

Abstract
This report details the enantioselective synthesis of β-amino-α-bromo nitroalkanes with β-alkyl substituents, using homogeneous catalysis to prepare either antipode. Use of a bifunctional Brønsted base/acid catalyst allows equal access to either enantiomer of the products, enabling the use of Umpolung Amide Synthesis (UmAS) to prepare the corresponding L- or D-α-amino amide bearing alkyl side chains – overall, in only 4 steps from aldehyde. The approach also addresses an underlying incompatibility between bromonitromethane and solid hydroxide bases.
Keywords: homogeneous catalysis, enantioselective catalysis, peptides, umpolung amide synthesis, organocatalysis
Great strides have been made in the development of stereoselective aza-Henry reactions, particularly over the past decade, as new metal-based1 and organic reagents2 have been discovered.3 These enantioselective variants have rendered the reaction a powerful entry to secondary monoamines and vic-diamines that are precursors to promising therapeutics.4,5 Most developments have focused on primary (RCH2NO2) and secondary nitroalkanes (R1R2CHNO2), while the chemistry of α-functionalized nitroalkanes bearing a heteroatom (O, S, halogen) has expanded only slowly.6,7,8 Enantioselective additions of α-sulfur and α-oxygen-bearing nitroalkanes are relatively limited, but bromonitromethane-based enantioselective aza-Henry reactions represent an area of growth, owing to their relevance to cyclopropane,9 β-amino alcohol,10 and aryl glycine α-amino amide synthesis.11,12 It was not until recently that the latter approach was first extended to N-Boc aldimines (Figure 1),13 since these electrophiles suffer from tautomerization to the unreactive N-acyl enamide isomer.14, 15 Unfortunately, the use of a heterogeneous catalyst was imperfect, owing to an underlying incompatibility between bromonitromethane and the solid hydroxide base, as well as a very low level of enantioselection when using the pseudoenantiomeric catalyst. This compelled us to develop a solution that addresses these shortcomings. In this report we describe the first enantioselective aza-Henry additions16 of bromonitromethane to N-Boc aliphatic aldimines using a homogeneous catalyst, one that is entirely compatible with bromonitromethane. This finding delivers either enantiomer in high yield, thereby extending the use of bromonitromethane as a carbonyl dianion synthon to include α-amino amides bearing aliphatic side chains in either D- or L-configuration.
Figure 1.

Phase transfer catalysis (top) provides efficient access to precursors only for D-amino amide homologation due to lower ee when using catalyst pseudoenantiomer, and depressed yield due to underlying incompatibility between bromonitromethane and (solid) cesium hydroxide. Homogeneous catalysis (bottom) has now addressed these limitations.
The α-amido sulfone derivatives (3) of commercially available aldehydes (1 step: BocNH2, NaSO2Tol, HCO2H, H2O) serve as bench stable precursors for N-Boc imines.17 Unlike N-Boc benzaldimines, N-Boc aliphatic aldimines formed from 3 are prone to tautomerization to their N-Boc enamides.15,18,19 Table 1 summarizes those experiments leading to efficient and selective conversion to 4a. Prolonged exposure to the elimination reaction conditions (>4 h, Cs2CO3) led to predominantly enamide formation, while shorter reaction times (<2 h) led to incomplete conversion. In the event, elimination of sulfinate with Cs2CO3 provided the imine (after filtration of solids, analysis by 1H NMR), which was carried forward without further purification. Addition of catalyst (R,R)-PBAM (5a) and bromonitromethane at −78 °C provided the desired α-bromo nitroalkane in 74% yield as a 1:1 mixture of diastereomers with 42% and 40 % ee, respectively (Table 1, entry 1). Diastereomers 4a are homochiral at the β-position, allowing the configuration at this position to translate to the α-amino amide carbon with complete conservation due to UmAS (vide infra). In an effort to increase the enantioselectivity, (R,R)-PBAM (5a) was first protonated with triflic acid, providing (R,R)-PBAM•HOTf (5b). This catalyst salt provided 4a in similar yield and increased ee up to 91/91% for each diastereomer (Table 1, entry 2). Replacement of the cyclohexane diamine backbone of 5 with a stilbene backbone20 resulted in lower enantioselection when using 6•HOTf and 7•HOTf (Table 1, entries 3–4). Electron–rich derivatives 9 and 10 provided α-bromo nitroalkane 4a in increased yield but diminished ee (Table 1, entries 5–6). Triflimidic acid, fluorosulfonic acid, and hexafluoroimidic (8) acid salts of PBAM were evaluated as co-acids for PBAM (5a) to interrogate the effect of the counterion on stereoselectivity,21 but no improvement was observed (Table 1, entries 7–9). PBAM•HOTf (5b) was therefore selected as the lead catalyst, and it was found that its loading could be lowered to 2 mol % while providing similar yield and slightly increased ee up to 93/93% (Table 1, entries 10–11).
