

Abstract
(S)-Equol, a gut bacterial isoflavone derivative, has drawn great attention because of its potent use for relieving female postmenopausal symptoms and preventing prostate cancer. Previous studies have reported on the dietary isoflavone metabolism of several human gut bacteria and the involved enzymes for conversion of daidzein to (S)-equol. However, the anaerobic growth conditions required by the gut bacteria and the low productivity and yield of (S)-equol limit its efficient production using only natural gut bacteria. In this study, the low (S)-equol biosynthesis of gut microorganisms was overcome by cloning the four enzymes involved in the biosynthesis from Slackia isoflavoniconvertens into Escherichia coli BL21(DE3). The reaction conditions were optimized for (S)-equol production from the recombinant strain, and this recombinant system enabled the efficient conversion of 200 μM and 1 mM daidzein to (S)-equol under aerobic conditions, achieving yields of 95% and 85%, respectively. Since the biosynthesis of trans-tetrahydrodaidzein was found to be a rate-determining step for (S)-equol production, dihydrodaidzein reductase (DHDR) was subjected to rational site-directed mutagenesis. The introduction of the DHDR P212A mutation increased the (S)-equol productivity from 59.0 mg/liter/h to 69.8 mg/liter/h in the whole-cell reaction. The P212A mutation caused an increase in the (S)-dihydrodaidzein enantioselectivity by decreasing the overall activity of DHDR, resulting in undetectable activity for (R)-dihydrodaidzein, such that a combination of the DHDR P212A mutant with dihydrodaidzein racemase enabled the production of (3S,4R)-tetrahydrodaidzein with an enantioselectivity of >99%.
INTRODUCTION
The daidzein metabolite (S)-equol is produced by the human gut microbiota, and it has drawn a significant amount of attention as a result of its estrogenic properties in the human body that lessen postmenopausal symptoms in female patients (1). (S)-Equol binds effectively to the type β estrogen receptor but not to the type α receptor, preventing menopausal symptoms without increasing the incidence rate of breast cancer. Since many other estrogenic chemicals that have been used to reduce menopausal symptoms have increased the incidence rate of breast cancer, such selectivity for the receptor subunit is critical to the development of any estrogenic reagents (2, 3).
Most (S)-equol-producing bacteria (4–6) that are known to convert daidzein to equol belong to the Coriobacteriaceae, including Slackia, Eggerthella, and Adlercreutzia (6–8). However, the first gene identification for equol metabolism was reported for the non-Coriobacteriaceae microbe Lactococcus sp. strain 20-92 (9–11). Similar identifications of equol metabolism in Slackia sp. have also been reported (12, 13). According to these reports, daidzein is converted to (S)-equol by four enzymes (Fig. 1): daidzein reductase (DZNR), dihydrodaidzein reductase (DHDR), tetrahydrodaidzein reductase (THDR), and dihydrodaidzein racemase (DDRC). DZNR reduces daidzein to (R)-dihydrodaidzein [(R)-DHD] enantioselectively (9, 11). (R)-DHD is subsequently racemized to (S)-DHD either through tautomerization at C-3 or by DDRC activity (9). DHDR is involved in the reduction of (S)- and (R)-DHD to trans- and cis-tetrahydrodaidzein (trans- and cis-THD), respectively (10). Only trans-THD can be converted enantioselectively to (S)-equol by THDR (7). In the case of THDR, a radical mechanism was suggested to produce (S)-equol from (3S,4R)-trans-tetrahydrodaidzein (14).
FIG 1.

Conversion of daidzein to (S)-equol by gut bacteria. The scheme shows the pathway from daidzein to (S)-equol as reported previously (9). DZNR, daidzein reductase; DDRC, dihydrodaidzein racemase; DHDR, dihydrodaidzein reductase; THDR, tetrahydrodaidzein reductase. The stereochemistry of tetrahydrodaidzein produced by DHDR was confirmed in this study.
The production of (S)-equol by use of gut bacteria was previously reported in some papers and patents (15–18). According to these reports, Slackia sp. YIT 11861 is more productive than any of the other strains, including Slackia sp. TM-30, Lactococcus garvieae strain 20-92, and Gram-positive bacterium do03 (15). However, the use of any equol-producing bacteria has a common disadvantage in that these bacteria exhibit oxygen-sensitive growth. Even though experiments have been carried out to overcome such oxygen-labile properties of cell growth to produce dihydrodaidzein by using a laboratory evolution method, the oxygen sensitivity is still serious and the cell growth rate is quite low, so production is difficult to achieve at an industrial scale (19). Recombinant strains have been constructed that are able to convert daidzein to (S)-equol or its precursors (11, 12). But previous studies identified and/or characterized only the enzymes involved in each step of the biosynthesis of (S)-equol, and since then, no reports have focused on producing (S)-equol by using recombinant strains.
In the present study, we developed a recombinant strain that can produce (S)-equol from daidzein under aerobic conditions. The enzyme expression and reaction conditions for (S)-equol production by whole-cell bioconversion were examined and optimized, and to increase the yield and productivity of (S)-equol, mutation of DHDR was attempted to improve its reactivity and/or to shift its enantioselectivity toward (S)-DHD and away from (R)-DHD. The P212A mutation was identified to improve the (S)-equol productivity, and the DHDR P212A mutant was characterized, revealing that the mutation improved not only (S)-equol productivity but also (S)-DHD selectivity. Using this unintentional advantage of the mutant, we suggested a novel preparation method for trans-THD and cis-THD with a high rate of enantioselectivity. The results were analyzed in terms of the pH dependency, enantioselectivity, and stereochemistry of the products. Furthermore, the improvement in productivity rather than in the final yield of (S)-equol implies that other enzyme reaction steps might be the rate-determining steps (RDS) in reactions with higher substrate concentrations.
MATERIALS AND METHODS
Chemicals.
Daidzein and dihydrodaidzein were purchased from Sigma-Aldrich and Toronto Research Chemicals Inc. (North York, Ontario, Canada), respectively. All other chemicals were purchased from Sigma-Aldrich. Dihydrodaidzein racemate was separated and prepared by using a high-pressure liquid chromatograph (HPLC) equipped with a Chiralcel OJ-H column (Daicel Chemical Industries, Japan). Each enantiomer was confirmed by chiral HPLC analysis, and no tautomerization or racemization of the separated DHD enantiomers was detected in hexane or ethanol. (S)-DHD and (R)-DHD had retention times of 10 min with a sharp peak and 39 min with a broad peak, respectively, in the eluent (hexane:ethanol [60:40]) at a 1-ml/min flow rate. The unusually large difference in their retention times arose from use of a different chiral column (Chiralcel OJ-H column) under normal-phase conditions, whereas most other researchers have utilized other chiral columns under reversed-phase conditions. It has been claimed that (S)-DHD is eluted first, followed by (R)-DHD (9, 20). Additionally, comparison with the retention times of (R)-DHD produced by purified DZNR and of (S)-DHD racemized from (R)-DHD by purified DDRC confirmed the assignment. Chiral analysis of THDs was also performed under the same conditions as those for DHD analysis.
Cloning and construction of recombinant strain.
Slackia isoflavoniconvertens DSM 22006 was cultured on Columbia blood agar plates with 5% sheep blood at 37°C under anaerobic conditions. S. isoflavoniconvertens genomic DNA was extracted by use of an Exgene Cell SV genomic DNA purification kit (Geneall, South Korea) and was used as a PCR template. Four genes, namely, dzr (gene ID 409131878; encoding DZNR), ddr (gene ID 409131876; encoding DHDR), tdr (gene ID 409131875; encoding THDR), and ifcA (gene ID 409131872; encoding DDRC), were amplified by PCR with a set of designed primers, and the PCR products were cloned into the pRSFDuet-1 (dzr and ifcA) and pCDFDuet-1 (ddr and tdr) expression vectors by use of specific restriction enzyme sites at multicloning sites 1 and 2, respectively (Table 1). All the cloned gene sequences were verified, and there were no errors compared to the gene sources. FastDigest restriction enzymes were purchased from Thermo Scientific. The two expression vectors were transformed simultaneously into Escherichia coli BL21(DE3) (Novagen) by heat shock, and the transformed colonies were selected on an LB agar (10.0 g/liter tryptone, 5.0 g/liter yeast extract, 10.0 g/liter sodium chloride, and 15.0 g/liter agar) plate containing kanamycin (50 μg/ml) and streptomycin (50 μg/ml). A single colony was inoculated with 3 ml of LB with two antibiotics and was grown overnight. The transformants were named tDDDT-WT or tDDDT-PA depending on the presence of the P212A mutation in the pCDFDuet-1 vectors. The transformants were then used for the whole-cell reaction. For the enzyme purification, ddr and ifcA were individually cloned into pCDFDuet-1 and pET24a, respectively, and transformed into BL21(DE3). The description and notation for all transformants are listed in Table 2, and the recombinant strains were stored at −80°C for further experiments.
