

Abstract
HCM is the most common inherited heart condition occurring in 1:500 individuals in the general population. Left ventricular outflow obstruction at rest or after provocation occurs in 2/3 of HCM patients and is a frequent cause of limiting symptoms. Pharmacologic therapy is the first-line treatment for obstruction, and should be aggressively pursued before application of invasive therapy. Beta-blockade is given first, and up-titrated to decrease resting heart rate to between 50 and 60 beats per minute. However, beta-blockade is not expected to decrease resting gradients; its effect rests on decreasing the rise in gradient that accompanies exercise. For patients who fail beta-blockade the addition of oral disopyramide in adequate dose often will decrease resting gradients and offer meaningful relief of symptoms. Disopyramide vagolytic side effects, if they occur, can be greatly mitigated by simultaneous administration of oral pyridostigmine. This combination allows adequate dosing of disopyramide to achieve therapeutic goals. Verapamil utility in obstructive HCM with high resting gradients is limited by its vasodilating effects that can, infrequently, worsen gradient and symptoms. As such, we tend to avoid it in patients with high gradients and limiting heart failure symptoms. In a head-to-head comparison of intravenous drug administration in individual obstructive HCM patients the relative efficacy for lowering gradient was disopyramide > beta-blockade > verapamil. Severe symptoms in non-obstructive HCM are caused by fibrosis or severe myocyte disarray, and often by very small LV chamber size. Severe symptoms caused by these anatomic and histologic abnormalities, in the absence of obstruction, are less amenable to current pharmacotherapy. New pharmacotherapeutic approaches to HCM are on the horizon, that are to be evaluated in formal therapeutic trials.
Keywords: Beta-blockade, HCM, obstruction, pharmacologic therapy, disopyramide
INTRODUCTION
In current practice symptom relief is the goal of pharmacologic therapy in hypertrophic cardiomyopathy (HCM). Prolonged survival will also be a fundamental goal, but randomized trials have not yet been organized to demonstrate survival benefit. Since symptoms have different causes in obstructed and non-obstructed HCM patients, the choice of pharmacotherapy differs between these 2 main categories. This review will first discuss the pathophysiology of symptoms and then how pharmacologic agents are thought to work; practical use will be discussed. Traditional HCM drugs will be reviewed as well as investigational agents. All patients with HCM should have risk-stratification to assess their vulnerability for sudden death (SCD). In patients with high risk, consideration and referral for ICD implantation should proceed in parallel with initial steps towards symptom relief [1, 2].
Gene sequencing has shown association between HCM and mutations that code for 9 cardiac sarcomeric proteins, with a variable yield between 30-60% depending on the population studied [3-7]. For example, higher yield is found in families with HCM than in sporadic cases, when HCM presents earlier in life, and with a characteristic morphology of the septum. Rarely non-sarcomeric variants have been found to segregate with HCM patients [5, 8] though the first high-throughput sequencing report has suggested that these variants may comprise a higher proportion of HCM patients than previously appreciated [5]. Research pertaining to how mutations predispose to development of phenotype will be discussed briefly below, in relation to novel therapeutic strategies.
PATHOLOGY COMMON TO ALL HCM PATIENTS
Patients with HCM have myocyte hypertrophy, highly variable thickening of the left ventricle (LV), and highly variable extent of myocyte fiber disarray and fibrosis [9]. These abnormalities impair LV diastolic relaxation and chamber compliance which result in increased LV filling pressures and contribute to dyspnea [10]. In many HCM patients LV diastolic volume is decreased due to encroachment by the hypertrophy [11] which may compromise stroke volume. A common feature of symptomatic HCM patients is limitation of the increase in stroke volume that should accompany exercise [12]. In 2015 there is no agent that has been convincingly demonstrated to directly improve diastolic function in HCM, [13, 14] regress hypertrophy or regress fibrosis. Improvement in diastolic function may be achieved in obstructed patients by pharmacologic relief of obstruction-systolic load, improving relaxation by decreasing LV systolic pressure [15]. Myocardial ischemia from narrowed intramural coronary arteries may cause exercise or rest myocardial ischemia in HCM [16, 17]. This represents an additional exacerbation on top of the abnormal myocardial substrate.
LV outflow obstruction (occurring at rest or after provocation in 2/3 of HCM patients) adds afterload to the abnormal myocardial substrate. The adverse effects of obstruction are listed in Table 1, consequences that may be improved by pharmacotherapy.
Table 1.
Adverse consequences of mitral-septal contact.
|
Latent gradients are common in HCM. Patients who appear clinically to be non-obstructed may develop profound SAM, mitral-septal contact and high gradients after physiologic stimuli that reduce preload, or afterload, or increase contractility such as Valsalva maneuver, standing, after eating, and particularly after exercise [18-22]. Therefore, Valsalva and standing acquisition of gradient to provoke obstruction should be performed on the first study when HCM is diagnosed. Once HCM is diagnosed an exercise echocardiogram is often essential to demonstrate gradient, the location of obstruction (mitral-septal contact vs. mid-LV obstruction), and to exclude ischemic wall motion abnormality due to either epicardial or intramural coronary narrowing [23]. Functional capacity and BP response are assessed too. It is apparent from this that detailed physician familiarity with echocardiographic variants, and sonographer training is essential to accomplish these multiplicity of goals [23-25]. On occasion, the clinician will suspect obstruction, even if all the above detailed tests are negative (because of long mitral leaflets, resting SAM, post-prandial symptoms, or syncope with no arrhythmia). Because making the differential diagnosis is essential, post-prandial exercise testing with the gradient acquired in the upright posture has been introduced [20]. This may be the strongest physiologic provocation available. Cardiac catheterization with catheter-induced premature beats may provoke gradient, as may isoproterenol infusion [26].
It is the provocable nature of gradients that makes many widely used vasodilator cardiac medications contraindicated in obstructive HCM (Table 2). These drugs may have been initiated before the correct diagnosis: for mild hypertension, to prevent renal failure, or to “regress hypertrophy”. The first recommended therapeutic change in obstructed patients at an HCM Center is to stop the vasodilators, often with dramatic therapeutic benefit both on gradient and symptoms. It is the provocable increase of obstruction that raises the tantalizing question as to whether the reverse course might be possible. Could gradients be reduced or abolished by altered loading or contractility? From the very beginning of the modern HCM era this 180 degree reverse in course was found to be feasible and clinically useful [27].
Table 2.
Drugs to avoid in obstructive HCM.
|
OBSTRUCTION IN HCM
The most common cause of obstruction in HCM is due to systolic anterior motion (SAM) and mitral-septal contact. SAM is caused by a crucial overlap of early LV systolic flow and the mitral valve; flow strikes the underside of the protruding mitral leaflets and sweeps them into the outflow tract and into the septum (Figs.1-3) [28, 29]. Once an orifice and an LV outflow (LVOT) pressure gradient is established, it is the pressure gradient itself that continues to push the mitral valve into the septum, further narrowing the orifice and increasing the gradient. Obstruction in this mid phase of systole is a classic amplification loop. This explains the concave-to-the left pattern observed on the CW Doppler tracing through the LVOT jet. Obstruction can be comprehensively conceptualized as a flow-drag triggered, amplifying feedback loop [28-30]. The extent of SAM and the gradient is modulated by a tug-of-war between anteriorly displacing forces (the pushing force of flow) and restraining posterior forces (the chordae and papillary muscles). There is a dynamic equilibrium between these forces that allows pharmacotherapic benefit.
Fig. (1).
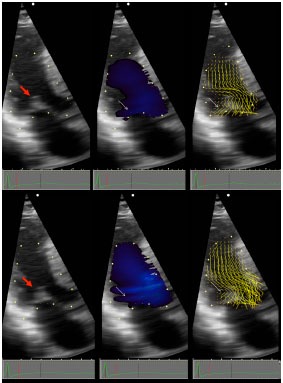
OHCM patient with ejection SAM. Vector Flow Mapping (VFM) is a novel method of processing Doppler information that demonstrates the vector of local blood flow velocity in intravascular structures. It differs from routine color Doppler: in VFM, a post-processing computational algorithm extracts information from the distribution of Doppler color flow in the beam direction and estimates the radial (perpendicular) component of the flow distribution, and displays it without angle dependence demonstrating direction and magnitude of blood flow velocity over 360 degrees. Local flow velocity is depicted as yellow lines proportional to, and in the direction of local velocity, indicated by red head of vector. Top panels are all pre-SAM frames; bottom panels are all post-SAM frames. Red arrow points to coapted MV. Early SAM is seen by comparing the 2D frames top and bottom left. White arrows point, in middle panels to blue color flow posterior to the MV, and in right panels to ricochet flow off the leaflet and into the cul-de-sac. Note that vector flow impacts the posterior surface of the mitral leaflets with high angle of attack and then ricochets off the leaflet into the cul-de-sac behind the valve. Neither blue flow posterior to leaflet, nor ricochet are seen in normals. Reproduced by permission from Ro R et al. Vector Flow Mapping in Obstructive Hypertrophic Cardiomyopathy to Assess the Relationship of Early Systolic Left Ventricular Flow and the Mitral Valve. J Am Coll Cardiol 2014; 64: 1984-95.