Table 1.
Initial Development of a Catalyzed aza-Henry using Bromonitromethane
 | |||
|---|---|---|---|
| entrya | catalyst (R,R) | yield (%)b | ee (%)c |
| 1 | PBAM (5a) | 74 | 42, 40 |
| 2 | PBAM•HOTf (5b) | 68 | 91, 91 |
| 3 | StilbPBAM(6)•HOTf | 71 | 53, 47 |
| 4 | 4MeO-StilbBAM (7)•HOTf | 50 | 25, 22 |
| 5 | 6MeO-PBAM(9)•HOTf | 79 | 86, 85 |
| 6 | 8MeO-PBAM(10)•HOTf | 74 | 57, 61 |
| 7 | PBAM•HNTf2 | 76 | 81, 81 |
| 8 | PBAM•HSO3F | 62 | 86, 86 |
| 9 | PBAM•8 | 61 | 66, 65 |
| 10d | PBAM•HOTf (5b) | 74 | 91, 90 |
| 11e | PBAM•HOTf (5b) | 70 | 93, 93 |
Reaction filtered through Celite after 3 h to remove Cs2CO3 prior to the second step. The reaction time for the second step was 24 h.
Isolated yields.
Adducts isolated as mixture of diastereomers (1:1), ee’s reported for each diastereomer.
5 mol % catalyst.
2 mol % catalyst.

The protocol established by was then evaluated against a wide range of alkyl α-amido sulfones derived from commercially available aliphatic aldehydes (3, Table 2). (S,S)-PBAM•HOTf (ent-5b) provided the β-amino-α-bromonitroalkane donor (ent-4) necessary for L-α-amino amide homologation. Straight–chain aliphatic substrates 3a–e performed well in the reaction with high yields (87–94%) and high enantioselectivity (81–90% ee) (Table 2, entries 2–5). Further diversity in the form of branching beta to the amido sulfone center (3f–h) was tolerated well with high ee’s up to 92% (Table 2, entries 6–8). Phenyl alanine donor 3i was obtained in significantly lower yield (39%) but with good enantioselection (89% ee) (Table 2, entry 9). The lower yield is due to the increased propensity for the imine intermediate to tautomerize to the unreactive, conjugated N-Boc enamide (vide supra). Branching alpha to the amido sulfone center was tolerated, although with only moderate yields (52–61%) and varying enantioselection (79–92% ee) (Table 2, entries 10–12). Unsaturation on the side chain in the form of both alkenes and an alkyne resulted in slightly lower enantioselection (84–91% ee) than their fully saturated counterparts (Table 2, entries 13–16). Trifluoromethyl derivative ent-4q was obtained in moderate yield and ee (Table 2, entry 17). The protocol also tolerated electron rich substrates with varying degrees of enantioselection (Table 2, entries 18–19).
Table 2.
