

Abstract
The mechanism of a recently reported Suzuki coupling reaction of quinoline-derived allylic N,O-acetals has been studied using a combination of structural, stereochemical, and kinetic isotope effect experiments. The data indicate that C–O activation is facilitated by Lewis acid assistance from the boronic acid coupling partner and an ionic SN1-like mechanism accounts for oxidative addition. In this context, we demonstrate the first direct observation of oxidative addition to a quinolinium salt. Notably, this mechanism is distinct from the more commonly described SN2(′)-type oxidative addition of low-valent transition metals to most allylic electrophiles.
Oxidative addition of a transition metal to an organic electrophile such as an aryl halide or vinyl triflate is a fundamental elementary step in cross-coupling catalysis. For allylic electrophiles, oxidative addition with a late transition metal generally occurs by a stereospecific mechanism with inversion or retention of configuration.1 We recently reported the first examples of Suzuki cross coupling with allylic N,O- and O,O-acetals.2 In these reactions, Csp3–O bond activation proceeds with a nickel catalyst under base-free conditions, enabling a mild and modular synthesis of various privileged heterocycles (eq 1). Coupling reactions with related electron-rich
 |
(1) |
electrophiles have all required harsh nucleophilic reaction partners such as Grignard reagents to achieve Csp3–O bond activation by stereospecific oxidative addition mechanisms.3 As such, we proposed that a distinct SN1-type mechanism was operative in the Suzuki couplings. Specifically, we envisioned that the Ni catalyst could undergo oxidative addition to a transient (iso)quinolinium or benzopyrylium cation accessed by boronic acid assisted ionization of the allylic ether leaving group (mechanism (i) in Scheme 1). While oxidative additions are traditionally understood as the reaction of a metal with an X–Y σ bond,4 limited precedent for an ionic mechanism is also available,5 including for the addition of nickel to iminium and oxocarbenium ions.6 However, this activation mode has not seen broad application in the design of synthetic methods.7 Arndtsen and Watson have reported Pd and Ni-catalyzed Lewis acid assisted C–C bond-forming reactions with N-acyliminium precursors;8 however, direct evidence for the oxidative addition of a metal to an iminium or oxocarbenium ion in the context of a catalytic bond-forming reaction has yet to be disclosed.
Scheme 1.
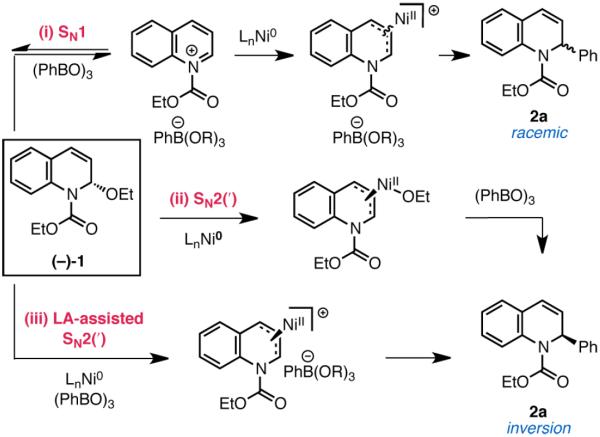
Possible Mechanisms for Csp3–O Activation
Since a better understanding of these processes could afford key insights for the development of new C–C bond-forming reactions with these classic organic electrophiles, we undertook mechanistic investigations of the reaction between aryl boroxines and 2-ethoxy-1-ethoxycarbonyl-1,2-dihydroquinoline (EEDQ) 1. We describe herein structural, isotopic, and stereochemical studies that offer direct evidence for the involvement of an ionic SN1-type mechanism for allylic Csp3–O activation. In this context, we demonstrate the first example of oxidative addition of a low valent metal to a quinolinium ion.
In our previously reported studies on the catalytic system,2a we observed the propensity of boroxines to racemize EEDQ. As such, it was difficult to distinguish the proposed stereoconvergent SN1-type oxidative addition (i) from the more classical mechanisms (ii) and (iii) in Scheme 1, wherein C–C bond formation takes place with overall inversion of configuration but is accompanied by an off-cycle racemization pathway. We conjectured that evaluation of a stoichiometric system would offer a solution to this problem as the rate of oxidative addition at high concentrations of nickel should be competitive with off-cycle boroxine-mediated racemization of enantio-enriched EEDQ.
