

Abstract
Mitomycin C (MC) and Decarbamoylmitomycin C (DMC) -a derivative of MC lacking the carbamate on C10- are DNA alkylating agents. Their cytotoxicity is attributed to their ability to generate DNA monoadducts as well as intrastrand and interstrand cross-links (ICLs). The major monoadducts generated by MC and DMC in tumor cells have opposite stereochemistry at carbon one of the guanine-mitosene bond: trans (or alpha) for MC and cis (or beta) for DMC. We hypothesize that local disruptions of DNA structure from trans or cis adducts are responsible for the different biochemical responses produced by MC and DMC. Access to DNA substrates bearing cis and trans MC/DMC lesions is essential to verify this hypothesis. Synthetic oligonucleotides bearing trans lesions can be obtained by bio-mimetic methods. However, this approach does not yield cis adducts. This report presents the first chemical synthesis of a cis mitosene DNA adduct. We also examined the stereopreference exhibited by the two drugs at the mononucleotide level by analyzing the formation of cis and trans adducts in the reaction of deoxyguanosine with MC or DMC using a variety of activation conditions. In addition, we performed Density Functional Theory calculations to evaluate the energies of these reactions. Direct alkylation under autocatalytic or bifunctional conditions yielded preferentially alpha adducts with both MC and DMC. DFT calculations showed that under bifunctional activation, the thermodynamically favored adducts are alpha, trans, for MC and beta, cis, for DMC. This suggests that the duplex DNA structure may stabilize/oriente the activated pro-drugs so that, with DMC, formation of the thermodynamically favored beta products are possible in a cellular environment.
Keywords: Mitomycin C, deoxyguanosine, post-oligomerization, stereo preference
Graphical abstract
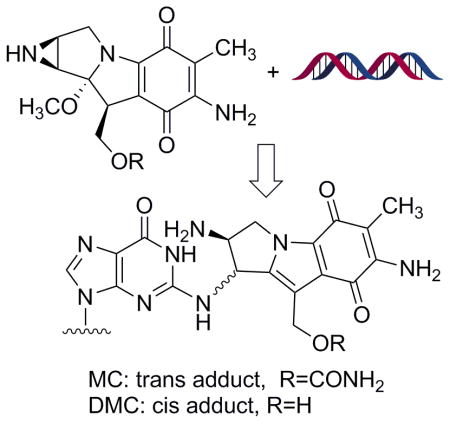
1. Introduction
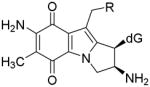
Mitomycin C (MC) [1] is a DNA alkylating agent [2] currently used to treat certain stomach, anal and lung cancers [3]. Its cytotoxicity is attributed to the formation of DNA-adducts, in particular to the formation of interstrand crosslinks (ICLs) [4]. 10-decarbamoylmitomycin C (DMC) is a derivative of MC lacking the carbamate group on C-10 (Fig. 1). As such, DMC was originally thought to only alkylate DNA monofunctionally. However, when EMT6 mouse mammary tumor cells were treated with DMC, both ICLs and DNA monoadducts were produced [4]. Major DNA adducts generated by treatment of EMT6 mouse mammary cells with MC or DMC have opposite stereochemistry at carbon one of the guanine-mitosene bond: trans (or alpha) for MC and cis (or beta) for DMC [4].
Fig. 1.

(2-column fitting image): Major DNA adducts generated by MC and DMC in cells.
In correlation with the stereopreference exhibited by DMC, it was found that DMC was more toxic to human cancer cells and that only DMC induces apoptosis efficiently in a p53 independent manner [5]. Access to mitosene-alpha and mitosene-beta DNA-adducts is essential to elucidate if and how the local DNA structure of these adducts is responsible for the different biochemical responses displayed by the two drugs. Individual structure-activity relationships of multiple DNA adducts generated by a single agent have been investigated mostly in the case of organic mutagens and carcinogens [6]. In general, such studies utilize synthetic oligonucleotides bearing a specific adduct at a unique position of their base sequences [7]. Similar investigations can be conducted on MC and DMC adducts, enabling direct comparisons of biological effects induced by a different stereochemistry at carbon 1 (Fig. 1). Trans adducts (or alpha) are available by reacting oligonucleotides with activated MC (bio-mimetic synthesis) [8], however, to this day, no site specific cis (or beta) adduct has been synthesized. Examination of the biochemical, structural, and biological effects of beta adducts has been severely limited by the inability to prepare sufficient quantities of this lesion. Our long term goal is to synthesize oligonucleotides bearing the beta ICL using a post-oligomerization strategy. Previously, we presented the synthesis of major mitomycin C-DNA adducts both at the nucleoside [9, 10] and at the oligonucleotide level [10] using post-oligomerization. This report presents the first chemical synthesis of a mitosene beta (cis) monoadduct; which is the first step toward the postoligomerization synthesis of site specific beta ICLs.
MC and DMC must be reduced in a monofunctional or bifunctional way before alkylating DNA [11]. In cells, the major mode of activation is bifunctional [11]. In this case, DMC produces mostly beta adducts and MC alpha adducts. A rationale for the opposite stereochemistry displayed by MC and DMC at CpG steps in DNA has been suggested [12]. On the other hand, the major adduct formed in the reaction of DMC with calf thymus DNA or poly(dG.dC)poly(dG.dC) is the alpha monoadduct, contrary to what happens in a cellular environment [4]. This apparent contradiction warrants some investigation. Factors responsible for the stereopreference exhibited by MC and DMC toward deoxyguanosine independently of the DNA structure have not been determined yet. Therefore, we also present here a study on the reactivity of deoxyguanosine with MC and DMC in an effort to understand the factors leading to the favored stereoselectivity. Different types of drug activation (monofunctional and bifunctional) have been considered along with different solvent conditions. Molecular calculations have also been performed to measure the energetics of all reactions examined.
2. Materials and methods
2.1 General Information
1H NMR and 13C NMR spectra were recorded using a JEOL ECX 300 (300 MHz), a Varian Inova 500 (500 MHz) or a Bruker AVANCE 500 (500 MHz) spectrometer. Spectra were recorded at 298 K and the residual solvent peak was used as the internal reference. Chemical shifts are reported in parts per million and coupling constants are in hertz (Hz). The conventional numbering system is used for the mitosene moiety and the purine carbons are numbered 1–6 also as per convention. The sugar carbons are numbered 1′–5′ beginning at the anomeric carbon and proceeding via the carbon chain to the primary carbinol center. Mitosene compounds with trans substituents at carbon 1 and 2 are labeled “a” and those with cis substituents at carbon 1 and 2 are labeled “b”.
