

Abstract
Streptomyces spp. are highly differentiated actinomycetes with large, linear chromosomes that encode an arsenal of biologically active molecules and catabolic enzymes. Members of this genus are well equipped for life in nutrient-limited environments and are common soil saprophytes. Out of the hundreds of species in the genus Streptomyces, a small group has evolved the ability to infect plants. The recent availability of Streptomyces genome sequences, including four genomes of pathogenic species, provided an opportunity to characterize the gene content specific to these pathogens and to study phylogenetic relationships among them. Genome sequencing, comparative genomics, and phylogenetic analysis enabled us to discriminate pathogenic from saprophytic Streptomyces strains; moreover, we calculated that the pathogen-specific genome contains 4,662 orthologs. Phylogenetic reconstruction suggested that Streptomyces scabies and S. ipomoeae share an ancestor but that their biosynthetic clusters encoding the required virulence factor thaxtomin have diverged. In contrast, S. turgidiscabies and S. acidiscabies, two relatively unrelated pathogens, possess highly similar thaxtomin biosynthesis clusters, which suggests that the acquisition of these genes was through lateral gene transfer.
INTRODUCTION
The genus Streptomyces has a huge coding capacity with hundreds of described species and large linear chromosomes (1–5). Large genomes are consistent with the saprophytic lifestyle, varied environmental niches, and developmentally complex growth exhibited by these filamentous actinobacteria. The linear chromosomes of Streptomyces spp. have syntenic central regions and less conserved chromosome arms (1–6). Most biosynthetic pathways for secondary metabolites reside in the chromosome arms. Streptomycetes synthesize structurally diverse secondary metabolites with antimicrobial, immunosuppressant, antitumor, and other pharmaceutically valuable properties (1, 7). In situ production of these molecules is believed to enhance growth, survival, and reproduction through antibiosis, signaling, metabolic homeostasis, and other mechanisms (1, 3–5).
The genus Streptomyces is overwhelmingly saprophytic, and to date, genomic analyses have been largely limited to such species. Research on model saprophytic Streptomyces species such as S. coelicolor, S. griseus, S. avermitilis, and others has provided a wealth of information on metabolic and regulatory processes (3–5). The pathogenic phenotype occurs in at least a dozen characterized species that infect plants (8) and in a few species that infect humans (9, 10). Molecular genetic analysis of pathogenicity in Streptomyces animal pathogens is lacking, but a substantial amount of information on determinants of virulence in plant pathogens is now available (8). The best-studied plant-pathogenic Streptomyces spp. are S. scabies, S. ipomoeae, S. turgidiscabies, and S. acidiscabies; all are economically important pathogens of tuber and/or root crops (8).
Plant-pathogenic Streptomyces species produce a dipeptide phytotoxin, thaxtomin, which is the main virulence factor of the group (11). While S. scabies, S. acidiscabies, and S. turgidiscabies produce thaxtomin A, S. ipomoeae synthesizes a slightly different toxin, called thaxtomin C (12). Moreover, isolation of S. ipomoeae has been limited to diseased sweet potato cultivars, suggesting a niche specificity distinct to this streptomycete (12). The emergence of plant pathogenicity in this genus has occurred multiple times in agricultural systems (8, 13). This process appears to involve LGT (lateral gene transfer) of pathogenicity islands (PAIs), including a large mobile island that has been characterized in S. turgidiscabies (PAISt) (14, 15).
Comparative genomics is a powerful strategy for revealing physiological, ecological, and evolutionary attributes of a taxon. In this study, we conducted comparative genomic analyses for the purpose of describing the set of genes that distinguish plant-pathogenic from saprophytic Streptomyces species (i.e., to define the pathogen-specific genome [PSG]). We also conducted phylogenetic analysis to probe the evolutionary history of plant pathogenicity within the genus.
MATERIALS AND METHODS
Genome data source.
S. turgidiscabies strain Car8 and S. ipomoeae strain 91-03 have been deposited in the USDA-ARS Culture Collection (Peoria, IL). We sequenced the genomes of S. turgidiscabies Car8 and S. ipomoeae 91-03 using Sanger technology. Sequencing of small-insert (4-5 kb) and medium-insert (10 to 12 kb) plasmid libraries was used to generate sequence reads. Sequences were assembled by using Celera Assembler v. 4.1 (16), while Glimmer v. 3.02 (17) was used to predict coding sequences (CDSs). Prediction of tRNAs was performed by using tRNAscan-SE v. 1.4 (18). Genome sequences of S. acidiscabies 104-84, S. scabies 87-22, and 10 saprophytic Streptomyces spp. were retrieved from GenBank. GenBank accession numbers and descriptions of these genome sequences are provided in Table 1.
TABLE 1.
Streptomyces genome sequences used in this study and their general features and accession numbers
| Organism | Length (Mb) | Sequence status | Reference(s) | GenBank accession no. |
|---|---|---|---|---|
| S. turgidiscabies Car8 | 10.8 | Draft | This study | NZ_AEJB00000000 |
| S. ipomoeae 91-03 | 10.4 | Draft | This study | NZ_AEJC00000000 |
| S. scabies 87.22 | 10.1 | Complete | 41, 51 | NC_013929 |
| S. acidiscabies 84-104 | 11.0 | Draft | 2 | NZ_AHBF00000000 |
| S. avermitilis MA 4680 | 9.1 | Complete | 5 | BA000030 |
| AP005645 | ||||
| S. griseus IFO 13350 | 8.5 | Complete | 3 | NC_010572 |
| S. bingchenggensis BCW-1 | 11.9 | Complete | 52 | CP002047 |
| S. coelicolor A3(2) | 9.0 | Complete | 4 | AL645882 |
| AL589148 | ||||
| AL645771 | ||||
| S. hygroscopicus subsp. jinggangensis | 10.4 | Complete | 53 | NC_017765 |
| NC_017766 | ||||
| NC_016972 | ||||
| S. violaceusniger Tu 4113 | 11.2 | Complete | —a | NC_015957 |
| NC_015951 | ||||
| NC_015952 | ||||
| S. flavogriseus ATCC33331 | 7.6 | Complete | —a | CP002475 |
| CP002477 | ||||
| S. cattleya DMS 46488 | 6.2 | Complete | 54 | FQ859185 |
| 1.8 | FQ859184 | |||
| S. venezuelae ATCC 10712 | 8.2 | Complete | FR845719 | |
| Streptomyces sp. strain sirexAA-E | 7.4 | Complete | —a | NC_015953 |
| Saccharopolyspora erythraea NRRL2338 | 8.2 | Complete | 55 | NC_009142 |
Genome sequences submitted directly to GenBank.