TABLE 1.
Primers for cloning and site-directed mutagenesisa
| Gene target or primer name | Restriction enzyme site | Primer sequence (5′ to 3′) |
|---|---|---|
| Primers for cloning | ||
| dzr | BamHI | ATAAGGATCCGATGCAGCACGCGAAATA |
| NotI | ATATAGCGGCCGCTACACCATGCGCGCTAC | |
| ifcA | NdeI | ATAATCATATGAAAGCTCAACTGAATC |
| EcoRV | ATAAGATATCCTAGTGGTGATGATGGTGATGCTCAGCGTCCACGTC | |
| ddr | BamHI | ATAAGGATCCGATGGCACAAGAGGTTAAG |
| NotI | ATATAGCGGCCGCTTAGGCGATTTCGCCCTG | |
| tdr | NdeI | ATAATCATATGGCAGAATTCGACGTTG |
| KpnI | ATAAGGTACCTCAGTGGTGGTGGTGGTGGTGCATGTTTGCAATCGCGTG | |
| Primers for site-directed mutagenesis of DHDR | ||
| P212A_F | CCGTGACCGCCGGCCTGGTGTG | |
| P212A_B | CCAGGCCGGCGGTCACGGAATTG | |
| H182Y_F | CAGGCATGCTACGCCGCCGCCAAG | |
| H182Y_B | GGCGGCGGCGTAGCATGCCTGCGG |
dzr and ifcA were cloned into the pRSFDuet-1 vector at multicloning sites 1 and 2, respectively. ddr and tdr were cloned into the pCDFDuet-1 vector at multicloning sites 1 and 2, respectively. dzr and ddr had an N-terminal 6×His tag sequence that was originally positioned in the plasmids, whereas ifcA and tdr had a C-terminal 6×His tag sequence followed by a stop codon. In the case of sole cloning of ifcA, the pET24a vector was used, and the C-terminal cloning primer was modified by replacing the EcoRV restriction site with a HindIII restriction site. All genes were expressed under the control of the T7 promoter.
TABLE 2.
Transformant notation
| Transformant | Plasmid(s) |
|---|---|
| tDDDT-WT | pRSFDuet-1 (dzr/ifcA) + pCDFDuet-1 (ddr/tdr) |
| tDDDT-PA | pRSF Duet-1 (dzr/ifcA) + pCDF Duet-1 [ddr(P212A)/tdr] |
| tDHDR-WT | pCDF (ddr) |
| tDHDR-PA | pCDF [ddr(P212A)] |
| tDDRC | pET24a (ifcA) |
Site-directed mutagenesis of DHDR.
A previously established protocol (21) was used to make amino acid mutations at residues P212 and H182 of DHDR. The mutations were introduced into the pCDFduet-1-ddr construct by PCR (Herculase II fusion DNA polymerase) using specific mutagenesis primers (Table 1), and all constructs were confirmed by sequencing.
Whole-cell reaction with (S)-equol-producing recombinant E. coli.
The tDDDT-WT or tDDDT-PA transformant was grown in LB broth medium (50 ml) containing 50 μg/ml of kanamycin and 50 μg/ml of streptomycin at 37°C until reaching an optical density at 600 nm (OD600) of 0.6 to 0.8. Isopropyl-thio-β-d-galactopyranoside (IPTG) was then added to a final concentration of 0.1 mM, along with 0.1 mM FeSO4 as the source of the iron-sulfur cluster of DZNR. After the IPTG induction, cells were grown at 18°C for 12 h and were subsequently harvested by centrifugation, washed with phosphate-buffered saline (PBS; 8.00 g/liter sodium chloride, 0.20 g/liter potassium chloride, 3.58 g/liter disodium hydrogen phosphate·12H2O, and 0.27 g/liter potassium dihydrogen phosphate, pH 7.2), and resuspended in 200 mM potassium phosphate buffer (KPB) (pH 8.0). The pH of the reaction buffer in the whole-cell reaction mixture was first optimized for the most equol production, because the whole-cell reaction includes four enzyme reactions and many other reaction parameters, e.g., substrate/product solubility, cell viability, and redox balance also depend on the pH of the reaction solution (see Fig. S1 in the supplemental material). After each whole-cell reaction, the pH of the reaction solution was measured, and no change was observed. The initial OD600 was set to 10, with 2% (wt/vol) glucose as a carbon source for cells and with an isoflavonoid substrate (daidzein or dihydrodaidzein), and biotransformation with the resting cells was executed in a 5-ml reaction volume in a 20-ml glass vial with gentle shaking (100 rpm) at 30°C. At different time points during the reaction, 200-μl aliquots were sampled from the reaction mixture, and the reaction was stopped by the addition of 200 μl of ethyl acetate (EA) with vigorous vortexing. After centrifugation at 14,000 × g for 5 min, 140 μl of the EA layer was evaporated using a centrifugal vacuum concentrator (Biotron, South Korea). The samples were then dissolved in 140 μl of methanol for HPLC analysis.
HPLC analysis.
The reaction samples prepared from the whole-cell reaction or enzyme assay were analyzed via an HPLC (YoungLin, South Korea) equipped with a C18 column (5-μm particle size; 4.6 by 250 mm; Vydac). Twenty microliters of reaction sample was injected for the HPLC analysis. The mobile phase was composed of a mixture of solvent A (0.1% trifluoroacetic acid-water) and solvent B (acetonitrile), and metabolites were separated by increasing the concentration of solvent B under the following conditions: an isocratic elution for 5 min with 10% solvent B, a linear gradient for 15 min with 10 to 25% solvent B, an isocratic elution for 9 min with 25% solvent B, a linear gradient for 2 min with 25 to 10% solvent B, and an isocratic elution for 5 min with 10% solvent B. The flow rate was 1 ml/min, and metabolites were detected with UV light at 280 nm.
For the statistical comparison of (S)-equol production levels between the wild-type and P212A mutant strains, the t test was used, and a difference was considered significant if the P value was <0.05.
Recombinant enzyme expression and purification.
The tDHDR-WT, tDHDR-PA, and tDDRC transformants were grown in LB broth medium until the OD600 reached 0.6, and IPTG was then added to a final concentration of 0.1 mM. After IPTG induction, the cells were cultured for 4 h at 30°C, and the cells were then harvested by centrifugation and subsequently washed with PBS. The prepared cells were resuspended in 100 mM KPB (pH 7.0) containing 1 mM phenylmethylsulfonyl fluoride and were disrupted using a sonicator (Misonix) in an ice-cooled bottle. The total pulsing time was 5 min (3 s on, 8 s off) with a 21-W powered microtip. The cell lysates were centrifuged at 14,000 × g for 30 min at 4°C, and supernatant samples were prepared for enzyme purification. The enzymes were purified using a Ni-nitrilotriacetic acid (Ni-NTA) His tag purification kit (Qiagen Korea Ltd., Seoul, South Korea), and purification was then carried out utilizing the Ni-NTA resin. The Ni-NTA-bound enzymes were washed twice with 50 mM sodium phosphate buffer (pH 8.0) containing 300 mM NaCl, 10 mM imidazole, and 0.1% (vol/vol) Tween 20 and were further washed with the same buffer containing 30 mM imidazole. The enzymes were eluted with the same buffer containing 250 mM imidazole, and finally, the eluents were dialyzed against 100 mM KPB (pH 8.0) to remove imidazole, NaCl, and Tween 20. Purified enzymes were subjected to 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) to confirm the purity of the enzymes (see Fig. S2 in the supplemental material), and the enzyme concentrations were determined according to the Bradford assay, with bovine serum albumin (BSA) as the standard (22).