(S,S)-PBAM·HOTf–Catalyzed Enantioselective aza-Henry Addition to Aliphatic N-Boc Imine Intermediates: Substrate Scope
 | ||||
|---|---|---|---|---|
| entrya | R | ent-4 | yieldb (%) | eec (%) |
| 1 | PhCH2CH2 | a | 70 | 93 |
| 2 | Et | b | 90 | 81 |
| 3 | nPr | c | 87 | 90 |
| 4 | nBu | d | 94 | 88 |
| 5 | C10H21 | e | 88 | 90 |
| 6 | (CH3)3CCH2 | f | 88 | 90 |
| 7 | iBu | g | 83 | 92 |
| 8 | (C6H11)CH2 | h | 82 | 85 |
| 9 | PhCH2 | i | 39 | 89 |
| 10 | iPr | j | 59 | 79 |
| 11 | cyclo-C3H5 | k | 61 | 90 |
| 12 | Cy | l | 61 | 92 |
| 13 | H2C=CHCH2CH2 | m | 77 | 84 |
| 14 | cis-EtCH=CH(CH2)2 | n | 80 | 89 |
| 15 | HC≡CCH2CH2 | o | 63 | 86 |
| 16 | HC≡CCH2CH2CH2 | p | 73 | 91 |
| 17 | CF3CH2 | q | 57 | 85 |
| 18 | p(MeO)PhCH2CH2 | r | 68 | 90 |
| 19 | EtOCH2CH2 | s | 68 | 76 |
Reaction filtered through Celite to remove Cs2CO3 prior to second step. The reaction time for the second step was 24 h.
Isolated yields.
Adducts isolated as a mixture of diastereomers (1:1), ee’s re-ported as an average of diastereomer ee’s (Δ% ee≤3).
Select α-bromo nitroalkanes were coupled to chiral non-racemic amines using UmAS conditions catalytic in NIS (Table 3).11a As expected, the β-homochirality of the bromo nitroalkane donors translated directly to a high diastereomeric ratio of the resulting α-amino amides. This feature is inherent to the UmAS reaction, since the key carbon-nitrogen bond-forming step occurs with a nucleophilic carbon; the β-chiral carbon is therefore never acidified. Homologation with D-N-Boc-Hph using donor 4a delivered α-amino amide 11 in 57% yield (Table 3, entry 1). Hph is a constituent residue of growth hormone secretagogue analogs,22 and peptides with a broad range of activity, including opioid receptor binding.23 Extension with D-Chg and D-Adod provided 12 and 13 in 56% and 65% yield, respectively (Table 3, entries 2–3). The incorporation of amino amides ‘tagged’ with an alkyne is almost equally effective, using D-homopropargyl glycine24 donor 4o (Table 3, entry 4). Finally, the sterically-hindered α-amino amide D-Npg can be homologated in 57% yield using donor 4f (Table 3, entry 5).
Table 3.
NIS-Catalyzed Umpolung Amide Synthesis to Pre-pare α-Amino Amides Bearing Aliphatic Substituents


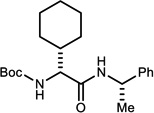

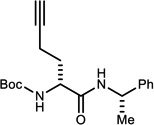

Reaction run in DME, 1.2 equiv. amine for 24 h at 0 °C under an atmosphere of O2
Isolated yields.
Measured by 1H NMR. α-Bromo nitroalkanes for entries 1,3,4 were recrystallized to 99% ee (see Supporting Information). α-Bromo nitroalkanes for entries 2 and 5 were 92 and 90% ee, respectively.
Insofar as our overarching goal is to provide rapid chemical access to peptides derived from unnatural α-amino acids, the tools described here were applied to the straightforward preparation of short peptide 18, a small molecule effective in the reversal of P-glycoprotein-mediated resistance to carfilzomib (Scheme 1).25 Donor ent-4a was prepared as described above, recrystallized (to 99% ee), and then coupled to amine 16 using UmAS in 54% isolated yield. Immediate Boc-deprotection and conventional amide synthesis from pyridyl-2-carboxylic acid delivered the product in 65% yield.
Scheme 1.

Preparation of 18, a Peptide Effective in Reversal of P-Glycoprotein-Mediated Resistance to Carfilzomib
In summary, the first enantioselective addition of bromonitromethane into alkyl imines using homogeneous catalysis has been developed, allowing access to structurally and configurationally diverse α-alkyl amino amides in four steps from aldehydes. The procedure developed here uses a catalyst that is commercially available,26 but also readily prepared on large scale in two steps from commercially available materials. This development significantly expands the array of non-proteinogenic α-amino amides available through expedient chemical synthesis, and draws closer a general solution to the efficient chemical synthesis of peptides. It is also notable that the approach described here further offers the benefits of metal-free chemical synthesis.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health (GM 063557 & GM 084333 (catalyst development)).
Footnotes
ASSOCIATED CONTENT
Experimental procedures and spectroscopic data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors.