Gene content analysis.
Protein-coding sequences were obtained from the genomes of the 14 Streptomyces species and the Saccharopolyspora erythraea strain NRRL2338 outgroup (Table 1). Ortholog groups were determined by using the OrthoMCL v. 1.4 program (19). The OrthoMCL program executes two main procedures. First, it carries out reciprocal comparisons of each predicted protein using the Basic Local Alignment Search Tool (BLAST) (20). In a second step, OrthoMCL uses the reciprocal E values generated from the BLAST output and creates a matrix that is analyzed by a Markov cluster algorithm (MCL) (19). As a result of this analysis, OrthoMCL detects ortholog and paralog genes and clusters them into groups (ortholog groups). OrthoMCL was run with a BLAST E value cutoff of 10−10, a percent match cutoff of 50, and an inflation rate of 1.5. The output was used to construct a gene content table that contains common and unique genes for each genome.
Proteins that were smaller than 50 amino acids were not included in the analysis. The gene content dendrogram was constructed in two steps. First, the R package Vegan (21) was used to calculate the similarity between genomes using the Jaccard coefficient (JC). To define the distance between genomes and conduct hierarchical clustering, we used 1 minus the JC, using the unweighted pair group method with arithmetic mean (UPGMA) algorithm, implemented in the fastcluster R package (22).
Phylogenetic tree reconstruction.
A total of 1,000 ortholog groups were selected for analysis based on the criteria that their members were present at a single copy per genome, were conserved in all 14 Streptomyces genomes, and were outgrouped within S. erythraea. Nucleotide sequences of each gene were aligned by using Probalign software (23). The Probalign program assigns a score based on the quality of the alignment; regions in the alignments with quality scores of <60% were excluded. The remaining regions were translated to amino acid sequences. A final back-translation of each gene was conducted to obtain alignments at the nucleotide level. These final alignments were used to reconstruct the phylogenetic trees.
Phylogenetic trees were constructed by maximum likelihood with PhyML software (24), using the general time-reversible plus gamma (GTR+G) substitution model (25). Bootstrap support (100 replicates) for each tree was determined by using PhyML with the nearest-neighbor interchange (NNI) branch-swapping method. We summarized all of the trees in a majority-rule consensus using the Sumtrees program in the DendroPy package (26). The branch lengths of the consensus tree were set to the mean of the branch lengths of all the common trees. For tree reconstruction of the thaxtomin biosynthetic cluster, we used the nucleotide sequence of each thaxtomin gene (or the portions of the txtA and txtB genes indicated in the text) and applied the same strategy described above for the common gene trees. Visualization and editing of trees were conducted with Dendroscope v. 3.0 software (27).
Recombination detection.
Alignments obtained from the common group of genes among the Streptomyces species were used to detect recombination signals. Recombination was detected by using three methods, maximum chi-square (MaxChi) (28), neighbor similarity score (NSS) (29), and pairwise homoplasy index (Phi), included in the PhiPack (30) software package (http://www.maths.otago.ac.nz/∼dbryant/software.html). We used a cutoff P value of ≤0.05 to consider a recombination signal significant.
Annotation and categorization of genes.
Both common and unique genes in pathogenic Streptomyces strains were annotated de novo by using Blast2Go software (31). The Gene Ontology database (AMIGO) (32), Pfam database (33), and Clusters of Orthologs Groups (COG) database (34) were used as references. SignalP v. 4.0 (35) was used to predict secreted proteins.
Nucleotide sequence accession numbers.
The draft genomes of S. turgidiscabies Car8 and S. ipomoeae 91-03 have been deposited in GenBank under accession numbers NZ_AEJB00000000 and NZ_AEJC00000000, respectively.
RESULTS AND DISCUSSION
Genome descriptions of S. turgidiscabies Car8 and S. ipomoeae 91-3.
The draft genome sequence of S. turgidiscabies Car8 comprises 10,825,282 bp, with 7-fold average coverage. The genome was assembled into 692 contigs with an N50 (i.e., median contig size) of 27 kb; the largest contig was 717,473 bp. With a GC content of 69.87%, the genome of S. turgidiscabies Car8 is predicted to harbor 10,069 genes. The draft genome sequence of S. ipomoeae 91-03 comprises 10,403,856 bp with 8-fold average coverage. The genome was assembled into 687 contigs with an N50 of 26 kb, and the largest contig was 130 kb. S. ipomoeae 91-03 is predicted to harbor 9,485 genes with an average GC content of 70.17% for the entire genome sequence.
Gene content analysis differentiates pathogenic from saprophytic Streptomyces spp.
In order to learn more about how genome content relates to pathogenicity, the genome sequences of S. turgidiscabies Car8 and S. ipomoeae 91-03 were compared with those of two previously sequenced pathogens, S. scabies 87-22 and S. acidiscabies 84-104, and 10 saprophytic Streptomyces spp. (Table 1). Interestingly, there was no evidence of genome reduction among pathogenic species; all four pathogen genomes are >10 Mb, while the genomes of nonpathogens range from 7.4 Mb to 11.9 Mb (Table 1). These data suggest that plant-pathogenic Streptomyces spp. have retained the sequences that allow them to live as saprophytes in the soil. This hypothesis is consistent with the fact that plant-pathogenic streptomycetes are not obligate intercellular pathogens and can survive in soil apart from their host plants (reviewed in reference 8).
The OrthoMCL program yielded 14,178 ortholog groups and 10,288 single taxon-specific genes (orphans) (see Data Set S1 in the supplemental material). Furthermore, the gene content table contains ortholog groups with many gene copies per genome (putative paralogs). These were identified as polyketide synthases, nonribosomal peptide synthases (NRPSs), and transposases (see Data Set S1 in the supplemental material).
Hierarchical clustering was used to construct a dendrogram based on the presence or absence of ortholog genes in saprophytes and pathogens. This analysis grouped the 14 strains into four clusters (Fig. 1). All four pathogens fell into cluster III and are clearly distinct from the saprophytic strains, which are distributed in the other three clusters (Fig. 1). Based on gene content, S. ipomoeae and S. scabies are more closely related to each other than they are to S. turgidiscabies and S. acidiscabies.
FIG 1.