Reagents were obtained from commercial sources and were used without further purification. All reactions were carried out under an atmosphere of Argon unless otherwise stated. Thin layer chromatographic analyses were carried out on 250 mm silica gel plates containing a fluorescent indicator and, if necessary, spots were visualized with I2. Column chromatographic purifications were performed using 200–300 mesh silica gel. Trans-2,7-diamino-1-hydroxymitosene and cis- 2,7-diamino-1-hydroxymitosene were synthesized by known methods [13]. Compounds 6a and 6b were synthesized according to a previously described procedure [9]. Adducts 1a, 1b, 2a and 2b generated via direct alkylation were isolated by chromatography using an Agilent 1200 HPLC system equipped with a Kromasil C18 column (4.6 mm*250 mm). Ratios of cis and trans adducts were calculated using the relative absorption of each adduct at λ=254 nm. HRMS spectra were recorded by direct infusion using Bruker’s micrOTOF-II ESI instrument at Notre Dame University Mass Spectrometry Facility. LC-ESI MS/MS experiments were performed at John Jay College using a Shimadzu LC-MS 8030 (Shimadzu Corporation, Kyoto, Japan). CD spectra were obtained with a Jasco 1500 CD spectrometer. UV spectra were obtained using a Shimadzu UV 2600 UV-vis spectrometer.
2.2 Synthesis of 7b
Compound 6b (33mg, 0.062 mmol) was dissolved in pyridine (3 mL) and ammonium hydroxide (1.5 mL). Trimethylphosphine (69 μL, 0.67 mmol) was added. The reaction was stirred at room temperature in a sealed vial flushed with Argon for 30 minutes. Solvents were evaporated and the red residue was purified by preparative thin layer chromatography (tlc) (SiO2: 1% NH4OH : 10% MeOH : 89% CH2Cl2) yielding 29 mg of 6b (93% yield). 1H NMR (methanol-d4) δ= 0.09 (s, CH3, 9H), 1.05 (t, CH2, J=8.3 Hz, 2H), 1.82 (s, CH3, 3H), 4.00 (dd, H3, J=8.2, 12.5 Hz, 1H), 4.22 (t, CH2, J=8.3 Hz, 2H), 4.46 (d, H1, J = 5.4 Hz, 1H), 4.54 (dd, H3, J=8.3, 12.2 Hz, 1H), 4.70 (m, H2, 1H), 5.23 and 5.30 (AB quartet, CH2, H10, J =12.7 Hz, 2H). 13C NMR (methanol-d4) δ= −1.4 (3C), 8.3 (1C), 18.6 (1C), 55.2 (1C), 57.4 (1C), 58.4 (1C), 64.2 (1C), 66.2 (1C), 106.4 (1C), 114.2 (1C), 123.5 (1C), 129.6 (1C), 141.1 (1C), 148.6 (1C), 154.3 (1C), 159.2 (1C), 178.5 (1C), 179.6 (1C). HRMS m/z calcd for C20H30N5O6Si [M+ H]+: 464.1965, found: 464.1958.
2.3 Synthesis of 9b
2-Fluoro-O6-(2-p-nitrophenylethyl)deoxyinosine (27 mg, 0.064 mmol) was dissolved in dry dimethylsulfoxide (200 μL). Compound 7b (15 mg, 0.032 mmol) and diisopropylethylamine (10 μL, 0.96 mmol) were added to the reaction mixture which was incubated at 45° for three weeks. The resulting crude material was dried with a flow of Argon to remove all solvents and further removal of trace of dimethylsulfoxide was performed under high vacuum. The desired product was isolated by preparative tlc (SiO2: 15% methanol : 6% NH4OH : 79% CH2Cl2) to give 15 mg (54% yield) of 9b (NMR spectrum shown in supporting information Fig. S12). 1H NMR (methanol-d4) : δ −0.10 (s, CH3, 9H), 0.70 (t, CH2, J=8.3 Hz, 2H), 1.84 (s, CH3, 3H), 2.38 (m, H2′, 1H), 2.82 (m, H2′, 1H), 3.75 (dd, H5′, J=12, 4 Hz, 1H), 3.82 (dd, H5″, J=12, 3 Hz, 1H), 3.98 (m, H4′ and CH2, 3H), 4.14 (dd, H3, J=13, 7 Hz, 1H), 4.55 (m, H3′, 1H), 4.58 (dd, H3, J=13, 7 Hz, 1H), 4.74 (m, CH2, 2H), 5.05 (m, H2, 1H), 5.07 and 5.10 (AB quartet, CH2, H10, J =7 Hz, 2H), 5.75 (d, H1, J = 6 Hz, 1H), 6.32 (t, H1′, J= 7Hz, 1H), 7.62 (d, Har, J=8 Hz, 2H), 8.10 (s, H8, 1H), 8.17 (d, Har, J=8 Hz, 2H), 8.55 (s, NH, 1H). HRMS m/z calcd for C38H47N10O12Si [M + H]+: 865.3139, found: 863.3147.
2.4 Synthesis of 1b
The N2- and O6-protected mitosene-nucleoside adduct 9b (12.0 mg, 0.017 mmol) was dissolved in dimethylsulfoxide (50 μL) and was treated with 1,8-diazabicyclo[5.4.0]undec-7-ene (10 μL, 0.06 mmol). All starting material disappeared after 2 h. The resulting crude material was diluted with water (1 mL) and lyophilized. The product was isolated by preparative tlc (SiO2: 3% NH4OH: 15% MeOH : 82% CHCl3) to give the N2-protected mitosene-nucleoside adduct 10b. Deprotection at the O6 position was confirmed by HRMS analysis: m/z calcd for C30H40N9O10Si [M+ H]+: 714.2662, found: 714.2641. The intermediate 10b was deprotected at the N2 position using tBuN4F (tetrabutylammonium fluoride, 1 M THF solution, 0.68 mmol, 68 μL) in 50 μL of DMSO or TAS-F (trisulfonium difluorotrimethylsilicate, 0.68 mmol, 18.7 mg) in 50 μL of DMF. Compound 1b (6.2 mg, 64 % yield) was isolated by precipitation in hexane followed by C18 chromatography (sep-pak Oasis© HLB, 12 cc, 500 mg; 15 % acetonitrile in water). Structure of 1b was confirmed by UV and CD spectroscopy (supporting information Fig. S13–S14) and coinjection with a sample obtained via the bio-mimetic route. HRMS m/z calcd for C24H28N9O8 [M+ H]+: 570.2055, found: 570.2029.