Glucose dehydrogenase (GDH) from Bacillus subtilis was also cloned, expressed, and purified, according to a method reported elsewhere (23), for regeneration of NADPH in the enzyme assay. All purified enzymes were stored at −80°C with 30% glycerol in the relevant buffer.
Enzyme assays.
Enzyme assays were performed at 37°C under aerobic conditions, with a final reaction volume of 200 μl. The solution contained 100 mM KPB (pH 6.0; the approximate optimum pH for reductase activity of DHDR), 0.5 mM NADPH, and purified enzyme at a final concentration of 0.5 μM. After 5 min of incubation at 37°C for enzyme activation, the reaction was started by adding 2 μl of a 10 mM stock solution of DHD racemate or each enantiomer of DHD in dimethyl sulfoxide (DMSO). The reaction was stopped by adding 200 μl of ethyl acetate, followed by vigorous vortexing. After 5 min of vortexing and 5 min of centrifugation at 14,000 × g, the solvent layer was evaporated using a centrifugal vacuum concentrator, and the products were finally redissolved in methanol for HPLC analysis.
For the stereospecific production of (3S,4R)-trans-tetrahydrodaidzein, final concentrations of 0.5 μM DHDR P212A and 5.0 μM DDRC were used for the assay. To estimate the pH optima of the reductase and dehydrogenase reactions, the initial rate activities were measured using different buffers for the enzyme assay (100 mM citrate buffer, pH 5.0 to 6.0; 100 mM KPB, pH 6.0 to 8.0; and 100 mM Tris-HCl buffer, pH 8.0 to 9.0). To determine the kinetic parameters (kcat and Km), product formation was measured by the increase in the peak area at 280 nm, using HPLC. The total reaction volume was set to 200 μl, with 100 mM KPB (pH 6.0) containing 100 nM His-tag-purified enzyme, 0.5 mM NADPH, and various concentrations (i.e., 5 to 500 μM) of each enantiomer of the DHD substrate. All the data were fitted to the Michaelis-Menten equation by using nonlinear curve fitting by the least-square regression method, all enzyme assays were performed more than three times each to obtain statistically meaningful values, and the mean values and standard deviations were recorded.
Enzymatic synthesis of trans- and cis-THD.
trans-THD and cis-THD were enzymatically synthesized by using (S)-DHD and (R)-DHD, respectively, as substrates, while His-tag-purified DHDR was used as a biocatalyst. The reaction conditions were almost the same as those for the enzymatic assay protocol. In addition, GDH from Bacillus subtilis was added at 0.25 U/ml to ensure efficient NADPH regeneration, with 20 mM glucose as a substrate (23, 24). The reaction samples were extracted with 2 volumes of ethyl acetate, and the extracts were concentrated using a rotary evaporator for further chromatographic separation. Enzymatically synthesized THD enantiomers were analyzed and confirmed via HPLC with a chiral column. The THD enantiomers were then derivatized with N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA) at 50°C for 5 min and were analyzed with a gas chromatograph-mass spectrometer (GC-MS) equipped with an electron impact (EI) ionization source for identification according to a previously described method (25), with some modifications. The GC oven temperature started at 65°C, was held for 5 min, and then increased by 3°C/min to 250°C, with holding for 10 min. The ionizing electron energy was 70 eV, and the collected mass range was 50 to 600 atomic mass units (amu). The retention times for trans-THD and cis-THD were 15.5 min and 15.9 min, respectively. The relative intensities of the fragmental peaks for each THD diastereomer were recorded and compared with previously reported data (26) (see Fig. S3 in the supplemental material). HPLC and GC-MS analyses confirmed the purity of each THD to be more than 95%. To determine the stereochemistry of the enzymatically produced THDs, each THD stereoisomer was dissolved in methanol, and circular dichroism (CD) spectra were obtained in the range of 220 to 300 nm by using a Chirascan-Plus CD spectrometer (Applied Photophysics, United Kingdom).
RESULTS
Biosynthesis of (S)-equol from daidzein by using recombinant E. coli BL21(DE3).
Soluble coexpression of four recombinant enzymes (i.e., DZNR, DDRC, DHDR, and THDR) was confirmed via SDS-PAGE. Despite their heterologous expression in E. coli, the four enzymes were successfully overexpressed at 18, 25, and 30°C (see Fig. S4A in the supplemental material). However, the expression levels of DZNR and DHDR were lower at 30°C than at 18 or 25°C, whereas DDRC expression showed the opposite case. Since less conversion of daidzein to (S)-equol was observed for the cells grown at 25 and 30°C, further induction experiments were carried out at 18°C as the optimum (see Fig. S4B in the supplemental material). The heterologous expression of DZNR of Lactococcus strain 20-92 in E. coli that was grown aerobically resulted in a somewhat low enzyme activity (11), and similarly, in our case, no conversion of daidzein was detected under vigorous shaking conditions (200 rpm with aeration) or in a whole-cell reaction at 37°C (data not shown), suggesting that the DZNR catalytic activity is vulnerable to oxygen or to high temperatures. However, no significant decrease in DZNR expression was observed in the recombinant strain under aerobic growth conditions during induction. To overcome the problem of the lack of DZNR activity, a biotransformation reaction was carried out by using resting cells at a fixed cell density (OD600 = 10) under gentle shaking conditions of 100 rpm at 30°C, which achieved effective reduction conditions for whole-cell conversion without the loss of DZNR activity.
Recombinant E. coli tDDDT-WT was used to convert between 0.2 mM and 5 mM daidzein to (S)-equol with a high conversion yield (Fig. 2A). The rate of conversion to (S)-equol was high (over 85%) at <1 mM daidzein. However, when the daidzein concentration went over 2 mM, the reaction was severely inhibited and the (S)-equol yield was quite low. Even when the reaction time was extended to 8 h for the reactions at 2 and 5 mM daidzein, the low-conversion problem was not changed, and daidzein was no longer converted into DHD or (S)-equol after 4 h. Furthermore, no DHD and THD were detected at the 4- to 8-h reaction time points, and the daidzein concentration was not decreased anymore, suggesting that cytoplasmic DZNR activity was inhibited by a high level of (S)-equol. The fact that DZNR activity is inhibited by high (S)-equol concentrations was confirmed with an experiment in which the recombinant strain expressed DZNR only (see Fig. S5 in the supplemental material). In this study, we focused only on increasing (S)-equol production at concentrations below 1 mM daidzein.
FIG 2.

(A) (S)-Equol production by a recombinant E. coli BL21(DE3) strain (tDDDT-WT), with initial daidzein concentrations ranging from 0.2 to 5 mM. (B) Concentrations of four metabolites, including daidzein (DZN), dihydrodaidzein (DHD), trans-tetrahydrodaidzein (trans-THD), and (S)-equol (EQL). Daidzein (1 mM) was used as the substrate, and the concentrations along the reaction time were recorded.
To determine a rate-determining step (RDS) at concentrations below 1 mM, the concentration profiles of the daidzein metabolites, i.e., daidzein, DHD, trans-THD, and (S)-equol, were recorded during the whole-cell biotransformation, and the profiles suggested that the DHDR reaction was the RDS among the series of enzymatic reactions (Fig. 2B). The trans-THD concentration was generally low (<50 μM) during the reaction time, and DHD appeared to have accumulated to a great extent and then gradually decreased. Moreover, trans-THD was rarely detected in the HPLC elution profiles of reactions done with daidzein concentrations of <500 μM, suggesting that DHDR catalyzing DHD to trans-THD was the RDS for (S)-equol production. In addition, the accumulated DHD was detected in its fully racemized form, with an enantiomeric excess (ee) of 0% (data not shown), indicating that DDRC was not a limiting step. The expression level of DHDR appeared to be lower than that of other enzymes (see Fig. S4A in the supplemental material), but its expression was optimized by using various induction temperatures. Additionally, the sole overexpression of DHDR in tDHDR-WT did not increase the THD level over the DHD level, suggesting that DHDR overexpression cannot simply increase THD production. After the findings on increasing the THD level in the E. coli cytoplasm, protein engineering was attempted to enhance the reductase catalytic activity of DHDR to increase the cytoplasmic THD level. The engineering results are described later.
pH effects and kinetics of DHDR.