Gene content dendrogram. Shown are phylogenetic relationships of 14 streptomycete strains based on gene content. The dendrogram was constructed by UPGMA clustering based on similarities in gene content for strains (cophenetic correlation coefficient of 0.95). Horizontal bars represent dissimilarities, measured by the Jaccard coefficient, which range from 0 to 1. Pathogenic Streptomyces spp. are highlighted in red. The gene content of Saccharopolyspora erythraea strain NRRL2338 was used as an outgroup.
We characterized the set of ortholog groups in individual pathogen genomes that were not shared across the saprophytic strains. These groups of orthologs comprise 1,528 orthologs in S. ipomoeae, 1,778 orthologs in S. turgidiscabies, 1,186 orthologs in S. scabies, and 1,332 orthologs in S. acidiscabies (Fig. 2). These combined genes comprise the pathogen-specific genome (PSG), which consists of 4,662 distinct ortholog groups. Within the PSG, there are only 63 orthologs shared by all four pathogens (Fig. 2), while 61 to 164 orthologs are shared by pairs of pathogenic strains. S. scabies and S. ipomoeae share the most orthologs (164), and this relatively large number is consistent with their overall shared gene content.
FIG 2.
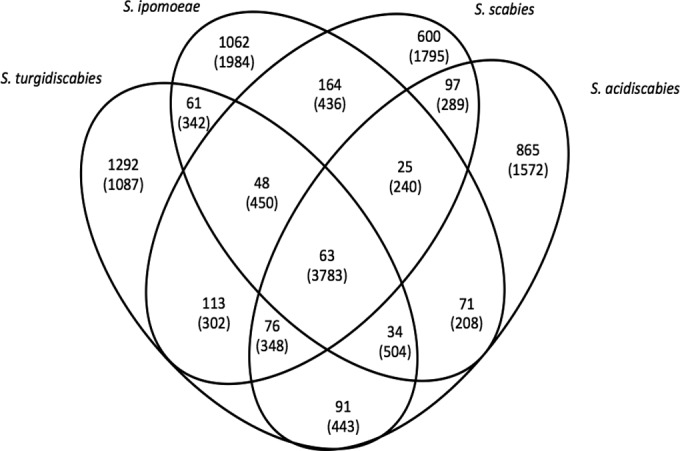
Distribution of pathogen-specific genes. The Venn diagram shows the distribution of the pathogen-specific ortholog groups among the four pathogenic Streptomyces spp. The numbers of ortholog groups not specific to the pathogens (i.e., present in the indicated pathogen[s] and one or more saprophytic species) are shown in parentheses.
Based on functional annotation using the COG database, 60% of the genes in the PSG do not have an identifiable function or protein motif. Most of these genes are taxon specific and are categorized as encoding hypothetical proteins (see Fig. S1 in the supplemental material). A further 2% of the genes in the PSG code for amino acid motifs or domains described in proteins in the COG database but lack a putative biological process. There are 1,097 functionally annotated genes in the PSG (24% of the total). Within this group, 191 genes were assigned to general function categories (17%), 175 genes code for proteins involved in transcriptional regulation (16%), 122 genes are related to carbohydrate transport and metabolism (11%), and 101 genes were assigned to signal transduction (9%) (see Fig. S1 in the supplemental material).
Signal sequence prediction suggests that 416 (9%) of the genes in the PSG code for secreted proteins. Among these are many catabolic proteins with functions consistent with the breakdown of plant polymers. All four pathogens contain pectate lyases that are specific to pathogens (STRIP9103_04917 in S. ipomoeae, STRTUCAR8_05454 in S. turgidiscabies, SCAB44901 in S. scabies, and Saci8_010100007963 in S. acidiscabies). Interestingly, S. turgidiscabies and S. acidiscabies contain pathogen-specific cellulases (STRTUCAR8_03570 and Saci8_010100000665, respectively). A group of pathogen-specific genes that encode lipases and esterases was found in the PSG (STRTUCAR8_09187, STRIP9103_00134, STRIP9103_00133, SCAB22851, Saci8_010100046927, Saci8_010100046932, and Saci8_010100014815). Another group of genes encoding cutinases is included in the PSG (STRIP9103_01052 in S. ipomoeae, STRTUCAR8_01086 in S. turgidiscabies, and SCAB78931 in S. scabies). Consistent with the inclusion of the cutinase group in the PSG, the SCAB78931 gene was recently shown to be present in numerous strains of S. scabies as well as one strain of the pathogen Streptomyces bottropensis but was absent in all of the nonpathogenic Streptomyces species analyzed (36).
Pectate lyases, cellulases, and lipases are virulence factors in other plant pathogens (37). Pectate lyases have been described in the plant-pathogenic bacterium Erwinia chrysanthemi. They play an important role in the process of infection and are able to macerate plant tissue, thereby creating conditions favoring colonization (38, 39). The role of lipases has been demonstrated in Xanthomonas oryzae pv. oryzae and in the emerging pathogen Burkholderia glumae, both pathogens of rice. Our data suggest that pathogenic Streptomyces species may also use a specific set of proteins to degrade plant cell components.
S. ipomoeae and S. acidiscabies share orthologs coding for a pertussis toxin subunit-like protein, STRIP9103_04682, and a rapid alkalization factor, Saci8_010100005203. The resulting proteins are predicted to be secreted, and they do not have a blast hit for related actinobacteria in GenBank. A protein with these motifs has been described in the plant-associated bacterium Pseudomonas synxantha strain BG33R (PsBG33R). The proteins in S. ipomoeae and S. acidiscabies share 31% identity with the ortholog in PsBG33R. Interestingly, in PsBG33R, the pertussis toxin subunit-like protein is encoded within a genomic island that resembles an integrative conjugative element (ICE) (40). The role of this protein in PsBG33R is unknown, and its function is an important topic of future research.
S. turgidiscabies Car8 contains two copies of the tomA cluster.
Previous studies determined that S. turgidiscabies encodes the α-tomatine-detoxifying enzyme TomA and that tomA is linked to a cluster of glycoside hydrolases (tomA cluster) (41). This tomA cluster is located within the mobile genomic island PAISt, and it has homologs within nonmobile yet syntenic PAIs in S. scabies and S. acidiscabies (2, 14). Furthermore, a homologous tomA cluster was previously identified in the plant pathogen Clavibacter michiganensis subsp. michiganensis (42). These data suggest that the tomA clusters in these Gram-positive organisms have a common origin and that they have been subjected to LGT.