2.5 HPLC conditions for isolation of Mitomycin C N2 deoxyguanosine adducts (1a and 1b) and decarbamoylmitomycin C N2 deoxyguanosine adducts (2a and 2b) from direct alkylation reactions
Analysis of MC and DMC adducts (1a, 1b, 2a, 2b) was performed using the following HPLC system: Buffer A: Ammonium Acetate; 20 mM, pH 5.5; Buffer B: Acetonitrile. Flow: 1mL/min. For MC adducts 1a and 1b: Gradient : From 0 min to 45 min, Isocratic 5% B; from 45min to 55 min, 5% to 8% B; from 55min to 90min, 8% to 11% B. Adducts appear at RT=56 min for 1a and RT=76 min for 1b. For DMC adducts 2a and 2b: Gradient: From 0 min to 45min, Isocratic 6% B; from 45min to 55 min, 6% to 9% B; from 55min to 70min, isocratic 9% B. Adducts appear at RT=30 min for 2a and RT=64 min for 2b. Examples of chromatograms can be seen in the supporting information (Fig. S1 and S2).
2.6 Autocatalytic or bifunctional activation of mitomycin C and decarbamoyl mitomycin C in Buffer
Potassium phosphate buffer (autocatalytic conditions: 34 mL, 0.01 M, pH 7.5 or bifunctional conditions: 34 mL, 0.01 M, pH 5.8) was deaerated 15 min with Argon. 2′-deoxyguanosine (336 mg, 1.36 mmol) was added with either MC (90 mg, 269 μmol) or DMC (78 mg, 269 μmol). The reaction mixture was stirred and deaerated for 30 min. Sodium dithionite (autocatalytic conditions: 1.79 mL of a 40 mM solution, 72 μmol, 0.27 equivalent or bifunctional conditions: 10.1 mL of a 40 mM solution, 403.5 μmol, 1.5 equivalent) in deaerated potassium phosphate buffer (autocatalytic conditions: 0.01 M, pH 7.5 or bifunctional conditions: 0.01 M, pH 5.8) was added in one portion to the reaction mixture. After stirring for one hour at room temperature under Argon, the reaction mixture was opened to air and stirred for another 30 min. The reaction mixture was then centrifuged (13 000 rpm, 10min) and prepurified using C18 chromatography (sep-pak Oasis© HLB, 12 cc, 500 mg; 5 to 20 % acetonitrile in water). Adducts were isolated in the 15% acetonitrile fraction. Further purification was performed by HPLC.
2.7 Autocatalytic or bifunctional activation of mitomycin C and decarbamoyl mitomycin C in DMSO
DMSO (1.88 mL) was first deaerated with Argon during 15 min. 2′-deoxyguanosine (150 mg, 562 μmol), MC (5 mg, 15 μmol) or DMC (4.3 mg, 15 μmol ) was then added. The reaction mixture was stirred and deaerated during 30min. Sodium dithionite (autocatalytic conditions: 10 μL of a 400 mM solution, 4 μmol, 0.27 eq. or bifunctional conditions: 56.2 μL of a 400 mM solution, 22.5 μmol, 1.5 eq or autocatalytic/bifunctional conditions: 37.5 μL of a 400 mM solution, 15 μmol, 1 eq ) was added in one portion. P-Toluenesulfonic acid (1 equivalent, 15 μmol, 2.58 mg) from a 80 mM solution in deaerated DMSO was added immediately afterward. After stirring for one hour at room temperature under Argon, the reaction mixture was opened to air and stirred for another 30 min. Solvents were evaporated and the residue was dissolved in water (1 mL). The solution was centrifuged (13 000 rpm, 10min) and prepurified using C18 chromatography (sep-pak Oasis© HLB, 12 cc, 500 mg; 5 to 20 % acetonitrile in water). Adducts were isolated in the 15% acetonitrile fraction. Further purification was performed by HPLC.
2.8 Yield of direct alkylation reactions
Estimated yields of direct alkylation reactions were obtained through HPLC analysis of crude reaction mixtures. A 5 point calibration curve was built where the area of the absorption at 312 nm of the mitosene chromophore (X axis) was plotted versus the amount of mitosene chromophore (Y axis) -supporting information, Fig. S11- [4]. For each reaction, a portion of the crude mixture was analyzed and the peak area from alpha and beta adducts was measured at 312 nm. The calibration curve was used to determine the quantity of adduct per injection.
2.9 Characterization of the MC and DMC adducts
All four adducts (1a, 1b, 2a, 2b) were isolated by HPLC and then characterized by LC-MS using a modified published procedure [15]. The column used was a Phenomenex Luna 3 μm C8, 100 Å, 150 mm X 2 mm. The LC column was maintained at 35°C. Mobile phase (MP) conditions were: MP A, 5 mM ammonium formate in water containing 0.1 % formic acid, and MP B, acetonitrile. Elution gradient: 95% A for 15 minutes, 5–30% B in 30 minutes, 30% B for 15 minutes, back to initial gradient conditions at min 61 and column equilibration for 4.5 min. The flow rate was 0.2 mL/min. The mass spectrometer (MS) was operating with dual ion source (DUIS) in positive mode and data was collected in SIM mode. The MS parameters were: interface voltage 4.5kV, nebulizer gas 2 L/min, desolvation line temperature 250°C, heat block temperature 400°C and drying gas flow 15 L/min. The m/z monitored for adducts 1a and 1b was 571, and for 2a and 2b was 528.
The chromatogram for each compound with the corresponding parent ion m/z are shown on Fig. S3 and S4 (supporting information). The retention time for each m/z is the following: adduct 1a: 7 min, adduct 1b: 18 min, adduct 2a: 6.3 min, adduct 2b: 18 min. In addition, alpha and beta adducts mixtures isolated from the sep-pak pre-purification were analyzed using optimized conditions. For each mixture of stereoisomers (1a and 1b, 2a and 2b) the parent ions and their two fragments were detected. These samples were injected using the LC conditions described above and monitoring 2 MRM transitions in the MS. The transitions for adduct 1a and 1b were 571.10>393.90 and 571.10>241.95, and for 2a and 2b were 528.10>393.85 and 528.10>243.20 (Fig. S5 and S6, supporting information). UV spectra of each adduct are shown in the supporting information section (Fig. S7–S10).
2.10 Density Functional Theory (DFT) calculations
Mitomycin C, Decarbamoylmitomycin C and their activated species were studied with quantum mechanics, specifically the Density Functional Theory (DFT), using the B3LP method, with the 6–31 G** basis set, as implemented by the Spartan program [16]. The energies of the different entities are obtained by geometry optimization in order to obtain global minima and the results are displayed in Table S2, supporting information. Optimized figures are shown in the supporting information section (Fig. S16–S31)
3. Results and Discussion
3.1 Chemical synthesis of a mitosene-deoxyguanosine beta adduct
Direct alkylation reactions between MC/DMC and deoxyguanosine are not efficient methods to synthesize nucleoside or nucleotide adducts. At best, alpha adducts can be obtained in short oligonucleotides via a bio-mimetic route, but the yield of adducts varies with the length and the sequence of the nucleotide [17, 18]. Our lab has been involved in alternative methods to access these DNA adducts. We successfully synthesized adduct 1a and a 2,7-diaminomitosene-DNA adduct using a post-oligomerization method [9, 10]. However, our previous efforts to access 1b failed [9]. Here, we finally report the first chemical synthesis of adduct 1b.
