were positive for rotavirus (RV) and adenovirus. However, only 80% of the growing water samples were positive for these pathogens. Viral titers were 400 times higher in the shellfish samples than in the growing water, indicating high levels of natural bioaccumulation (3). In the Galician Rias area, the largest shellfish production area in the European Union, 55% of mussel, clam, and cockle samples were contaminated by HuNoV genogroup I (GI) and GII and HAV (4). Thus, understanding of the ecology and persistence of enteric viruses in shellfish is needed to help prevent future outbreaks.
The consumption of uncooked contaminated bivalve shellfish continues to pose a public health risk. These bivalve filter feeders sieve many gallons of water a day through their gills, which can lead to the bioaccumulation of both bacterial and viral pathogens within shellfish tissues. Human enteric viruses have been reported to be among the main causative agents associated with bivalve mollusk outbreaks. HuNoV and HAV account for the majority of these shellfish outbreaks (2, 5–8). Pioneering studies have shown that these enteric viruses persist and can bioaccumulate to high titers within shellfish tissues (9–12). Infectious HAV has been detected in oysters held in HAV-contaminated water for as long as 3 weeks, and viral RNA has been detected in oysters for 6 weeks (13). It has also been shown that acid-stable enteric viruses persist in the hemocytes of Eastern oysters (Crassostrea virginica) (11). Transfer of virus-containing hemocytes to naïve oysters leads to virus detection for as long as 3 weeks (11).
HuNoV causes severe gastroenteritis characterized by vomiting, diarrhea, and stomach cramps (14). In the United States, it is estimated that HuNoV accounts for as much as 60% of foodborne illnesses, and it is the second leading cause of gastroenteritis-related mortality, causing 797 deaths annually (15, 16). It has been a challenge to study HuNoV, because this virus cannot be grown in a cell culture system and lacks a small-animal model. Therefore, cultivable viral surrogates that are closely related to HuNoV, such as murine norovirus (MNV) and Tulane virus (TV), have been used to study the survival of HuNoVs in foods and the environment (17–21). HAV causes gastroenteritis, liver damage, and jaundice. HAV outbreaks have declined in frequency due to improvements in drinking water quality and sanitation practices, as well as the availability of effective vaccines in developed countries. However, HAV remains endemic in developing countries (22). Although shellfish imported into the United States is often cooked, and importation is limited to a few countries, there is still a potential for HAV contamination in imported shellfish.
RVs are the major etiological agent of acute gastroenteritis in infants worldwide and account for 27% of deaths in children, according to the WHO (23). RV is highly infectious among children due to its low infectious dose (<10 particles can lead to disease) and the fact that the virus is shed at a very high titer (1010 to 1012 virus particles/g of stool) (24, 25). All five of these viruses are highly stable in foods, water, and the environment. In addition, all three are transmitted mainly via the fecal-oral route, so the consumption of contaminated food or water often leads to disease.
The current standards used to monitor the safety of shellfish and the quality of growing waters depend on the levels of Escherichia coli or total fecal coliforms (26). Historically, these standards (27–30) have succeeded in reducing many types of shellfish-related bacterial outbreaks; however, it has been shown that the bacterial standards are inadequate for estimating the presence of enteric viruses in shellfish and growing waters. Moreover, the depuration process for shellfish harvested from conditionally approved (or category B) growing areas has been shown to be less effective at eliminating enteric viruses than bacteria from contaminated shellfish (31–33). Therefore, it is necessary to understand the interaction of viruses and shellfish and also to develop efficient methods for inactivating/removing the viruses from this food commodity.
Previous work has evaluated the distribution of bacterial pathogens in shellfish; however, information about the bioaccumulation and distribution of viral pathogens is more limited (34–36). In general, viral localization in shellfish has been shown to differ based on the exposure time and the virus type (37–41). It has also been demonstrated that the type of shellfish and the season affect the rate of viral bioaccumulation (10, 42). Previous work has demonstrated that TV, a primate calicivirus, can bioaccumulate in shellfish and can serve as a potential surrogate mimicking norovirus behavior in oysters (17). However, no study has directly compared the bioaccumulation profiles of the three most prevalent viruses (HuNoV, HAV, and RV) causing shellfish outbreaks.
Thermal treatment is still regarded as one of the most effective means of delaying spoilage and inactivating pathogenic microorganisms. Most thermal inactivation methods for shellfish have been standardized to target bacterial species. However, these inactivation parameters (e.g., time, temperature) are not optimized for the inactivation of foodborne viruses. Therefore, there is an urgent need to identify effective thermal processing parameters for the inactivation of foodborne viruses in shellfish (43). To date, no standard method for study of the thermal stability of enteric viruses has been established (44). Treatment conditions, such as the temperature and holding time, the sample matrix, and the data-modeling method, as well as the type of virus tested, have been found to influence the kinetics of thermal inactivation (44).
Additionally, it has been shown that virus inactivation by thermal processing does not follow a linear inactivation model (first-order kinetics), which assumes a linear logarithmic reduction of the quantity of the treated virus with time (45). A virus thermal inactivation curve usually shows shouldering and tailing at the beginning and end of the survival curve, respectively, limiting the ability to use decimal reduction times (D values) to establish the inactivation parameters. Therefore, alternative models, such as the biphasic reduction model and the Weibull model, are being used to overcome the issues with the nonlinear inactivation curve (46). Although these models are helpful in estimating virus survival during thermal processing, their application requires comparable data on the stabilities of different viruses in order to build the models and appropriately describe the inactivation kinetics (46).
The aim of this study was to obtain comprehensive data on the localization, bioaccumulation, and heat stabilities of the predominant enteric viruses, specifically HuNoV surrogates (MNV-1 and TV), HAV, and RV, in oyster tissues. Comparable data on the thermal stabilities of different enteric viruses will provide insight into the kinetics of inactivation and aid in establishing new standards that are effective against all enteric viruses.
MATERIALS AND METHODS
Cell culture and virus propagation.
MNV-1, a generous gift from Herbert W. Virgin IV, Washington University School of Medicine (47), was propagated in confluent flasks of the murine macrophage cell line RAW 264.7 (ATCC, Manassas, VA) grown in high-glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (Invitrogen), as described by Lou et al. (48). TV was provided by Xi Jiang of the Children's Hospital Medical Center, Cincinnati, OH, and was propagated in monkey kidney cells (MK2-LLC), which were cultured in Opti-MEM supplemented with l-glutamine and 2% FBS as described previously (49). HAV (strain HM-175) was cultivated in fetal rhesus monkey kidney cells (FRhK-4). FRhK-4 cells were grown in Eagle's minimal essential medium (EMEM) containing 10% FBS. Human RV (Wa strain; provided by John Hughes, The Ohio State University Medical Center) was propagated in rhesus monkey kidney cells (MA-104) cultured in EMEM supplemented with 6 μg/ml trypsin.
Viral plaque assays.
MNV-1, TV, HAV, and RV plaque assays were performed in 6-well plates containing RAW 264.7, LLC-MK2, FRhK-4, or MA-104 cells, respectively, as described by Lou et al. (50). A monolayer of 2 × 106 cells/well in 6-well plates (Corning Life Sciences, Wilkes-Barre, PA) was infected with 400 μl of serial 10-fold dilutions of the virus and was incubated at 37°C under a 5% CO2 atmosphere for 1 h for MNV-1, TV, and RV and for 90 min for HAV, with hand agitation every 10 to 15 min. An overlay solution containing 2× EMEM, 1% agarose, 2% FBS, 1% sodium bicarbonate, 0.1 mg of kanamycin/ml, 0.05 mg of gentamicin/ml, 15 mM HEPES (pH 7.7), and 2 mM l-glutamine (Invitrogen) was added to each well for MNV-1, TV, and HAV, and the plates were incubated for 48 h (MNV-1 and TV plates) or for 9 days (HAV plates). For RV, the overlay solution contained 2.5 μl/ml trypsin and no FBS, and plates were incubated for 72 h. Following incubation, the cell monolayer was fixed with 10% formaldehyde and was stained with 0.05% crystal violet to visualize viral plaques. Viral titers were expressed in mean PFU per milliliter ± 1 standard deviation.
Virus bioaccumulation and distribution in oysters.
Live Eastern oysters (Crassostrea virginica) were obtained from a local grocery store (Kroger Inc.) and were cultivated in a tank containing 4 liters of artificial seawater (1.5% sea salt) (Instant Ocean Aquarium Salt; Spectrum Brands, Blacksburg, VA) under aeration conditions at room temperature. Phytoplankton (Phyto Feast; Reef Mariculture Inc., Campbell, CA) was added to the tank to feed the oysters. Prior to the experiments, three oysters from each batch were shucked, homogenized, and examined for the presence of enteric viruses. Five batches of 25 oysters each were grown separately as described above, and the salt water was artificially contaminated with either TV, MNV-1, HAV, or RV at a virus level of 104 PFU/ml or with HuNoV GII.4 at 104 RNA copies/ml. The oysters were held for 72 h in the contaminated tank at room temperature. At 24, 48, and 72 h, three oysters from each tank were aseptically dissected in a biosafety cabinet. The oysters were frozen at −80°C before dissection to limit cross-contamination when individual tissues were harvested. Different portions of the oyster tissue, including the gills, stomach, and adductor muscles, were isolated using autoclaved forceps and were examined for the presence of virus by plaque assays or reverse transcription-quantitative PCR (RT-qPCR).