Upon analysis of the draft genome of S. turgidiscabies here, we detected two copies of the tomA cluster (Fig. 3). Interestingly, the previously described (41) copy (i.e., tomA cluster 1) in the PAI in S. turgidiscabies shares higher identity with the tomA cluster in S. scabies (98%) than does the second copy (tomA cluster 2), which is not harbored by the PAI (87%). It is noteworthy that tomA cluster 2 lacks any remnant of open reading frames (ORFs) 1374 and 1375 (Fig. 3), which encode a transcriptional regulator and a cutinase, respectively; instead, this region of the cluster encodes a putative hypothetical protein.
FIG 3.

Phylogenetic relationships and synteny analysis of the tomA clusters in plant-pathogenic Streptomyces spp. On the left is the topology of the tree constructed from the concatenated nucleotide sequences of genes in the 15-kb tomA cluster. The Clavibacter michiganensis subsp. michiganensis tomA cluster was used as the outgroup. Labels in the genes indicate the names of the genes or the locus tags for each species. Orange, tomA (tomatinase) and blgA (beta-glucosidase); green, tetR (transcriptional regulator); red, ABC transporter system; blue (from left to right), beta-glucosidase and glycosyl hydrolase. Gray open reading frames 1374 and 1375 in tomA cluster 1 encode a transcriptional regulator and a putative cutinase, respectively. The gray arrow in tomA cluster 2 represents a hypothetical protein. The gray arrow in the tomA cluster in Clavibacter michiganensis subsp. michiganensis represents a transcriptional regulator. Pink areas designate syntenic gene regions, and only these regions were used in the phylogenetic analysis.
Sequence comparison and phylogenetic analysis of all of the tomA clusters found in plant-pathogenic streptomycetes (Fig. 3) suggest two possible explanations for the appearance of multiple copies in S. turgidiscabies. One possibility is that the two copies of the tomA cluster in S. turgidiscabies were acquired at different times by independent LGT events. Alternatively, it is possible that a duplication of tomA cluster 1 occurred at some point and that the duplicated copy, tomA cluster 2, has been subjected to a higher mutation rate. If the latter scenario is accurate, then it is interesting that a similar duplication of the tomA cluster has not been observed for either S. scabies or S. acidiscabies. Meanwhile, we found no evidence here of a tomA cluster within the S. ipomoeae 91-03 genome.
Phylogeny of plant-pathogenic Streptomyces.
The gene content table indicates that there are 1,984 orthologs that are conserved among the 14 Streptomyces species (core genome). Among this group, 1,000 ortholog groups were selected to reconstruct phylogenetic trees. The selection rationale is described in Materials and Methods. A majority-rule consensus tree shows five well-supported clades (i.e., at least 70% of the individual trees): clades STRA, STRA1, STRB, STRB1, and STRC (Fig. 4). Notably, the pathogenic Streptomyces species are clustered with three saprophytic species (S. coelicolor, S. hygroscopicus, and S. avermitilis) in clade STRB. However, the only subcluster that is strongly supported is clade STRB1, which contains the pathogenic species S. scabies and S. ipomoeae. Our analysis suggests that despite the weak resolution of some clades of the phylogenetic tree, it is likely that S. scabies and S. ipomoeae have a common phylogenetic history. A previous study (43) was not able to resolve this phylogenetic relationship using 16S rRNA and several housekeeping gene sequences. However, we demonstrate here that 77% of the genes in the core genome tree support the hypothesis that S. scabies and S. ipomoeae are in a single cluster with a common ancestor (Fig. 4).
FIG 4.
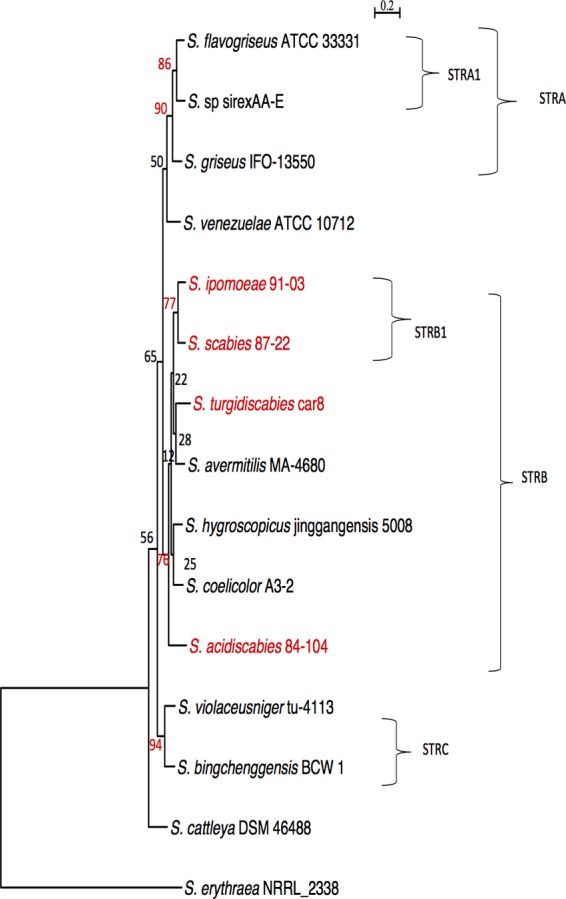
Consensus supertree inferred from common genes. The 1,000 individual gene trees are summarized in this consensus supertree. The percentages of genes that support the branches of the tree are indicated in red. The horizontal bar at the top represents substitutions per nucleotide site. Pathogenic Streptomyces strains are highlighted in red. Sequences of Saccharopolyspora erythraea NRRL2338 were used as outgroups in the analyses. Five well-supported clades, designated STRA, STRA1, STRB, STRB1, and STRC, are indicated.
Recombination may cause incongruent topologies in comparisons of phylogenetic trees and may preclude the resolution of the consensus tree. To determine if this is the case here, we tested the level of recombination affecting the Streptomyces species used in this study. We used three algorithms that are based on nucleotide substitution: MaxChi, NSS, and Phi (see Materials and Methods for more details). When a P value cutoff of 0.05 was used, recombination signals were detected in 1,598 genes with at least one of the three methods (see Fig. S2 in the supplemental material). Moreover, recombination was detected in 1,061 genes with two or more of the methods. This finding indicates that recombination has occurred in at least 53% of the 1,984 orthologs composing the core genome.
Gene content analysis reveals complex evolution of the thaxtomin biosynthetic pathway.