REFERENCES
- 1.(a) Yamada K, Harwood SJ, Groger H, Shibasaki M. Angew. Chem. Int. Ed. 1999;38:3504–3506. doi: 10.1002/(sici)1521-3773(19991203)38:23<3504::aid-anie3504>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]; (b) Nishiwaki N, Knudsen KR, Gothelf KV, Jorgensen KA. Angew. Chem. Int. Ed. 2001;40:2992–2995. doi: 10.1002/1521-3773(20010817)40:16<2992::AID-ANIE2992>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 2.Selected examples: Nugent BM, Yoder RA, Johnston JN. J. Am. Chem. Soc. 2004;126:3418–3419. doi: 10.1021/ja031906i. Okino T, Nakamura S, Furukawa T, Takemoto Y. Org. Lett. 2004;6:625–627. doi: 10.1021/ol0364531. Oyaizu K, Uraguchi D, Ooi T. Chem. Commun. 2015;51:4437–4439. doi: 10.1039/c4cc10261d. Uraguchi D, Oyaizu K, Noguchi H, Ooi T. Chem. - Asian J. 2015;10:334–337. doi: 10.1002/asia.201402943.
- 3.(a) Noble A, Anderson JC. Chem. Rev. 2013;113:2887–2939. doi: 10.1021/cr300272t. [DOI] [PubMed] [Google Scholar]; (b) Rueping M, Antonchick AP. Org. Lett. 2008;10:1731–1734. doi: 10.1021/ol8003589. [DOI] [PubMed] [Google Scholar]
- 4.Review: Arrayas RG, Carretero JC. Chem. Soc. Rev. 2009;38:1940–1948. doi: 10.1039/b820303b.
- 5.Applications of bis(amidine) complexes: Vilgelm AE, Pawlikowski JS, Liu Y, Hawkins OE, Davis TA, Smith J, Weller KP, Horton LW, McClain CM, Ayers GD, Turner DC, Essaka DC, Stewart CF, Sosman JA, Kelley MC, Ecsedy JA, Johnston JN, Richmond A. Cancer Res. 2015;75:181–193. doi: 10.1158/0008-5472.CAN-14-2405. Vara BA, Mayasundari A, Tellis JC, Danneman MW, Arredondo V, Davis TA, Min J, Finch K, Guy RK, Johnston JN. J. Org. Chem. 2014;79:6913–6938. doi: 10.1021/jo501003r. Villalta F, Dobish MC, Nde PN, Kleshchenko YY, Hargrove TY, Johnson CA, Waterman MR, Johnston JN, Lepesheva GI. J. Infect. Dis. 2013;208:504–511. doi: 10.1093/infdis/jit042. Davis TA, Vilgelm AE, Richmond A, Johnston JN. J. Org. Chem. 2013;78:10605–10616. doi: 10.1021/jo401321a. Dobish MC, Villalta F, Waterman MR, Lepesheva GI, Johnston JN. Org. Lett. 2012;14:6322–6325. doi: 10.1021/ol303092v. Davis TA, Danneman MW, Johnston JN. Chem. Commun. 2012;48:5578–5580. doi: 10.1039/c2cc32225k. Davis TA, Johnston JN. Chem. Sci. 2011;2:1076–1079. doi: 10.1039/C1SC00061F.
- 6.Ballini R, Petrini M. Tetrahedron. 2004;60:1017–1047. [Google Scholar]
- 7.For example, α-benzyloxy: Barrett AGM, Flygare JA, Spilling CD. J. Org. Chem. 1989;54:4723–4726. α-Phenylthio: Barrett AGM, Cheng MC, Spilling CD, Taylor SJ. J. Org. Chem. 1989;54:992–994.
- 8.Barrett AGM. Chem. Soc. Rev. 1991;20:95–127. [Google Scholar]
- 9.Selected examples: Hansen HM, Longbottom DA, Ley SV. Chem. Commun. 2006:4838–4840. doi: 10.1039/b612436b. Lv J, Zhang J, Lin Z, Wang Y. Chem. - Eur. J. 2009;15:972–979. doi: 10.1002/chem.200801651.