Plaque assays and virus extraction from contaminated oyster tissues.
TV, MNV-1, HAV, and RV were extracted, and plaque assays were conducted, as described previously (13), with some modifications. In brief, three contaminated oysters were harvested after 24, 48, or 72 h and were shucked, and each targeted tissue (gills, stomach, and adductor muscles) was aseptically separated in a weigh boat. Two grams of each separated tissue was homogenized in 5 ml of Hanks' balanced salt solution (HBSS) using a mortar and pestle, and the homogenized tissues were centrifuged at 2,300 × g for 10 min at 4°C. The supernatant was collected, and 10-fold serial dilutions were made in the suitable medium for each virus. Plaque assays were conducted as described above.
Thermal treatment of viruses in culture medium.
To minimize the effect of the heating vessel on the temperature come-up time, capillary tubes were used to conduct the heat treatment. Eighty microliters of a viral suspension of HAV, RV, MNV-1, or TV in the culture medium was inserted into capillary tubes (diameter, 1.5 to 1.8 mm; length, 100 mm; Kimble Chase), which were then sealed with a vinyl plastic cover (Critoseal; Leica). The sealed tubes were heated in a circulating thermostatically controlled water bath at a treatment temperature of 62, 72, or 80°C for different treatment times (ranging from 2 s to 30 min). The water bath temperature was monitored. The come-up times at each temperature were determined using a thermocouple probe inserted inside a capillary tube filled with culture medium. The come-up time was less than 1 s at all treatment temperatures; therefore, it was ignored in the calculations of the decimal reduction time (D value) or time to first log10 reduction (TFL). Following the heat treatment, the capillary tubes were immediately cooled on ice. The treated viruses were transferred to sterilized Eppendorf tubes, and 50 μl of each treated virus was used for the plaque assay.
Heat inactivation of MNV-1, TV, HAV, and RV in oyster tissues.
Before the experiment, oysters were held in the salt water for 24 h. For the inoculated-oyster experiment, 25 oysters were cultivated in 4 liters of artificially contaminated salt water (1.5% sea salts and phytoplankton) containing 1 × 105 PFU of MNV-1, TV, HAV, or RV/ml for 24 h. The water was aerated and was kept at room temperature. The contaminated oysters were harvested, and three oysters were randomly assigned to each heat treatment duration. Contaminated oysters were treated in an 80°C water bath for periods ranging from 1 to 6 min. The water bath temperature was monitored. The heat-treated oysters were aseptically shucked, and the oyster tissues were homogenized in 5 ml of HBSS. Virus survival was determined by a standard plaque assay. The thermal inactivation kinetics for each virus in oyster tissues was determined, and the TFL for each virus was calculated as described above.
Production of HuNoV GII.4 VLPs.
The capsid VP1 gene of a human NoV GII.4 strain (HS66) was amplified by high-fidelity PCR and was cloned into a pFastBac Dual expression vector (Invitrogen) at SmaI and XhoI sites under the control of the p10 promoter, resulting in the construction of the pFastBac Dual-VP1 expression vector. The correct insertion of the VP1 gene was confirmed by DNA sequencing. Subsequently, the pFastBac Dual-VP1 vector was transformed into DH10Bac competent cells, and the baculovirus expressing VP1 protein was generated by the transfection of bacmids into Spodoptera frugiperda (Sf9) cells (ATCC CRL-1711; ATCC, Manassas, VA) using a Cellfectin transfection kit (Invitrogen) according to the manufacturer's instructions. HuNoV virus-like particles (VLPs) were purified from insect cells as described previously, with minor modifications (51). Briefly, Sf9 cells were infected with the baculovirus at a multiplicity of infection (MOI) of 10, and the infected Sf9 cells and cell culture supernatants were harvested at 6 days postinoculation. The VLPs were purified from cell culture supernatants and cell lysates by ultracentrifugation through a 40% (wt/vol) sucrose cushion, followed by isopycnic CsCl gradient (0.39 g/cm3) ultracentrifugation. Purified VLPs were analyzed by SDS-PAGE, Western blotting, and electron microscopy. The protein concentrations of the VLPs were determined using the Bradford reagent (Sigma Chemical Co., St. Louis, MO).
Preparation of PGM-MBs.
Porcine gastric mucin conjugated to magnetic beads (PGM-MBs) was prepared as described previously (52–54). In short, 1 ml of MagnaBind carboxyl-derivatized beads (Thermo Scientific, Rockford, IL) was washed three times with 1 ml of phosphate-buffered saline (PBS) on a bead attractor (EMD Millipore) to separate the beads. A porcine gastric mucin (PGM) solution was prepared by dissolving 10 mg of type III mucin from porcine stomach (Sigma, St. Louis, MO) in 1 ml of conjugation buffer [0.1 M 2-(N-morpholino)ethanesulfonic acid (MES), 0.9% NaCl (pH 4.7)]. The suspension was shaken well, and 0.1 ml of 10-mg/ml 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) in the same conjugation buffer was added to the type III mucin solution. The mixture was added to the washed beads, which were shaken for 30 min at room temperature with rotation at 8 rpm. After rotation for 30 min, the beads were washed three times with 1 ml of PBS and were resuspended in 1 ml PBS containing 0.05% sodium azide. The beads were stored in the refrigerator until use.
PGM-MBs binding assay.
A porcine gastric mucin—magnetic bead binding assay followed by real-time PCR was used to distinguish between infectious and noninfectious particles of HuNoV. The PGM-MBs binding assay was carried out as described previously (53). Fifty microliters of heat-treated or untreated TV, HuNoV GII.4, or HuNoV GII.4 VLPs was added to 100 μl of PGM-MBs, and the volume was brought up to 1 ml by adding 850 μl of PBS in a low-adhesion 1.5-ml centrifuge tube. The mixture was shaken for 30 min at room temperature. After incubation, the beads were separated from the mixture by the magnetic bead attractor. The beads were washed three times with 1 ml of PBS. The beads were resuspended in 100 μl PBS and were used for RNA extraction for the detection of viruses by RT-qPCR or were tested by SDS-PAGE for the detection of VLPs.
Quantification of viral RNA by real-time RT-PCR.
Total RNAs were extracted from HuNoV GII.4 and TV, either untreated or heat treated at 62, 72, or 80°C for holding times ranging from 2 s to 36 min, using the RNeasy kit (Qiagen). Primers for HuNoV GII.4 and TV were designed to target the VP1 gene of each virus. The first strand of cDNA of the VP1 gene of HuNoV GII.4 or TV was synthesized with SuperScript III reverse transcriptase (Invitrogen) by RT-PCR. Two different primers (5′-TTATAATACACGTCTGCGCCC-3′ and 5′-AATTCCACCTTCAACCCAAGTG-3′) were used to amplify the targeted VP1 genes of HuNoV GII.4 and TV, respectively. The first-strand cDNA of HuNoV GII.4 or TV was quantified by real-time PCR using custom TaqMan primers and probes (for HuNoV, the forward primer was 5′-CACCGCCGGGAAAATCA-3′, the reverse primer was 5′-GCCTTCAGTTGGGAAATTTGG-3′, and the reporter was 5′-ATTTGCAGCAGTCCC-nonfluorescent quencher [NFQ]-3′; for TV, the forward primer was 5′-TTGCAGGAGGGTTTCAAGATG-3′, the reverse primer was 5′-CACGGTTTCATTGTCCCCATA-3′, and the probe was 5′-6-carboxyfluorescein [FAM]-TGATGCACACATGTGGGA-NFQ-3′) on a StepOne real-time PCR machine (Applied Biosystems, Foster City, CA). PCR and cycling parameters were carried out according to the manufacturer's protocol (Invitrogen). Cycling parameters were as follows: a holding stage at 95°C was maintained for 20 s prior to cycling, followed by 50 cycles of 95°C for denaturation, 1 s at 57°C for annealing, and 60°C for 20 s for extension. Standard curves were used to convert threshold cycle (CT) values into log10 RNA copies. The RNA copies are expressed as genomic RNA copies per milliliter. Error bars represent means for three replicates ± 1 standard deviation.
Thermal treatment of HuNoV GII.4 VLPs.
To investigate the effect of heat treatment on the ability of HuNoV to bind to PGM-MBs, 20 μl of VLPs of HuNoV GII.4 was treated either at 80°C for 10 s, 30 s, 60 s, or 5 min or at 100°C for 5 s in a capillary tube sealed with a vinyl plastic cover (Critoseal; Leica), followed by rapid cooling in ice. Treated or untreated VLPs were bound to the beads for 30 min at room temperature. The beads were washed on the bead attractor 3 times with 1 ml of PBS and were resuspended in 10 μl of PBS. Treated and untreated VLPs were mixed (1:4, vol/vol) with SDS-PAGE loading buffer, which consists of 1% SDS, 2.5% β-mercaptoethanol, 6.25 mM Tris-HCl (pH 6.8), and 5% glycerol. The mixture of each sample with the buffer was boiled for 5 min and was loaded onto a 15% polyacrylamide gel. The protein bands were visualized by Coomassie blue staining.
Purification of TV.