The synthesis of 1b requires the amino precursor 7b (Fig. 2). Briefly, the amino hydrin precursors were obtained from Mitomycin C using HCl (Fig. 2). The 2-amino position was protected using the trimethylsilylethoxycarbonyl (teoc) group and the hydroxy group was converted to an azido group in two steps via a mesylate to give a mixture of stereomers 6a and 6b. At that stage, both stereomers can be separated by chromatography. Reduction to the amino group progressed efficiently in the case of the alpha (trans) amino precursor using triphenylphosphine -Staudinger reaction- [9]. However, the reaction was challenged in the case of the beta (cis) amino mitosene 7b. Our previous work has shown that steric hindrance was likely the cause for the sluggish conversion [9]. Indeed, the conversion proceeded rapidly when triphenyl phosphine was replaced by trimethyl phosphine and the final amino mitosene 7b was obtained in less than a hour in 93% yield.
Fig. 2.

(2-column fitting image): Synthesis of 7b
Coupling of precursor 7b (cis) with 2-fluoro-O6-(2-p-nitrophenylethyl)deoxyinosine [19] afforded intermediate 9b and subsequent deprotection at the O6 position using DBU yielded 10b (Fig. 3). We previously reported that ZnBr2 was a good reagent for the complete and rapid deprotection of the teoc group [10]. However, in the case of adduct 9b, these conditions also cleaved the 10-carbamate group resulting in a mixture of products. Several other deprotection conditions were tried. Both tetrabutylammonium fluoride and tris(dimethylamino)sulfonium difluorotrimethylsilicate successfully deprotected the teoc group. The pure desired beta adduct 1b was finally isolated by hexane precipitation followed by C18 purification (15% acetonitrile in water). UV [11] CD [2b] and HRMS spectra were recorded (supporting information, Fig. S13–S15) and its structure was confirmed via co-elution with an authentic sample prepared by direct alkylation.
Fig. 3.

(1-column fitting image): Synthesis of 1b
Circular dichroism is a published and reliable method to assign the configuration of mitosene adducts at C1. As can be observed in the CD spectra of the cis and trans amine (Fig. S32 and S33), compounds 7a and 7b display an opposite cotton effect around around 530 nm. The cis amine (7b, beta) displays a positive CE while the diastereomer (7a, alpha) displays a negative CE. The sign is dependent only of the cis/trans configuration and is not affected by the substituent at C1 [20]. This effect is also observed for nucleoside-mitosene adduct pairs [20]. The guanosine chromophore attached at C-1 absorbs at 250–280 nm and the 2 chromophors (mitosene and guanosine) constitute a coupled oscillator and give rise to coupled CD in the 200–400 nm. Fortunately, the 530 nm maximum is too far removed from the purine maxima so that the bands cannot efficiently couple hence this longest wavelength CE is dominated by the cis/trans (alpha /beta) configuration of the mitosene moiety. Thus, the presence of a positive CE for adduct 1b around 530 nm confirms the cis/beta stereochemistry.
This is the first chemical synthesis of a beta mitosene-DNA adduct. This method paves the way to the beta ICL via the post-oligomerization method. In this case, the amino precursor 7b will be coupled to a DNA amonoadduct bearing a fluoroinosine at a specific site. After annealing, crosslinking under reductive activation of the mono DNA adduct should afford the desired beta ICL.
3.2 Direct alkylation of deoxyguanosine by MC and DMC
An important feature of the molecular mechanism of action of the mitomycins is that their original quinone forms are inactive towards DNA. A reductive activation to the hydroquinone is required to trigger the monoalkylating and cross-linking activities of the prodrugs. Spontaneous elimination of methanol generates a leuco-aziridinomitosene (12) which can follow two paths to guanine N2 alkylation. Path one, an autocatylic process, generates a 7-aminoaziridine (13) and leads to DNA monoalkylation. Path 2 involves protonation of the leuco-aziridinomitosene (12) to give a C-1 carbonium ion (14). This intermediate can yield ICLs. The activated species generated (13 or 14) determine the outcome of DNA alkylation both with DNA extract and within cells [11]. Although all mitomycin C-DNA adducts obtained upon reductive activation have been isolated and characterized, questions remain regarding the activation and the reactivity of both mitomycins [21] and aziridinomitosenes [22].
When DMC is chemically reduced in the presence of calf thymus DNA or poly(dG.dC)poly(dG-dC), the major adduct obtained is the alpha (trans) monoadduct, whereas in a cellular environment, the beta (cis) isomer is produced [4]. Bio-mimetic conditions favor the autocatalytic pathway -path 1- because of high concentration of DMC i.e. the major alkylating species is the 7-aminoaziridine (13). Under intracellular conditions, however, the alkylating species is believed to be the protonated leucoaziridinomitosene -(14), from path 2- since low concentrations of drug disfavor the autocatalytic pathway. Concomitantly, when bifunctional activation -path 2- is favored in cancer cells treated with MC -under hypoxic conditions for instance- higher yields of ICLs and 2,7 DAM adducts -from path 2- are observed [23].
In order to gain insight into the reactivity of MC and DMC -independently of the surrounding DNA structure-, we generated either the leuco-aziridinomitosene (14) or the 7-aminoaziridine (13) in the presence of deoxyguanosine and analyzed the products obtained via HPLC and LC-MS. We reduced MC and DMC using sodium dithionite for two reasons: 1) reduction is rapid and 2) it is the reducing agent of choice for the bio-mimetic synthesis of MC and DMC DN-Aadducts. Reactions were run in potassium phosphate buffer and in DMSO. Activation type was favored by varying the amount of sodium dithionite used, hence promoting either the autocatalytic or the bifunctional pathway.
The relative proportion of alpha and beta adducts (Ra/b) generated in water or DMSO are presented in table 1. Under autocatalytic conditions - with 16 or 17 as the activated species, Fig. 4-, reactions in aqueous buffer generate preferentially the alpha adduct with both MC and DMC. The ratio alpha/beta (Ra/b) is larger with DMC. This indicates that a trans attack prevails leading to the formation of the kinetically favored alpha (trans) products rather than the more stable beta (cis) adducts. The yield of adducts is higher with DMC; which suggests it’s a more efficient deoxyguanosine alkylating agent, as was previously observed [4]. This result may partially explain why the alpha adduct is the major adduct when DMC is reacted with poly(dG.dC)poly(dG-dC) under autocatalytic conditions [4].