To evaluate these possibilities, we studied a system comprising Ni, DPEPhos, and EEDQ. DPEPhos was selected as a ligand because it shares electronic and structural similarities to PPh3 but is rigid and chelating, which we reasoned should stabilize the putative allyl complex and facilitate characterization of relevant catalytic intermediates.9 For pathway (ii), oxidative addition should occur in the absence of the boronate nucleophile. Combining Ni(cod)2, DPEPhos, and EEDQ (1:1:1) in the absence of phenylboroxine led to formation of a yellow crystalline compound 3 in nearly quantitative yield (Scheme 2). However, the 1H, 13C, and 31P NMR spectra of 3 were consistent with complexation of (DPEPhos)Ni to the styrenyl olefin of EEDQ rather than an oxidative adduct. X-ray crystallographic analysis provided further confirmation of this structural assignment. Although olefin complexes have been invoked as intermediates in numerous allylic substitution reactions,10 complex 3 represents a rare example of a structurally characterized late transition metal olefin complex to an allylic electrophile (Figure 1).11
Scheme 2.

Stoichiometric Studies of Oxidative Addition
Figure 1.
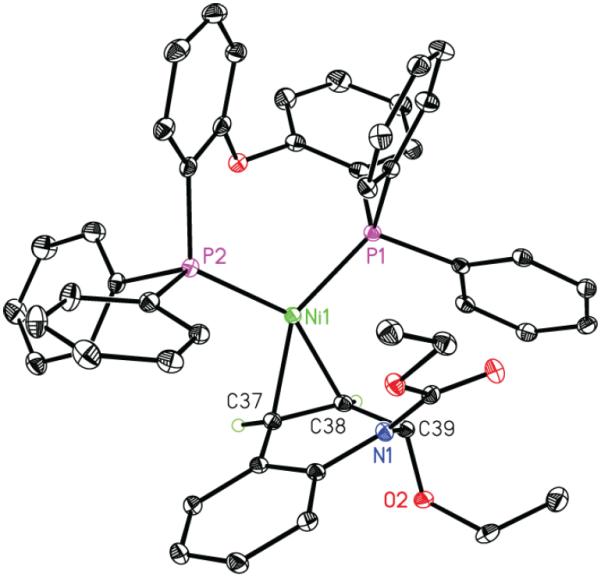
Solid-state structure of olefin-complex 3 at 30% probability ellipsoids. Toluene solvent molecule omitted for clarity. Hydrogen atoms omitted for clarity, except those attached to C37 and C38.
When complex 3 was heated at 60 °C in dioxane/t-AmOH over 12 h, no oxidative addition was observed to take place as determined by 1H and 31P NMR spectroscopy. This result is in agreement with the observation that the C39–O2 bond in 3 exhibits negligible elongation compared to that in free EEDQ (1.434(4) vs 1.417(4) Å).12 By contrast, 2-phenyl dihydroquinoline 2a was produced in 81% yield within 5 h upon treatment of 3 with phenyl boroxine at 23 °C. Monitoring this reaction by 31P NMR spectroscopy revealed clean formation of olefin complex 4 with no detectable build-up of intermediates.13 Moreover, complex 3 was found to be a competent catalyst for the phenylation of EEDQ, delivering 2a in 54% isolated yield at 10 mol % loading.