Fig. (3).
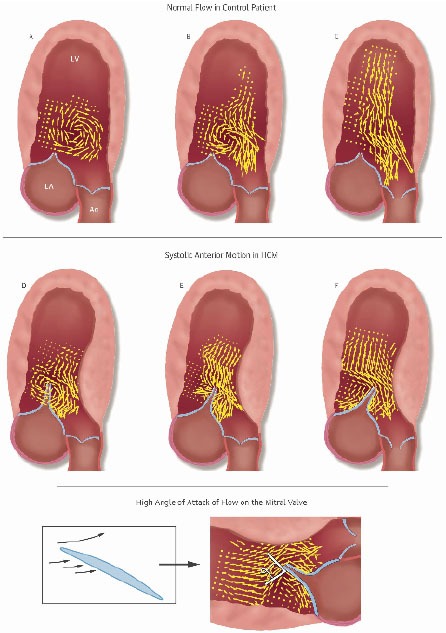
Vector Flow Maps in Normal and Obstructive HCM Patients. Top: Progression of early systolic vector flow maps in a normal patient. A: Isovolumic vortex. Note that aortic valve is closed. B: Confluence of isovolumic vortex and beginning of ejection flow. C: Ejection begins. There is orderly flow in the outflow tract; the MV does not protrude and there is no anteriorly directed flow behind it.
Middle: Two patients with obstructive HCM. D and E are sequential frames from a patient in whom vortical flow initiates SAM. F is another patient, who instead, has ejection SAM. Note the overlap of early systolic flow and the mitral leaflets. In obstructive HCM patients flow gets behind the mitral leaflets and sweeps them into the septum.
D: In 40% of patients isovolumic vortical flow initiates the anterior motion. The elongated protruding mitral leaflets extend past the center of the clockwise isovolumic vortex. The anteriorly directed limb of the clockwise vortex impacts the posterior surface of the mitral leaflet tip with high angle of attack and initiates SAM. E: Same patient as D but ejection flow has now begun. The mitral valve has been pre-postitioned into the outflow tract. Early ejection flow impacts the MV with high angle of attack. The aortic valve is still closed. F: Another patient, who instead, has ejection SAM. In 60% of cases SAM is initiated by early ejection; flow is first deflected posteriorly by the septal bulge; it then must course from a posterior to anterior direction. En route to the outflow tract it catches the posterior surface of the protruding MV with resulting high angle of attack. In both E and F note the ricochet of flow off the leaflet into the cul-de-sac.
Bottom: Even in airfoils specifically designed for lift, lifting force declines after the angle of attack exceeds 15°. In obstructive HCM patients we found that the anteriorly directed flow impacts the protruding leaflet tip with angles of attack of >60º before and after SAM begins. At these angles of attack, drag, the pushing force of flow, is the dominant hydrodynamic force on the leaflets. Reproduced by permission from Ro R et al. J Am Coll Cardiol 2014 same as Fig. (1).
Pharmacologic decrease of ejection acceleration displaces the equilibrium point towards restraint. Since the force on the mitral valve is proportional to the square of the velocity, even small changes in ejection velocity yield large changes in the force on the valve. Negative inotropes decrease LV ejection acceleration and thus the force on the mitral valve (Fig. 4) [31]. The consequent delay in mitral-septal contact decreases the time in systole of mitral-septal apposition, decreasing the duration of the feedback loop and the magnitude of the final gradient (Fig. 5). Thus, 2 pathologic features of HCM magnify the effect of negative inotropes for gradient reduction: 1) the force on the mitral valve is exponentially reduced by lower early ejection velocities, and 2) prolonging the time to mitral-septal contact interrupts of the self-amplifying rise of gradient. This explains how potent negative inotropes may substantially reduce or eliminate obstruction (Figs.6and7) [32, 33]. An analogy: The mitral valve is like an open door in a breezy corridor. As it is pushed closed it presents an ever greater face to a gust of wind. Negative inotropes decrease ejection acceleration - gentling the gust of wind – slamming the door later, or allowing it to remain open altogether. Reduction of obstructing intracardiac gradients by pharmacologic means is unique in clinical cardiology, and is thoroughly gratifying when it occurs.
Fig. (4).
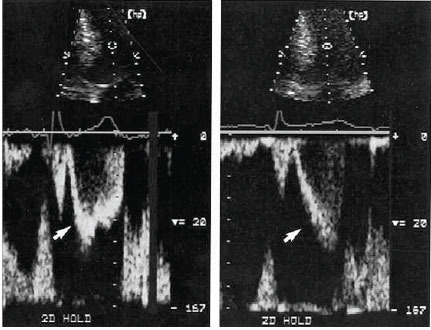
Comparison of left ventricular pulsed Doppler tracings before treatment (left) and after successful medical treatment (right) for patient 2. The sample volume was 2.5 cm apical of mitral valve coaptation point. Before treatment, ejection acceleration was rapid (arrowhead), and velocity peaked in the first half of systole. After treatment, ejection acceleration was slowed (arrowhead), and velocity peaked in the second half of systole. Systolic anterior mitral motion was delayed, and a 96 mm Hg gradient was eliminated. Note that although acceleration slowed, peak velocity remained virtually unchanged. This contrast highlights the importance of acceleration and the timing of ejection in successful medical therapy. The velocity calibration is identical in both panels. The scale is 20 cm/s between white marks. Reproduced by permission from Sherrid, M.V. et al. Mechanism of benefit of negative inotropes in obstructive hypertrophic cardiomyopathy. Circulation 1998; 97: 41-7.
Fig. (5).
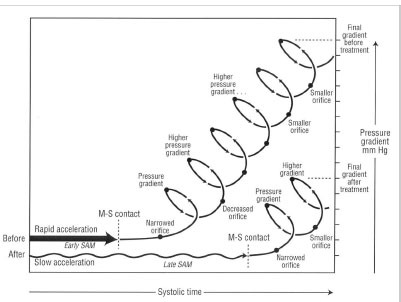
Proposed explanation of pressure gradient development before and after treatment of obstruction. Before treatment (top tracing), rapid left ventricular acceleration apical of the mitral valve, shown as a horizontal thick arrow, triggers early systolic anterior motion (SAM) and early mitral-septal (M-S) contact. Once mitral-septal contact occurs, a narrowed orifice develops, and a pressure difference results. The pressure difference forces the leaflet against the septum, which decreases the orifice size and further increases the pressure difference. An amplifying feedback loop is established, shown as a rising spiral. The longer the leaflet is in contact with the septum, the higher the pressure gradient. After treatment (bottom tracing), negative inotropes slow early SAM (shown as a horizontal wavy arrow) and may thereby decrease the force on the mitral leaflet, delaying SAM. Mitral - septal contact would occur later, leaving less time in systole for the feedback loop to narrow the orifice. This would reduce the final pressure difference. Delaying SAM may also allow more time for papillary muscle shortening to provide countertraction. In the figure, for clarity, the "before" arrow is positioned above the "after" arrow, although at the beginning of systole they both actually begin with a pressure gradient of 0 mm Hg. Reproduced by permission from Sherrid, M.V. et al. Mechanism of benefit of negative inotropes in obstructive hypertrophic cardiomyopathy. Circulation 1998; 97: 41-7.
Fig. (6).

Simultaneous left ventricle (LV) and aortic (AO) pressures before (control) and 20 Minutes after disopyramide (100 mg intravenously) in patient 1 (upper panel) and before and 10 Minutes after disopyramide in patient 2 (lower panel). Reproduced by permission from Pollick C. Muscular subaortic stenosis: hemodynamic and clinical improvement after disopyramide. N Engl J Med 1982; 307: 997-9.
Fig. (7).
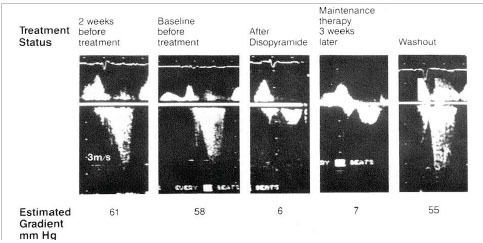
A representative series of Doppler tracings performed on 1 patient over a period of 5 weeks, showing left ventricular outflow tract flow before and after treatment with disopyramide. The first 2 tracings are before treatment. The third tracing is 2.5 hours after the first oral dose of 300mg disopyramide. The fourth tracing is 3 weeks later, on maintenance oral disopyramide. The fifth tracing is after drug washout, 72 hours after discontinuing disopyramide. Because of day to day differences in technique, there are minor differences in the calibration of the Doppler velocities. However, in every case, the distance between white markers is 1m/s. The calculated pressure gradient is shown beneath each tracing. Reproduced by permission from Sherrid M. et al. Oral disopyramide therapy for obstructive hypertrophic cardiomyopathy. Am J Cardiol 1988; 62: 1085-8.
There are no large randomized trials to compare efficacy and safety of the available medications for gradient reduction. No drug has been approved by the FDA for HCM; thus, reports of efficacy and safety depend on observational studies.