Table 1.
Ratio of monoadducts: alpha versus beta (Ra/b) for direct alkylation reactions. The relative proportion of alpha versus beta deoxyguanosine-N2 monoadducts was calculated using the ratio of the absorbance at 253 nm of the DNA-adducts. (ε253= 23900 in 0.01 M potassium phosphate [24a] or ε252= 24360 in water [24b]). The ratios were determined by direct HPLC analysis of the reaction mixtures and measurement of the area under the individual isomer peaks. Average values were determined from triplicate experiments.
| Solvent | Na2S2O4 (equivalents) | Activation Type | MC | DMC | ||
|---|---|---|---|---|---|---|
| Yield (%) | Ra/b | Yield | Ra/b | |||
| Buffer pH 7.4 | 0.27 | Autocatalytic | 0.21 | 1.70 | 0.58 | 2.01 |
| Buffer pH 5.8 | 1.5 | Bifunctional | - | - | - | - |
| DMSO | 0.27 | Autocatalytic | 0.70 | 7.00 | 1.32 | 7.77 |
| DMSO | 1.5 | Bifunctional | 0.004 | only alpha | - | - |
| DMSO | 1 | Bifunctional | 1.65 | 6.9 | 0.16 | 1.94 |
Fig. 4.

(2-column fitting image): Molecular mechanisms of activation of MC and DMC
In DMSO, autocatalytic reactions required addition of acid to protonate the aziridine -one equivalent of p-toluenesulfonic acid was found to be the optimal amount, table S1, supporting information-. The overall yield of adducts improved in DMSO versus water (3.5 times for MC and 2.3 times for DMC) and the ratio Ra/b increased (MC: Ra/b=1.70 in water and 7.00 in DMSO; DMC: Ra/b=2.01 in water and 7.77 in DMSO). This suggests that the nucleophilic trans attack of either the protonated aziridine (16) or the open carbocation (17) is enhanced. As was observed in aqueous conditions, yields of alkylation are higher with DMC than with MC -2.7 times higher in buffer and 1.9 times higher in DMSO-, confirming that DMC is a more efficient alkylating agent.
Under bifunctional activation conditions -when 15 is the activated species- no adduct formation was detected with DMC either in buffer or DMSO. Other kinetically favored products were formed (from hydrolysis or reduction). A minute amount of alpha adduct was detected in DMSO with MC. A plausible explanation for the extremely low yield is that 15 is so reactive that hydrolysis and/or reduction products are formed preferentially.
Finally, we examined the proportion of adducts generated when we treated MC and DMC in DMSO with 1 equivalent of sodium dithionite. Under these conditions, the mechanism of alkylation is expected to be mainly bifunctional. For MC, we observed a similar alpha/beta ratio of products as was found under monofunctional activation conditions (Ra/b=6.94 versus 7.00). However, for DMC, the alpha/beta ratio drops dramatically (Ra/b=1.94 versus 7.77). This evidences that under bifunctional conditions DMC has a higher stereopreference for the beta adduct, even in reactions that do not involve duplex DNA. This may partially explain the formation of beta adducts in a cellular environment. However, with DMC, the DNA structure seems essential to provide either stabilization and/or favorable orientation of the reduced species to generate the beta adduct as the major product.
3.3 DFT calculations
Next, we investigated the energies of the direct reactions between deoxyguanosine and the prodrugs 15, 16 or 17 (Fig. 4) to appreciate if these different outcomes can be explained by energetics. Energies of the formation of the adducts were calculated by subtracting the sum of the energies of the reactants from the sum of the energies of the products -pictures and energies of the different entities involved are shown in the supporting information section: Fig. S16 to S31 and table S2-. After optimization, DFT calculations established that the product resulting from the alpha attack of bifunctionally activated MC (15, R=COONH2) with deoxyguanosine (18a, Fig. 5) is more stable than the beta one (18b, Fig. 6) and that, in the DMC case, the beta adduct (19b, Fig. 7) is more stable than the alpha one (19a, Fig. 8) -table S2, supporting information-.
Fig. 5.

(1-column fitting image): Trans guanosine adduct from bifunctionally activated MC (18a, alpha)
Fig. 6.

(1-column fitting image): Cis guanosine adduct from bifunctionally activated MC (18b, beta)
Fig. 7.

(1-column fitting image): Trans guanosine adduct from bifunctionally activated DMC (19a, alpha)
Fig. 8.
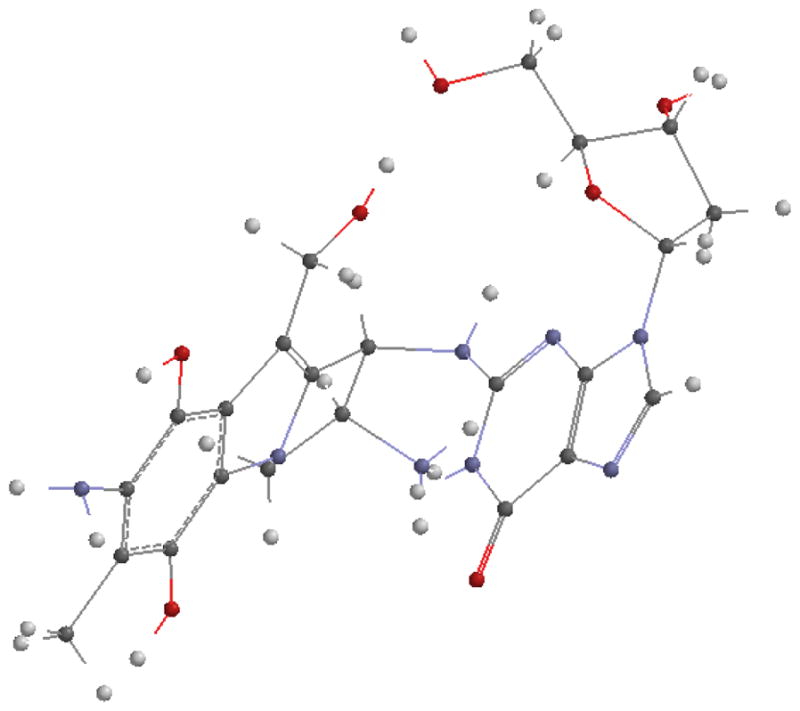
(1-column fitting image): Cis guanosine adduct from bifunctionally activated DMC (19b, beta)
In addition, during bifunctional activation i.e. when compound 15 is the activated species, formation of the alpha adduct (18a, Fig. 5) is thermodynamically favored by 2.45 kcal/mol for MC and formation of the beta adduct (18b, Fig. 8) is favored by 5.08 kcal/mol for DMC -table 2. These results correlate with the stereopreference exhibited by MC and DMC in a cellular environment, when 15 is believed to be the major alkylating species.