Prior to DHDR engineering, the effects of pH on DHDR activity, as well as DHDR's kinetic parameters and enantioselectivity, were characterized. Since DHDR belongs to the short-chain dehydrogenase/reductase (SDR) family, the reaction was expected to be reversible, and the pH optima of the reduction and dehydrogenation reactions were expected to be different, as for other SDR enzymes. In order to determine the optimum pH for the reductase activity, an enzyme assay was performed in the range of pH 5 to 7, with intervals of 0.5 unit. DHDR showed the highest reductase activity at a pH of around 6.0 (Fig. 3). To measure the dehydrogenase activity, purified DHDR was incubated with (3S,4R)-trans-THD as the substrate in the pH range from 6.5 to 8.5, and its optimum was identified as pH 7.5. In the pH range from 5.0 to 6.5, the reductase activity of DHDR was dominant over its dehydrogenase activity, and the dehydrogenase activity dominated at pHs above 7 (Fig. 3).
FIG 3.
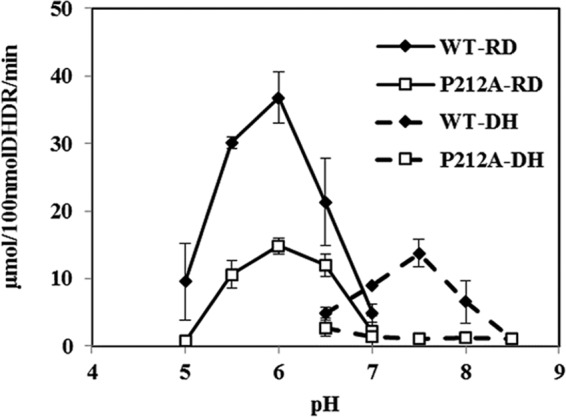
Effect of pH on DHDR activity. The reduction reactions (RD) of the wild-type (WT) and P212A mutant enzymes were performed at pHs of 5.0 to 7.0. The dehydrogenation reactions (DH) of the wild-type (WT) and P212A mutant enzymes were performed at pHs of 6.5 to 8.5.
According to several previous studies, the cytoplasmic pH of E. coli is maintained between 7.2 and 7.8 owing to its pH homeostasis enacted by the prokaryotic primary proton pumps on the membrane (27–30). Rapid measurement of the pH of the cytoplasm and periplasm of Escherichia coli by green fluorescent protein fluorimetry enabled the observation that pH perturbation by HCl addition was recovered in several minutes to again reach the initial pH of 7.6 (31). Therefore, it was expected that DHDR would be a dehydrogenase rather than a reductase under the conditions of the E. coli cytoplasm. This inference was confirmed by use of an E. coli transformant, tDHDR-WT, to see which reaction, reduction or dehydrogenation, was more favorable in the cytoplasm. tDHDR-WT showed a low level of conversion of DHD to trans- and cis-THD (Fig. 4A). When 100 μM DHD racemate was used, tDHDR-WT yielded 10 μM trans-THD from 50 μM (S)-DHD [20% yield for (S)-DHD] and about 4 μM cis-THD from 50 μM (R)-DHD [8% yield for (R)-DHD].
FIG 4.

DHDR reduction reaction by use of a whole-cell biocatalyst. tDHDR-WT (A) and tDHDR-PA (B) were examined in the presence of 100 μM (initial concentration) dihydrodaidzein racemate. Metabolite concentrations were recorded along the reaction time. Data points show means and standard deviations.
The kinetics of DHDR was analyzed at pH 6, i.e., the optimum pH for reductase activity. Since it was reported that DHDR could convert the (S) and (R) enantiomers of DHD to trans- and cis-THD, respectively, the kinetic parameters were measured separately with each enantiomer (Table 3). The kcat for (S)-DHD was about 9.5 times larger than that for (R)-DHD, and the Km value for (S)-DHD was 3.6 times smaller than that for (R)-DHD. Wild-type DHDR has a catalytic efficiency that is higher by a factor of about 23.5 for (S)-DHD than for (R)-DHD in terms of the kcat/Km ratio. We confirmed that DHDR prefers (S)-DHD to (R)-DHD as its substrate (9, 13).
TABLE 3.
Kinetic parameters of purified wild-type DHDR and the P212A mutanta
| Strain | (S)-DHD |
(R)-DHD |
||||
|---|---|---|---|---|---|---|
| Km, (S) (μM) | kcat, (S) (min−1) | kcat, (S)/Km, (S) (min−1 μM−1) | Km, (R) (μM) | kcat, (R) (min−1) | kcat, (R)/Km, (R) (min−1 μM−1) | |
| Wild type | 28.3 ± 6.9 | 328.1 ± 23.5 | 12.00 ± 3.31 | 102.4 ± 7.6 | 34.7 ± 1.2 | 0.51 ± 0.05 |
| P212A mutant | 60.7 ± 4.9 | 216.4 ± 44.7 | 3.60 ± 0.95 | ND | ND | ND |
(S)- and (R)-dihydrodaidzein enantiomers were used as substrates. All enzyme assays were performed at 37°C and pH 6.0 in 100 mM KPB. The results are presented as means ± standard deviations for triplicates. ND, no detection of any product formation by the DHDR P212A mutant.
Construction and characterization of the DHDR P212A mutant.
In the previous section, we identified DHDR as an RDS in the recombinant whole-cell reaction system for (S)-equol production from daidzein. Based on this fact, enzyme engineering of DHDR was attempted in order to increase the DHDR activity in the whole-cell reaction. For this purpose, we chose a site-directed mutagenesis method based on a previous report about an SDR family enzyme. Previous mutations of the 3α-hydroxysteroid dehydrogenase/carbonyl reductase (3αHSD) from Comamonas testosteroni (32), which belongs to the SDR family, like DHDR, were mimicked to obtain mutants with changes in substrate/product binding properties or enantioselectivity. The reference suggested that when Pro185 (which was aligned to Pro212 in DHDR) was replaced with glycine or alanine, with a small alkyl group side chain, the substrate binding loop in 3αHSD appeared to have more flexibility as measured by the fluorescence of tryptophan on the substrate binding loop. Intrigued by the previous research, we constructed the DHDR P212A mutant by using site-directed mutagenesis, performed enzyme characterization, and evaluated the whole-cell reaction system with the overexpressed mutant.
Similarly to wild-type DHDR, the DHDR P212A mutant showed the highest reductase activity at pH 6. However, its catalytic activity (kcat) was reduced to 66% and its Km increased by 214%, resulting in a 30% decrease in the kcat/Km value for (S)-DHD (Table 3). Interestingly, the P212A mutant showed no activity toward (R)-DHD, even with 1 μM purified enzyme and up to 500 μM substrate, suggesting that the DHDR P212A mutant is completely inactive on (R)-DHD. However, such an observation was inconsistent with the results of the whole-cell reaction, which indicated that tDHDR-PA still showed some reductase activity for both (S)- and (R)-DHD (Fig. 4B). One possible explanation is that the enzyme concentration in the cell after IPTG induction was much higher than 1 μM to be able to show any remaining reductase activity, even for (R)-DHD. Time course reactions with wild-type and P212A mutant DHDR revealed that wild-type DHDR consumed primarily (S)-DHD, and then (R)-DHD, which corresponds to its kinetics of having a lower Km value for (S)-DHD than for (R)-DHD, while the DHDR P212A mutant consumed (S)-DHD only (Fig. 5A and B).