TV was purified using a protocol described previously (50). Approximately 180 ml of TV stock (1.5 × 107 PFU/ml) was centrifuged at 82,000 × g through a 40% (wt/vol) sucrose cushion at 4°C for 3 h in a Ty50.2 rotor (Beckman Coulter, Fullerton, CA). The virus pellet was resuspended in 300 μl of TNC buffer (100 mM NaCl, 10 mM Tris, 1 mM CaCl2) on ice overnight. An isopycnic CsCl gradient (1.37 g/ml) was used to purify TV in an SW50.1 rotor (Beckman Coulter) by centrifugation at 115,000 × g and 4°C for 18 h. The TV band was collected (close to a 1.73-g/ml density) and was resuspended in TNC buffer (0.05 M Tris-HCl, 0.15 M NaCl, 15 mM CaCl2 [pH 6.5]). The final TV pellets were resuspended in 300 μl of TNC buffer.
TEM.
Ten microliters of purified TV was heated at 80°C for 5 s, 30 s, 5 min, or 10 min. The samples were then fixed to copper grids (Electron Microscopy Sciences, Inc.) and were subjected to negative staining using uranyl acetate. Virus particles were then visualized by a FEI Tecnai G2 Spirit transmission electron microscope (TEM) at the Microscopy and Imaging Facility at the Ohio State University.
RT-PCR.
RT-PCR and RNase treatment were combined in order to determine whether the viral capsid of TV was degraded by heat at 80°C for 5 s, 30 s, 5 min, or 10 min. Eighty microliters of TV was heated at 80°C for 5 s, 30 s, 5 min, or 10 min in capillary tubes, followed by treatment with 10 μl (0.5 μg/μl) of RNase (Invitrogen) for 30 min at 37°C. Total viral RNA was extracted from heat-treated and untreated TV using the RNeasy minikit (Qiagen, Valencia, CA). Two primers were used to amplify the VP1 gene (5′-TTATAATACACGTCTGCGCCC-3′ and 5′-GCCAGCCATTATCTAAAGA-3′). Bands were visualized using 1% gel electrophoresis.
Statistical analysis.
All experiments were performed in triplicate. Virus titers were expressed as mean log PFU per milliliter ± 1 standard deviation. Statistical analysis was performed by one-way multiple comparisons using SPSS statistical analysis software (version 8.0; SPSS Inc., Chicago, IL). A P value of <0.05 was considered statistically significant.
RESULTS
Virus bioaccumulation and distribution in oysters.
To monitor the uptake and localization of enteric viruses within oyster tissues, three caliciviruses (HuNoV GII.4 and two of its surrogates, MNV-1 and TV), HAV, and RV (Wa strain) were compared. Different portions of oyster tissue, including the gills, stomach, and muscles, were isolated and examined for the presence of virus. The observed accumulation patterns of the three caliciviruses (the HuNoV GII.4 strain, MNV-1, and TV) are presented in Fig. 1.
FIG 1.

Bioaccumulation of caliciviruses in oyster tissues. Shown is the bioaccumulation of MNV-1 (A), TV (B), and HuNoV GII.4 (C) in oyster gills, stomach, and muscles after 24, 48, and 72 h, as determined by plaque assays (A and B) or RT-qPCR (C). Each bar represents the mean for three replicates ± 1 standard deviation. Asterisks indicate statistically significant differences (P < 0.05).
In general, HuNoV GII.4, MNV-1, and TV bioaccumulated and were detected very efficiently in all the oyster tissues throughout the 3-day experimental period. At 24 h, HuNoV GII.4, TV, and MNV-1 had accumulated at a significantly higher level (P < 0.05) in the digestive gland than in the gills and adductor muscles. The viral titers in the digestive gland were 4.3 and 4.6 log10 PFU/g for MNV-1 and TV, respectively, and 5.0 log10 RNA copies/g for HuNoV. In the gills and muscles, the titers of MNV-1 and TV ranged between 3 and 4.5 log10 PFU/g, and those of HuNoV were 4.3 and 3.8 log10 RNA copies/g. At the 48- and 72-h time points, HuNoV GII.4, TV, and MNV-1 were distributed equally in the oyster tissues, with no significant differences in the levels of the viruses between the different oyster tissues (P > 0.05).
The results for HAV bioaccumulation in oysters are presented in Fig. 2A. HAV was detected at high titers in all oyster tissues after 24 h; the highest level was detected in the digestive gland. The virus titer in the stomach reached 4.7 log10 PFU/g, compared to 3.7 and 3.6 log10 PFU/g in gills and muscles, respectively. After 48 and 72 h, HAV was detected at similar levels in all oyster tissues, with no significant differences in the HAV titer between different oyster tissues (P > 0.05).
FIG 2.

Bioaccumulation of HAV and RV in oyster tissues. Shown is the bioaccumulation of HAV (A) and RV (B) in oyster gills, stomach, and muscles after 24, 48, and 72 h, as determined by plaque assays. Each bar represents the mean for three replicates ± 1 standard deviation. The asterisk indicates a statistically significant difference (P < 0.05).
RV (Fig. 2B) accumulated at lower titers than TV, MNV-1, HuNoV GII.4, and HAV and showed a different distribution pattern. After 24 h, RV was detected in the gills at a level of 3 log10 PFU/g and in the muscles at a titer of 2.3 log10 PFU/g. At the 24-h time point, no RV was detected in the digestive gland. At 48 h, RV was detected in the digestive gland; however, the titer was less than 2 log10 PFU/g. After 72 h, the levels of RV detected in the gills and muscles were 2.5 and 3.5 log10 PFU/g, respectively, and the level of RV detected in the stomach was 1.7 log10 PFU/g.
Thermal inactivation of virus in cell culture medium.
To provide more-precise data about the thermal stability of enteric viruses, the thermal inactivation of HAV, RV, TV, and MNV-1 was systematically investigated at 62, 72, and 80°C in culture medium. The concentration of surviving virus, expressed as log10 PFU per milliliter, was plotted against the treatment time at each temperature, as shown in Fig. 3.
FIG 3.

Effects of heat treatment at 62, 72, and 80°C on MNV-1, TV, HAV, and RV in culture medium. The effects of heat treatment at 62°C (A), 72°C (B), and 80°C (C) on the four viruses were determined by plaque assays. Each data point represents the mean for three replicates; error bars, ±1 standard deviation.
The log linear, Weibull distribution, and biphasic models were compared in order to describe survival curve kinetics (46, 55, 56). The regression coefficients (R2) and root mean square error (RMSE) values were used to evaluate model fitness. The D values or the TFL value was calculated from the best-fit model.
The data on inactivation at 62°C are presented in Table 1. The log linear model showed the worst fit of the experimental data, with the lowest R and the highest RMSE. Following mild heat treatment, the viral inactivation curve showed two reduction phases with two different slopes (the first for the most heat sensitive quasispecies and the second for the more resistant quasispecies). The biphasic reduction model was used to calculate two different D-like values, one for each distinct reduction phase in the inactivation curve. In our results, D value 1 is reported for the heat-sensitive quasispecies and D value 2 is reported for the more-resistant quasispecies. At this temperature, the D-like values, as calculated from the biphasic model, for the low-resistance and highly resistant subsets (quasispecies) of each the four viruses were 0.13 and 7.58 min for HAV, 0.36 and 2.81 min for RV, 0.48 and 2.07 min for TV, and 0.55 and 1.81 min for MNV-1. Since the biphasic model had a higher R2 and a lower RMSE value than the log linear and Weibull distribution models (Table 2), it can be concluded that the biphasic model showed the best fit to the experimental data for the four viruses at this treatment temperature.
TABLE 1.
D values and TFL for HAV, RV, TV, and MNV-1 after heat treatment at 62°C in culture medium
| Virus | Value (min) for the following model: |
|||
|---|---|---|---|---|
| Log linear (D value) | Weibull (TFL) | Biphasic reduction |
||
| D value 1 | D value 2 | |||
| HAV | 5.46 | 0.09 | 0.13 | 7.58 |
| RV | 5.43 | 0.25 | 0.36 | 2.81 |
| TV | 2.38 | 0.49 | 0.48 | 2.07 |
| MNV-1 | 2.17 | 1.53 | 0.55 | 1.81 |
TABLE 2.
Comparison of the log linear, Weibull distribution, and biphasic models for best fit to the survival curves of TV, MNV-1, HAV, and RV at 62, 72, and 80°C in culture medium
| Temp and virus | Log linear model |
Weibull model |
Biphasic modela |
||||
|---|---|---|---|---|---|---|---|
| RMSE | R2 | RMSE | R2 | P | RMSE | R2 | |
| 62°C | |||||||
| HAV | 0.74 | 0.583 | 0.18 | 0.980 | 0.21 | 0.09 | 0.996 |
| RV | 0.96 | 0.459 | 0.43 | 0.910 | 0.23 | 0.17 | 0.988 |
| TV | 0.87 | 0.844 | 0.42 | 0.969 | 0.44 | 0.38 | 0.980 |
| MNV-1 | 0.92 | 0.851 | 0.65 | 0.939 | 0.59 | 0.38 | 0.983 |
| 72°C | |||||||
| HAV | 0.91 | 0.734 | 0.54 | 0.923 | 0.51 | 0.26 | 0.986 |
| RV | 0.65 | 0.898 | 0.22 | 0.987 | 0.50 | 0.05 | 0.999 |
| TV | 0.90 | 0.861 | 0.62 | 0.943 | 0.58 | 0.46 | 0.979 |
| MNV-1 | 1.14 | 0.791 | 0.64 | 0.951 | 0.39 | 0.25 | 0.995 |
| 80°C | |||||||
| HAV | 0.95 | 0.865 | 0.83 | 0.910 | 0.57 | 0.81 | 0.945 |
| RV | 0.72 | 0.925 | 0.634 | 0.958 | 0.67 | 0.68 | 0.960 |
| TV | 0.85 | 0.892 | 0.78 | 0.937 | 0.59 | ND | ND |
| MNV-1 | 0.60 | 0.964 | 0 | 1 | 0.52 | ND | ND |
ND, not determined.