β-blockers are the first line therapeutic agents used for gradient reduction and symptom relief [27, 34]. However, the negative inotropic effects of β-blockade are relatively mild. (This is why they can be used even in systolic heart failure patients with profound reduction in ejection fraction). β-blockers cause no acute change in resting ejection fraction in normals. β-blockade is not expected to reduce resting LVOT gradients; rather, their beneficial effect on symptoms stems from a reduction in LVOT gradient rise after exercise [35]. HCM guidelines recommend a trial of β-blockade for symptomatic obstructed patients, and recent literature offers support for treating patients with latent exercise-induced gradients even with mild or no symptoms [35]. We currently begin bisoprolol till heart rate of 50-60 bpm or clinical improvement. We choose bisoprolol because we have anecdotally observed better patient tolerance than for metoprolol, and relief of non-specific β-blocker side effects (fatigue, somnolence) at comparable heart rate reduction. There is also basic research that supports this choice; bisoprolol is more β-1 selective than atenolol or metoprolol [36]. Among the β-1 adrenoceptor blockers, selectivity varies, with β-1 / β-2 -affinity ratios ranging from 13.5 for bisoprolol, to 4.7 for atenolol, and 2.3 for metoprolol. β-blockers with vasodilating effects (i.e. carvedilol, nebivolol) should be avoided in obstructive HCM but can be used if obstruction is excluded (best by stress exercise echocardiography).
Verapamil has been used for obstructive HCM but its benefit is limited by its vasodilating effects, that in an individual patient may outweigh its negative inotropic effect [37]. It is therefore generally reserved for patients with mild to moderate obstruction (resting gradients<50 mm Hg). Epstein and Rosing recommend not administering verapamil to patients with high resting gradients, suspicion of high left atrial pressure or evidence of AV block, to avoid complications [37]. (Fig. 8). This is problematic since the most ill patients, with the most dyspnea, are those with elevated left atrial pressure. We have been reluctant to treat patients >65 years old with a combination of β-blockade and verapamil because of the potential for severe bradycardia or heart block. For younger patients this is a potentially viable combination, but doses of verapamil should be started low 180mg/day and titrated up slowly with observation of heart rate response and PR interval.
Fig. (8).
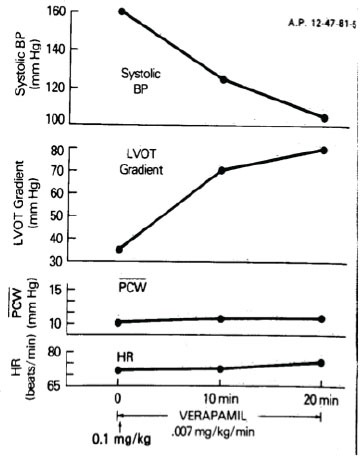
Paradoxical increase in left ventricular outflow obstruction after verapamil. Increase in the left ventricular outflow tract (LVOT) gradient after a verapamil-induced marked fall in systemic arterial blood pressure (BP). PCW = mean pulmonary capillary wedge pressure; HR = heart rate. Reproduced with permission from Epstein SE, Rosing DR. Verapamil: Its potential for causing serious complications in patients with hypertrophic cardiomyopathy. Circulation 1981; 64: 437-41.
Disopyramide is considered the most reliable agent for resting gradient reduction [1, 2, 38-43]. Disopyramide is a type I anti-arrhythmic drug that has marked negative inotropic effects. It reduces ejection fraction in normals [41]. In a direct head-to-head comparison of its acute effect on resting gradient in the same HCM patients, it is more potent than verapamil or β-blockade (Fig. 9) [44]. Disopyramide is usually given in combination with β-blockade to blunt the exercise-related rise in gradient, and for synergistic negative inotropic effect, and to provide AV delay should atrial fibrillation occur. If there is a contraindication to β-blockade, disopyramide can be given with verapamil.
Fig. (9).
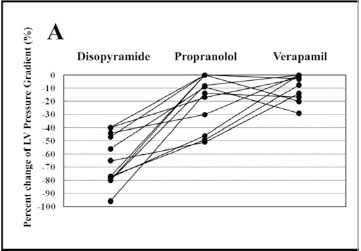
Individual percentage of changes in LV pressure gradient at rest after intravenous administration of disopyramide, propranolol, or verapamil. Reproduced by permission from Kajimoto K. et al. Comparison of acute reduction in left ventricular outflow tract pressure gradient in obstructive hypertrophic cardiomyopathy by disopyramide versus pilsicainide versus cibenzoline. Am J Cardiol 2010; 106: 1307-12.
There is a prominent dose-response to disopyramide; higher doses yield better gradient reduction [33, 43, 45]. However, disopyramide’s principal side effect - its vagolytic effects - dry mouth, and urinary hesitancy may prevent adequate dosing. Recently pyridostigmine controlled release (Mestinon Timespan) has been added to disopyramide to improve tolerance of disopyramide and allow higher, more adequate disopyramide dosing, averaging 500 mg/day in 2 divided doses [43, 46]. Pyridostigmine is a cholinesterase inhibitor with a very favorable safety profile and very infrequent side effects. We offer it to every patient, in the control release preparation, either as a prn medication or every day, 90-180mg 2x/day. The safety of this approach in HCM has been documented in a large cohort [43]. Organ toxicity from disopyramide is very rare. We have observed no hematologic, central nervous system, renal, hepatic, or adverse effects; this makes it suitable for long-term use. Disopyramide has a IIa recommendation from the 2011 AHA/ACC guidelines and a Ib recommendation from the European Society Guidelines [1, 2].
In a multicenter retrospective registry study of disopyramide from 4 HCM programs we found that 2/3 of patients could be successfully managed without the need for septal reduction. In these patients resting gradients were reduced by half with a concomitant relief of symptoms. In contrast, 1/3 of patients needed intervention because they had persistent gradients or drug side effects. There was a trend toward improved survival and lower sudden death incidence in the disopyramide-treated patients compared to obstructed patients from the same institutions not treated with disopyramide (Fig. 10). We believe this was because of the pharmacologic gradient reduction with disopyramide though we cannot exclude a direct beneficial intracellular metabolic effect. There had been the theoretic risk that disopyramide, as a type I antiarrhythmic might be pro-arrhythmic. The survival results of this study, to a great extent, alleviated this concern. Disopyramide is not a dangerous drug in obstructive HCM [1, 2, 39, 42].
Fig. (10).
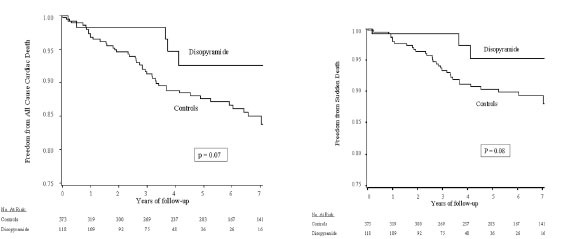
(Left) Kaplan-Meier survival plot for all-cause cardiac mortality in disopyramide-treated and non–disopyramide-treated patients with obstructive hypertrophic cardiomyopathy. (Right) Kaplan-Meier survival plot for sudden cardiac death in disopyramide-treated and non–disopyramide-treated patients with obstructive hypertrophic cardiomyopathy. Reproduced by persission from Sherrid M et al. Multicenter Study of the Efficacy and Safety of Disopyramide in Obstructive Hypertrophic Cardiomyopathy. Multicenter study of the efficacy and safety of disopyramide in obstructive hypertrophic cardiomyopathy. J Am Coll Cardio 2005; 45: 1251-8.
Two recent studies have provided further evidence of dispyramide’s important role for patients with obstruction. Ball and colleagues reported a disopyramide/β-blockade experience for HCM patients with resting obstruction [38]. In their report >150 patients achieved symptom relief with disopyramide but without septal reduction. Patients who responded to conservative pharmacologic therapy with symptom relief had an 87% HCM-related survival at 10 years [38]. Another report, from our center, described the management of 299 patients who had obstruction resistant to β-blockade or verapamil. This discrete subgroup of patients have a relatively common dilemma [47]; their symptoms and gradient often prompt referral to an HCM center for advanced care (Fig. 11). These patients can be managed with a stepped approach in which disopyramide plays a central role; patients who respond favorably to disopyramide/β-blockade are continued on pharmacologic therapy; those who fail disopyramide/β-blockade (40% in this report) are promptly referred for septal reduction, in our center generally with surgical myectomy [48]. Such an approach can result in a meaningful improvement in symptoms, very low sudden death mortality, and overall mortality that did not differ from that expected in a matched cohort of the general U.S. population [43] (Figs. 12-14). A chart showing how we actually approached the stepped management of patients resistant to β-blockade or verapamil is shown in (Fig. 12). Fig. (13) show reductions in resting gradient, by the treatment selected; and Fig. (14) shows survival data compared with age and gender-matched normal US population. There was only one sudden death in the 299 patients in the obstructed advanced-care group, a very low annual incidence of 0.06% / year. The combined annual rate of sudden death, resuscitated ventricular tachycardia and appropriate primary-prevention ICD discharge was low, 0.4% /year. The disopyramide treated patients had a very low SCD mortality rate, 0.1% /year.
Fig. (11).