FIG 5.

Time course reactions of purified wild-type DHDR (A) and DHDR P212A (B). Dihydrodaidzein racemate (100 μM) was used as the substrate, with 0.5 μM purified enzyme. Data are means and standard deviations.
tDHDR-PA converted 50 μM (S)-DHD to 50 μM trans-THD [100% yield for (S)-DHD)] and 50 μM (R)-DHD to 14 μM cis-THD [28% yield for (R)-DHD] (Fig. 4B). The preference for (S)-DHD over (R)-DHD was maintained in the tDHDR-PA mutant, and the ratio of product concentrations for trans-THD and cis-THD at the 1-h time point was 2.3 for the wild type and 3.8 for the P212A mutant. These results appear to be inconsistent with the fact that DHDR P212A showed a poorer catalytic efficiency than that of wild-type DHDR (Table 3). However, it is in fact a reasonable result, since the kinetic study was performed at pH 6, at which no dehydrogenase activity was displayed. As a result, at pH 6.0, wild-type DHDR surely showed more reductase activity than the DHDR P212A mutant did, whereas all the reactions of DHDs and THDs in the cytoplasm by tDHDR-WT or tDHDR-PA would be affected by the pH of the cytoplasm.
In addition, the DHDR H182Y mutant was also constructed by site-directed mutagenesis to see the function or exchangeability of His182, which was expected to be a catalytic hydrogen donor in the case of DHDR, because most SDR family enzymes have tyrosine as a catalytic hydrogen donor. The S. isoflavoniconvertens DHDR H182Y mutant showed no catalytic reductase and dehydrogenase activities, suggesting that His182 is the main catalytic functional residue and cannot be replaced (data not shown).
Enantioselectivity of DHDR and production of (3S,4R)-trans-THD.
Chiral analysis revealed that the biosynthetic products of DHDR consisted of two forms of diastereomeric THD, trans- and cis-THD, whereas chemical synthesis generated four forms of THD: (3R,4S)- and (3S,4R)-THD for trans-isomers and (3R,4R)- and (3S,4S)-THD for cis-isomers (Fig. 6). As shown in Fig. 6, chiral separation of a chemical preparation of THD that yields about 30% trans- and 70% cis-THD generated two small peaks first and then two large peaks (33). Additionally, trans-THD had a shorter elution time than cis-THD on the C18 HPLC column. Based on such facts, the first two peaks were assigned to trans-isomers and the last two peaks to cis-isomers. The DHDR P212A mutant also produced one enantiomer of trans-THD that had the same stereochemistry as that of a trans-isomer produced by wild-type DHDR. However, it was difficult to identify the exact stereochemistry by using only the HPLC retention time. To elucidate the stereochemistry of the THDs produced by DHDR, CD spectroscopy data were exploited (Fig. 7). According to the previous CD spectra of THDs, trans-THD and cis-THD were (3S,4R)-trans-THD and (3R,4R)-cis-THD, respectively (34). Direct observation of the stereochemistry of THDs produced by DHDR is first reported in the present study, and our results show that (3S,4R)-trans-THD rather than (3R,4S)-trans-THD is a direct precursor of (S)-equol, which is the same as that verified in a previous report (14), and that only (3R,4R)-cis-THD is generated by DHDR, as a by-product from (R)-DHD.
FIG 6.
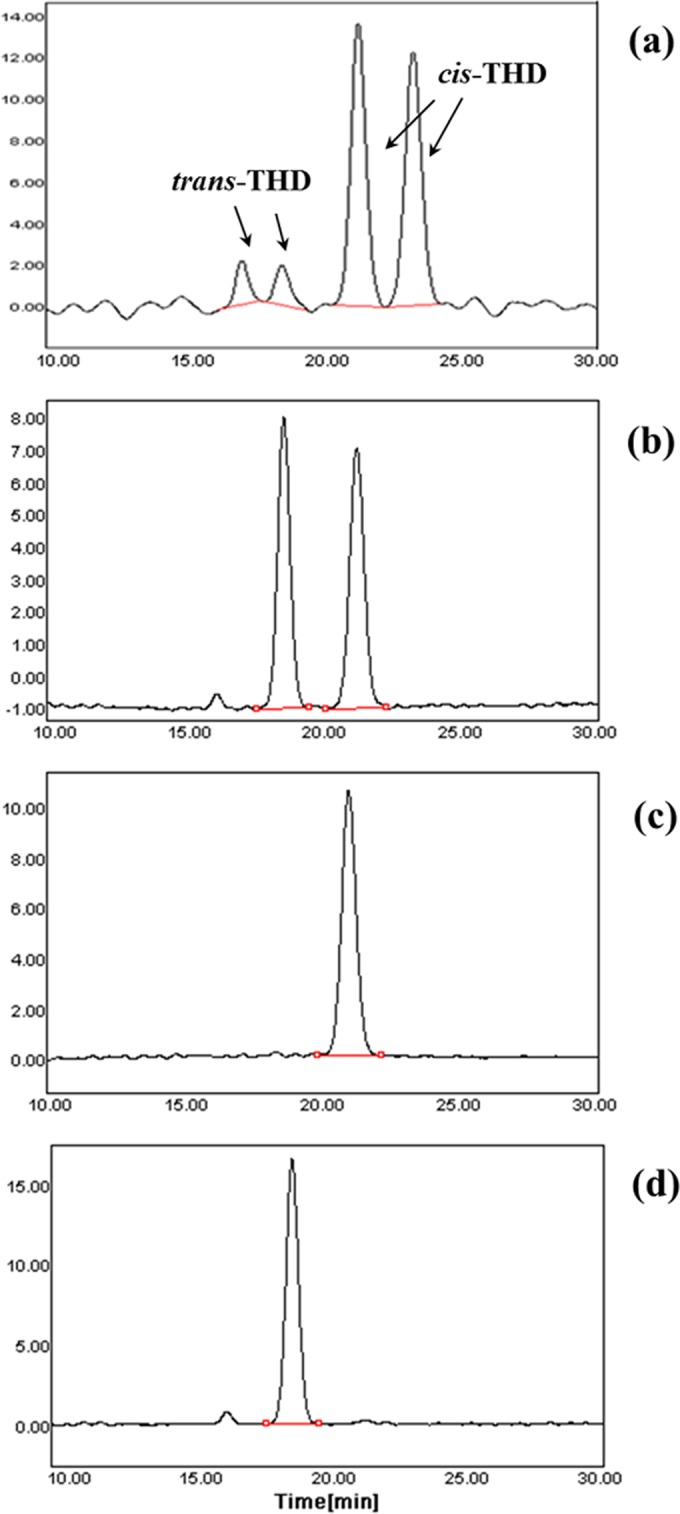
Elution profiles of four THDs. Profiles are shown for four chemically synthesized THDs (a) and enzymatically synthesized THDs (b) produced by purified DHDR using (R,S)-DHDs as the substrate. Profiles are also shown for THDs produced by DHDR using (R)-DHD (c) and (S)-DHD (d) as substrates. Each THD was separated and detected on a chiral column (Chiralcel OJ-H) under elution conditions (hexane:ethanol [60:40]) and at a 1-ml/min flow rate.
FIG 7.

Circular dichroism spectra for trans- and cis-THD produced by DHDR were generated using a CD spectrometer. Each THD was dissolved in methanol, and CD spectra were recorded in the range of 220 to 300 nm.
Enantioselective (3S,4R)-trans-THD production was also confirmed by utilizing the stereospecificity of the DHDR P212A mutant. To achieve a 100% conversion using DHD racemate as the substrate, DDRC should be included in the enzyme reaction system. Due to the enantioselectivity of DHDR P212A, (S)-DHD was preferentially consumed, and then the remaining (R)-DHD was racemized to (S)-DHD by DDRC. Gradually, both enantiomers of DHD could be converted to (3S,4R)-trans-THD, and as a result, when 0.5 μM DHDR P212A was used with 5 μM DDRC, 100 μM DHD racemate was converted to (3S,4R)-trans-THD, with a 93% yield within 5 min, and no (3R,4R)-cis-THD was detected during the reaction (Fig. 8).
