

Abstract
Abstract
Recessive dystrophic epidermolysis bullosa (RDEB) is a rare genodermatosis with severe blistering. No curative treatment is available. Scientific data indicated that epigallocatechin-3-gallate (EGCG), a green tea extract, might improve the phenotype of RDEB patients. In a multicentre, randomized, crossover, double-blind, placebo-controlled clinical trial, we evaluated a 4-month oral EGCG treatment regimen in 17 RDEB patients. We found that EGCG treatment was not more effective than placebo in modified intention to treat and per protocol analysis (n = 16; p = 0.78 and n = 10; p = 1 respectively). Tolerance was good. Specific organizational and technical difficulties of controlled randomized double-blind trials in EB patients are discussed.
Trial registration
US National Institutes of Health Clinical Trial Register (NCT00951964).
Electronic supplementary material
The online version of this article (doi:10.1186/s13023-016-0411-5) contains supplementary material, which is available to authorized users.
Keywords: Dystrophic epidermolysis bullosa, Polyphenon, Green tea, Cathechin, Metalloproteinase
Recessive dystrophic epidermolysis bullosa (RDEB) is a rare genodermatosis characterized by skin and mucosal fragility due to mutations in the COL7A1 gene [1]. No curative treatment is available [2]. It has been shown that the level of activation of dermal metalloproteinases (MMP) could modulate the phenotype in RDEB patients [3–5] and that epigallocatechin-3-gallate (EGCG), a green tea extract [6–8], is able to regulate this activity in vitro and ex vivo [9].
We then evaluated the efficacy of oral EGCG to improve skin impairment in RDEB patients in a multicentre, randomized, crossover, double-blind, placebo-controlled clinical trial. The trial was approved by the local ethics committee and was registered in the Clinical Trial Register (NCT00951964). Patients of both sexes, over 2 years of age, with generalized severe or intermediate RDEB, confirmed by immunohistological analysis of skin biopsy, were recruited. Patients received treatment or placebo for 4 months, followed by a 2-month wash-out period and then by the other treatment for 4 months (Fig. 1). Dosage of EGCG treatment depended on the patient’s weight (from 400 to 800 mg a day) (Additional file 1, Supplementary Methods).
Fig. 1.
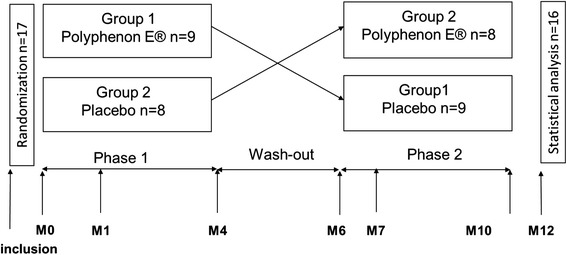
Study design. Each patient has a 4 month period of treatment separated by a 2 month period of wash-out
The main outcome was binary: success, defined as a decrease ≥ 20 % in the number of new blisters per day counted by patients at each dressing, upon 7 consecutive dressings before the initial and final visit of each treatment period, or failure of treatment. Secondary outcomes were the affected cutaneous surface area, the severity of mucosal impairment, skin fragility, itch and the mean duration of healing measured on 3 new blisters selected by patients in the first week of each period of treatment (Additional file 2: Table S1). Adverse events were collected by investigators at each visit. Assuming an expected success rate of 30 % in the EGCG group and 5 % in the placebo group, with 80 % power and 5 % type I error, we planned to include 22 patients. The main outcome was analysed in a modified intention to treat (mITT) and per protocol, secondary outcomes in a mITT.
Findings
Seventeen patients were included in this study, mean age 19.4 years (±16.2 SD). One patient did not start treatment and was not included in the mITT population (n = 16). Only 10/16 patients were included in the per protocol analysis (available data for each visit in both treatment periods for the main outcome). Eight patients/16 (50 %) had a decrease of at least 20 % in the mean number of new blisters per day with EGCG and 5/16 (31 %) with placebo in the mITT analysis. This difference was not statistically significant (Prescott’s test, p = 0.78). Results were similar in per protocol analysis (p = 1). Analyses of secondary outcomes showed no difference between the 2 treatment periods (Table 1) despite a dramatic reduction of the mean duration of wound in the EGCG group (-14.62 days ± 18.76) compared to the placebo group (1.78 ± 14.65). Tolerance was good with 26 and 16 adverse events in the EGCG and placebo group respectively (p = 0.47) (Additional file 3: Table S2).
Table 1.