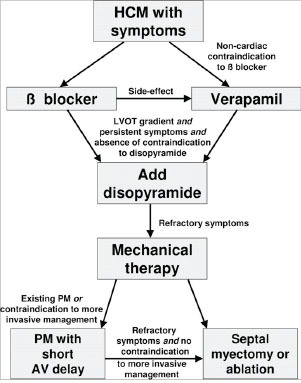
Proposed treatment path for patients with obstructive hypertrophic cardiomyopathy. Note that patients generally should receive aggressive pharmacologic therapy before referral to invasive therapy. Patients resistant to beta-blockade or verapamil form a discrete patient group who are candidates for advanced care: treatments available include disopyramide, surgical septal myectomy, alcohol septal ablation and DDD pacing with short AV delay. Reproduced by permission Fifer MA, Vlahakes GJ. Management of symptoms in hypertrophic cardiomyopathy. Circulation 2008; 117: 429-39.
Fig. (12).
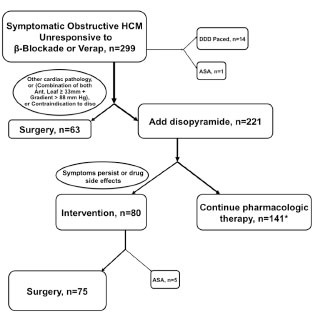
Treatment paths in 299 obstructed advanced-care HCM patients who were resistant to beta-blockade or verapamil. ASA=alcohol septal ablation; Ant. Leaf=anterior leaflet of mitral valve; DDD paced=AV sequential paced with short AV delay; Diso = diso; verap = verapamil; *43 (30%) of the 141 patients who were successfully treated with disopyramide were DDD paced as well. Reproduced by permission from Sherrid MV et al. Treatment of obstructive hypertrophic cardiomyopathy symptoms and gradient resistant to first-line therapy with beta-blockade or verapamil. Circ Heart Fail 2013; 6: 694-702.
Fig. (13).
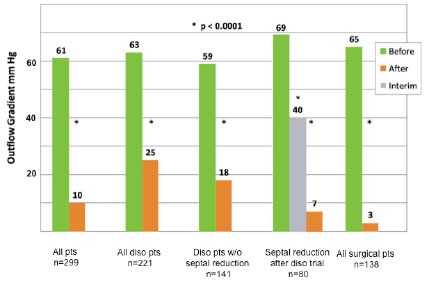
Echocardiographic resting LVOT gradients in 299 patients in the obstructed advanced-care group at initial evaluation and at last measurement after treatment. In the patients who received a disopyramide trial, but then required septal reduction, interim gradients obtained just before the invasive intervention are shown as well. Reproduced by permission Sherrid et al. Circ Heart Fail 2013 same as Fig. (13).
Fig. (14).
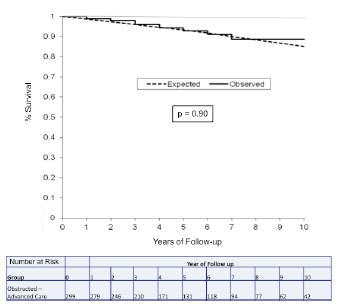
Kaplan-Meier plot comparing observed survival in the obstructed advanced-care group (n=299, solid line) versus the expected survival based on 2005 US survival matched for age, sex and race (dashed line). Reproduced by permission Sherrid et al. Circ Heart Fail 2013 same as Fig. (13).
After initiation maintainance disopyramide dosing of 400-600 mg/day is continued, with an average dose 500mg/day, given as 250mg Q12 H of the controlled released preparation, if available. We recommend monitoring patient ECG every 4 months. Disopyramide is continued at the same dose unless QTc is prolonged greater than 525 msec in patients with normal QRS initially, and greater than 550 msec in patients with underlying initial bundle branch block. The safety data in the previously cited study was obtained using these criteria [43]. Disopyramide in obstructive HCM appears similar to other cardiovascular agents where QT prolongation does not increase risk of SCD [49]. It is important to alert patients to avoid other drugs that can prolong QTc. As with any pharmacologic agent with potential for side effects, thorough physician familiarity by review of the literature enhances safety; with this in mind a practical primer of disopyramide use in obstructive HCM is published [39].
HCM AND HYPERTENSION
Hypertension is exceedingly common in the United States population, with an estimated prevalence close to 30% in adult population, while HCM has a prevalence of 0.2% (1:500) in the general population. By coincidence alone one might expect that close to a third of HCM patients may have hypertension. Moreover, there are hemodynamic explanations for this association, as well as sympathetic activation [50]. As previously mentioned the potent vasodilator medications, often successfully used to manage hypertension, cannot be applied in obstructive HCM. (They can be given without restriction in non-obstructive HCM). In a study in which the diagnosis of both obstructive HCM and hypertension were confirmed by strict criteria we found that judicious administration of pharmacotherapy could control both problems in the majority of cases. In symptomatic obstructive HCM patients the initial treatment consisted of discontinuation of vasodilators (if the patient was on a vasodilator) and administration of maximal tolerated doses of β-blockers, verapamil, or both. β-blockade was first-line therapy. In patients in whom hypertension was the main current problem, verapamil was started as heart rate permitted. If symptoms and elevated gradients persisted after β-blockade or verapamil, disopyramide controlled release was generally added as previously reported. A combination of all 3 negatively inotropic agents was generally avoided unless the patient had a permanent pacemaker.
Patients with heart failure symptoms refractory to pharmacologic management, and resting or provoked gradients greater than 50 mm Hg were referred for surgical myectomy. In patients with persistent hypertension after the initial therapy, clonidine 0.1 mg once or twice per day or a clonidine patch was given. Spironolactone is a useful adjunct. In patients with good control of gradient by disopyramide low dose hydrochlorthiazide can be administered with triampterene without exacerbation of symptoms. Despite optimal pharmacologic management, symptoms persisted in 22% of obstructed patients necessitating septal reduction therapy, while in 16% DDD pacing with short AV delay was used for gradient reduction. Septal reduction was undertaken for uncontrolled obstructive symptoms, not to control hypertension in the face of obstructive HCM [50].
NONOBSTRUCTIVE HCM
β-blockade and verapamil are the mainstays of therapy for heart failure symptoms in non-obstructive HCM. These are given as empiric trials with symptom relief as the end-point. β-blockade is uptitrated until HR is between 50 and 60. If ineffective, we begin verapamil control release monotherapy starting at 180mg/day and titrating up to 360 mg/day. For patients less than 65 years we may administer lower doses of both agents with surveillance to detect severe bradycardia<40 bpm and early signs of heart block (long PR interval). Patients with severe symptoms will often have very small left ventricular volumes [11] that impair stroke volume. Such patients are often refractory to pharmacotherapy. Potassium sparing diuretics may be used for pulmonary congestion and rales. Predominant right heart failure with edema as the presenting feature is uncommon in HCM; search for another etiology is indicated (amyloid). Anginal chest pain may respond to verapamil [51] which should be empirically tried, in increasing doses.
About 2% of patients will experience a decline in LV systolic function, often associated with severe exacerbation of symptoms. Such patients will often have extensive fibrosis on CMR. Prognosis is guarded in these patients. Pharmacotherapy requires complete reevaluation at this time, and therapy similar to that administered to dilated cardiomyopathy is administered: angiotensin converting enzyme inhibitors or angiotensin receptor blockade (ARB); β-blockade; diuretics. Implanted defibrillators are indicated; for patients with LBBB resynchronizaton therapy is recommended. Progressive heart failure, and hospitalization should prompt consideration of referral for transplantation listing [52-54].
NOVEL PHARMACOTHERAPY FOR HCM
Reports of efficacy and safety in HCM depend on observational studies. Multiple novel therapeutic strategies are being pursued for symptom relief in HCM patients [55]
ALTERNATE ENERGY
Inefficient use of intracellular energy has been identified as a potential pathophysiologic link between mutations and development of the HCM phenotype and symptoms. The development of metabolic magnetic resonance imaging (CMR) now allows in-vivo assay of myocardial energetics, comparison of patients and normals, and objective assessment of therapeutic efficacy [56].
Phosphocreatine (PCr) is part of the heart’s energetic store; it is the reserve energy supply of rapidly hydrolysable ATP during periods of high energy demand. The ratio of PCr/ATP is considered one of the best noninvasive markers of energy stores and is measurable with metabolic CMR. In animal and human heart failure PCr levels decrease by up to 70% irrespective of the etiology of heart failure [57-59]. In HCM PCr/ATP ratios are decreased [60]. Energy depletion in HCM may contribute to inability to increase cardiac output with exercise [12]. We hypothesize that energy depletion may also contribute to the left ventricle’s inability to maintain mid-systolic ejection in the face of severe obstruction, the echocardiographic finding of a mid-systolic drop in LV ejection velocities [61-64].
The energy needs of the heart are enormous. The daily turnover of ATP, an astonishing 6-35kg, is very many times the mass of the heart itself, and its myocardial ATP pool. In light of this exceedingly high demand, subtle variations in the efficiency of energy generation by a switch in fuel type may have a major impact on cellular energy levels [59] (Table 3).
Table 3.