The direct oxidative addition pathway (ii) in Scheme 1 is inconsistent with these initial studies, which clearly demonstrate a role for the boroxine in the oxidative addition step. Boronic acids have been used as Lewis acid catalysts for a wide variety of transformations,14 but a role for them in oxidative addition is less precedented.15 Circumstantial evidence in favor of a role as Lewis acid includes: (a) when 3 was treated with the less Lewis acidic phenyl pinacol boronate ester, no reaction was observed; (b) in competition experiments between aryl boroxines of differing electronic properties (p-CF3, p-H, and p-OMe), more electron-deficient boroxines reacted faster with olefin complex 3 in a 9.5:2.2:1.0 ratio.16
Unfortunately, in none of these reactions was the presumed Ni-allyl intermediate observed directly. In an attempt to detect such a species, we sought to identify a system that would be resistant to reductive elimination. Addition of hindered and electronically deactivated boroxines to 3 resulted in a color change from yellow to dark orange without concomitant C–C bond formation. However, our attempts to characterize any intermediates by 1H, 13C, and 31P NMR spectroscopy were hampered by the presence of multiple species with broadened spectra. This problem was addressed by using B(C6F5)3, which induced precipitation of a dark orange solid, 5-borate, when combined with equimolar 3 (Scheme 2). Characterization of 5-borate by NMR spectroscopy and single crystal X-ray diffraction were both consistent with its formulation as a nickel π-allyl complex.17 Notably, association of the ethoxide leaving group to the Lewis acidic borane in 5-borate offers compelling structural evidence for a Lewis acid assisted oxidative addition and may have relevance to the course of other allylic substitution reactions with unactivated ether substrates.18
With access to 3 and 5, we were poised to evaluate the stereochemical outcome of the Lewis acid assisted oxidative addition in an effort to distinguish mechanisms (i) and (iii). Exposing enantioenriched complex 3 (99% ee)19 to 0.17 equiv of phenyl boroxine resulted in formation of racemic 2a, consistent with the stereochemical experiments conducted in the catalytic system (eq 2). However, unlike in the catalytic system, recovered
 |
(2) |
EEDQ showed high levels of enantioenrichment (72% ee). This result is a significant because it establishes that C–C bond formation is stereoconvergent. It also suggests that Ni must dissociate from 3 prior to ionization. Consistent with this proposal, 31P and 19F NMR experiments revealed fast ligand exchange between the two distinct (DPEPhos)Ni(EEDQ) complexes 6 and 7 (eq 3).17
 |
(3) |
To test the likelihood that racemization instead occurs post oxidative addition by a Ni(0)-mediated displacement of the Ni-allyl complex,20 we carried out the same reaction at low [3]. Notably, the concentration of Ni(0) had no impact on the stereochemical outcome. Moreover, C–C bond formation occurred with conservation of ee (95%) when the stereochemical experiment was performed with MeMgBr as nucleophile,17 suggesting that a Ni-allyl complex of EEDQ can be configurationally stable. Taken together, these data are most consistent with the fact that the oxidative addition step in the Suzuki coupling is SN1-like in character (i).
A kinetic isotope effect experiment provided further evidence against mechanism (iii). In particular, we observed a secondary kinetic isotope effect of 1.13 ± 0.02 when 3 and its isotopologue prepared from 2-d1-EEDQ were treated with 0.8 equiv of B(C6F5)3. This value is intriguing because it is most in line with reported KIE values for solvolysis reactions of activated electrophiles like benzhydryl halides.21 By comparison, previously reported secondary kinetic isotope values for SN2-type oxidative additions typically range from 0.94 to 1.05.22 In isolated cases, including for oxidative addition to electrophiles with highly stabilized leaving groups, isotope effects as high as 1.53 have been observed.23 Either an SN1 or radical chain mechanism has been proposed to account for these data. Although the stereochemical outcome of the arylation in eq 2 would be consistent with a radical intermediate, the fact that radical traps such as TEMPO and dihydroanthracene do not impede efficient formation of 2a under otherwise standard reaction conditions argues against such a mechanism.17
These studies support the stepwise mechanism (i) wherein a Lewis acidic boroxine abstracts the alkoxide leaving group of EEDQ to generate a quinolinium intermediate that can be intercepted by Ni from either of its two prochiral faces. To our knowledge, no structural evidence for oxidative addition of a low-valent transition metal to a quinolinium or related heteroaromatic ion has been reported. To interrogate the feasibility of this step, we prepared quinolinium triflate 8 from EEDQ and TBSOTf. Gratifyingly, addition of Ni(cod)2 and DPEPhos to 8 resulted in rapid precipitation of 5-triflate in 93% yield as a bright red-orange crystalline solid (Scheme 3). As determined by single crystal X-ray diffraction, the metrical parameters of 5-triflate (Figure 2) are very similar to the Ni-allyl complex 5-borate, with the nickel center lying directly above the central carbon of the allyl functionality. Furthermore, exposing allyl complexes 5-borate or 5-triflate to excess phenyl boroxine in the presence of cesium fluoride furnished 2a in 65 and 70% isolated yield, respectively (eq 4). These data
 |
(4) |
demonstrate that both complexes are competent in the C–C bond-forming step of the coupling reaction.