By increasing the treatment temperature to 72°C, the inactivation curve gained more linearity and the tailing effect became less apparent, as shown by the increase in the P value in the Weibull model (Table 2). The biphasic model showed the best curve fitness. However, the log linear regression had the lowest R2 and the highest RMSE values among the three models used. The D values at 72°C for the low-resistance and highly resistant quasispecies of the four viruses were calculated from the biphasic model and were 1.26 and 13.85 s for HAV, 1.87 and 11.83 s for RV, 1.46 and 9.73 s for TV, and 1.13 and 7.29 s for MNV-1 (Table 3).
TABLE 3.
Comparison of the log linear, Weibull distribution, and biphasic models for best fit to the survival curves of TV, MNV-1, HAV, and RV at 72°C in culture medium
| Virus | Value (s) with the following model: |
|||
|---|---|---|---|---|
| Log linear (D value) | Weibull (TFL) | Biphasic |
||
| D value 1 | D value 2 | |||
| HAV | 7.57 | 3.27 | 1.26 | 13.85 |
| RV | 5.3 | 3.11 | 1.87 | 11.83 |
| TV | 4.58 | 2.71 | 1.46 | 9.73 |
| MNV-1 | 4.63 | 0.72 | 1.13 | 7.29 |
At 80°C, application of the biphasic model was not suitable for MNV-1 and TV, because it required more data points. The results showed that the Weibull distribution model was more appropriate for modeling virus inactivation than the log linear model. In general, within 12 s, all of the viruses were completely inactivated at 80°C. D values, as calculated from the Weibull model, were 0.46, 0.72, 0.32, and 0.32 s for HAV, RV, TV, and MNV-1, respectively (Table 4). Based on the D values or TFL values for the four viruses, they can be ranked as follows, from the most heat resistant to the least stable: HAV, RV, TV, MNV-1.
TABLE 4.
Comparison of the log linear, Weibull distribution, and biphasic models for best fit to the survival curves of TV, MNV-1, HAV, and RV at 80°C in culture medium
| Virus | Value (s) with the following model: |
|||
|---|---|---|---|---|
| Log linear (D value) | Weibull (D value) | Biphasica |
||
| TFL | D value | |||
| HAV | 0.89 | 0.46 | 0.44 | 1.79 |
| RV | 0.87 | 0.72 | 0.39 | 1.53 |
| TV | 0.84 | 0.32 | ND | ND |
| MNV-1 | 0.39 | 0.32 | ND | ND |
ND, not determined.
Thermal inactivation of viruses within oyster tissues.
The survival of HAV, RV, TV, and MNV-1 in oyster tissues is presented in Table 5. The starting titer of each virus in oysters was approximately 1 × 104 PFU/g of oyster tissue. The Weibull model showed the best fit for virus inactivation in oyster tissues. The TFL values for the viruses tested were calculated from the Weibull model and are presented in Table 5. TV, MNV-1, and RV showed declines in titers commensurate with increases in treatment time. TV and MNV-1 titers were at undetectable levels (a 4.8-log10 reduction for TV and a 4.7-log10 reduction for MNV-1) in the oysters following treatment for 3 min at 80°C. A holding time of 4 min at 80°C was required to eliminate 3.1 log units of RV (below the detection limit). However, no significant decrease in the HAV titer was observed even after 6 min of treatment at 80°C. The highest TFL value was that for HAV, and the lowest was that for MNV-1. Based on the TFL, the four viruses can be ranked from the most to the least heat stable as follows: HAV, RV, TV, MNV-1.
TABLE 5.
TFL values for HAV, RV, TV, and MNV-1 at 80°C in oyster tissues
| Virus | TFL (min) | R2 | RMSE |
|---|---|---|---|
| MNV-1 | 0.61 ± 0.47 | 0.946 | 0.66 |
| RV | 2.99 ± 0.38 | 0.975 | 0.24 |
| TV | 1.46 ± 0.26 | 0.989 | 0.27 |
| HAV | 19.99 ± 1.0 | 0.73 | 0.14 |
Effect of thermal treatment on HuNoV GII.4 VLPs.
To evaluate the PGM-MBs binding assay for the survival of HuNoV treated by heat, we first used HuNoV VLPs as a model, because VLPs possess the same authentic receptor binding activity as native virions. For this purpose, VLPs of HuNoV GII.4 suspended in TNC buffer were treated either at 80°C, for 10 s, 30 s, 60 s, or 5 min, or at 100°C for 5 s. Treated and untreated VLPs were incubated with PGM-MBs for 30 min at room temperature. The beads were washed 3 times with PBS and were resuspended in 10 μl of PBS. The beads and the bound VLPs were loaded onto a 15% SDS-PAGE gel and were visualized by Coomassie blue staining. The protein density of VLPs bound to the beads after treatment at 80°C for 10, 30, or 60 s was similar to that of untreated VLPs (Fig. 4A and D). After heat treatment at 80°C for 5 min, the protein density of the VLPs binding to the PGM-MBs was slightly reduced from that of the original input VLPs (Fig. 4B and D). The VLPs completely lost their ability to bind to the PGM-MBs after treatment at 100°C for 5 s (Fig. 4D).
FIG 4.

Effects of heat treatment at 80 and 100°C on the receptor binding abilities of HuNoV VLPs. Heat-treated or untreated VLPs were either loaded directly onto the gel (Input) or were first subjected to the PGM-MBs binding assay. Beads and bound VLPs (Bound) were then loaded directly onto the gel, as was the portion of the heat-treated or untreated VLPs that did not bind to the PGM-MBs (Unbound). VP1, human NoV VP1 protein; cVP1, the cleaved form of VP1. (A) Purified VLPs treated at 80°C for 10 or 30 s. (B) VLPs treated at 80°C for 1 or 5 min. (C) Purified VLPs treated at 100°C for 5 s. (D) VLP binding to PGM-MBs after heat treatment, expressed as a percentage of the level of binding of the untreated control.
The data indicate that thermal treatment conditions that led to reductions in the levels of HuNoV surrogates (TV and MNV-1) below the detection limit were not sufficient to inactivate the receptor binding activity of HuNoV VLPs. However, a higher temperature (100°C for 5 s) or a longer holding time (80°C for 5 min) did damage VLP receptor binding activity.
Effects of heat treatment on HuNoV GII.4 and TV.
The result presented above showed that HuNoV VLPs were much more heat stable than HuNoV surrogates (TV and MNV-1). One possibility is that the VLPs lacked the genome component of the native virus and therefore were more difficult to inactivate than the complete viruses, which are composed of the capsid and genomic RNA. The results for TV are shown in Fig. 5. At 62°C (Fig. 5A), 72°C (Fig. 5B), and 80°C (Fig. 5C), the reduction in the number of RNA copies of TV was less than 1 log10 RNA copies/ml.
FIG 5.

Heat inactivation of TV at 62, 72, and 80°C, as determined by the PGM-MBs binding assay followed by RT-qPCR. TV was heated at 62°C (A), 72°C (B), or 80°C (C). The receptor binding ability of the heat-treated or untreated virus was determined by the PGM-MBs binding assay, and the RNA from the viral particles retaining receptor binding ability was quantified using RT-qPCR. Each data point represents the mean for three replicates ± 1 standard deviation.
No change in the number of RNA copies was observed for TV after heat treatment at 62°C, while treatment at 72°C for 1 min resulted in a 0.7-log10 reduction in the number of TV RNA copies. Increasing the temperature to 80°C (Fig. 5C) resulted in a 2.5-log reduction in the number of TV RNA copies after 10 s.
For HuNoV GII.4, heat treatment at 62°C (Fig. 6A) resulted in a 0.7-log10 reduction in the number of viral RNA copies, while treatment at 80°C led to a ∼1-log reduction (Fig. 6C). Comparison of the results of the plaque assay and the PGM-MBs binding assay for TV led to the conclusion that the PGM-MBs binding assay overestimated the survival of TV and, by inference, may not provide a precise evaluation of the survival of HuNoV after heat treatment.
FIG 6.

Heat inactivation of HuNoV at 62, 72, and 80°C, as determined using the PGM-MBs binding assay followed by RT-qPCR. HuNoV was heated at 62°C (A), 72°C (B), and 80°C (C). The receptor binding ability of the heat-treated or untreated virus was determined by the PGM-MBs binding assay, and the RNA from the viral particles retaining receptor binding ability was quantified using RT-qPCR. Each data point represents the mean for three replicates ± 1 standard deviation.
Mechanism of TV inactivation by heat.