FIG 8.
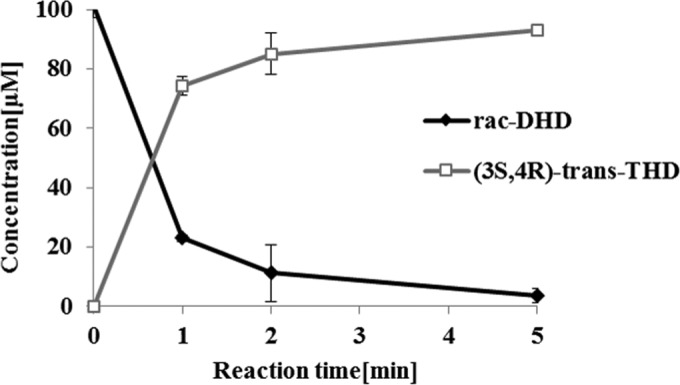
Time course production of (3S,4R)-trans-THD from (R,S)-DHD by purified DHDR P212A and the DDRC enzyme cocatalyzing the reaction. Dihydrodaidzein racemate (100 μM) was used as the substrate, with 0.5 μM DHDR P212A and 5.0 μM DDRC added to the reaction mixture. Data are means and standard deviations.
Efficient (S)-equol production by use of the DHDR P212A mutant.
Integrating all the above-described findings, (S)-equol production was performed by using two transformants, i.e., tDDDT-WT and tDDDT-PA. With 500 μM daidzein, the control strain, tDDDT-WT, exhibited an 83% (413 μM) yield of (S)-equol and a productivity of 59.0 mg/liter/h. On the other hand, the mutant strain, tDDDT-PA, exhibited a 93% (466 μM) yield of (S)-equol and a productivity of 69.8 mg/liter/h, which were 13% and 18% higher, respectively, than those of the control (Fig. 9). Except for the 0.5-h time point, the higher yield was statistically significant at all time points. However, the improved productivity was observed only at initial daidzein concentrations of <500 μM, and the higher productivity of (S)-equol in the mutant strain seems to have been caused by the fast conversion of (S)-DHD to (3S,4R)-trans-THD, without (S)- or (R)-DHD accumulation (Fig. 9A and B). At daidzein concentrations above 2 mM, however, DHD did not accumulate, and daidzein consumption was severely reduced. In such a case, the control tDDDT-WT and mutant tDDDT-PA strains showed almost similar profiles of (S)-equol production, suggesting that a step other than DHDR is likely to become a new rate-limiting step for (S)-equol biosynthesis (data not shown).
FIG 9.

Concentrations of four metabolites, including DZN, DHD, (3S,4R)-trans-THD, and (S)-EQL. Daidzein (500 μM) was used as the substrate, and the concentrations were recorded along the reaction time. (A) tDDDT-WT (control strain); (B) tDDDT-PA mutant; (C) comparison of (S)-equol production levels between the control strain (WT) and the mutant strain (P212A). Graphs A and B show data representative of each data set. To confirm the statistical difference between tDDDT-WT and the tDDDT-PA mutant, more than three repeated biotransformations were performed independently, and data shown are average values with standard deviations.
DISCUSSION
In this study, DHDR from S. isoflavoniconvertens was fully characterized in terms of its pH optimum, kinetic parameters, and stereoselectivity. Although DHDRs from S. isoflavoniconvertens (13) and Lactococcus sp. strain 20-82 (10) were previously characterized based on their kinetic parameters, cofactor specificity, and preference for (S)-DHD, the analyses were restricted only to neutral pH, which prohibited a complete understanding of DHDR's characteristics. Moreover, previous studies (10, 13) did not assess any of the kinetics of DHDR toward each DHD enantiomer or the exact stereochemistry of trans- and cis-THD produced from each DHD enantiomer. It is worth mentioning that we confirmed only that (3S,4R)-THD was transformed to (3S)-equol by THDR in a biological system (14). This study also reports the first direct measurement of the stereochemistry of THDs produced by DHDR and the quantification of its (S)-DHD preference by kinetic values. The stereochemistry confirmed for the THDs revealed that no stereochemical inversion at C-3 occurs by the DHDR reaction and that DHDR introduces 4R stereochemistry at C-4 to both (S)- and (R)-DHD.
DHDR is a novel enzyme that belongs to the short-chain dehydrogenase/reductase (SDR) family, and it has a unique catalytic tetrad pattern (Asn, Ser, and HXXXK) that is somewhat different from that of general SDR enzymes (Asn, Ser, and YXXXK) (10, 35). A catalytic tyrosine as a proton donor is a well-conserved residue in SDR enzymes, but its replacement by histidine is observed in some SDR family proteins. According to the Basic Local Alignment Search Tool (BLAST [http://www.ncbi.nlm.nih.gov/blast]) result, more than 100 proteins were aligned to have a histidine as a potential catalytic residue when the DHDR protein sequence (protein ID AFV15451.1) was used as a template.
However, there are no scientific reports about those SDR proteins having a catalytic histidine residue. In the case of DHDR from S. isoflavoniconvertens, the tyrosine of most of the known SDR family enzymes seems to be replaced with histidine, for which imidazole ring (de)protonation would be engaged in substrate reduction or oxidation. The histidine is likely a catalytic residue that can affect the optimal pH values of DHDR for its reductase and dehydrogenase activities. The optimal values are near pH 7 (i.e., 6 for reduction and 7.5 for dehydrogenation), which is similar to one of the pKa values of the amine groups in the imidazole ring of histidine. In the case of other SDR family enzymes, such as mannitol dehydrogenase from Candida magnolia or d-arabitol dehydrogenase from Gluconobacter oxydans, the optimal pHs were around 6.5 to 7.5 for reduction and 8.5 to 10 for dehydrogenation (36, 37). Therefore, the histidine residue would lower the pH optimum for dehydrogenase activity.
Interestingly, DHDR P212A lost its dehydrogenase activity at pH values above 7.0. Pro212 is likely to be positioned on the substrate binding loop, according to its alignment to Pro185 in 3αHSD from Comamonas testosteroni (32). A P185A or P185G mutation in 3αHSD makes the loop flexible, increasing product release and overall catalytic activity for 3αHSD (i.e., an increase in kcat, Km, and Ki NAD), but there was no mention of any change in enzyme stereospecificity. As a result, the P185A and P185G mutations changed the rate-limiting step from product release to hydride transfer. In the case of DHDR, the corresponding substitution resulted in different changes in the enzyme properties. The introduction of the P212A mutation caused DHDR to have decreased catalytic activity for both (R)- and (S)-DHD, resulting in no (3R,4R)-THD formation. The underlying reason for the decreased activity is not clear, as it is the opposite of the conclusion of the previous report. One possible explanation is that the hydride transfer from NADPH to DHD can be prohibited by the P212A substitution. In contrast to the case with 3αHSD, perhaps hydride transfer is the primary rate-limiting step in DHDR, and then the P212A mutation might reduce the transfer rate even further.
As a result of the improved enantioselectivity of DHDR P212A, rapid enantioselective production of (3S,4R)-THD was possible with an enzymatic method using a combination of DHDR P212A and DDRC. In addition, the enantioselective production of (3R,4R)-cis-THD was also possible by using only purified (R)-DHD substrate and the DHDR wild-type enzyme (Fig. 6C). Until now, there has been no way to prepare chiral isoflavan-4-ols through chemical and/or enzymatic synthesis (38). Therefore, this is the first report to produce (3S,4R)- and (3R,4R)-isoflavan-4-ols with a high enantiomeric excess (both above an ee of 99%).