The production of a member of the thaxtomin family is a hallmark of pathogenesis in the genus Streptomyces, and plant-pathogenic streptomycetes are the only organisms known to produce this family of molecules (11). Phylogenetic analysis of individual genes in the thaxtomin biosynthetic cluster allowed us to infer the relation of the clusters in the four pathogens. Trees constructed from the TxtE, TxtD, and TxtR sequences, together with the condensation and AMP-binding domains of TxtA and TxtB, show robust bootstrap support (see Fig. S3 in the supplemental material). A majority consensus tree of the thaxtomin components (Txt tree) indicates that the thaxtomin clusters in S. scabies 87-22, S. acidiscabies 84-104, and S. turgidiscabies Car8, but not S. ipomoeae 91-03, are highly similar (Fig. 5 and 6). Furthermore, the Txt tree contradicts the close relationship of S. ipomoeae 91-3 and S. scabies 87-22 observed in the core genome consensus tree and the gene content dendrogram, a result which suggests that the thaxtomin clusters in S. scabies and S. ipomoeae have diverged. It is possible that such divergence has been driven, at least in part, by the development of the niche specificity described above for S. ipomoeae.
FIG 5.

Gene content analysis of the thaxtomin biosynthetic cluster. The thaxtomin biosynthetic operon of each pathogen is depicted. Orthologs of the txtD and txtE genes are found in S. lavendulae and Streptomyces sp. Mg1. The GC content is depicted at the top of each cluster (upward peaks in gold indicate above-average GC content, and downward peaks in purple designate below-average GC content). S. scabies and S. acidiscabies clusters are identical and depicted only once. Genes in blue are predicted insertion sequences; truncated coding regions that might represent pseudogenes are in gray.
FIG 6.

Comparison of topologies derived from the common gene phylogenic tree (core tree) and the gene content (Gcont) tree with the thaxtomin tree (Txt tree). The top diagram shows the general structure of the thaxtomin biosynthesis cluster. Depicted within the txtA and txtB genes are the relative positions of the encoded AMP-binding adenylation (A), methyltransferase (M), peptidyl carrier protein (P), and condensation (C) domains. Asterisks denote the entire txt genes or domain regions of txtA and txtB used to construct the trees. The txtC sequence was not included in the analysis because it is absent in S. ipomoeae.
The high similarity of the thaxtomin cluster in three pathogenic taxa that are more phylogenetically distant from each other strongly suggests a process of LGT that included S. scabies, S. acidiscabies, and S. turgidiscabies but not S. ipomoeae. This model is supported by previous studies that demonstrated the presence of a large mobile pathogenicity island in S. turgidiscabies Car8, which contains the thaxtomin cluster and which is located at a chromosomal site that is different from the location of the thaxtomin locus in S. scabies (15, 44). Moreover, the excision of the entire thaxtomin cluster from the chromosome of S. scabies 87-22 has been demonstrated experimentally (45). It is possible that the acquisition of the thaxtomin clusters by S. acidiscabies and S. turgidiscabies on a contemporary time scale explains their relatively recent emergence as plant pathogens in the United States and Japan, respectively (46, 47).
Gene content analysis also reveals that the current structure of the thaxtomin cluster is the result of multiple recombination processes. The presence of insertion sequences within the thaxtomin pathway of all the pathogens suggests that transposition events have been critical to the evolution of some of the elements of the pathway (Fig. 5). This is clearly the case in the analysis of the sequence of the pathway-specific regulator txtR. Orthologs of txtR do not occur in the genomes of saprophytic Streptomyces species; the best hit found in GenBank corresponds to the distantly related actinobacterium Arthrobacter sp. strain FB24 (WP_011693329), with 34% identity. Furthermore, the average GC content of txtR is 56%, in contrast to an average of 72% in Streptomyces genomes. The presence of a putative ortholog in Arthrobacter sp. FB24 and the low GC content of the txtR gene suggest acquisition of txtR by LGT.
The domain organization of the NRPSs TxtA and TxtB follows a canonical pattern for such proteins. Both synthetases belong to a large ortholog group that is conserved within all 14 Streptomyces strains analyzed in this study (see Data Sets S1 and S2 in the supplemental material). Additional amino acid sequence inspection, however, reveals that TxtA and TxtB differ from most NRPSs evaluated in this study in that TxtA and TxtB contain a methyltransferase domain nested within the C terminus of the AMP-binding domain (Fig. 7). All four pathogens have TxtA containing methyltransferase domain type 12 (InterPro accession number IPR013217), while three of the four pathogens, S. scabies, S. turgidiscabies, and S. acidiscabies, have TxtB containing methyltransferase domain type 11 (InterPro accession number IPR013216). In S. ipomoeae, TxtB contains methyltransferase domain type 12 (Fig. 7; see also Data Set S2 in the supplemental material).
FIG 7.

Architecture of domains in the NRPSs TxtA and TxtB. NRPSs contain four conserved domains: adenylation (AMP), methyltransferase (MET), peptidyl carrier protein (PP), and condensation (COND). In the cases of TxtA (top) and TxtB (bottom), the MET domain is embedded within the C-terminal region of the AMP domain. TxtA of all 4 Streptomyces plant pathogens and TxtB of S. ipomoeae contain the MET12 (methyltransferase type 12) domain, while TxtB in S. scabies, S. acidiscabies, and S. turgidiscabies contains the methyltransferase type 11 domain. Alignments and conserved residues are shown in blue.
Inspection of domains of NRPSs in the saprophytic group indicates that while the nested methyltransferase type 12 domain is found in NRPSs of other saprophytic Streptomyces species, methyltransferase domain type 11 is almost entirely absent in these saprophytes. Furthermore, a BLAST search of the GenBank nonredundant database with TxtB as a query indicates that very few NRPSs show methyltransferase domain type 11. Among the BLAST hits were TxtB orthologs (GenBank accession numbers WP_046706290.1 and WP_046912674.1) for two additional recently sequenced plant pathogens, Streptomyces europaeiscabiei (GenBank accession number NZ_LCTL00000000.1) and Streptomyces stelliscabiei (GenBank accession number NZ_LBNW00000000.1), respectively. Also, Streptomyces sp. strain AA1529 has a NRPS (accession number WP_020699829.1) with this nested domain; however, the product of the NRPS in this saprophyte is unknown. It is tempting to speculate that the presence of the rare methylation domain found in TxtB of S. scabies, S. acidiscabies, and S. turgidiscabies (and S. europaeiscabiei and S. stelliscabiei) versus the more common methylation domain found in S. ipomoeae TxtB may be related to differences in thaxtomin production, since all of the former species produce thaxtomin A, which is more methylated than the thaxtomin C congener produced by S. ipomoeae.