Table 2.
Energies of reduction and alkylation reactions.
| Reactants | Products | Energy of formation |
|---|---|---|
|
| ||
| MC + 2H+ + 2e− Or DMC + 2H + + 2e− Reduction |
 15, R=OCONH2 or R=OH
15, R=OCONH2 or R=OH |
R=OCONH2 (MC case) |
| E=−278.19 kcal/mol
| ||
| R=OH (DMC case) | ||
| E=−267.69 kcal/mol | ||
|
| ||
| 15 + deoxyguanosine |
 Trans, alpha |
R=OCONH2 (MC case, 18 a) |
| E=−11.30 Kcal/mol
| ||
| R=OH (DMC case, 19 a) | ||
| E=−9.73 Kcal/mol | ||
|
|
||
| Bifunctional activation |
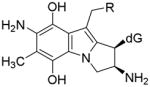 Cis, beta |
R=OCONH2 (MC case, 18 b) |
| E=−8.85 kcal/mol
| ||
| R=OH (DMC case, 19 b) | ||
| E=−14.81 kcal/mol | ||
|
| ||
| 16 + deoxyguanosine |
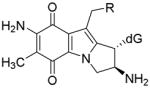 Trans, alpha |
R=OCONH2 (MC case, 1a) |
| E=−25.53 kcal/mol
| ||
| R=OH (DMC case, 2a) | ||
| E=−18.57 kcal/mol | ||
|
|
||
| Autocatalytic activation |
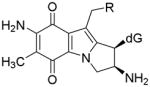 Cis, beta |
R=OCONH2 (MC case, 1b) |
| E=−51.82 kcal/mol
| ||
| R=OH (DMC case, 2b) | ||
| E=−51.29 kcal/mol | ||
|
| ||
| 17 + deoxyguanosine |
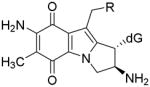 Trans, alpha |
R=OCONH2 (MC case, 1a) |
| E=−24.1 kcal/mol
| ||
| R=OH (DMC case, 2a) | ||
| E=−8.5 kcal/mol | ||
|
|
||
| Autocatalytic activation |
 Cis, beta |
R=OCONH2 (MC case, 1b) |
| E=−50.3 kcal/mol
| ||
| R=OH (DMC case, 2b) | ||
| E=−41.2 kcal/mol | ||
Beta attack by deoxyguanosine may be disfavored in the MC case because of steric hindrance from the carbamate; which results in the preferential formation of the alpha adduct. With DMC, which lacks the carbamate at C10, there is no such steric hindrance and the beta attack is favored. Formation of additional hydrogen bonds may also favor the beta attack. Alternatively, differences in the atomic charges as obtained by Mulliken Population analysis may be responsible for the observed stereopreference. The energies of the reactions of activation for both MC and DMC are quite large -table 2-. This is due to the relieving of the strain present in the aziridine which opens upon activation.
Figures depicting intermediate complexes between MC/DMC and deoxyguanosine formed during bifunctional activation (path 2) are also shown in Fig. 9, 10, 11 and 12. Energies of the complexes are shown in table 3. These structures represent pseudo-transition states. The energy of a hydrogen molecule has to be subtracted from their energy when compared to the final products.
Fig. 9.

(1-column fitting image): Pseudo transition state between MC and guanosine toward the formation of 1a.
Fig. 10.

(1-column fitting image): Pseudo transition state between MC and guanosine toward the formation of 1b.
Fig. 11.

(1-column fitting image): Pseudo transition state between DMC and guanosine toward the formation of 2a.
Fig. 12.
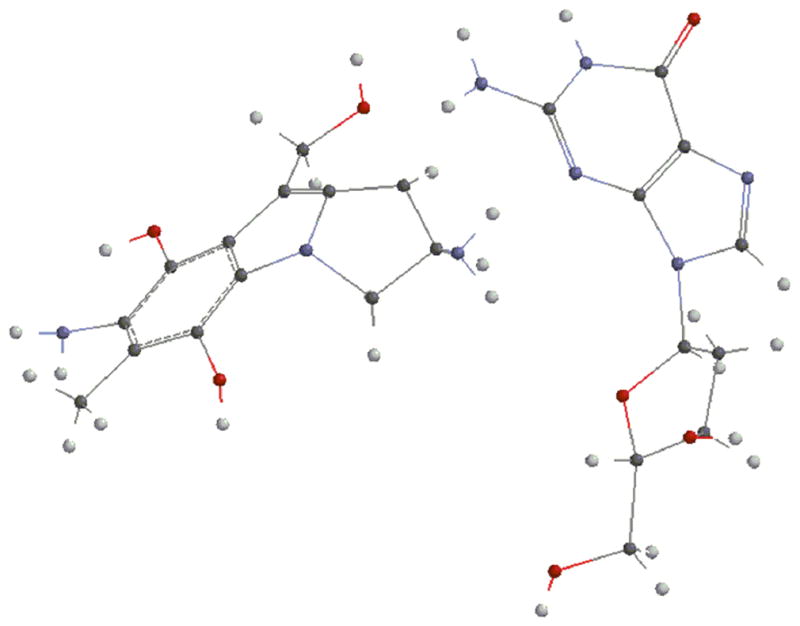
(1-column fitting image): Pseudo transition state between DMC and guanosine toward the formation of 2b.
Table 3.
Energies of pseudo transition states between MC/DMC and guanosine.
| Pseudo transition state: | Energies (ua): |
|---|---|
| MC-guanosine alpha | −2026.5257 |
| MC-guanosine beta | −2026.8514 |
| DMC-guanosine alpha | −1858.1394 |
| DMC-guanosine beta | −1858.0312 |
The 1a and 1b intermediates as well as the 2a and 2b intermediates should not be compared to each other, since they represent different points on the path. However, it is clear that they feature higher energies than the final products.
Finally, SN2 type reactions of the protonated aziridine (Path 1) predominantly produces alpha stereochemistry in a kinetic-control manner as was observed experimentally. From our calculations, formation of the beta adducts 1b and 2b seems to be favored thermodynamically in the case of both MC and DMC under autocatalytic conditions -when 16 or 17 are the reactive species, table 2-.
However, because the adduct formation is not reversible, the adducts’ energy can not be compared to explain the favored stereochemistry.