Scheme 3.

Studies with Quinolinium Triflate
Figure 2.
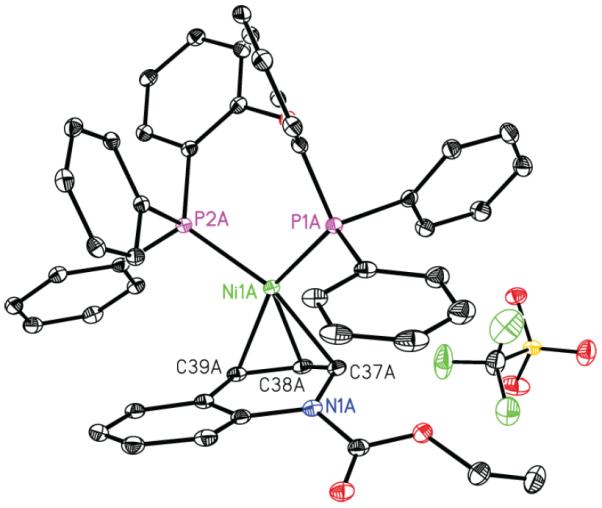
Solid-state structure of Ni-allyl complex 5-triflate at 30% probability ellipsoids. Dichloromethane molecules, disorder on ethyl group of carbamate, and all hydrogen atoms omitted for clarity.
In summary, we have presented a series of experiments that probe the mechanism of allylic N,O-acetal Csp3–O oxidative addition with Ni. Our data are consistent with a Lewis acid assisted oxidative addition mechanism wherein the boroxine nucleophile serves as the Lewis acid. The C–O activation reaction appears to proceed by an SN1-like mechanism. As evidence for this proposal, we demonstrate oxidative addition of Ni into a quinolinium ion. Future work will be directed toward evaluating the generality of these results with other iminium and oxocarbenium ion partners. We anticipate that this mode of activation will ultimately enable the development of sp3-cross-coupling reactions complementary to the much more common transformations of alkyl halides.
Supplementary Material
ACKNOWLEDGMENTS
We thank Scott Semproni for X-ray crystallographic structure determination of 3, 5-borate, and 5-triflate, Derek Ahneman and Jason Shields for preparing the 6-CF3-EEDQ, Thomas Graham and Daniel Nielsen for helpful discussions, and Lotus Separations for chiral separations of 1. Financial support provided by Princeton University is gratefully acknowledged.
Footnotes
Supporting Information Experimental procedures and spectroscopic data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1).For examples see: Trost BM, Verhoeven TR. J. Am. Chem. Soc. 1980;102:4730. doi: 10.1021/ja00418a063. Hayashi T, Hagihara T, Konishi M, Kumada M. J. Am. Chem. Soc. 1983;105:7767. Sheffy FK, Godschalx JP, Stille JK. J. Am. Chem. Soc. 1984;106:4833. Kurosawa H, Kajimaru H, Ogoshi S, Yoneda H, Miki K, Kasai N, Murai S, Ikeda I. J. Am. Chem. Soc. 1992;114:8417. In this paper, we designate stereospecific allylic oxidative addition that proceeds with inversion as an SN2(′) mechanism.