The products of txtD and txtE conduct the nitration of the l-tryptophan moiety in thaxtomin biosynthesis (48, 49). Although orthologs of txtD also occur in the nonpathogens S. avermitilis and S. venezuelae, this gene is not linked to txtE in these genomes. However, orthologs of txtD and txtE are linked in S. lavendulae and Streptomyces sp. strain Mg1, which were not included in this study. In S. lavendulae, the cluster txtD-txtE is associated with genes that are predicted to be involved in sodium/potassium transport, while in Streptomyces strain Mg1, the txtD-txtE cluster is linked to genes of unknown function (Fig. 5). Studies of S. lavendulae have ruled out the role of txtD-txtE in the biosynthesis of the nitrated antibiotic d-cycloserine (50). These observations indicate that although txtD and txtE are not unique to thaxtomin-producing streptomycetes, their only described function is the nitration of thaxtomin.
Conclusions.
Here we present the genome sequences of S. ipomoeae 91-03 and S. turgidiscabies Car8 and a comparative analysis of four pathogenic Streptomyces species and 10 saprophytic Streptomyces species. Gene contents differed between genomes of pathogenic and saprophytic strains, which allowed us to define a PSG consisting of thousands of distinct ortholog groups. We also described the presence of multiple copies of the tomA cluster in S. turgidiscabies, which might have occurred by independent LGT events or via duplication. Our findings further emphasize the importance of LGT in the emergence of plant-pathogenic species while also providing evidence that S. scabies and S. ipomoeae are derived from a common ancestor.
Most of the individual genes of the thaxtomin cluster have orthologs in the saprophytic group; indeed, only the txtR regulator gene seems to be unique to the pathogenic group and thus a member of the PSG. Based on gene content and phylogenetic analyses, it is possible to postulate that the thaxtomin biosynthetic cluster is a composite structure and is the result of a combination of genes that are evolving and mobilizing within other Streptomyces species. The presence of similar thaxtomin clusters in S. scabies, S. acidiscabies, and S. turgidiscabies is consistent with the notion that LGT is involved in the emergence of new pathogenic species.
Although Streptomyces species are well known as successful soil saprophytes, the plant-pathogenic group has not been studied in as much detail. This study has helped to delineate their phylogenetic relationships and genome composition, with the caveat that only one strain each of the four pathogenic species was analyzed. Additional genome sequences of Streptomyces pathogens will undoubtedly lead to a better understanding of the evolution of pathogenicity in this large genus.
Supplementary Material
Funding Statement
The National Research Initiative of the U.S. Department of Agriculture (USDA) Cooperative State Research, Education, and Extension Service provided funding to Gregg S. Pettis under grant number 2007-35600-17813 and to Rosemary Loria under grant number 2010-65110-20416. The USDA National Institute of Food and Agriculture (NIFA) provided funding to Gregg S. Pettis under Hatch project number LAB94112.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03504-15.
REFERENCES
- 1.Hopwood DA. 2006. Soil to genomics: the Streptomyces chromosome. Annu Rev Genet 40:1–23. doi: 10.1146/annurev.genet.40.110405.090639. [DOI] [PubMed] [Google Scholar]
- 2.Huguet-Tapia JC, Loria R. 2012. Draft genome sequence of Streptomyces acidiscabies 84-104, an emergent plant pathogen. J Bacteriol 194:1847. doi: 10.1128/JB.06767-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ohnishi Y, Ishikawa J, Hara H, Suzuki H, Ikenoya M, Ikeda H, Yamashita A, Hattori M, Horinouchi S. 2008. Genome sequence of the streptomycin-producing microorganism Streptomyces griseus IFO 13350. J Bacteriol 190:4050–4060. doi: 10.1128/JB.00204-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bentley SD, Chater KF, Cerdeño-Tárraga A-M, Challis GL, Thomson NR, James KD, Harris DE, Quail MA, Kieser H, Harper D, Bateman A, Brown S, Chandra G, Chen CW, Collins M, Cronin A, Fraser A, Goble A, Hidalgo J, Hornsby T, Howarth S, Huang C-H, Kieser T, Larke L, Murphy L, Oliver K, O'Neil S, Rabbinowitsch E, Rajandream M-A, Rutherford K, Rutter S, Seeger K, Saunders D, Sharp S, Squares R, Squares S, Taylor K, Warren T, Wietzorrek A, Woodward J, Barrell BG, Parkhill J, Hopwood DA. 2002. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417:141–147. doi: 10.1038/417141a. [DOI] [PubMed] [Google Scholar]
- 5.Omura S, Ikeda H, Ishikawa J, Hanamoto A, Takahashi C, Shinose M, Takahashi Y, Horikawa H, Nakazawa H, Osonoe T, Kikuchi H, Shiba T, Sakaki Y, Hattori M. 2001. Genome sequence of an industrial microorganism Streptomyces avermitilis: deducing the ability of producing secondary metabolites. Proc Natl Acad Sci U S A 98:12215–12220. doi: 10.1073/pnas.211433198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ventura M, Canchaya C, Tauch A, Chandra G, Fitzgerald GF, Chater KF, van Sinderen D. 2007. Genomics of Actinobacteria: tracing the evolutionary history of an ancient phylum. Microbiol Mol Biol Rev 71:495–548. doi: 10.1128/MMBR.00005-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Challis GL, Hopwood DA. 2003. Synergy and contingency as driving forces for the evolution of multiple secondary metabolite production by Streptomyces species. Proc Natl Acad Sci U S A 100(Suppl 2):14555–14561. doi: 10.1073/pnas.1934677100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loria R, Kers J, Joshi M. 2006. Evolution of plant pathogenicity in Streptomyces. Annu Rev Phytopathol 44:469–487. doi: 10.1146/annurev.phyto.44.032905.091147. [DOI] [PubMed] [Google Scholar]