- 10.Blay G, Hernandez-Olmos V, Pedro JR. Chem. Commun. 2008:4840–4842. doi: 10.1039/b809739a. [DOI] [PubMed] [Google Scholar]
- 11.(a) Schwieter KE, Shen B, Shackleford JP, Leighty MW, Johnston JN. Org. Lett. 2014;16:4714–4717. doi: 10.1021/ol502089v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Makley DM, Johnston JN. Org. Lett. 2014;16:3146–3149. doi: 10.1021/ol501297a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Shackleford JP, Shen B, Johnston JN. Proc. Natl. Acad. Sci. U. S. A. 2012;109:44–46. doi: 10.1073/pnas.1113553108. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Shen B, Makley DM, Johnston JN. Nature. 2010;465:1027–1032. doi: 10.1038/nature09125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.See also α-oxy amides: Leighty MW, Shen B, Johnston JN. J. Am. Chem. Soc. 2012;134:15233–15236. doi: 10.1021/ja306225u.
- 13.Schwieter KE, Johnston JN. Chem. Sci. 2015;6:2590–2595. doi: 10.1039/c5sc00064e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Successful examples with primary aliphatic nitroalkanes: Robak MT, Trincado M, Ellman JA. J. Am. Chem. Soc. 2007;129:15110–15111. doi: 10.1021/ja075653v. Wang C-J, Dong X-Q, Zhang Z-H, Xue Z-Y, Teng H-L. J. Am. Chem. Soc. 2008;130:8606–8607. doi: 10.1021/ja803538x. Palomo C, Oiarbide M, Laso A, Lopez R. J. Am. Chem. Soc. 2005;127:17622–17623. doi: 10.1021/ja056594t. Fini F, Sgarzani V, Pettersen D, Herrera RP, Bernardi L, Ricci A. Angew. Chem., Int. Ed. 2005;44:7975–7978. doi: 10.1002/anie.200502646.
- 15.Use of N-acyl enamides as precursors to N-acyl imines has relatively limited success in enantioselective catalysis: Terada M, Sorimachi K. J. Am. Chem. Soc. 2007;129:292–293. doi: 10.1021/ja0678166. Jia Y-X, Zhong J, Zhu S-F, Zhang C-M, Zhou Q-L. Angew. Chem. Int. Ed. 2007;46:5565–5567. doi: 10.1002/anie.200701067. See also: Kataja AO, Masson G. Tetrahedron. 2014;70:8783–8815. Hashimoto T, Nakatsu H, Takiguchi Y, Maruoka K. J. Am. Chem. Soc. 2013;135:16010–16013. doi: 10.1021/ja407501h.
- 16.Selected reviews: Palomo C, Oiarbide M, Lopez R. Chem. Soc. Rev. 2009;38:632–653. doi: 10.1039/b708453f. Connon SJ. Chem--Eur. J. 2006;12:5419–5427. Shen J, Tan CH. Org. Biomol. Chem. 2008;6:3229–3236. doi: 10.1039/b809505c. Akiyama T. Chem. Rev. 2007;107:5744–5758. doi: 10.1021/cr068374j. Terada M. Synthesis. 2010:1929–1982. Rueping M, Kuenkel A, Atodiresei I. Chem. Soc. Rev. 2011;40:4539–4549. doi: 10.1039/c1cs15087a.