Effect on ATP-to-oxygen ratio of a total change from glucose to FFA utilization by myocardium.
| Molecule | ATP Yield per Molecule |
ATP Yield per Carbon Atom |
ATP Yield per Oxygen Atom Taken Up, P/O Ratio |
Relative Decreased ATP Production Efficiency per Unit Oxygen if Glucose is Substituted by FFA, % |
|---|---|---|---|---|
| Glucose (C6) | 38 | 6.33 | 3.17 | 0 |
| Palmitate (C16) | 129 | 8.06 | 2.80 | 11.7 |
| Oleate (C18) | 144 | 8.00 | 2.82 | 11.0 |
From: Ashrafian H, Frenneaux MP, Opie LH. Metabolic mechanisms in heart failure. Circulation 2007; 116: 434-48.
Fatty acid oxidation requires greater oxygen utilization for a given quantity of ATP synthesis than do carbohydrates. In pigs a switch from maximum glucose to maximum free fatty acid utilization increased oxygen requirement by about 50% [65, 66].
Perhexiline is an effective antianginal agent that was introduced in 1970’s. Perhexiline inhibits the metabolism of free fatty acids by the heart by suppressing function of carnitine palmitoyltransferase 1 and 2 transporters that mediate the uptake of long chain free fatty acids into the mitochondria. By blocking uptake of long fatty acid chains perhexiline shifts substrate utilization toward carbohydrate. Since carbohydrate metabolism utilizes less oxygen per unit of energy produced, overall efficiency of the myocardium is enhanced [67].
In 56 systolic heart failure patients, randomized to perhexiline or placebo resulted in an increase in peak exercise O2 uptake and LV function in perhexiline patients after 6 weeks [68]. In HCM patients, investigators from the University of Birmingham and University College, London administered perhexiline 100 mg/day adjusted to keep serum level from .15 to .6 mg per liter to avoid drug toxicity [60]. Perhexiline improved the myocardial ratios of phosphocreatine to ATP from 1.27 to 1.73 compared to no change in placebo groups. There was improvement in the primary endpoint, peak oxygen consumption, VO2 increasing from 22 ± .2 to 24 ± .2 vs. no change in the placebo group. Improvement in exercise related diastolic function was shown and New York Heart Association and quality of life improved in parallel.
The toxicity of perhexiline has been studied; it is now understood that genetic heterogeneity in drug metabolism resulted in cases of neuropathy and hepatotoxicity reported soon after the drug's introduction into clinical practice [67, 69-75]. Genotype analysis and dose adjustment decreases the incidence of toxicity.
LATE SODIUM CURRENT BLOCKADE
Abnormally prolonged action potential has been shown in isolated HCM cardiomyocytes harvested at myectomy [76]. This abnormality has been related to an increase in the late sodium current (not observed in normals), and prolonged calcium transients with diastolic calcium overload and myocyte tension development. These abnormalities in turn lead to prolonged action duration, early afterdepolarizations and enhanced arrhythmogenicity. Sustained activation of calcium calmodulin-dependent protein kinase II is implicated as a potential underlying mechanism. Ranolazine inhibits the late sodium current and improves calcium handling and has been shown in isolated cardiomyocytes to reduce sodium and calcium overload and interrupt this pathophysiologic cycle [76]. There is, thus, the potential to both improve diastolic function and ventricular irritability with this agent or its congeners [49, 77]. In isolated HCM cardiomyocytes ranolazine shortened action potential duration, decreased early and delayed after depolarizations, and accelerated the contraction-relaxation cycle of HCM and improved diastolic function [76]. A randomized trial of a novel agent that blocks the late inward sodium current in HCM is in progress, NCT02291237. End point is maximal treadmill oxygen consumption.
Fibrotardive therapy: Fibrosis in HCM is an important cause of diastolic dysfunction and is thought to contribute to arrhythmic burden and sudden death [9]. Fibrosis can be interstitial, replacment and perivascular [9]; its presence and extent can now be imaged with CMR. Preventing fibrosis or reversing fibrosis has emerged as a goal in HCM patients. Mouse models have indicated that sarcomeric mutations promote trophic mediators of fibrosis especially TGf- β [78]. Animal models have indicated the potential of ARB to prevent fibrosis in mouse HCM [78, 79], ARB effect appears mediated by blockade of angiotensin II and decreased expression of transforming growth factor β [78]. Preliminary trials in humans have suggested benefit [80]. ARB cannot be used in HCM patients with obstruction.
A 2 year multicenter clinical trial, organized by Ho and colleagues, comparing valsartan treated and placebo treated individuals is recruiting patients, NCT 01912534. Sarcomere mutation carriers with asymptomatic or mildly symptomatic overt disease (NYHA class I-II), and mutation carriers without left ventricular hypertrophy (LVH) will be studied. Endpoints are: left atrial size; assessment of left ventricle (LV) mass by cardiac magnetic resonance imaging(CMR); LV systolic and diastolic function; the degree of interstitial myocardial fibrosis as assessed by novel CMR sequences; additional parameters from metabolic exercise testing and additional serum biomarkers that reflect fibrosis, injury and stress [81].
The effect of aldosterone blockade with spironolactone has been evaluated and shown to be ineffective [82]. Maron randomized 53 HCM patients to 50 mg of spironolactone or placebo for one year and found no difference in serum markers of collagen synthesis, fibrosis by CMR, peak VO2, diastolic function by echo and heart failure symptoms. Whether spironolactone or similar aldosterone blockade might prevent HCM in gene+/LVH- patients has not been determined.
CONCLUSION
Pharmacotherapy of HCM symptoms is often effective for reducing symptoms in obstructive HCM. Patients who fail advanced drug therapy should be referred expeditiously to septal reduction. In patients with severe symptoms from non-obstructive HCM there are fewer options available.
The curtain has come down on Act I of pharmacotherapy of HCM, reports of observational trials. We are currently in intermission. The second Act will feature randomized trials of novel agents, designed specifically for HCM.
Fig. (2).
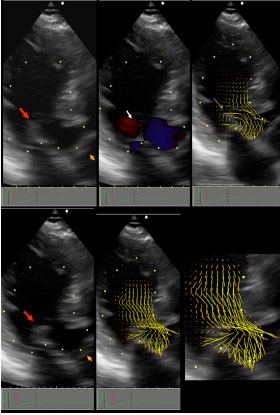
OHCM patient with vortical SAM: Top panels are pre-SAM frames; bottom panels are post-SAM frames. In left panel red arrow points to coapted MV, orange arrow points to the closed aortic valve. In top middle panel white arrowhead points to Doppler color flow that forms the isovolumic vortex. In top right panel, the thin white arrow points to the vector flow maps of the isovolumic vortex. Note that a limb of the clockwise anteriorly directed vortex flow impacts the posterior surface of the mitral leaflet tip with high angle of attack (thin white arrow). The bottom panel show that the aortic leaflets are still closed. There is now a confluence of vortical flow and early ejection flow that impacts the MV with high angle of attack (lower right enlargement). Reproduced by permission from Ro R et al. J Am Coll Cardiol 2014 same as Fig. (1).
ACKNOWLEDGEMENT
Declared none.
CONFLICT OF INTEREST
Dr. Sherrid has provided consultation for Heart Metabolics Limited. There are no other potential conflicts of interest.