Statistical analysis of secondary outcomes
| Evolution of score (end of the period - beginning of the period) | Polyphenon E® Mean ± SD (N) | Placebo Mean ± SD (N) | p value |
|---|---|---|---|
| Surface area | - 4.07 ± 7.62 (12) | - 4.42 ± 9.84 (14) | 0.93 |
| Skin fragility | - 0.90 ± 2.46 (12) | - 0.64 ± 2.06 (14) | 0.75 |
| Mucosal involvement | 0.55 ± 1.12 (8) | 1.97 ± 1.64 (6) | 0.07 |
| Itch | - 1.17 ± 3.53 (12) | 0 ± 2.16 (14) | 0.38 |
| Mean duration of wound healing (days) | -14.62 ± 18.76 (7) | 1.78 ± 14.65 (9) | 0.21 |
N number of patients, SD standard deviation
Generalized DEB is a rare and severe genodermatosis. Hence, evaluation of a new treatment in a controlled randomized and double-blind trial is challenging. In this study, even if fewer new blisters per day were observed in the EGCG arm as compared with the placebo and the mean duration of wound healing was shorter, we failed to show a statistically significant difference. These disappointing results can be explained by several limitations of our study. First, under-enrolment and the high rate of missing data for the main outcome are of important concern. Low enrolment is a major drawback for studies on all rare and severe diseases [10–13]. Indeed, despite the active involvement of the DEBRA France patients’ support group and the main French centres for EB care, together with the reimbursement of the patient’s travel expenses, only 17 patients instead of the 22 planned could be enrolled and only 10 completed the study. Shorter studies with less visits and/or home evaluation by a study nurse and/or international studies could improve the patient recruitment and protocol adherence. Moreover, factors influencing the severity of phenotype in DEB are complex and not only related to the MMP activity as recently shown [3, 14, 15]. Finally the high rate of therapeutic success in the placebo group is intriguing, but seems to be frequent in the few controlled versus placebo published studies on DEB [10–13]. The variable course of DEB, depending on numerous factors such as the weather, associated diseases and or/secondary complications or trauma, is well known. We tried to minimize the impact of these factors by counting the number of new blisters per day averaged on seven consecutive dressings before each visit. However other outcome measures like a validated EB severity score may be more relevant. Analysis of the inclusion date of each patient did not support an influence of seasonal variation. EGCG is a potentially interesting and safe treatment for DEB patients. An international randomized, double-blinded and placebo-controlled trial with targeted subpopulation is necessary.
Acknowledgment
Authors thank DEBRA France association for its implication in patient recruitment for this study and their financial help for immunohistochemical and molecular analyses.
Abbreviations
- DEB
dystrophic epidermolysis bullosa
- EGCG
epigallocatechin-3-gallate
- mITT
modified intention to treat
- MMP
metalloproteinases
- RDEB
recessive dystrophic epidermolysis bullosa
- SD
standard deviation
Additional files
Supplementary methods. (DOC 22 kb)
Primary and secondary outcomes assessments. (DOC 31 kb)
Detailed adverse events for EGCG and placebo treatment. * indicates the adverse events reported as severe by investigators. (DOC 41 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Participation in designing study: CC, CR, EF, CB and J-PL. Participation in generating data for the study: CC, EB, EB-L, CL, JM, PV, CB and J-PL. Participation in gathering the data for the study: CC, CR, EF. Participation in the analysis of the data: CC, CR, EF, CB and J-PL. Writing the majority of the original draft of the paper: CC, EF, Participation in writing the paper: CC, CR, EF, EB, EB-L, CL, JM, PV, CB and J-PL. Review the pertinent raw data on which the results and conclusions of this study are based: CC, CR, EF. Approval the final version of this paper: CC, CR, EF, EB, EB-L, CL, JM, PV, CB and J-PL.
Contributor Information
Christine Chiaverini, Phone: +33 492036107, Email: chiaverini.c@chu-nice.fr.
Coralie Roger, Email: roger.c2@chu-nice.fr.
Eric Fontas, Email: fontas.e@chu-nice.fr.
Emmanuelle Bourrat, Email: emmanuelle.bourrat@sls.ap-hop-paris.fr.
Eva Bourdon-Lanoy, Email: eva.bourdon-lanoy@nck.aphp.fr.
Christine Labrèze, Email: christine.labreze@chu-bordeaux.fr.
Juliette Mazereeuw, Email: mazereeuw-hautier.j@chu-toulouse.fr.
Pierre Vabres, Email: pierre.vabres@chu-dijon.fr.
Christine Bodemer, Email: christine.bodemer@nck.aphp.fr.
Jean-Philippe Lacour, Email: lacour@unice.fr.