- 9.Kirby R, Sangal V, Tucker NP, Zakrzewska-Czerwinska J, Wierzbicka K, Herron PR, Chu C-J, Chandra G, Fahal AH, Goodfellow M, Hoskisson PA. 2012. Draft genome sequence of the human pathogen Streptomyces somaliensis, a significant cause of actinomycetoma. J Bacteriol 194:3544–3545. doi: 10.1128/JB.00534-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quintana ET, Wierzbicka K, Mackiewicz P, Osman A, Fahal AH, Hamid ME, Zakrzewska-Czerwinska J, Maldonado LA, Goodfellow M. 2008. Streptomyces sudanensis sp. nov., a new pathogen isolated from patients with actinomycetoma. Antonie Van Leeuwenhoek 93:305–313. doi: 10.1007/s10482-007-9205-z. [DOI] [PubMed] [Google Scholar]
- 11.Loria R, Bignell DRD, Moll S, Huguet-Tapia JC, Joshi MV, Johnson EG, Seipke RF, Gibson DM. 2008. Thaxtomin biosynthesis: the path to plant pathogenicity in the genus Streptomyces. Antonie Van Leeuwenhoek 94:3–10. doi: 10.1007/s10482-008-9240-4. [DOI] [PubMed] [Google Scholar]
- 12.Guan D, Grau BL, Clark CA, Taylor CM, Loria R, Pettis GS. 2012. Evidence that thaxtomin C is a pathogenicity determinant of Streptomyces ipomoeae, the causative agent of Streptomyces soil rot disease of sweet potato. Mol Plant Microbe Interact 25:393–401. doi: 10.1094/MPMI-03-11-0073. [DOI] [PubMed] [Google Scholar]
- 13.Bukhalid RA, Takeuchi T, Labeda D, Loria R. 2002. Horizontal transfer of the plant virulence gene, nec1, and flanking sequences among genetically distinct Streptomyces strains in the Diastatochromogenes cluster. Appl Environ Microbiol 68:738–744. doi: 10.1128/AEM.68.2.738-744.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huguet-Tapia JC, Badger JH, Loria R, Pettis GS. 2011. Streptomyces turgidiscabies Car8 contains a modular pathogenicity island that shares virulence genes with other actinobacterial plant pathogens. Plasmid 65:118–124. doi: 10.1016/j.plasmid.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 15.Kers JA, Cameron KD, Joshi MV, Bukhalid RA, Morello JE, Wach MJ, Gibson DM, Loria R. 2005. A large, mobile pathogenicity island confers plant pathogenicity on Streptomyces species. Mol Microbiol 55:1025–1033. [DOI] [PubMed] [Google Scholar]
- 16.Myers EW, Sutton GG, Delcher AL, Dew IM, Fasulo DP, Flanigan MJ, Kravitz SA, Mobarry CM, Reinert KH, Remington KA, Anson EL, Bolanos RA, Chou HH, Jordan CM, Halpern AL, Lonardi S, Beasley EM, Brandon RC, Chen L, Dunn PJ, Lai Z, Liang Y, Nusskern DR, Zhan M, Zhang Q, Zheng X, Rubin GM, Adams MD, Venter JC. 2000. A whole-genome assembly of Drosophila. Science 287:2196–2204. doi: 10.1126/science.287.5461.2196. [DOI] [PubMed] [Google Scholar]
- 17.Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. 1999. Improved microbial gene identification with GLIMMER. Nucleic Acids Res 27:4636–4641. doi: 10.1093/nar/27.23.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li L, Stoeckert CJ, Roos DS. 2003. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 21.Oksanen J, Kindt R, Legendre P, O'Hara B. 2007. Vegan: community ecology package. Institute for Statistics and Mathematics, Vienna University of Economics and Business, Vienna, Austria: http://CRAN.R-project.org/package=vegan. [Google Scholar]
- 22.Müllner D. 2013. fastcluster: fast hierarchical, agglomerative clustering routines for R and Python. J Stat Softw 53(9):1–18. [Google Scholar]
- 23.Roshan U, Livesay DR. 2006. Probalign: multiple sequence alignment using partition function posterior probabilities. Bioinformatics 22:2715–2721. doi: 10.1093/bioinformatics/btl472. [DOI] [PubMed] [Google Scholar]
- 24.Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 25.Lanave C, Preparata G, Saccone C, Serio G. 1984. A new method for calculating evolutionary substitution rates. J Mol Evol 20:86–93. doi: 10.1007/BF02101990. [DOI] [PubMed] [Google Scholar]
- 26.Sukumaran J, Holder MT. 2010. DendroPy: a Python library for phylogenetic computing. Bioinformatics 26:1569–1571. doi: 10.1093/bioinformatics/btq228. [DOI] [PubMed] [Google Scholar]
- 27.Huson DH, Scornavacca C. 2012. Dendroscope 3: an interactive tool for rooted phylogenetic trees and networks. Syst Biol 61:1061–1067. doi: 10.1093/sysbio/sys062. [DOI] [PubMed] [Google Scholar]
- 28.Smith JM. 1992. Analyzing the mosaic structure of genes. J Mol Evol 34:126–129. [DOI] [PubMed] [Google Scholar]
- 29.Jakobsen IB, Easteal S. 1996. A program for calculating and displaying compatibility matrices as an aid in determining reticulate evolution in molecular sequences. Comput Appl Biosci 12:291–295. [DOI] [PubMed] [Google Scholar]
- 30.Bruen TC, Philippe H, Bryant D. 2006. A simple and robust statistical test for detecting the presence of recombination. Genetics 172:2665–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conesa A, Götz S. 2008. Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics 2008:619832. doi: 10.1155/2008/619832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carbon S, Ireland A, Mungall CJ, Shu S, Marshall B, Lewis S, AmiGO Hub, Web Presence Working Group. 2009. AmiGO: online access to ontology and annotation data. Bioinformatics 25:288–289. doi: 10.1093/bioinformatics/btn615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, Khanna A, Marshall M, Moxon S, Sonnhammer ELL, Studholme DJ, Yeats C, Eddy SR. 2004. The Pfam protein families database. Nucleic Acids Res 32:D138–D141. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tatusov RL. 2000. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res 28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petersen TN, Brunak S, von Heijne G, Nielsen H. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 36.Komeil D, Simao-Beaunoir A-M, Beaulieu C. 2013. Detection of potential suberinase-encoding genes in Streptomyces scabiei strains and other actinobacteria. Can J Microbiol 59:294–303. doi: 10.1139/cjm-2012-0741. [DOI] [PubMed] [Google Scholar]