REFERENCES
- 1.Gersh B.J., Maron B.J., Bonow R.O., Dearani J.A., Fifer M.A., Link M.S., Naidu S.S., Nishimura R.A., Ommen S.R., Rakowski H., Seidman C.E., Towbin J.A., Udelson J.E., Yancy C.W., American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. American Association for Thoracic Surgery. American Society of Echocardiography. American Society of Nuclear Cardiology. Heart Failure Society of America. Heart Rhythm Society. Society for Cardiovascular Angiography and Interventions. Society of Thoracic Surgeons 2011 accf/aha guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: A report of the american college of cardiology foundation/american heart association task force on practice guidelines. Circulation. 2011;124(24):e783–e831. doi: 10.1161/CIR.0b013e318223e2bd. [DOI] [PubMed] [Google Scholar]
- 2.Elliott PM, Anastasakis A, Authors/Task Force m 2014 esc guidelines on diagnosis and management of hypertrophic cardiomyopathy: The task force for the diagnosis and management of hypertrophic cardiomyopathy of the european society of cardiology (esc). . Eur Heart J . 2014;35 (39 ):2733–79. doi: 10.1093/eurheartj/ehu284. [DOI] [PubMed] [Google Scholar]
- 3.Bos J.M., Will M.L., Gersh B.J., Kruisselbrink T.M., Ommen S.R., Ackerman M.J. Characterization of a phenotype-based genetic test prediction score for unrelated patients with hypertrophic cardiomyopathy. Mayo Clin. Proc. 2014;89(6):727–737. doi: 10.1016/j.mayocp.2014.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lopes L.R., Rahman M.S., Elliott P.M. A systematic review and meta-analysis of genotype-phenotype associations in patients with hypertrophic cardiomyopathy caused by sarcomeric protein mutations. Heart. 2013;99(24):1800–1811. doi: 10.1136/heartjnl-2013-303939. [DOI] [PubMed] [Google Scholar]
- 5.Lopes L.R., Zekavati A., Syrris P., Hubank M., Giambartolomei C., Dalageorgou C., Jenkins S., McKenna W., Plagnol V., Elliott P.M., Uk10k Consortium Genetic complexity in hypertrophic cardiomyopathy revealed by high-throughput sequencing. J. Med. Genet. 2013;50(4):228–239. doi: 10.1136/jmedgenet-2012-101270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olivotto I., Girolami F., Ackerman M.J., Nistri S., Bos J.M., Zachara E., Ommen S.R., Theis J.L., Vaubel R.A., Re F., Armentano C., Poggesi C., Torricelli F., Cecchi F. Myofilament protein gene mutation screening and outcome of patients with hypertrophic cardiomyopathy. Mayo Clin. Proc. 2008;83(6):630–638. doi: 10.1016/S0025-6196(11)60890-2. [DOI] [PubMed] [Google Scholar]
- 7.Van Driest S.L., Ellsworth E.G., Ommen S.R., Tajik A.J., Gersh B.J., Ackerman M.J. Prevalence and spectrum of thin filament mutations in an outpatient referral population with hypertrophic cardiomyopathy. Circulation. 2003;108(4):445–451. doi: 10.1161/01.CIR.0000080896.52003.DF. [DOI] [PubMed] [Google Scholar]
- 8.Arad M., Maron B.J., Gorham J.M., Johnson W.H., Jr, Saul J.P., Perez-Atayde A.R., Spirito P., Wright G.B., Kanter R.J., Seidman C.E., Seidman J.G. Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N. Engl. J. Med. 2005;352(4):362–372. doi: 10.1056/NEJMoa033349. [DOI] [PubMed] [Google Scholar]
- 9.Shirani J., Pick R., Roberts W.C., Maron B.J. Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J. Am. Coll. Cardiol. 2000;35(1):36–44. doi: 10.1016/S0735-1097(99)00492-1. [DOI] [PubMed] [Google Scholar]
- 10.Geske J.B., Sorajja P., Nishimura R.A., Ommen S.R. Evaluation of left ventricular filling pressures by Doppler echocardiography in patients with hypertrophic cardiomyopathy: correlation with direct left atrial pressure measurement at cardiac catheterization. Circulation. 2007;116(23):2702–2708. doi: 10.1161/CIRCULATIONAHA.107.698985. [DOI] [PubMed] [Google Scholar]
- 11.Shah A., Duncan K., Winson G., Chaudhry F.A., Sherrid M.V. Severe symptoms in mid and apical hypertrophic cardiomyopathy. Echocardiography. 2009;26(8):922–933. doi: 10.1111/j.1540-8175.2009.00905.x. [DOI] [PubMed] [Google Scholar]
- 12.Lele S.S., Thomson H.L., Seo H., Belenkie I., McKenna W.J., Frenneaux M.P. Exercise capacity in hypertrophic cardiomyopathy. Role of stroke volume limitation, heart rate, and diastolic filling characteristics. Circulation. 1995;92(10):2886–2894. doi: 10.1161/01.CIR.92.10.2886. [DOI] [PubMed] [Google Scholar]
- 13.Nishimura R.A., Schwartz R.S., Holmes D.R., Jr, Tajik A.J. Failure of calcium channel blockers to improve ventricular relaxation in humans. J. Am. Coll. Cardiol. 1993;21(1):182–188. doi: 10.1016/0735-1097(93)90735-J. [DOI] [PubMed] [Google Scholar]
- 14.Kass D.A., Wolff M.R., Ting C.T., Liu C.P., Chang M.S., Lawrence W., Maughan W.L. Diastolic compliance of hypertrophied ventricle is not acutely altered by pharmacologic agents influencing active processes. Ann. Intern. Med. 1993;119(6):466–473. doi: 10.7326/0003-4819-119-6-199309150-00004. [DOI] [PubMed] [Google Scholar]
- 15.Matsubara H., Nakatani S., Nagata S., Ishikura F., Katagiri Y., Ohe T., Miyatake K. Salutary effect of disopyramide on left ventricular diastolic function in hypertrophic obstructive cardiomyopathy. J. Am. Coll. Cardiol. 1995;26(3):768–775. doi: 10.1016/0735-1097(95)00229-W. [DOI] [PubMed] [Google Scholar]
- 16.Maron B.J., Wolfson J.K., Epstein S.E., Roberts W.C. Intramural (“small vessel”) coronary artery disease in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 1986;8(3):545–557. doi: 10.1016/S0735-1097(86)80181-4. [DOI] [PubMed] [Google Scholar]
- 17.Maron B.J., Epstein S.E., Roberts W.C. Hypertrophic cardiomyopathy and transmural myocardial infarction without significant atherosclerosis of the extramural coronary arteries. Am. J. Cardiol. 1979;43(6):1086–1102. doi: 10.1016/0002-9149(79)90139-5. [DOI] [PubMed] [Google Scholar]
- 18.Maron M.S., Olivotto I., Zenovich A.G., Link M.S., Pandian N.G., Kuvin J.T., Nistri S., Cecchi F., Udelson J.E., Maron B.J. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation. 2006;114(21):2232–2239. doi: 10.1161/CIRCULATIONAHA.106.644682. [DOI] [PubMed] [Google Scholar]
- 19.Joshi S., Patel U.K., Yao S.S., Castenada V., Isambert A., Winson G., Chaudhry F.A., Sherrid M.V. Standing and exercise Doppler echocardiography in obstructive hypertrophic cardiomyopathy: the range of gradients with upright activity. J. Am. Soc. Echocardiogr. 2011;24(1):75–82. doi: 10.1016/j.echo.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 20.Feiner E., Arabadjian M., Winson G., Kim B., Chaudhry F., Sherrid M.V. Post-prandial upright exercise echocardiography in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2013;61(24):2487–2488. doi: 10.1016/j.jacc.2013.02.079. [DOI] [PubMed] [Google Scholar]
- 21.Alhaj E.K., Kim B., Cantales D., Uretsky S., Chaudhry F.A., Sherrid M.V. Symptomatic exercise-induced left ventricular outflow tract obstruction without left ventricular hypertrophy. J. Am. Soc. Echocardiogr. 2013;26(5):556–565. doi: 10.1016/j.echo.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 22.Shah J.S., Esteban M.T., Thaman R., Sharma R., Mist B., Pantazis A., Ward D., Kohli S.K., Page S.P., Demetrescu C., Sevdalis E., Keren A., Pellerin D., McKenna W.J., Elliott P.M. Prevalence of exercise-induced left ventricular outflow tract obstruction in symptomatic patients with non-obstructive hypertrophic cardiomyopathy. Heart. 2008;94(10):1288–1294. doi: 10.1136/hrt.2007.126003. [DOI] [PubMed] [Google Scholar]
- 23.Argulian E., Chaudhry F.A. Stress testing in patients with hypertrophic cardiomyopathy. Prog. Cardiovasc. Dis. 2012;54(6):477–482. doi: 10.1016/j.pcad.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 24.Sherrid M.V., Arabadjian M. Echocardiography to individualize treatment for hypertrophic cardiomyopathy. Prog. Cardiovasc. Dis. 2012;54(6):461–476. doi: 10.1016/j.pcad.2012.04.007. [DOI] [PubMed] [Google Scholar]
- 25.Halpern D.G., Sherrid M.V. Echocardiography in hypertrophic cardiomyopathy. Comprehensive Textbook of Echocardiography; 2013. pp. 1348–1368. [Google Scholar]