Chiral THDs have the potential to be used as precursors for useful isoflavonoid compounds, such as (S)-equol or dehydroequol (7, 26, 39). (S)-Equol is well known for its estrogenic activity in the human body to treat postmenopausal symptoms, and a synthetic dehydroequol (or phenoxodiol) is currently undergoing phase II clinical trials as an anticancer drug for prostate cancer (40). In addition, the THDs themselves have anticancer activity toward prostate cancer cells, and trans-THD was reported to be more effective than the cis form (33). Four stereoisomers of THD, i.e., (3R,4S)-trans, (3S,4R)-trans, (3R,4R)-cis, and (3S,4S)-cis forms, have been known to be effective at suppressing vascular disease. trans-THD was reported to suppress neointimal hyperplasia by capturing superoxide anions (41), and similarly, cis-THD inhibits ERK-1 activation and proliferation in human vascular smooth muscle cells (42). However, these results were acquired with chemically synthesized THDs that had both enantiomers of each diastereomer. Therefore, the biological method to prepare enantio-pure THDs presented here might help us to understand their individual effects on vascular disease or prostate cancer in further detail.
Our goal was to develop a recombinant E. coli strain with an improved (S)-equol production capacity under aerobic conditions. Compared to native anaerobic bacterial culture and reaction systems, this recombinant system enables aerobic microbial growth and reaction, with a higher (S)-equol productivity enhanced by a DHDR mutation. The improvement in (S)-equol productivity by introducing the DHDR P212A mutation can be explained in several ways. First, the DHDR P212A mutation almost removed the dehydrogenase activity, and the reduction in DHD became dominant with the cytoplasmic pH. Second, the decrease in reductase activity toward both (S)- and (R)-DHD increased the selectivity for (S)-DHD over (R)-DHD. The increased selectivity can save the NADPH pool that should be used in DZNR or DHDR reactions for (S)-equol production.
Even though the improvements in the yield and productivity of (S)-equol were minor, this is the first report of the construction of recombinant strains for (S)-equol production. Several strategies can be suggested to further improve the (S)-equol production by recombinant strains. First, protein engineering to reverse the enantioselectivity of DZNR from the (R)-DHD preference to an (S)-DHD preference would diminish the necessity for DDRC, which would eventually relieve the metabolic burden of the heterologous expression of four recombinant genes and increase the expression levels of the other three enzymes. Second, the maximization and balancing of reducing powers in cell cytoplasm might possibly increase (S)-equol production. Since the full reactions largely depend on NADPH supplied by the cell, the coexpression of NADPH-regenerating enzymes, such as glucose dehydrogenase, or metabolic engineering for strain development to increase the NADPH concentration or to balance the concentration ratio of NAD(H) to NADP(H) would be helpful (43, 44). In addition, the identification of the rate-determining step at high daidzein concentrations and resolution of the product inhibition by systematic approaches or protein engineering should be the next goals for future work.
Supplementary Material
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03584-15.
REFERENCES
- 1.Jenks BH, Iwashita S, Nakagawa Y, Ragland K, Lee J, Carson WH, Ueno T, Uchiyama S. 2012. A pilot study on the effects of S-equol compared to soy isoflavones on menopausal hot flash frequency. J Womens Health 21:674–682. doi: 10.1089/jwh.2011.3153. [DOI] [PubMed] [Google Scholar]
- 2.Muthyala RS, Ju YH, Sheng S, Williams LD, Doerge DR, Katzenellenbogen BS, Helferich WG, Katzenellenbogen JA. 2004. Equol, a natural estrogenic metabolite from soy isoflavones. Bioorg Med Chem 12:1559–1567. doi: 10.1016/j.bmc.2003.11.035. [DOI] [PubMed] [Google Scholar]
- 3.Setchell KD, Clerici C, Lephart ED, Cole SJ, Heenan C, Castellani D, Wolfe BE, Nechemias-Zimmer L, Brown NM, Lund TD. 2005. S-Equol, a potent ligand for estrogen receptor β, is the exclusive enantiomeric form of the soy isoflavone metabolite produced by human intestinal bacterial flora. Am J Clin Nutr 81:1072–1079. [DOI] [PubMed] [Google Scholar]
- 4.Yokoyama S-I, Suzuki T. 2008. Isolation and characterization of a novel equol-producing bacterium from human feces. Biosci Biotechnol Biochem 72:2660–2666. doi: 10.1271/bbb.80329. [DOI] [PubMed] [Google Scholar]
- 5.Uchiyama S, Ueno T, Suzuki T. 2007. Identification of a newly isolated equol-producing lactic acid bacterium from the human feces. J Intest Microbiol 21:217. [Google Scholar]
- 6.Tsuji H, Moriyama K, Nomoto K, Miyanaga N, Akaza H. 2010. Isolation and characterization of the equol-producing bacterium Slackia sp. strain NATTS. Arch Microbiol 192:279–287. doi: 10.1007/s00203-010-0546-z. [DOI] [PubMed] [Google Scholar]
- 7.Kim M, Kim SI, Han J, Wang XL, Song DG, Kim SU. 2009. Stereospecific biotransformation of dihydrodaidzein into (3S)-equol by the human intestinal bacterium Eggerthella strain Julong 732. Appl Environ Microbiol 75:3062–3068. doi: 10.1128/AEM.02058-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matthies A, Blaut M, Braune A. 2009. Isolation of a human intestinal bacterium capable of daidzein and genistein conversion. Appl Environ Microbiol 75:1740–1744. doi: 10.1128/AEM.01795-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shimada Y, Takahashi M, Miyazawa N, Abiru Y, Uchiyama S, Hishigaki H. 2012. Identification of a novel dihydrodaidzein racemase essential for biosynthesis of equol from daidzein in Lactococcus sp. strain 20-92. Appl Environ Microbiol 78:4902–4907. doi: 10.1128/AEM.00410-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shimada Y, Takahashi M, Miyazawa N, Ohtani T, Abiru Y, Uchiyama S, Hishigaki H. 2011. Identification of two novel reductases involved in equol biosynthesis in Lactococcus strain 20-92. J Mol Microbiol Biotechnol 21:160–172. doi: 10.1159/000335049. [DOI] [PubMed] [Google Scholar]
- 11.Shimada Y, Yasuda S, Takahashi M, Hayashi T, Miyazawa N, Sato I, Abiru Y, Uchiyama S, Hishigaki H. 2010. Cloning and expression of a novel NADP(H)-dependent daidzein reductase, an enzyme involved in the metabolism of daidzein, from equol-producing Lactococcus strain 20-92. Appl Environ Microbiol 76:5892–5901. doi: 10.1128/AEM.01101-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsuji H, Moriyama K, Nomoto K, Akaza H. 2012. Identification of an enzyme system for daidzein-to-equol conversion in Slackia sp. strain NATTS. Appl Environ Microbiol 78:1228–1236. doi: 10.1128/AEM.06779-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schroder C, Matthies A, Engst W, Blaut M, Braune A. 2013. Identification and expression of genes involved in the conversion of daidzein and genistein by the equol-forming bacterium Slackia isoflavoniconvertens. Appl Environ Microbiol 79:3494–3502. doi: 10.1128/AEM.03693-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim M, Marsh ENG, Kim S-U, Han J. 2010. Conversion of (3S,4R)-tetrahydrodaidzein to (3S)-equol by THD reductase: proposed mechanism involving a radical intermediate. Biochemistry 49:5582–5587. doi: 10.1021/bi100465y. [DOI] [PubMed] [Google Scholar]
- 15.Tsuji H, Nomoto K, Moriyama K, Akaza H. April 2013. Equol-producing bacterium and use thereof. US patent 8420073.
- 16.Minamida K, Tanaka M, Abe A, Sone T, Tomita F, Hara H, Asano K. 2006. Production of equol from daidzein by gram-positive rod-shaped bacterium isolated from rat intestine. J Biosci Bioeng 102:247–250. doi: 10.1263/jbb.102.247. [DOI] [PubMed] [Google Scholar]
- 17.Elghali S, Mustafa S, Amid M, Manap MYABD, Ismail A, Abas F. 2012. Bioconversion of daidzein to equol by Bifidobacterium breve 15700 and Bifidobacterium longum BB536. J Funct Foods 4:736–745. doi: 10.1016/j.jff.2012.04.013. [DOI] [Google Scholar]
- 18.Uchiyama S, Ueno T, Suzuki T. February 2014. Equol-producing lactic acid bacteria-containing composition. US patent 20140050703 A1.