References
- 1.Bruckner-Tuderman L. Dystrophic epidermolysis bullosa: pathogenesis and clinical features. Dermatol Clin. 2010;28:107–114. doi: 10.1016/j.det.2009.10.020. [DOI] [PubMed] [Google Scholar]
- 2.Hsu CK, Wang SP, Lee JY, et al. Treatment of hereditary epidermolysis bullosa: updates and future prospects. Am J Clin Dermatol. 2014;15:1–6. doi: 10.1007/s40257-013-0059-z. [DOI] [PubMed] [Google Scholar]
- 3.Titeux M, Pendaries V, Tonasso L, et al. A frequent functional SNP in the MMP1 promoter is associated with higher disease severity in recessive dystrophic epidermolysis bullosa. Hum Mutat. 2008;29:267–276. doi: 10.1002/humu.20647. [DOI] [PubMed] [Google Scholar]
- 4.Sawamura D, Sugawara T, Hashimoto I, et al. Increased gene expression of matrix metalloproteinase-3 (stromelysin) in skin fibroblasts from patients with severe recessive dystrophic epidermolysis bullosa. Biochem Biophys Res Commun. 1991;174:1003–1008. doi: 10.1016/0006-291X(91)91518-H. [DOI] [PubMed] [Google Scholar]
- 5.Bodemer C, Tchen SI, Ghomrasseni S, et al. Skin expression of metalloproteinases and tissue inhibitor of metalloproteinases in sibling patients with recessive dystrophic epidermolysis and intrafamilial phenotypic variation. J Invest Dermatol. 2003;121:273–279. doi: 10.1046/j.1523-1747.2003.12325.x. [DOI] [PubMed] [Google Scholar]
- 6.Singh BN, Shankar S, Srivastava RK. Green tea catechin, epigallocatechin-3-gallate (EGCG): mechanisms, perspectives and clinical applications. Biochem Pharmacol. 2011;82:1807–1821. doi: 10.1016/j.bcp.2011.07.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu G, Zhang L, Rong Y, et al. Downstream carcinogenesis signaling pathways by green tea polyphenols: a translational perspective of chemoprevention and treatment for cancers. Curr Drug Metab. 2014;15:14–22. doi: 10.2174/1389200214666131211155613. [DOI] [PubMed] [Google Scholar]
- 8.Rhodes LE, Darby G, Massey KA, et al. Oral green tea catechin metabolites are incorporated into human skin and protect against UV radiation-induced cutaneous inflammation in association with reduced production of pro-inflammatory eicosanoid 12-hydroxyeicosatetraenoic acid. Br J Nutr. 2013;110:891–900. doi: 10.1017/S0007114512006071. [DOI] [PubMed] [Google Scholar]
- 9.Changotade SI, Assoumou A, Gueniche F, et al. Epigallocatechin gallate’s protective effect against MMP7 in recessive dystrophic epidermolysis bullosa patients. J Invest Dermatol. 2007;127:821–828. doi: 10.1038/sj.jid.5700645. [DOI] [PubMed] [Google Scholar]
- 10.Wally V, Kitzmueller S, Lagler F, et al. Topical diacerein for epidermolysis bullosa: a randomized controlled pilot study. Orphanet J Rare Dis. 2013;8:69. doi: 10.1186/1750-1172-8-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lara-Corrales I, Parkin PC, Stephens D, et al. The efficacy of trimethoprim in wound healing of patients with epidermolysis bullosa: a feasibility trial. J Am Acad Dermatol. 2012;66:264–270. doi: 10.1016/j.jaad.2010.01.047. [DOI] [PubMed] [Google Scholar]
- 12.Caldwell-Brown D, Stern RS, Lin AN, et al. Lack of efficacy of phenytoin in recessive dystrophic epidermolysis bullosa. Epidermolysis Bullosa Study Group. N Engl J Med. 1992;327:163–167. doi: 10.1056/NEJM199207163270305. [DOI] [PubMed] [Google Scholar]
- 13.Weiner M, Stein A, Cash S, et al. Tetracycline and epidermolysis bullosa simplex: a double-blind, placebo-controlled, crossover randomized clinical trial. Br J Dermatol. 2004;150:613–614. doi: 10.1046/j.1365-2133.2004.05816.x. [DOI] [PubMed] [Google Scholar]
- 14.Garza-Gomez J, Cerda-Flores RM, Gomez-Flores M, et al. An investigation into the MMP1 gene promoter region polymorphism--1607 2G with recessive dystrophic epidermolysis bullosa disease severity in northeastern Mexican patients. Int J Dermatol. 2014;53:985–990. doi: 10.1111/ijd.12499. [DOI] [PubMed] [Google Scholar]
- 15.Kern JS, Gruninger G, Imsak R, et al. Forty-two novel COL7A1 mutations and the role of a frequent single nucleotide polymorphism in the MMP1 promoter in modulation of disease severity in a large European dystrophic epidermolysis bullosa cohort. Br J Dermatol. 2009;161:1089–1097. doi: 10.1111/j.1365-2133.2009.09333.x. [DOI] [PubMed] [Google Scholar]