- 26.Elesber A., Nishimura R.A., Rihal C.S., Ommen S.R., Schaff H.V., Holmes D.R., Jr Utility of isoproterenol to provoke outflow tract gradients in patients with hypertrophic cardiomyopathy. Am. J. Cardiol. 2008;101(4):516–520. doi: 10.1016/j.amjcard.2007.09.111. [DOI] [PubMed] [Google Scholar]
- 27.Cohen L.S., Braunwald E. Amelioration of angina pectoris in idiopathic hypertrophic subaortic stenosis with beta-adrenergic blockade. Circulation. 1967;35(5):847–851. doi: 10.1161/01.CIR.35.5.847. [DOI] [PubMed] [Google Scholar]
- 28.Ro R., Halpern D., Sahn D.J., Homel P., Arabadjian M., Lopresto C., Sherrid M.V. Vector flow mapping in obstructive hypertrophic cardiomyopathy to assess the relationship of early systolic left ventricular flow and the mitral valve. J. Am. Coll. Cardiol. 2014;64(19):1984–1995. doi: 10.1016/j.jacc.2014.04.090. [DOI] [PubMed] [Google Scholar]
- 29.Sherrid M.V., Chu C.K., Delia E., Mogtader A., Dwyer E.M., Jr An echocardiographic study of the fluid mechanics of obstruction in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 1993;22(3):816–825. doi: 10.1016/0735-1097(93)90196-8. [DOI] [PubMed] [Google Scholar]
- 30.Sherrid M.V., Gunsburg D.Z., Moldenhauer S., Pearle G. Systolic anterior motion begins at low left ventricular outflow tract velocity in obstructive hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2000;36(4):1344–1354. doi: 10.1016/S0735-1097(00)00830-5. [DOI] [PubMed] [Google Scholar]
- 31.Sherrid M.V., Pearle G., Gunsburg D.Z. Mechanism of benefit of negative inotropes in obstructive hypertrophic cardiomyopathy. Circulation. 1998;97(1):41–47. doi: 10.1161/01.CIR.97.1.41. [DOI] [PubMed] [Google Scholar]
- 32.Pollick C. Muscular subaortic stenosis: hemodynamic and clinical improvement after disopyramide. N. Engl. J. Med. 1982;307(16):997–999. doi: 10.1056/NEJM198210143071607. [DOI] [PubMed] [Google Scholar]
- 33.Sherrid M., Delia E., Dwyer E. Oral disopyramide therapy for obstructive hypertrophic cardiomyopathy. Am. J. Cardiol. 1988;62(16):1085–1088. doi: 10.1016/0002-9149(88)90553-X. [DOI] [PubMed] [Google Scholar]
- 34.Flamm M.D., Harrison D.C., Hancock E.W. Muscular subaortic stenosis. Prevention of outflow obstruction with propranolol. Circulation. 1968;38(5):846–858. doi: 10.1161/01.CIR.38.5.846. [DOI] [PubMed] [Google Scholar]
- 35.Nistri S., Olivotto I., Maron M.S., Ferrantini C., Coppini R., Grifoni C., Baldini K., Sgalambro A., Cecchi F., Maron B.J. β Blockers for prevention of exercise-induced left ventricular outflow tract obstruction in patients with hypertrophic cardiomyopathy. Am. J. Cardiol. 2012;110(5):715–719. doi: 10.1016/j.amjcard.2012.04.051. [DOI] [PubMed] [Google Scholar]
- 36.Baker J.G. The selectivity of beta-adrenoceptor antagonists at the human beta1, beta2 and beta3 adrenoceptors. Br. J. Pharmacol. 2005;144(3):317–322. doi: 10.1038/sj.bjp.0706048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Epstein S.E., Rosing D.R. Verapamil: its potential for causing serious complications in patients with hypertrophic cardiomyopathy. Circulation. 1981;64(3):437–441. doi: 10.1161/01.CIR.64.3.437. [DOI] [PubMed] [Google Scholar]
- 38.Ball W., Ivanov J., Rakowski H., Wigle E.D., Linghorne M., Ralph-Edwards A., Williams W.G., Schwartz L., Guttman A., Woo A. Long-term survival in patients with resting obstructive hypertrophic cardiomyopathy comparison of conservative versus invasive treatment. J. Am. Coll. Cardiol. 2011;58(22):2313–2321. doi: 10.1016/j.jacc.2011.08.040. [DOI] [PubMed] [Google Scholar]
- 39.Sherrid M.V., Arabadjian M. A primer of disopyramide treatment of obstructive hypertrophic cardiomyopathy. Prog. Cardiovasc. Dis. 2012;54(6):483–492. doi: 10.1016/j.pcad.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 40.Pollick C. Disopyramide in hypertrophic cardiomyopathy. II. Noninvasive assessment after oral administration. Am. J. Cardiol. 1988;62(17):1252–1255. doi: 10.1016/0002-9149(88)90269-X. [DOI] [PubMed] [Google Scholar]
- 41.Pollick C., Giacomini K.M., Blaschke T.F., Nelson W.L., Turner-Tamiyasu K., Briskin V., Popp R.L. The cardiac effects of d- and l-disopyramide in normal subjects: a noninvasive study. Circulation. 1982;66(2):447–453. doi: 10.1161/01.CIR.66.2.447. [DOI] [PubMed] [Google Scholar]
- 42.Sherrid M.V., Barac I., McKenna W.J., Elliott P.M., Dickie S., Chojnowska L., Casey S., Maron B.J. Multicenter study of the efficacy and safety of disopyramide in obstructive hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2005;45(8):1251–1258. doi: 10.1016/j.jacc.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 43.Sherrid M.V., Shetty A., Winson G., Kim B., Musat D., Alviar C.L., Homel P., Balaram S.K., Swistel D.G. Treatment of obstructive hypertrophic cardiomyopathy symptoms and gradient resistant to first-line therapy with β-blockade or verapamil. Circ Heart Fail. 2013;6(4):694–702. doi: 10.1161/CIRCHEARTFAILURE.112.000122. [DOI] [PubMed] [Google Scholar]
- 44.Kajimoto K., Imai T., Minami Y., Kasanuki H. Comparison of acute reduction in left ventricular outflow tract pressure gradient in obstructive hypertrophic cardiomyopathy by disopyramide versus pilsicainide versus cibenzoline. Am. J. Cardiol. 2010;106(9):1307–1312. doi: 10.1016/j.amjcard.2010.06.059. [DOI] [PubMed] [Google Scholar]
- 45.Kimball B.P., Bui S., Wigle E.D. Acute dose-response effects of intravenous disopyramide in hypertrophic obstructive cardiomyopathy. Am. Heart J. 1993;125(6):1691–1697. doi: 10.1016/0002-8703(93)90760-7. [DOI] [PubMed] [Google Scholar]
- 46.Teichman S.L., Ferrick A., Kim S.G., Matos J.A., Waspe L.E., Fisher J.D. Disopyramide-pyridostigmine interaction: selective reversal of anticholinergic symptoms with preservation of antiarrhythmic effect. J. Am. Coll. Cardiol. 1987;10(3):633–641. doi: 10.1016/S0735-1097(87)80207-3. [DOI] [PubMed] [Google Scholar]
- 47.Fifer M.A., Vlahakes G.J. Management of symptoms in hypertrophic cardiomyopathy. Circulation. 2008;117(3):429–439. doi: 10.1161/CIRCULATIONAHA.107.694158. [DOI] [PubMed] [Google Scholar]
- 48.Balaram SK, Ross RE, Sherrid MV, et al. Role of mitral valve plication in the surgical management of hypertrophic cardiomyopathy. 2012. [DOI] [PubMed]
- 49.Scirica B.M., Morrow D.A., Hod H., Murphy S.A., Belardinelli L., Hedgepeth C.M., Molhoek P., Verheugt F.W., Gersh B.J., McCabe C.H., Braunwald E. Effect of ranolazine, an antianginal agent with novel electrophysiological properties, on the incidence of arrhythmias in patients with non ST-segment elevation acute coronary syndrome: results from the Metabolic Efficiency With Ranolazine for Less Ischemia in Non ST-Elevation Acute Coronary Syndrome Thrombolysis in Myocardial Infarction 36 (MERLIN-TIMI 36) randomized controlled trial. Circulation. 2007;116(15):1647–1652. doi: 10.1161/CIRCULATIONAHA.107.724880. [DOI] [PubMed] [Google Scholar]
- 50.Argulian E., Messerli F.H., Aziz E.F., Winson G., Agarwal V., Kaddaha F., Kim B., Sherrid M.V. Antihypertensive therapy in hypertrophic cardiomyopathy. Am. J. Cardiol. 2013;111(7):1040–1045. doi: 10.1016/j.amjcard.2012.12.026. [DOI] [PubMed] [Google Scholar]
- 51.Udelson J.E., Bonow R.O., O’Gara P.T., Maron B.J., Van Lingen A., Bacharach S.L., Epstein S.E. Verapamil prevents silent myocardial perfusion abnormalities during exercise in asymptomatic patients with hypertrophic cardiomyopathy. Circulation. 1989;79(5):1052–1060. doi: 10.1161/01.CIR.79.5.1052. [DOI] [PubMed] [Google Scholar]
- 52.Harris K.M., Spirito P., Maron M.S., Zenovich A.G., Formisano F., Lesser J.R., Mackey-Bojack S., Manning W.J., Udelson J.E., Maron B.J. Prevalence, clinical profile, and significance of left ventricular remodeling in the end-stage phase of hypertrophic cardiomyopathy. Circulation. 2006;114(3):216–225. doi: 10.1161/CIRCULATIONAHA.105.583500. [DOI] [PubMed] [Google Scholar]
- 53.Thaman R., Gimeno J.R., Murphy R.T., Kubo T., Sachdev B., Mogensen J., Elliott P.M., McKenna W.J. Prevalence and clinical significance of systolic impairment in hypertrophic cardiomyopathy. Heart. 2005;91(7):920–925. doi: 10.1136/hrt.2003.031161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kato T.S., Takayama H., Yoshizawa S., Marboe C., Schulze P.C., Farr M., Naka Y., Mancini D., Maurer M.S. Cardiac transplantation in patients with hypertrophic cardiomyopathy. Am. J. Cardiol. 2012;110(4):568–574. doi: 10.1016/j.amjcard.2012.04.030. [DOI] [PubMed] [Google Scholar]