The fact that TV RNA levels were not significantly reduced when TV was subjected to a lethal heat treatment (80°C for 10 s) suggests that heat treatment did not physically damage viral genomic RNA. Since real-time RT-PCR detects only a small portion of the genomic RNA, it remains possible that other portions of the genomic RNA were degraded following a lethal dose of heat. To rule out this possibility, the full-length VP1 gene was amplified by one-step RT-PCR. As shown in Fig. 7, there was no change in the abundance of the VP1 gene even after 5 min of treatment at 80°C, indicating that the heat treatment did not degrade the viral RNA.
FIG 7.
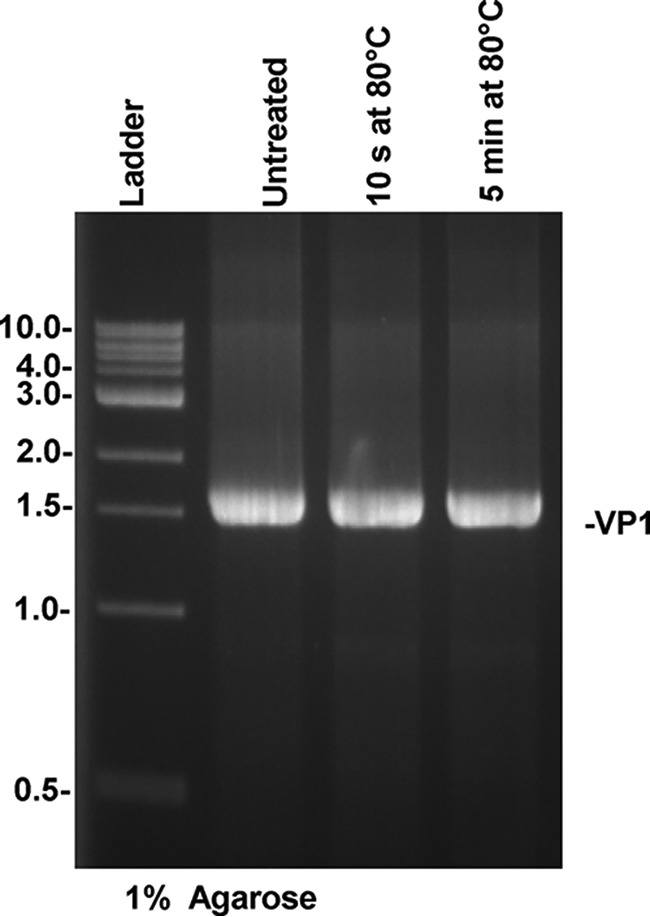
Effect of heat treatment at 80°C on the capsid gene (VP1) of TV. TV was treated at 80°C for 10 s or 5 min. The VP1 gene was amplified by one-step RT-PCR, and the DNA band was visualized by 1% agarose gel electrophoresis.
To investigate the effect of heat treatment on the integrity of the viral capsid, purified TV particles suspended in TNC buffer were heated at 80°C for 5 s, 10 s, or 5 min. The treated and untreated particles were negatively stained by 1% uranyl acetate and were visualized by TEM. After the shortest treatment time (5 s), there was a clear change in virion appearance from that of the untreated virus, including a loss of the normal round structure, with rough edges observed on the particle (Fig. 8). More-severe damage was observed following 10-s and 5-min treatment times (Fig. 8). These results indicate that the TV capsid lost its integrity following 5 s of thermal treatment at 80°C.
FIG 8.

Damage to TV particles after heat treatment at 80°C for 5 s, 10 s, or 5 min. TV was treated at 80°C for 5 s, 10 s, or 5 min, and damage to viral particles was visualized by electron microscopy.
DISCUSSION
In this study, we explored the localization, bioaccumulation patterns, and heat inactivation parameters of enteric viruses frequently associated with shellfish. Data available for the heat inactivation of enteric viruses differ among research studies due to differences in thermal treatment conditions, such as the temperature and holding time, as well as differences in the sample matrix, data modeling, and virus type (44). Therefore, comprehensive studies investigating the natural bioaccumulation patterns of the viruses in shellfish and the thermal inactivation of viruses in shellfish tissues are useful for industry and for regulatory purposes, to improve seafood safety.
Virus bioaccumulation and distribution in oysters.
This study aimed to directly assess the bioaccumulation of the most important enteric viruses that are frequently associated with shellfish outbreaks, including HuNoV, HAV, and RV. Our results demonstrate that all enteric viruses can bioaccumulate efficiently in oysters, to high titers, from the growing water and can be detected in all oyster tissues within 72 h.
Although all the viruses examined were detected in different oyster tissues, the pattern of viral localization differed based on the exposure time and the virus. For instance, TV, MNV-1, and HAV were detected in the digestive gland at high titers early after viral exposure (24 h), while RV was more likely to be detected in the gills and muscles. Specific and nonspecific binding may be responsible for the localization of the virus within the oyster tissues. Both HuNoV and TV recognize histo-blood group antigens (HBGAs) as a cellular receptor or coreceptor (57–59). Recently, both HuNoV and TV have also been shown to bind sialic acid (60, 61). Maalouf et al. (9) showed that different oyster and clam tissues express carbohydrate ligands similar to the HBGAs, the cellular receptor of HuNoVs. The expression of these ligands differs with the season and the organ (10). Higher rates of bioaccumulation of the virus in oyster tissues were observed in the months in which the oysters expressed higher levels of HBGAs (10). The finding that many members of the Caliciviridae, such as HuNoV and TV, accumulate to high levels in the digestive tissues of oysters within 24 h suggests that a specific interaction of HuNoV and TV with the HGBA-like carbohydrates expressed in these oyster tissues may play a role in virus bioaccumulation.
It has been shown that the viral genotype and the season have an impact on virus localization. For instance, Maalouf et al. (9) compared the bioaccumulation of three HuNoV strains belonging to genotypes GI.1, GII.4, and GII.3 in Eastern oyster tissues. The GI.1 strain had a higher bioaccumulation level in the digestive organs than the GII.4 strain. This effect was increased in the winter, when the HBGA-like carbohydrate ligand is expressed at higher levels in the oyster digestive organs. In addition to the digestive tissues, the GII.4 strain was detected in all other oyster tissues. This result may be due to the fact that the GII.4 strain can recognize a sialic acid-containing ligand as an alternative receptor, which allows for bioaccumulation in other shellfish tissues not expressing the HGBA-like moiety (61). The GII.3 strain accumulated most in the gills and mantle and was subsequently concentrated in the digestive gland (27). Drouaz et al. (17) compared the distributions of HuNoV GI, TV, and mengovirus (MgV) in oyster tissues after 24 h of oyster cultivation in contaminated seawater. The results showed that HuNoV GI and TV accumulated more in digestive glands, while MgV was more concentrated in gills, and the three viruses persisted in oyster tissues for more than 50 days (17). These data, together with our findings on HuNoV GII.4, TV, MNV-1, HAV, and RV, demonstrate that different enteric viruses have different distribution patterns in oyster organs based on the virus, virus genotype, and shellfish type.
Thermal inactivation of viruses.
In this study, we systemically compared the stabilities of four viruses following heat treatment and found that they can be ranked from the most to the least heat stable as follows: HAV, RV, TV, MNV-1. Treatment temperatures of <60°C required longer holding times to achieve high viral inactivation. At higher temperatures—for example, 80°C—most human enteric viruses are rapidly inactivated. However, HAV and RV showed more heat resistance than other enteric viruses. In our study, we found that MNV-1 was more sensitive to heat treatment than the other viruses examined. Previously, it was shown that the quantity of MNV-1 was reduced by 1 log unit at 63°C in <25 s and at 72°C in <10 s (62). Similarly, we found that the D value for MNV-1 at 62°C ranged from 0.55 to 1.81 min and the D value at 72°C ranged from 1.13 to 7.29 s. In another study, the thermal stabilities of MNV-1 and TV were compared, and both viruses were inactivated, and were below the detection limit (ca. 6-log-unit reduction), at temperatures of 70°C and 75°C in cell culture medium (63). At lower temperatures, the thermal inactivation kinetics of MNV-1 and TV were similar, indicating that both viruses are sensitive to heat treatment (62, 63). We found that MNV-1 and TV had very similar D values at temperatures of 62, 72, and 80°C, suggesting that these viruses are equally sensitive to heat treatment.
We found that thermal inactivation of viruses does not follow a linear logarithmic model. In a linear logarithmic model, the slope of the inactivation curve can be used to calculate the decimal reduction value (D value), the time required to reach a 1-log reduction of the quantity of virus at a specific temperature (46, 64). Several other models have been used to describe virus inactivation by heat. For example, the biphasic reduction model is used to calculate the time to the first log10 reduction (TFL value), instead of calculating the D value, from the inactivation curve based on two different rates of inactivation (21, 46, 55). The Weibull model was described by van Boekel (55) and has been shown to be the best fit for the expression of virus inactivation in many studies (55). In the Weibull model, the inactivation parameters, α and β, and the scale and shape of the probability density function are used to describe the time required for a desired amount of inactivation at a specific temperature (21). In our study, we found that at temperatures of 62 and 72°C, the biphasic reduction model was the best fit to the data. This is due to the shouldering and tailing effects observed in the curve. The four viruses showed monotonic upward concave (tailing) behavior, which may indicate that the thermally sensitive members of the viral quasispecies were quickly inactivated while the more resistant quasispecies survived the mild treatment. The RNA-dependent RNA polymerases (RdRp) of RNA viruses have poor fidelity, and therefore, during virus replication, viral progeny often are not identical to the parental virus or other progeny viruses. This leads to a viral population containing quasispecies (viral subsets that are not genetically identical), and some of the quasispecies may harbor mutations that enable them to be more resistant to heat than other viruses. In addition to quasispecies, the tailing effect may also be due to clumping of viral particles, leading to a protective effect for a subset of viruses during heat treatment. However, at a treatment temperature of 80°C (with culture medium or oyster tissue), the Weibull model was the best fit for the data. This is likely due to the increased linearity of the inactivation curve due to the increased temperature. These results demonstrate the necessity of applying appropriate models to accurately determine the D values.