- 37.Aparna G, Chatterjee A, Sonti RV, Sankaranarayanan R. 2009. A cell wall-degrading esterase of Xanthomonas oryzae requires a unique substrate recognition module for pathogenesis on rice. Plant Cell 21:1860–1873. doi: 10.1105/tpc.109.066886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shevchik VE, Robert-Baudouy J, Hugouvieux-Cotte-Pattat N. 1997. Pectate lyase PelI of Erwinia chrysanthemi 3937 belongs to a new family. J Bacteriol 179:7321–7330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tardy F, Nasser W, Robert-Baudouy J, Hugouvieux-Cotte-Pattat N. 1997. Comparative analysis of the five major Erwinia chrysanthemi pectate lyases: enzyme characteristics and potential inhibitors. J Bacteriol 179:2503–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loper JE, Hassan KA, Mavrodi DV, Davis EW, Lim CK, Shaffer BT, Elbourne LDH, Stockwell VO, Hartney SL, Breakwell K, Henkels MD, Tetu SG, Rangel LI, Kidarsa TA, Wilson NL, van de Mortel JE, Song C, Blumhagen R, Radune D, Hostetler JB, Brinkac LM, Durkin AS, Kluepfel DA, Wechter WP, Anderson AJ, Kim YC, Pierson LS, Pierson EA, Lindow SE, Kobayashi DY, Raaijmakers JM, Weller DM, Thomashow LS, Allen AE, Paulsen IT. 2012. Comparative genomics of plant-associated Pseudomonas spp.: insights into diversity and inheritance of traits involved in multitrophic interactions. PLoS Genet 8:e1002784. doi: 10.1371/journal.pgen.1002784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seipke RF, Loria R. 2008. Streptomyces scabies 87-22 possesses a functional tomatinase. J Bacteriol 190:7684–7692. doi: 10.1128/JB.01010-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gartemann KH, Abt B, Bekel T, Burger A, Engemann J, Flugel M, Gaigalat L, Goesmann A, Grafen I, Kalinowski J, Kaup O, Kirchner O, Krause L, Linke B, McHardy A, Meyer F, Pohle S, Ruckert C, Schneiker S, Zellermann EM, Puhler A, Eichenlaub R, Kaiser O, Bartels D. 2008. The genome sequence of the tomato-pathogenic actinomycete Clavibacter michiganensis subsp. michiganensis NCPPB382 reveals a large island involved in pathogenicity. J Bacteriol 190:2138–2149. doi: 10.1128/JB.01595-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Labeda DP. 2011. Multilocus sequence analysis of phytopathogenic species of the genus Streptomyces. Int J Syst Evol Microbiol 61:2525–2531. doi: 10.1099/ijs.0.028514-0. [DOI] [PubMed] [Google Scholar]
- 44.Huguet-Tapia JC, Bignell DRD, Loria R. 2014. Characterization of the integration and modular excision of the integrative conjugative element PAISt in Streptomyces turgidiscabies Car8. PLoS One 9:e99345. doi: 10.1371/journal.pone.0099345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chapleau M, Guertin JF, Farrokhi A, Lerat S, Burrus V, Beaulieu C. 14 July 2015. Identification of genetic and environmental factors stimulating excision from Streptomyces scabiei chromosome of the toxicogenic region responsible for pathogenicity. Mol Plant Pathol doi: 10.1111/mpp.12296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lambert DH, Loria R. 1989. Streptomyces acidiscabies sp. nov. Int J Syst Bacteriol 39:393–396. doi: 10.1099/00207713-39-4-393. [DOI] [Google Scholar]
- 47.Miyajima K, Tanaka F, Takeuchi T, Kuninaga S. 1998. Streptomyces turgidiscabies sp. nov. Int J Syst Bacteriol 48(Part 2):495–502. [DOI] [PubMed] [Google Scholar]
- 48.Kers JA, Wach MJ, Krasnoff SB, Widom J, Cameron KD, Bukhalid RA, Gibson DM, Crane BR, Loria R. 2004. Nitration of a peptide phytotoxin by bacterial nitric oxide synthase. Nature 429:79–82. doi: 10.1038/nature02504. [DOI] [PubMed] [Google Scholar]
- 49.Barry SM, Kers JA, Johnson EG, Song L, Aston PR, Patel B, Krasnoff SB, Crane BR, Gibson DM, Loria R, Challis GL. 2012. Cytochrome P450-catalyzed l-tryptophan nitration in thaxtomin phytotoxin biosynthesis. Nat Chem Biol 8:814–816. doi: 10.1038/nchembio.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kumagai T, Takagi K, Koyama Y, Matoba Y, Oda K, Noda M, Sugiyama M. 2012. Heme protein and hydroxyarginase necessary for biosynthesis of d-cycloserine. Antimicrob Agents Chemother 56:3682–3689. doi: 10.1128/AAC.00614-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bignell DRD, Seipke RF, Huguet-Tapia JC, Chambers AH, Parry RJ, Loria R. 2010. Streptomyces scabies 87-22 contains a coronafacic acid-like biosynthetic cluster that contributes to plant-microbe interactions. Mol Plant Microbe Interact 23:161–175. doi: 10.1094/MPMI-23-2-0161. [DOI] [PubMed] [Google Scholar]
- 52.Wang X-J, Yan Y-J, Zhang B, An J, Wang J-J, Tian J, Jiang L, Chen Y-H, Huang S-X, Yin M, Zhang J, Gao A-L, Liu C-X, Zhu Z-X, Xiang W-S. 2010. Genome sequence of the milbemycin-producing bacterium Streptomyces bingchenggensis. J Bacteriol 192:4526–4527. doi: 10.1128/JB.00596-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu H, Qu S, Lu C, Zheng H, Zhou X, Bai L, Deng Z. 2012. Genomic and transcriptomic insights into the thermo-regulated biosynthesis of validamycin in Streptomyces hygroscopicus 5008. BMC Genomics 13:337. doi: 10.1186/1471-2164-13-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barbe V, Bouzon M, Mangenot S, Badet B, Poulain J, Segurens B, Vallenet D, Marlière P, Weissenbach J. 2011. Complete genome sequence of Streptomyces cattleya NRRL 8057, a producer of antibiotics and fluorometabolites. J Bacteriol 193:5055–5056. doi: 10.1128/JB.05583-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Oliynyk M, Samborskyy M, Lester JB, Mironenko T, Scott N, Dickens S, Haydock SF, Leadlay PF. 2007. Complete genome sequence of the erythromycin-producing bacterium Saccharopolyspora erythraea NRRL23338. Nat Biotechnol 25:447–453. doi: 10.1038/nbt1297. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.