- 55.Spoladore R., Maron M.S., D’Amato R., Camici P.G., Olivotto I. Pharmacological treatment options for hypertrophic cardiomyopathy: high time for evidence. Eur. Heart J. 2012;33(14):1724–1733. doi: 10.1093/eurheartj/ehs150. [DOI] [PubMed] [Google Scholar]
- 56.Neubauer S. The failing heart--an engine out of fuel. N. Engl. J. Med. 2007;356(11):1140–1151. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 57.Ingwall J.S., Weiss R.G. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circ. Res. 2004;95(2):135–145. doi: 10.1161/01.RES.0000137170.41939.d9. [DOI] [PubMed] [Google Scholar]
- 58.Knaapen P., Germans T., Knuuti J., Paulus W.J., Dijkmans P.A., Allaart C.P., Lammertsma A.A., Visser F.C. Myocardial energetics and efficiency: current status of the noninvasive approach. Circulation. 2007;115(7):918–927. doi: 10.1161/CIRCULATIONAHA.106.660639. [DOI] [PubMed] [Google Scholar]
- 59.Ashrafian H., Frenneaux M.P., Opie L.H. Metabolic mechanisms in heart failure. Circulation. 2007;116(4):434–448. doi: 10.1161/CIRCULATIONAHA.107.702795. [DOI] [PubMed] [Google Scholar]
- 60.Abozguia K., Elliott P., McKenna W., Phan T.T., Nallur-Shivu G., Ahmed I., Maher A.R., Kaur K., Taylor J., Henning A., Ashrafian H., Watkins H., Frenneaux M. Metabolic modulator perhexiline corrects energy deficiency and improves exercise capacity in symptomatic hypertrophic cardiomyopathy. Circulation. 2010;122(16):1562–1569. doi: 10.1161/CIRCULATIONAHA.109.934059. [DOI] [PubMed] [Google Scholar]
- 61.Sherrid M.V., Gunsburg D.Z., Pearle G. Mid-systolic drop in left ventricular ejection velocity in obstructive hypertrophic cardiomyopathy--the lobster claw abnormality. J. Am. Soc. Echocardiogr. 1997;10(7):707–712. doi: 10.1016/S0894-7317(97)70112-3. [DOI] [PubMed] [Google Scholar]
- 62.Barac I., Upadya S., Pilchik R., Winson G., Passick M., Chaudhry F.A., Sherrid M.V. Effect of obstruction on longitudinal left ventricular shortening in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2007;49(11):1203–1211. doi: 10.1016/j.jacc.2006.10.070. [DOI] [PubMed] [Google Scholar]
- 63.Sherrid M.V., Balaran S.K., Korzeniecki E., Chaudhry F.A., Swistel D.G. Reversal of acute systolic dysfunction and cardiogenic shock in hypertrophic cardiomyopathy by surgical relief of obstruction. Echocardiography. 2011;28(9):E174–E179. doi: 10.1111/j.1540-8175.2011.01459.x. [DOI] [PubMed] [Google Scholar]
- 64.Sherrid M.V., Wever-Pinzon O., Shah A., Chaudhry F.A. Reflections of inflections in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2009;54(3):212–219. doi: 10.1016/j.jacc.2009.03.052. [DOI] [PubMed] [Google Scholar]
- 65.Jeffrey F.M., Alvarez L., Diczku V., Sherry A.D., Malloy C.R. Direct evidence that perhexiline modifies myocardial substrate utilization from fatty acids to lactate. J. Cardiovasc. Pharmacol. 1995;25(3):469–472. doi: 10.1097/00005344-199503000-00018. [DOI] [PubMed] [Google Scholar]
- 66.Korvald C., Elvenes O.P., Myrmel T. Myocardial substrate metabolism influences left ventricular energetics in vivo. Am. J. Physiol. Heart Circ. Physiol. 2000;278(4):H1345–H1351. doi: 10.1152/ajpheart.2000.278.4.H1345. [DOI] [PubMed] [Google Scholar]
- 67.Ashrafian H., Horowitz J.D., Frenneaux M.P. Perhexiline. Cardiovasc. Drug Rev. 2007;25(1):76–97. doi: 10.1111/j.1527-3466.2007.00006.x. [DOI] [PubMed] [Google Scholar]
- 68.Lee L., Campbell R., Scheuermann-Freestone M., Taylor R., Gunaruwan P., Williams L., Ashrafian H., Horowitz J., Fraser A.G., Clarke K., Frenneaux M. Metabolic modulation with perhexiline in chronic heart failure: a randomized, controlled trial of short-term use of a novel treatment. Circulation. 2005;112(21):3280–3288. doi: 10.1161/CIRCULATIONAHA.105.551457. [DOI] [PubMed] [Google Scholar]
- 69.Horowitz J.D., Button I.K., Wing L. Is perhexiline essential for the optimal management of angina pectoris? Aust. N. Z. J. Med. 1995;25(2):111–113. doi: 10.1111/j.1445-5994.1995.tb02820.x. [DOI] [PubMed] [Google Scholar]
- 70.Horowitz J.D., Sia S.T., Macdonald P.S., Goble A.J., Louis W.J. Perhexiline maleate treatment for severe angina pectoris--correlations with pharmacokinetics. Int. J. Cardiol. 1986;13(2):219–229. doi: 10.1016/0167-5273(86)90146-4. [DOI] [PubMed] [Google Scholar]
- 71.Singlas E., Goujet M.A., Simon P. Pharmacokinetics of perhexiline maleate in anginal patients with and without peripheral neuropathy. Eur. J. Clin. Pharmacol. 1978;14(3):195–201. doi: 10.1007/BF02089960. [DOI] [PubMed] [Google Scholar]
- 72.Evans W.E., Relling M.V., Rahman A., McLeod H.L., Scott E.P., Lin J.S. Genetic basis for a lower prevalence of deficient CYP2D6 oxidative drug metabolism phenotypes in black Americans. J. Clin. Invest. 1993;91(5):2150–2154. doi: 10.1172/JCI116441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gardiner S.J., Begg E.J. Pharmacogenetics, drug-metabolizing enzymes, and clinical practice. Pharmacol. Rev. 2006;58(3):521–590. doi: 10.1124/pr.58.3.6. [DOI] [PubMed] [Google Scholar]
- 74.Cole P.L., Beamer A.D., McGowan N., Cantillon C.O., Benfell K., Kelly R.A., Hartley L.H., Smith T.W., Antman E.M. Efficacy and safety of perhexiline maleate in refractory angina. A double-blind placebo-controlled clinical trial of a novel antianginal agent. Circulation. 1990;81(4):1260–1270. doi: 10.1161/01.CIR.81.4.1260. [DOI] [PubMed] [Google Scholar]
- 75.Alderman C.P., Hundertmark J.D., Soetratma T.W. Interaction of serotonin re-uptake inhibitors with perhexiline. Aust. N. Z. J. Psychiatry. 1997;31(4):601–603. doi: 10.3109/00048679709065084. [DOI] [PubMed] [Google Scholar]
- 76.Coppini R., Ferrantini C., Yao L., Fan P., Del Lungo M., Stillitano F., Sartiani L., Tosi B., Suffredini S., Tesi C., Yacoub M., Olivotto I., Belardinelli L., Poggesi C., Cerbai E., Mugelli A. Late sodium current inhibition reverses electromechanical dysfunction in human hypertrophic cardiomyopathy. Circulation. 2013;127(5):575–584. doi: 10.1161/CIRCULATIONAHA.112.134932. [DOI] [PubMed] [Google Scholar]
- 77.Antzelevitch C., Belardinelli L., Zygmunt A.C., Burashnikov A., Di Diego J.M., Fish J.M., Cordeiro J.M., Thomas G. Electrophysiological effects of ranolazine, a novel antianginal agent with antiarrhythmic properties. Circulation. 2004;110(8):904–910. doi: 10.1161/01.CIR.0000139333.83620.5D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Teekakirikul P., Eminaga S., Toka O., Alcalai R., Wang L., Wakimoto H., Nayor M., Konno T., Gorham J.M., Wolf C.M., Kim J.B., Schmitt J.P., Molkentin J.D., Norris R.A., Tager A.M., Hoffman S.R., Markwald R.R., Seidman C.E., Seidman J.G. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-β. J. Clin. Invest. 2010;120(10):3520–3529. doi: 10.1172/JCI42028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lim D.S., Lutucuta S., Bachireddy P., Youker K., Evans A., Entman M., Roberts R., Marian A.J. Angiotensin II blockade reverses myocardial fibrosis in a transgenic mouse model of human hypertrophic cardiomyopathy. Circulation. 2001;103(6):789–791. doi: 10.1161/01.CIR.103.6.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shimada Y.J., Passeri J.J., Baggish A.L., O’Callaghan C., Lowry P.A., Yannekis G., Abbara S., Ghoshhajra B.B., Rothman R.D., Ho C.Y., Januzzi J.L., Seidman C.E., Fifer M.A. Effects of losartan on left ventricular hypertrophy and fibrosis in patients with nonobstructive hypertrophic cardiomyopathy. JACC Heart Fail. 2013;1(6):480–487. doi: 10.1016/j.jchf.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ho C. Vanish: Valsartan for attenuating disease evolution in early sarcomeric HCM. 2014. 2014. [DOI] [PMC free article] [PubMed]
- 82.Maron M., Kerur B., Chan R.H., et al. Can spironolactone mitigate myocardial fibrosis and alter suddendeath risk and heart failure symptoms in patients with hypertrophic cardiomyopathy? A prospective, randomized trial. Circulation. 2013;128:A16910. [Google Scholar]