The complexity of the sample matrix has been shown to affect the efficacy of heat at inactivating enteric viruses. In our study, we found that the oyster provided a protective effect to the virus during heat treatment at 80°C. A treatment time of 3 min was required to inactivate TV (4.8-log reduction) and MNV-1 (4.7-log reduction) in oyster tissues, while 4 min was required to inactivate RV to below the detection limit (3.1-log reduction) in oysters. Even after 6 min of treatment at 80°C, there was no significant reduction in the titer of HAV in oyster tissues. Previously, Croci et al. (65) showed that heat treatment of HAV at 60°C for 30 min and at 80°C for 10 min was not sufficient to eliminate 5 log units of the virus from contaminated mussel tissues. In fact, treatment at 100°C was required to inactivate HAV in the mussels (65). HAV was also more resistant to thermal inactivation in dried mussels than in culture medium (66). Only 3.16- and 4.38-log reductions in HAV titers were achieved at 60 and 85°C after 30 and 10 min of heat treatment, respectively. The D values at 60, 85, and 100°C were 6.3, 0.98, and 0.2 min, respectively, in culture medium, whereas in dried mussels, the D values were increased to 7.93, 3.05, and 0.85 min at 60, 85, and 100°C, respectively (66). Soft-shell clams were artificially contaminated with either HAV or MNV-1, and the efficacy of thermal treatment at viral inactivation was evaluated. It was found that a treatment at 90°C for 180 s was required to completely inactivate the viruses in shucked clams (67).
Insight into the mechanism underlying the thermal inactivation of viruses.
Currently, nucleic acid-based detection methods are used to test for HuNoV in foods. These methods do not distinguish between RNA detected from an infectious or a noninfectious viral particle, and therefore, positive results may not accurately reflect the risk of disease development upon consumption. Recently, porcine gastric mucin conjugated to magnetic beads (PGM-MBs) has been developed to capture HuNoV particles capable of binding to their cellular receptor, the histo-blood group antigens (HBGAs) (52, 53, 68, 69). HuNoV bound to PGM-MBs can then be collected, excluding particles that have lost receptor binding ability and theoretically allowing for RNA detection from intact HuNoV particles only. PGM-MBs has been used to evaluate HuNoV inactivation by thermal, high-pressure processing (HPP), and UV treatments (53), as well as by treatments with chlorine, chlorine dioxide, peroxyacetic acid, hydrogen peroxide, and trisodium phosphate (70). TV also utilizes HBGAs as cellular receptors (57), so there is the potential to use this cultivatable virus as a side-by-side control to determine the ability of the PGM-MBs binding assay to distinguish between infectious and noninfectious virus particles.
In this study, we attempted to utilize the PGM-MBs binding assay to distinguish between infectious and noninfectious HuNoV GII.4 strain particles following heat treatment. TV was used as a critical control, since it is cultivable and utilizes the same receptors as HuNoV. However, the log reduction of TV obtained using the PGM-MBs binding assay was not equivalent to the survival data obtained from the plaque assay, suggesting that lethal thermal damage to TV generally occurs prior to the loss of PGM (receptor) binding ability. For example, the number of TV RNA copies was not significantly reduced when TV was treated with a lethal dose of heat (80°C for 10 s). Thus, the PGM-MBs binding assay does not provide a precise estimate of the thermal inactivation of TV following heat treatment.
The PGM-MBs binding assay is a receptor binding assay that requires the inactivation technologies to efficiently inactivate receptor binding ability and as such does not directly measure inactivation. Thus, the applicability of the PGM-MBs binding assay depends on the mechanism of the inactivation induced by a particular thermal or nonthermal processing technology. Given the difference observed in TV inactivation and PGM binding with temperature, it should be noted that the PGM-MBs binding assay may also not provide a precise estimate of HuNoV inactivation in response to heat treatment. Perhaps other technologies (such as HPP, UV, and chlorine) may damage receptor binding more efficiently than heat treatment.
Based on the heat treatment of VLPs, which may behave similarly to HuNoV, we found that the receptor binding of HuNoV was severely damaged only at a high temperature (100°C for 5 s) or a longer holding time (80°C for 5 min). Perhaps heat treatment first damaged the interaction between the capsid and genomic RNA, which was lethal to the virus. It is possible that the genomic RNA was still partially or fully bound to the capsid, which allowed for viral RNA detection using RT-qPCR. Using purified TV, we found that the integrity of the viral capsid was disrupted by a lethal heat dose. TV infectivity was lost quickly (<10 s) during treatment at 80°C, which correlates with the change in the particles observed by TEM. However, it appears that the viral particles still bound efficiently to the PGM-MBs and that the viral RNA was minimally impacted by thermal treatment. No significant RNA reduction was observed at a lethal dose of heat. Using the PGM-MBs binding assay followed by RT-qPCR to detect HuNoV inactivation by thermal treatment at 80°C for 1 min, a maximum 1-log reduction was detected. Similarly, TV treated at 80°C for 1 min showed a maximum 2.5-log reduction. Direct RT-PCR detection of the TV VP1 gene also confirmed that viral RNA remained intact following the most severe thermal treatments.
HuNoV GII.4 VLPs retained the ability to bind to PGM-MBs following treatment at 80°C, as detected by SDS-PAGE analysis. In fact, a treatment of 100°C for 5 s was required to inhibit HuNoV VLP binding to PGM-MBs. Taken together, these results indicate that disruption of the integrity of the viral capsid and denaturation of viral protein, but not degradation of viral RNA, are the primary mechanisms of viral inactivation by heat. The fact that a lethal dose of heat was not sufficient to damage viral receptor binding activity suggests that heat may disrupt the capsid-genome interaction or other capsid functions and thus inhibit virus infectivity.
Overall, this study demonstrated that (i) enteric viruses can efficiently bioaccumulate in oyster tissues within 72 h; (ii) differences in the major bioaccumulation site in oysters can be observed between different viruses early after virus exposure (24 h); (iii) enteric viruses can be ranked, from most resistant to most sensitive to heat treatment, as HAV, RV, TV, and MNV-1; (iv) oyster tissue provides protection to viruses during heat treatment; and (v) thermal treatment at 80°C for more than 6 min is capable of reducing the levels of major foodborne viruses from oysters, with the exception of HAV. These results can be used by industry professionals to design and implement prevention and control measures to limit shellfish virus-associated outbreaks.
ACKNOWLEDGMENTS
J.L. acknowledges the support of the National Institute of Food and Agriculture, U.S. Department of Agriculture (USDA) (grant 2011-68003-30005).
Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the USDA.