4. Conclusion
Our lab has been involved in the synthesis of MC and DMC-DNA adducts in order to understand how/if the isomeric crosslinks generated by MC and DMC are responsible for the compounds’ different cytotoxicity. We successfully synthesized the first deoxyguanosine mitosene beta adduct. This is the first time a mitosene beta adduct is synthesized using organic chemistry. In the future, we will study the application of this methodology to the synthesis of oligonucleotides bearing a cis-adduct. In addition, we identified and quantified the MC and DMC deoxyguanosine adducts -trans or alpha versus cis or beta- generated through direct alkylation to examine the stereopreference exhibited by the MC and DMC outside the duplex DNA. Yields of deoxyguanosine adducts were higher with DMC than with MC -except when one equivalent of sodium dithionite in DMSO was used- and this may partially explain why DMC is a more efficient alkylating agent in cells. Direct alkylation reactions in buffer yielded preferentially alpha adducts with both MC and DMC. This correlates with previous observations i.e. only alpha alkylation was observed when oligonucleotides are reacted with DMC under autocatalytic conditions, contrary to what happens in cells [4]. Bifunctional activation is believed to be prevalent in a cellular environment and under these conditions, DMC generates beta adducts preferentially. We did not observe formation of beta adducts when MC and DMC were activated bifunctionally and reacted with deoxyguanosine in buffer. This suggests that the duplex DNA structure may stabilize/oriente the activated pro-drugs so that formation of the thermodynamically favored products is possible [5]. DFT calculations showed that under bifunctional activation, the thermodynamically favored adducts are alpha, trans, for MC and beta, cis, for DMC.
Supplementary Material
Highlights.
We synthesized the first cis mitosene deoxyguanosine adduct via chemical synthesis
We activated MC and DMC through an autocatalytic or bifunctional pathway
We measured the ratio of cis and trans adducts from direct alkylation reactions
We examined the stereopreference exhibited by the two drugs
We evaluated the energies of direct alkylation reactions using DFT
Acknowledgments
This work was supported by NIH grant 5SC3GM105460 to Elise Champeil as well as by the Program for Research Initiatives for Science Majors (PRISM) at John Jay College. We also want to thank Dr. Padmanava Pradhan for his help in running CD experiments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Elise Champeil, Email: echampeil@jjay.cuny.edu.
Shu-Yuan Cheng, Email: shcheng@jjay.cuny.edu.
Bik Tzu Huang, Email: biktzu.huang@jjay.cuny.edu.
Marta Conchero-Guisan, Email: mconcheiro-guisan@jjay.cuny.edu.
Thibaut Martinez, Email: thibaut.martinez@etu.chimie-paristech.fr.
Manuel Paz, Email: manuel.paz@usc.es.
Anne-Marie Sapse, Email: acransg6@aol.com.
References
- 1.Hata T, Sano Y, Sugawara R, Matsumae A, Kanamorei K, Shima T, Hoshi T. Mitomycin, a New Antibiotic from Streptomyces. J Antibiotics, Ser A. 1956;9:141–146. [PubMed] [Google Scholar]
- 2.(a) Tomasz M. Mitomycin C: small, fast and deadly (but very selective) Chem Biol. 1995;2:575–579. doi: 10.1016/1074-5521(95)90120-5. [DOI] [PubMed] [Google Scholar]; (b) Palom Y, Belcourt MF, Musser SM, Sartorelli AC, Rockwell S, Tomasz M. Structure of adduct X, the last unknown of the six major DNA adducts of mitomycin C formed in EMT6 mouse mammary tumor cells. Chem Res Toxicol. 2000;13:479–488. doi: 10.1021/tx000024j. [DOI] [PubMed] [Google Scholar]
- 3.(a) Verweij J, Pinedo H. In: Cancer chemotherapy and biological modifiers, Annual 11. Pinedo HM, Chabner BA, Longo DL, editors. Elsevier Science Publishers B. V; Amsterdam: 1990. p. 67. [Google Scholar]; (b) Chabner BA, Amrein PC, Druker BJ, Michaelson MD, Mitsiades CS, Goss PE, Ryan DP, Ramachandra S, Richardson PJ, Supko JG, Wilson WH. Antineoplastic Agents. In: Brunton LL, Lazo JS, Parker KL, editors. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. McGraw-Hill Publishers; New York: 2005. pp. 1315–1403. [Google Scholar]
- 4.Palom Y, Suresh Kumar G, Tang LQ, Paz MM, Musser SM, Rockwell S, Tomasz M. Relative toxicities of DNA cross-links and monoadducts: new insights from studies of decarbamoyl mitomycin C and mitomycin C. Chem Res Toxicol. 2002;15:1398–1406. doi: 10.1021/tx020044g. [DOI] [PubMed] [Google Scholar]
- 5.(a) Boamah EK, Breckman A, Tomasz M, Natura Myeku M, Figueiredo-Pereira M, Hunter S, Meyer J, Bhosle RC, Bargonetti J. DNA adducts of decarbamoyl mitomycin C efficiently kill cells without wild-type p53 resulting from proteasome-mediated degradation of checkpoint protein 1. Chem Res Toxicol. 2010;23:1151–1162. doi: 10.1021/tx900420k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Boamah EK, White DE, Talbott KE, Arva NC, Berman D, Tomasz M, Bargonetti J. Mitomycin-DNA adducts induce p53-dependent and p53-independent cell death pathways. ACS Chem Biol. 2007;2:399–407. doi: 10.1021/cb700060t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Abbas T, Olivier M, Lopez J, Houser S, Xiao G, Kumar GS, Tomasz M, Bargonetti J. Differential activation of p53 by the various adducts of Mitomycin C. J Biol Chem. 2002;277:40513–40519. doi: 10.1074/jbc.M205495200. [DOI] [PubMed] [Google Scholar]
- 6.Basu AK, Essigmann JM. Site-specifically alkylated oligodeoxynucleotides: probes for mutagenesis, DNA repair and the structural effects of DNA damage. Mutat Res. 1990;233:189–201. doi: 10.1016/0027-5107(90)90162-w. [DOI] [PubMed] [Google Scholar]