- 17.(a) Petrini M. Chem. Rev. 2005;105:3949–3977. doi: 10.1021/cr050528s. [DOI] [PubMed] [Google Scholar]; (b) Petrini M, Torregiani E. Synthesis. 2007:159–186. [Google Scholar]; (c) Vesely J, Rios R. Chem. Soc. Rev. 2014;43:611–630. doi: 10.1039/c3cs60321k. [DOI] [PubMed] [Google Scholar]; (d) Yin B, Zhang Y, Xu L-W. Synthesis. 20102010:3583–3595. [Google Scholar]
- 18.For aza-Henry reactions using aliphatic aldimines and primary nitroalkanes (des-bromo), see: Wang B, Liu Y, Sun C, Wei Z, Cao J, Liang D, Lin Y, Duan H. Org. Lett. 2014;16:6432–6435. doi: 10.1021/ol503264n. Cao D, Chai Z, Zhang J, Ye Z, Xiao H, Wang H, Chen J, Wu X, Zhao G. Chem. Commun. 2013;49:5972–5974. doi: 10.1039/c3cc42864h. Palomo C, Oiarbide M, Laso A, López R. J. Am. Chem. Soc. 2005;127:17622–17623. doi: 10.1021/ja056594t. Gomez-Bengoa E, Linden A, López R, Múgica-Mendiola I, Oiarbide M, Palomo C. J. Am. Chem. Soc. 2008;130:7955–7966. doi: 10.1021/ja800253z. Fini F, Sgarzani V, Pettersen D, Herrera RP, Bernardi L, Ricci A. Angew. Chem. Int. Ed. 2005;44:7975–7978. doi: 10.1002/anie.200502646. Robak MT, Trincado M, Ellman JA. J. Am. Chem. Soc. 2007;129:15110–15111. doi: 10.1021/ja075653v. Takada K, Nagasawa K. Adv. Synth. Catal. 2009;351:345–347. Błaszczyk R, Gajda A, Zawadzki S, Czubacka E, Gajda T. Tetrahedron. 2010;66:9840–9848. Handa S, Gnanadesikan V, Matsunaga S, Shibasaki M. J. Am. Chem. Soc. 2010;132:4925–4934. doi: 10.1021/ja100514y. Johnson KM, Rattley MS, Sladojevich F, Barber DM, Nuñez MG, Goldys AM, Dixon DJ. Org. Lett. 2012;14:2492–2495. doi: 10.1021/ol300779x. Núñez MG, Farley AJM, Dixon DJ. J. Am. Chem. Soc. 2013;135:16348–16351. doi: 10.1021/ja409121s. Wei Y, He W, Liu Y, Liu P, Zhang S. Org. Lett. 2012;14:704–707. doi: 10.1021/ol203170x. Li H, Zhang X, Shi X, Ji N, He W, Zhang S, Zhang B. Adv. Synth. Catal. 2012;354:2264–2274. Uraguchi D, Koshimoto K, Ooi T. J. Am. Chem. Soc. 2008;130:10878–10879. doi: 10.1021/ja8041004.
- 19.Aliphatic aldimines are represented in other enantioselective Mannich reactions, often with lower enantioselectivity relative to aryl aldimines: Neuvonen AJ, Pihko PM. Org. Lett. 2014;16:5152–5155. doi: 10.1021/ol5025025. Wang Q, Leutzsch M, van Gemmeren M, List B. J. Am. Chem. Soc. 2013;135:15334–15337. doi: 10.1021/ja408747m. Yan H, Suk Oh J, Lee J-W, Eui Song C. Nat. Commun. 2012;3:1212. doi: 10.1038/ncomms2216. Lou S, Dai P, Schaus SE. J. Org. Chem. 2007;72:9998–10008. doi: 10.1021/jo701777g.
- 20.Kim H, Yen C, Preston P, Chin J. Org. Lett. 2006;8:5239–5242. doi: 10.1021/ol062000v. [DOI] [PubMed] [Google Scholar]
- 21.Dobish MC, Johnston JN. J. Am. Chem. Soc. 2012;134:6068–6071. doi: 10.1021/ja301858r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin P, Pisano JM, Schoen WR, Kang C, Chan WWS, Butler BS, Smith RG, Fisher MH, Wyvratt MJ. Bioorg. Med. Chem. Lett. 1999;9:3237–3242. doi: 10.1016/s0960-894x(99)00568-5. [DOI] [PubMed] [Google Scholar]
- 23.Gao Y, Liu X, Liu W, Qi Y, Liu X, Zhou Y, Wang R. Bioorg. Med. Chem. Lett. 2006;16:3688–3692. doi: 10.1016/j.bmcl.2006.04.063. [DOI] [PubMed] [Google Scholar]
- 24.Dong S, Merkel L, Moroder L, Budisa N. J. Pept. Sci. 2008;14:1148–1150. doi: 10.1002/psc.1065. [DOI] [PubMed] [Google Scholar]
- 25.Ao L, Wu Y, Kim D, Jang ER, Kim K, Lee D-m, Kim KB, Lee W. Mol. Pharm. 2012;9:2197–2205. doi: 10.1021/mp300044b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sigma-Aldrich catalog number 799599
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.