REFERENCES
- 1.Butt AA, Aldridge KE, Sanders CV. 2004. Infections related to the ingestion of seafood. Part I. Viral and bacterial infections. Lancet Infect Dis 4:201–212. doi: 10.1016/S1473-3099(04)00969-7. [DOI] [PubMed] [Google Scholar]
- 2.Bellou M, Kokkinos P, Vantarakis A. 2013. Shellfish-borne viral outbreaks: a systematic review. Food Environ Virol 5:13–23. doi: 10.1007/s12560-012-9097-6. [DOI] [PubMed] [Google Scholar]
- 3.Keller R, Justino JF, Cassini ST. 2013. Assessment of water and seafood microbiology quality in a mangrove region in Vitoria, Brazil. J Water Health 11:573–580. doi: 10.2166/wh.2013.245. [DOI] [PubMed] [Google Scholar]
- 4.Polo D, Varela MF, Romalde JL. 2015. Detection and quantification of hepatitis A virus and norovirus in Spanish authorized shellfish harvesting areas. Int J Food Microbiol 193:43–50. doi: 10.1016/j.ijfoodmicro.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 5.Brake F, Ross T, Holds G, Kiermeier A, McLeod C. 2014. A survey of Australian oysters for the presence of human noroviruses. Food Microbiol 44:264–270. doi: 10.1016/j.fm.2014.06.012. [DOI] [PubMed] [Google Scholar]
- 6.Loury P, Le Guyader FS, Le Saux JC, Ambert-Balay K, Parrot P, Hubert B. 2015. A norovirus oyster-related outbreak in a nursing home in France, January 2012. Epidemiol Infect 143:2486–2493. doi: 10.1017/S0950268814003628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boxman IL, Tilburg JJ, Te Loeke NA, Vennema H, Jonker K, de Boer E, Koopmans M. 2006. Detection of noroviruses in shellfish in the Netherlands. Int J Food Microbiol 108:391–396. doi: 10.1016/j.ijfoodmicro.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 8.Le Guyader FS, Bon F, DeMedici D, Parnaudeau S, Bertone A, Crudeli S, Doyle A, Zidane M, Suffredini E, Kohli E, Maddalo F, Monini M, Gallay A, Pommepuy M, Pothier P, Ruggeri FM. 2006. Detection of multiple noroviruses associated with an international gastroenteritis outbreak linked to oyster consumption. J Clin Microbiol 44:3878–3882. doi: 10.1128/JCM.01327-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maalouf H, Schaeffer J, Parnaudeau S, Le Pendu J, Atmar RL, Crawford SE, Le Guyader FS. 2011. Strain-dependent norovirus bioaccumulation in oysters. Appl Environ Microbiol 77:3189–3196. doi: 10.1128/AEM.03010-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maalouf H, Zakhour M, Le Pendu J, Le Saux JC, Atmar RL, Le Guyader FS. 2010. Distribution in tissue and seasonal variation of norovirus genogroup I and II ligands in oysters. Appl Environ Microbiol 76:5621–5630. doi: 10.1128/AEM.00148-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Provost K, Dancho BA, Ozbay G, Anderson RS, Richards GP, Kingsley DH. 2011. Hemocytes are sites of enteric virus persistence within oysters. Appl Environ Microbiol 77:8360–8369. doi: 10.1128/AEM.06887-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ueki Y, Shoji M, Suto A, Tanabe T, Okimura Y, Kikuchi Y, Saito N, Sano D, Omura T. 2007. Persistence of caliciviruses in artificially contaminated oysters during depuration. Appl Environ Microbiol 73:5698–5701. doi: 10.1128/AEM.00290-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kingsley DH, Richards GP. 2003. Persistence of hepatitis A virus in oysters. J Food Prot 66:331–334. [DOI] [PubMed] [Google Scholar]
- 14.Atmar RL, Opekun AR, Gilger MA, Estes MK, Crawford SE, Neill FH, Graham DY. 2008. Norwalk virus shedding after experimental human infection. Emerg Infect Dis 14:1553–1557. doi: 10.3201/eid1410.080117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scallan E, Griffin PM, Angulo FJ, Tauxe RV, Hoekstra RM. 2011. Foodborne illness acquired in the United States—unspecified agents. Emerg Infect Dis 17:16–22. doi: 10.3201/eid1701.P21101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hall AJ, Wikswo ME, Manikonda K, Roberts VA, Yoder JS, Gould LH. 2013. Acute gastroenteritis surveillance through the National Outbreak Reporting System, United States. Emerg Infect Dis 19:1305–1309. doi: 10.3201/eid1908.130482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Drouaz N, Schaeffer J, Farkas T, Le Pendu J, Le Guyader FS. 2015. Tulane virus as a potential surrogate to mimic norovirus behavior in oysters. Appl Environ Microbiol 81:5249–5256. doi: 10.1128/AEM.01067-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Predmore A, Sanglay GC, DiCaprio E, Li J, Uribe RM, Lee K. 2015. Electron beam inactivation of Tulane virus on fresh produce, and mechanism of inactivation of human norovirus surrogates by electron beam irradiation. Int J Food Microbiol 198:28–36. doi: 10.1016/j.ijfoodmicro.2014.12.024. [DOI] [PubMed] [Google Scholar]
- 19.Li X, Ye M, Neetoo H, Golovan S, Chen H. 2013. Pressure inactivation of Tulane virus, a candidate surrogate for human norovirus and its potential application in food industry. Int J Food Microbiol 162:37–42. doi: 10.1016/j.ijfoodmicro.2012.12.016. [DOI] [PubMed] [Google Scholar]
- 20.Bozkurt H, D'Souza DH, Davidson PM. 2013. Determination of the thermal inactivation kinetics of the human norovirus surrogates, murine norovirus and feline calicivirus. J Food Prot 76:79–84. doi: 10.4315/0362-028X.JFP-12-327. [DOI] [PubMed] [Google Scholar]
- 21.Bozkurt H, Leiser S, Davidson PM, D'Souza DH. 2014. Thermal inactivation kinetic modeling of human norovirus surrogates in blue mussel (Mytilus edulis) homogenate. Int J Food Microbiol 172:130–136. doi: 10.1016/j.ijfoodmicro.2013.11.026. [DOI] [PubMed] [Google Scholar]
- 22.Jacobsen KH, Koopman JS. 2004. Declining hepatitis A seroprevalence: a global review and analysis. Epidemiol Infect 132:1005–1022. doi: 10.1017/S0950268804002857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lanata CF, Fischer-Walker CL, Olascoaga AC, Torres CX, Aryee MJ, Black RE, Child Health Epidemiology Reference Group of the World Health Organization and UNICEF. 2013. Global causes of diarrheal disease mortality in children <5 years of age: a systematic review. PLoS One 8:e72788. doi: 10.1371/journal.pone.0072788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ward RL, Bernstein DI, Young EC, Sherwood JR, Knowlton DR, Schiff GM. 1986. Human rotavirus studies in volunteers: determination of infectious dose and serological response to infection. J Infect Dis 154:871–880. doi: 10.1093/infdis/154.5.871. [DOI] [PubMed] [Google Scholar]
- 25.Bishop RF. 1996. Natural history of human rotavirus infection. Arch Virol Suppl 12:119–128. [DOI] [PubMed] [Google Scholar]
- 26.Lee RJ, Reese RA. 2014. Relating the bivalve shellfish harvesting area classification criteria in the United States and European Union programmes. J Water Health 12:280–287. doi: 10.2166/wh.2013.128. [DOI] [PubMed] [Google Scholar]
- 27.Finance C, Brigaud M, Lucena F, Aymard M, Bosch A, Schwartzbrod L. 1982. Viral pollution of seawater at Barcelona. Zentralbl Bakteriol Mikrobiol Hyg B 176:530–536. [PubMed] [Google Scholar]
- 28.Skraber S, Gassilloud B, Gantzer C. 2004. Comparison of coliforms and coliphages as tools for assessment of viral contamination in river water. Appl Environ Microbiol 70:3644–3649. doi: 10.1128/AEM.70.6.3644-3649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ebdon JE, Sellwood J, Shore J, Taylor HD. 2012. Phages of Bacteroides (GB-124): a novel tool for viral waterborne disease control? Environ Sci Technol 46:1163–1169. doi: 10.1021/es202874p. [DOI] [PubMed] [Google Scholar]
- 30.McQuaig S, Griffith J, Harwood VJ. 2012. Association of fecal indicator bacteria with human viruses and microbial source tracking markers at coastal beaches impacted by nonpoint source pollution. Appl Environ Microbiol 78:6423–6432. doi: 10.1128/AEM.00024-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Abreu Corrêa A, Souza DS, Moresco V, Kleemann CR, Garcia LA, Barardi CR. 2012. Stability of human enteric viruses in seawater samples from mollusc depuration tanks coupled with ultraviolet irradiation. J Appl Microbiol 113:1554–1563. doi: 10.1111/jam.12010. [DOI] [PubMed] [Google Scholar]
- 32.Doré WJ, Henshilwood K, Lees DN. 2000. Evaluation of F-specific RNA bacteriophage as a candidate human enteric virus indicator for bivalve molluscan shellfish. Appl Environ Microbiol 66:1280–1285. doi: 10.1128/AEM.66.4.1280-1285.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garcia LA, Nascimento MA, Barardi CR. 2015. Effect of UV light on the inactivation of recombinant human adenovirus and murine norovirus seeded in seawater in shellfish depuration tanks. Food Environ Virol 7:67–75. doi: 10.1007/s12560-014-9177-x. [DOI] [PubMed] [Google Scholar]
- 34.Marino A, Lombardo L, Fiorentino C, Orlandella B, Monticelli L, Nostro A, Alonzo V. 2005. Uptake of Escherichia coli, Vibrio cholerae non-O1 and Enterococcus durans by, and depuration of mussels (Mytilus galloprovincialis). Int J Food Microbiol 99:281–286. doi: 10.1016/j.ijfoodmicro.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 35.Powell A, Baker-Austin C, Wagley S, Bayley A, Hartnell R. 2013. Isolation of pandemic Vibrio parahaemolyticus from UK water and shellfish produce. Microb Ecol 65:924–927. doi: 10.1007/s00248-013-0201-8. [DOI] [PubMed] [Google Scholar]