- 7.Krishnegowda G, Sharma AK, Krzeminski J, Gowda AS, Lin JM, Desai D, Spratt TE, Amin S. Facile syntheses of O(2)-[4-(3-pyridyl)-4-oxobut-1-yl]thymidine, the major adduct formed by tobacco specific nitrosamine 4-methylnitrosamino-1-(3-pyridyl)-1-butanone (NNK) in vivo, and its site-specifically adducted oligodeoxynucleotides. Chem Res Toxicol. 2011;24:960–967. doi: 10.1021/tx200127j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Kumar S, Lipman R, Tomasz M. Recognition of specific DNA sequences by mitomycin for alkylation. Biochemistry. 1992;31:1399–1407. doi: 10.1021/bi00120a016. [DOI] [PubMed] [Google Scholar]; (b) Borowy-Borowski H, Lipman R, Tomasz M. Recognition between mitomycin C and specific DNA sequences for cross-link formation. Biochemistry. 1990;29:2999–3006. doi: 10.1021/bi00464a016. [DOI] [PubMed] [Google Scholar]; (c) Kumar S, Musser SM, Cummings J, Tomasz M. 2,7-Diaminomitosene, a monofunctional mitomycin C derivative, alkylates DNA in the major groove. Structure and base-sequence specificity of the DNA adduct and mechanism of the alkylation. J Am Chem Soc. 1996;118:9209–9217. [Google Scholar]
- 9.Champeil E, Paz M, Lukasiewicz E, Kong W, Watson S, Sapse AM. Synthesis of a major mitomycin C DNA adduct via a triaminomitosene. Bioorg Med Chem Lett. 2012;22:7198–7200. doi: 10.1016/j.bmcl.2012.09.052. [DOI] [PubMed] [Google Scholar]
- 10.Champeil E, Paz M, Ladwa S, Clement C, Zatorski A, Tomasz M. Synthesis of an oligodeoxyribonucleotide adduct of mitomycin C by the postoligomerization method via a tiamino mitosene. J Am Chem Soc. 2008;130:9556–9565. doi: 10.1021/ja802118p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suresh Kumar G, Lipman R, Cummings J, Tomasz M. Mitomycin C-DNA adducts generated by DT-diaphorase. Revised mechanism of the enzymatic reductive activation of mitomycin C. Biochemistry. 1997;36:1428–14136. doi: 10.1021/bi971394i. [DOI] [PubMed] [Google Scholar]
- 12.Bueren-Calabuig JA, Negri A, Morreale A, Gago F. Rationale for the opposite stereochemistry of the major monoadducts and interstrand crosslinks formed by mitomycin C and its decarbamoylated analogue at CpG steps in DNA and the effect of cytosine modification on reactivity. Org Biomol Chem. 2012;10:1543–1552. doi: 10.1039/c1ob06675g. [DOI] [PubMed] [Google Scholar]
- 13.Taylor WG, Remers WA. Structure and stereochemistry of some 1,2-disubstituted mitosenes from solvolysis of mitomycin C and Mitomycin A. J Med Chem. 1975;18:307–311. doi: 10.1021/jm00237a020. [DOI] [PubMed] [Google Scholar]
- 14.Verdine GL, Nakanish K. p-Dimethylaminocinnamate, a new red-shifted chromophore for use in the exciton chirality method. Its application to mitomycin C. Chem Commun. 1985;16:1093–1095. [Google Scholar]
- 15.Paz MM, Ladwa S, Champeil E, Liu Y, Rockwell S, Boamah EK, Bargonetti J, Callahan J, Roach J, Tomasz M. Mapping DNA adducts of mitomycin C and decarbamoyl mitomycin C in cell lines using liquid chromatography/electrospray tandem mass spectrometry. Chem Res Toxicol. 2008;21:2370–2378. doi: 10.1021/tx8002615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spartan 06. Irvine; California: p. 91612. [Google Scholar]
- 17.Warren AJ, Hamilton JW. Synthesis and structural characterization of the N2G—Mitomycin C—N2G interstrand cross-link in a model synthetic 23 base pair oligonucleotide DNA duplex. Chem Res Toxicol. 1996;9:1063–1071. doi: 10.1021/tx960070c. [DOI] [PubMed] [Google Scholar]
- 18.Borowy-Borowski H, Lipman R, Tomasz M. Recognition between mitomycin C and specific DNA sequences for cross-link formation. Biochemistry. 1990;29:2999–3006. doi: 10.1021/bi00464a016. [DOI] [PubMed] [Google Scholar]
- 19.Lee H, Hinz M, Stezowski JJ, Harvey RG. Syntheses of polycyclic aromatic hydrocarbon-nucleoside and oligonucleotide adducts specifically alkylated on the amino functions of deoxyguanosine and deoxyadenosine. Tetrahedron Lett. 1990;31:6773–6776. [Google Scholar]
- 20.Tomasz M, Jung M, Verdine G, Nakanishi K. Circular dichroism spectroscopy as a probe for the stereochemistry of aziridine cleavage reactions of mitomycin C. Application to adducts of mitomycin with DNA constituents. J Am Chem Soc. 1984;106:7367–7370. [Google Scholar]
- 21.(a) Bass PD, Gubler DA, Judd TC, Williams RM. Mitomycinoid alkaloids: mechanism of action, biosynthesis, total syntheses, and synthetic approaches. Chem Rev. 2013;113:6816–6863. doi: 10.1021/cr3001059. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Paz MM, Zhang X, Lu J, Holmgren A. A new mechanism of action for the anticancer drug Mitomycin C: Mechanism-based inhibition of thioredoxin reductase. Chem Res Toxicol. 2012;25:1502–1511. doi: 10.1021/tx3002065. [DOI] [PubMed] [Google Scholar]; (c) Paz MM. Reductive activation of mitomycins A and C by vitamin C. Bioorg Chem. 2013;48:1–7. doi: 10.1016/j.bioorg.2013.03.002. [DOI] [PubMed] [Google Scholar]; (d) Park HJ, Kim JJ, Kim HR, Lee EK, Kim ES, Jeong CS, Moon A, Lee SH. Synthesis and mechanistic studies of a mitomycin dimer containing an eight-membered cyclic disulfide. Bioorg Med Chem. 2015;19:4004–4013. doi: 10.1016/j.bmc.2011.05.020. [DOI] [PubMed] [Google Scholar]
- 22.(a) Wiedner SD, Vedejs E. Reactivity of aziridinomitosene derivatives related to FK317 in the presence of protic nucleophiles. J Org Chem. 2012;77:1045–1055. doi: 10.1021/jo202286a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mallory CM, Carfi RP, Moon SP, Cornell KA, Warner DL. Modification of cellular DNA by synthetic aziridinomitosenes. Bioorg Med Chem. 2015;23:7378–7385. doi: 10.1016/j.bmc.2015.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bizanek R, Chowdary D, Arai H, Kasai M, Hughes CS, Sartorelli AC, Rockwell S, Tomasz M. Adducts of mitomycin C and DNA in EMT6 mouse mammary tumor cells: effects of hypoxia and dicumarol on adduct patterns. Cancer res. 1993;53:5127–5134. [PubMed] [Google Scholar]
- 24.(a) Tomasz M, Chowdary D, Lipman R, Shimotakahara S, Veiro D, Walker V, Verdine GL. Reaction of DNA with chemically or enzymatically activated mitomycin C: isolation and structure of the major covalent adduct. Proc Natl Acad Sci. 1986;83:6702–6706. doi: 10.1073/pnas.83.18.6702. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tomasz M, Lipman R, Verdine GL, Nakanishi K. Reassignment of the guanine-binding mode of reduced mitomycin C. Biochemistry. 1986;25:4337–4344. doi: 10.1021/bi00363a024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.