- 19.Zhao H, Wang XL, Zhang HL, Li CD, Wang SY. 2011. Production of dihydrodaidzein and dihydrogenistein by a novel oxygen-tolerant bovine rumen bacterium in the presence of atmospheric oxygen. Appl Microbiol Biotechnol 92:803–813. doi: 10.1007/s00253-011-3278-3. [DOI] [PubMed] [Google Scholar]
- 20.Park H-Y, Kim M, Han J. 2011. Stereospecific microbial production of isoflavanones from isoflavones and isoflavone glucosides. Appl Microbiol Biotechnol 91:1173–1181. doi: 10.1007/s00253-011-3310-7. [DOI] [PubMed] [Google Scholar]
- 21.Zheng L. 2004. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res 32:e115. doi: 10.1093/nar/gnh110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 23.Yun H, Choi HL, Fadnavis NW, Kim BG. 2005. Stereospecific synthesis of (R)-2-hydroxy carboxylic acids using recombinant E. coli BL21 overexpressing YiaE from Escherichia coli K12 and glucose dehydrogenase from Bacillus subtilis. Biotechnol Prog 21:366–371. [DOI] [PubMed] [Google Scholar]
- 24.Yun H, Yang Y-H, Cho B-K, Hwang B-Y, Kim B-G. 2003. Simultaneous synthesis of enantiomerically pure (R)-1-phenylethanol and (R)-α-methylbenzylamine from racemic α-methylbenzylamine using ω-transaminase/alcohol dehydrogenase/glucose dehydrogenase coupling reaction. Biotechnol Lett 25:809–814. doi: 10.1023/A:1023500406897. [DOI] [PubMed] [Google Scholar]
- 25.Bae J, Park B, Jung E, Lee P-G, Kim B-G. 2014. fadD deletion and fadL overexpression in Escherichia coli increase hydroxy long-chain fatty acid productivity. Appl Microbiol Biotechnol 98:8917–8925. doi: 10.1007/s00253-014-5974-2. [DOI] [PubMed] [Google Scholar]
- 26.Joannou GE, Kelly GE, Reeder AY, Waring M, Nelson C. 1995. A urinary profile study of dietary phytoestrogens. The identification and mode of metabolism of new isoflavonoids. J Steroid Biochem Mol Biol 54:167–184. doi: 10.1016/0960-0760(95)00131-I. [DOI] [PubMed] [Google Scholar]
- 27.Padan E, Zilberstein D, Rottenberg H. 1976. The proton electrochemical gradient in Escherichia coli cells. Eur J Biochem 63:533–541. doi: 10.1111/j.1432-1033.1976.tb10257.x. [DOI] [PubMed] [Google Scholar]
- 28.Salmond CV, Kroll RG, Booth IR. 1984. The effect of food preservatives on pH homeostasis in Escherichia coli. J Gen Microbiol 130:2845–2850. [DOI] [PubMed] [Google Scholar]
- 29.Slonczewski JL, Rosen BP, Alger JR, Macnab RM. 1981. pH homeostasis in Escherichia coli: measurement by 31P nuclear magnetic resonance of methylphosphonate and phosphate. Proc Natl Acad Sci U S A 78:6271–6275. doi: 10.1073/pnas.78.10.6271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zilberstein D, Agmon V, Schuldiner S, Padan E. 1984. Escherichia coli intracellular pH, membrane potential, and cell growth. J Bacteriol 158:246–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilks JC, Slonczewski JL. 2007. pH of the cytoplasm and periplasm of Escherichia coli: rapid measurement by green fluorescent protein fluorimetry. J Bacteriol 189:5601–5607. doi: 10.1128/JB.00615-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hwang CC, Chang YH, Lee HJ, Wang TP, Su YM, Chen HW, Liang PH. 2013. The catalytic roles of P185 and T188 and substrate-binding loop flexibility in 3α-hydroxysteroid dehydrogenase/carbonyl reductase from Comamonas testosteroni. PLoS One 8:e63594. doi: 10.1371/journal.pone.0063594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wähälä K, Koskimies JK, Mesilaakso M, Salakka AK, Leino TK, Adlercreutz H. 1997. The synthesis, structure, and anticancer activity of cis- and trans-4′,7-dihydroxyisoflavan-4-ols. J Org Chem 62:7690–7693. doi: 10.1021/jo970892u. [DOI] [Google Scholar]
- 34.Kim M, Won D, Han J. 2010. Absolute configuration determination of isoflavan-4-ol stereoisomers. Bioorg Med Chem Lett 20:4337–4341. doi: 10.1016/j.bmcl.2010.06.074. [DOI] [PubMed] [Google Scholar]
- 35.Filling C, Berndt KD, Benach J, Knapp S, Prozorovski T, Nordling E, Ladenstein R, Jörnvall H, Oppermann U. 2002. Critical residues for structure and catalysis in short-chain dehydrogenases/reductases. J Biol Chem 277:25677–25684. doi: 10.1074/jbc.M202160200. [DOI] [PubMed] [Google Scholar]
- 36.Lee J-K, Koo B-S, Kim S-Y, Hyun H-H. 2003. Purification and characterization of a novel mannitol dehydrogenase from a newly isolated strain of Candida magnoliae. Appl Environ Microbiol 69:4438–4447. doi: 10.1128/AEM.69.8.4438-4447.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheng H, Jiang N, Shen A, Feng Y. 2005. Molecular cloning and functional expression of d-arabitol dehydrogenase gene from Gluconobacter oxydans in Escherichia coli. FEMS Microbiol Lett 252:35–42. doi: 10.1016/j.femsle.2005.08.023. [DOI] [PubMed] [Google Scholar]
- 38.Jokela T. 2011. Synthesis of reduced metabolites of isoflavonoids, and their enantiomeric forms. Ph.D. thesis. University of Helsinki, Department of Chemistry, Laboratory of Organic Chemistry, Helsinki, Finland. [Google Scholar]
- 39.Wang X-L, Shin K-H, Hur H-G, Kim S-I. 2005. Enhanced biosynthesis of dihydrodaidzein and dihydrogenistein by a newly isolated bovine rumen anaerobic bacterium. J Biotechnol 115:261–269. doi: 10.1016/j.jbiotec.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 40.Gibney G, Elfiky A, Bussom S, Hoimes C, Burns A, McDonough J, Rowen E, Cheng Y, Kelly W. 2010. A phase II study of oral phenoxodiol in castrate and noncastrate prostate cancer patients with associated cytokine changes, poster. 2010 ASCO Annu Meet, abstr no. 4661. [Google Scholar]
- 41.Kanellakis P, Nestel P, Bobik A. 2004. Angioplasty-induced superoxide anions and neointimal hyperplasia in the rabbit carotid artery: suppression by the isoflavone trans-tetrahydrodaidzein. Atherosclerosis 176:63–72. doi: 10.1016/j.atherosclerosis.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 42.Ling S, Dai A, Williams MR, Husband AJ, Nestel PJ, Komesaroff PA, Sudhir K. 2004. The isoflavone metabolite cis-tetrahydrodaidzein inhibits ERK-1 activation and proliferation in human vascular smooth muscle cells. J Cardiovasc Pharmacol 43:622–628. doi: 10.1097/00005344-200405000-00003. [DOI] [PubMed] [Google Scholar]
- 43.Kataoka M, Sri Rohani LP, Wada M, Kita K, Yanase H, Urabe I, Shimizu S. 1998. Escherichia coli transformant expressing the glucose dehydrogenase gene from Bacillus megaterium as a cofactor regenerator in a chiral alcohol production system. Biosci Biotechnol Biochem 62:167–169. doi: 10.1271/bbb.62.167. [DOI] [PubMed] [Google Scholar]
- 44.Chemler JA, Fowler ZL, McHugh KP, Koffas MAG. 2010. Improving NADPH availability for natural product biosynthesis in Escherichia coli by metabolic engineering. Metab Eng 12:96–104. doi: 10.1016/j.ymben.2009.07.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.