- 36.Wang D, Shi X. 2011. Distribution and detection of pathogens in shellfish—a review. Wei Sheng Wu Xue Bao 51:1304–1309. (In Chinese.) [PubMed] [Google Scholar]
- 37.Corrêa Ade A, Rigotto C, Moresco V, Kleemann CR, Teixeira AL, Poli CR, Simões CM, Barardi CR. 2012. The depuration dynamics of oysters (Crassostrea gigas) artificially contaminated with hepatitis A virus and human adenovirus. Mem Inst Oswaldo Cruz 107:11–17. doi: 10.1590/S0074-02762012000100002. [DOI] [PubMed] [Google Scholar]
- 38.Gabrieli R, Macaluso A, Lanni L, Saccares S, Di Giamberardino F, Cencioni B, Petrinca AR, Divizia M. 2007. Enteric viruses in molluscan shellfish. New Microbiol 30:471–475. [PubMed] [Google Scholar]
- 39.Le Guyader F, Loisy F, Atmar RL, Hutson AM, Estes MK, Ruvoen-Clouet N, Pommepuy M, Le Pendu J. 2006. Norwalk virus-specific binding to oyster digestive tissues. Emerg Infect Dis 12:931–936. doi: 10.3201/eid1206.051519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Le Guyader F, Dubois E, Menard D, Pommepuy M. 1994. Detection of hepatitis A virus, rotavirus, and enterovirus in naturally contaminated shellfish and sediment by reverse transcription-seminested PCR. Appl Environ Microbiol 60:3665–3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tian P, Engelbrektson AL, Jiang X, Zhong W, Mandrell RE. 2007. Norovirus recognizes histo-blood group antigens on gastrointestinal cells of clams, mussels, and oysters: a possible mechanism of bioaccumulation. J Food Prot 70:2140–2147. [DOI] [PubMed] [Google Scholar]
- 42.Grodzki M, Schaeffer J, Piquet JC, Le Saux JC, Cheve J, Ollivier J, Le Pendu J, Le Guyader FS. 2014. Bioaccumulation efficiency, tissue distribution, and environmental occurrence of hepatitis E virus in bivalve shellfish from France. Appl Environ Microbiol 80:4269–4276. doi: 10.1128/AEM.00978-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bertrand I, Schijven JF, Sanchez G, Wyn-Jones P, Ottoson J, Morin T, Muscillo M, Verani M, Nasser A, de Roda Husman AM, Myrmel M, Sellwood J, Cook N, Gantzer C. 2012. The impact of temperature on the inactivation of enteric viruses in food and water: a review. J Appl Microbiol 112:1059–1074. doi: 10.1111/j.1365-2672.2012.05267.x. [DOI] [PubMed] [Google Scholar]
- 44.Arthur SE, Gibson KE. 2015. Comparison of methods for evaluating the thermal stability of human enteric viruses. Food Environ Virol 7:14–26. doi: 10.1007/s12560-014-9178-9. [DOI] [PubMed] [Google Scholar]
- 45.Bozkurt H, D'Souza DH, Davidson PM. 2015. Thermal inactivation of foodborne enteric viruses and their viral surrogates in foods. J Food Prot 78:1597–1617. doi: 10.4315/0362-028X.JFP-14-487. [DOI] [PubMed] [Google Scholar]
- 46.Tuladhar E, Bouwknegt M, Zwietering MH, Koopmans M, Duizer E. 2012. Thermal stability of structurally different viruses with proven or potential relevance to food safety. J Appl Microbiol 112:1050–1057. doi: 10.1111/j.1365-2672.2012.05282.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Karst SM, Wobus CE, Lay M, Davidson J, Virgin HW IV. 2003. STAT1-dependent innate immunity to a Norwalk-like virus. Science 299:1575–1578. doi: 10.1126/science.1077905. [DOI] [PubMed] [Google Scholar]
- 48.Lou F, Neetoo H, Chen H, Li J. 2011. Inactivation of a human norovirus surrogate by high-pressure processing: effectiveness, mechanism, and potential application in the fresh produce industry. Appl Environ Microbiol 77:1862–1871. doi: 10.1128/AEM.01918-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dicaprio E, Ma Y, Purgianto A, Hughes J, Li J. 2012. Internalization and dissemination of human norovirus and animal caliciviruses in hydroponically grown romaine lettuce. Appl Environ Microbiol 78:6143–6152. doi: 10.1128/AEM.01081-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lou F, Neetoo H, Li J, Chen H. 2011. Lack of correlation between virus barosensitivity and the presence of a viral envelope during inactivation of human rotavirus, vesicular stomatitis virus, and avian metapneumovirus by high-pressure processing. Appl Environ Microbiol 77:8538–8547. doi: 10.1128/AEM.06711-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ma Y, Li J. 2011. Vesicular stomatitis virus as a vector to deliver virus-like particles of human norovirus: a new vaccine candidate against an important noncultivable virus. J Virol 85:2942–2952. doi: 10.1128/JVI.02332-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tian P, Yang D, Jiang X, Zhong W, Cannon JL, Burkhardt W III, Woods JW, Hartman G, Lindesmith L, Baric RS, Mandrell R. 2010. Specificity and kinetics of norovirus binding to magnetic bead-conjugated histo-blood group antigens. J Appl Microbiol 109:1753–1762. doi: 10.1111/j.1365-2672.2010.04812.x. [DOI] [PubMed] [Google Scholar]
- 53.Dancho BA, Chen H, Kingsley DH. 2012. Discrimination between infectious and non-infectious human norovirus using porcine gastric mucin. Int J Food Microbiol 155:222–226. doi: 10.1016/j.ijfoodmicro.2012.02.010. [DOI] [PubMed] [Google Scholar]
- 54.Li X, Chen H, Kingsley DH. 2013. The influence of temperature, pH, and water immersion on the high hydrostatic pressure inactivation of GI.1 and GII.4 human noroviruses. Int J Food Microbiol 167:138–143. doi: 10.1016/j.ijfoodmicro.2013.08.020. [DOI] [PubMed] [Google Scholar]
- 55.van Boekel MA. 2002. On the use of the Weibull model to describe thermal inactivation of microbial vegetative cells. Int J Food Microbiol 74:139–159. doi: 10.1016/S0168-1605(01)00742-5. [DOI] [PubMed] [Google Scholar]
- 56.Schielke A, Filter M, Appel B, Johne R. 2011. Thermal stability of hepatitis E virus assessed by a molecular biological approach. Virol J 8:487. doi: 10.1186/1743-422X-8-487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Farkas T, Cross RW, Hargitt E III, Lerche NW, Morrow AL, Sestak K. 2010. Genetic diversity and histo-blood group antigen interactions of rhesus enteric caliciviruses. J Virol 84:8617–8625. doi: 10.1128/JVI.00630-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tan M, Jiang X. 2005. Norovirus and its histo-blood group antigen receptors: an answer to a historical puzzle. Trends Microbiol 13:285–293. doi: 10.1016/j.tim.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 59.Zhang D, Huang P, Zou L, Lowary TL, Tan M, Jiang X. 2015. Tulane virus recognizes the A type 3 and B histo-blood group antigens. J Virol 89:1419–1427. doi: 10.1128/JVI.02595-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tan M, Wei C, Huang P, Fan Q, Quigley C, Xia M, Fang H, Zhang X, Zhong W, Klassen JS, Jiang X. 2015. Tulane virus recognizes sialic acids as cellular receptors. Sci Rep 5:11784. doi: 10.1038/srep11784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Han L, Tan M, Xia M, Kitova EN, Jiang X, Klassen JS. 2014. Gangliosides are ligands for human noroviruses. J Am Chem Soc 136:12631–12637. doi: 10.1021/ja505272n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cannon JL, Papafragkou E, Park GW, Osborne J, Jaykus LA, Vinje J. 2006. Surrogates for the study of norovirus stability and inactivation in the environment: a comparison of murine norovirus and feline calicivirus. J Food Prot 69:2761–2765. [DOI] [PubMed] [Google Scholar]
- 63.Hirneisen KA, Kniel KE. 2013. Inactivation of internalized and surface contaminated enteric viruses in green onions. Int J Food Microbiol 166:201–206. doi: 10.1016/j.ijfoodmicro.2013.07.013. [DOI] [PubMed] [Google Scholar]
- 64.Chick H. 1908. An investigation of the laws of disinfection. J Hyg 8:92–158. doi: 10.1017/S0022172400006987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Croci L, Ciccozzi M, De Medici D, Di Pasquale S, Fiore A, Mele A, Toti L. 1999. Inactivation of hepatitis A virus in heat-treated mussels. J Appl Microbiol 87:884–888. doi: 10.1046/j.1365-2672.1999.00935.x. [DOI] [PubMed] [Google Scholar]
- 66.Park SY, Ha S-D. 2015. Thermal inactivation of hepatitis A virus in suspension and in dried mussels (Mytilus edulis). Int J Food Sci Technol 50:717–722. doi: 10.1111/ijfs.12674. [DOI] [Google Scholar]
- 67.Sow H, Desbiens M, Morales-Rayas R, Ngazoa SE, Jean J. 2011. Heat inactivation of hepatitis A virus and a norovirus surrogate in soft-shell clams (Mya arenaria). Foodborne Pathog Dis 8:387–393. doi: 10.1089/fpd.2010.0681. [DOI] [PubMed] [Google Scholar]
- 68.Tian P, Brandl M, Mandrell R. 2005. Porcine gastric mucin binds to recombinant norovirus particles and competitively inhibits their binding to histo-blood group antigens and Caco-2 cells. Lett Appl Microbiol 41:315–320. doi: 10.1111/j.1472-765X.2005.01775.x. [DOI] [PubMed] [Google Scholar]
- 69.Tian P, Engelbrektson AL, Mandrell RE. 2008. Seasonal tracking of histo-blood group antigen expression and norovirus binding in oyster gastrointestinal cells. J Food Prot 71:1696–1700. [DOI] [PubMed] [Google Scholar]
- 70.Kingsley DH, Vincent EM, Meade GK, Watson CL, Fan X. 2014. Inactivation of human norovirus using chemical sanitizers. Int J Food Microbiol 171:94–99. doi: 10.1016/j.ijfoodmicro.2013.11.018. [DOI] [PubMed] [Google Scholar]