- (2).(a) Graham TJA, Shields JD, Doyle AG. Chem. Sci. 2011;2:980. [Google Scholar]; (b) Graham TJA, Doyle AG. Org. Lett. 2012;14:1616. doi: 10.1021/ol300364s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).For reactions with allylic ethers or acetals: Didiuk MT, Morken JP, Hoveyda AH. J. Am. Chem. Soc. 1995;117:7273. Nomura N, RajanBabu TV. Tetrahedron Lett. 1997;38:1713. Didiuk MT, Morken JP, Hoveyda AH. Tetrahedron. 1998;54:1117. Weatherhead GS, Houser JH, Ford JG, Jamieson JY, Schrock RR, Hoveyda AH. Tetrahedron Lett. 2000;41:9553. For reactions with benzylic ethers, see: Taylor BLH, Swift EC, Waetzig JD, Jarvo ER. J. Am. Chem. Soc. 2011;133:389. doi: 10.1021/ja108547u. Greene MA, Yonova IM, Williams FJ, Jarvo ER. Org. Lett. 2012;14:4293. doi: 10.1021/ol300891k. Taylor BLH, Jarvo ER. Synlett. 2011:2761.
- (4).Hartwig JF. Organotransition Metal Chemistry. University Science Books; Sausalito, CA: 2010. [Google Scholar]
- (5).(a) Louw WJ, de Waal DJA, Chapman JE. Chem. Commun. 1977:845. [Google Scholar]; (b) Crabtree RH, Quirk JM, Fillebeen-Khan T, Morris GE. J. Organomet. Chem. 1979;181:203. [Google Scholar]; Singleton and co-workers have reported a secondary carbon isotope effect for the oxidative addition of a tertiary allylic acetate to Pd that is consistent with an SN1-like mechanism: Singleton DA, Christian CF. Tetrahedron Lett. 2005;46:1631.
- (6).(a) Sepelak DJ, Pierpont CG, Barefield EK, Budz JT, Poffenberger CA. J. Am. Chem. Soc. 1976;98:6178. [Google Scholar]; (b) Ogoshi S, Oka M.-a., Kurosawa H. J. Am. Chem. Soc. 2004;126:11802. doi: 10.1021/ja0460716. [DOI] [PubMed] [Google Scholar]; (c) Barefield EK, Carrier AM, Sepalek DJ, Derveer DG. Organometallics. 1985;4:1395. [Google Scholar]
- (7).Beller M, Eckert W. Angew. Chem., Int. Ed. 2000;39 doi: 10.1002/(sici)1521-3773(20000317)39:6<1010::aid-anie1010>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- (8).(a) Davis JL, Dhawan R, Arndtsen BA. Angew. Chem., Int. Ed. 2004;43:590. doi: 10.1002/anie.200352123. [DOI] [PubMed] [Google Scholar]; (b) Lu Y, Arndtsen BA. Org. Lett. 2007;9:4395. doi: 10.1021/ol7021017. [DOI] [PubMed] [Google Scholar]; (c) Lu Y, Arndtsen BA. Angew. Chem., Int. Ed. 2008;47:5430. doi: 10.1002/anie.200801385. [DOI] [PubMed] [Google Scholar]; (d) Shacklady-McAtee DM, Dasgupta S, Watson MP. Org. Lett. 2011;13:3490. doi: 10.1021/ol201248c. [DOI] [PubMed] [Google Scholar]
- (9).DPEPhos is a competent ligand for the catalytic arylation of EEDQ; however, coupling reactions with DPEPhos proceed with depressed rates compared with those using PPh3. See SI for further details.
- (10).(a) Trost BM. Chem. Rev. 1996;96:395. doi: 10.1021/cr9409804. [DOI] [PubMed] [Google Scholar]; (b) Hagelin H, Scensson M, Åkermark B, Norrby P-O. Organometallics. 1999;18:4574. [Google Scholar]
- (11).For an example of a Pd-olefin complex to an allylic substitution product see: Steinhagen H, Reggelin M, Helmchen G. Angew. Chem., Int. Ed. Engl. 1997;36:2108. For an example of a complex between Ni and diallylether that does not undergo oxidative addition see: Yamamoto T, Ishizu J, Yamamoto A. J. Am. Chem. Soc. 1981;103:6863. A Ni-allyl nitrile olefin complex has been characterized by NMR spectroscopy: Acosta-Ramírez A, Flores-Gaspar A, Muñoz-Herńndez M, Arévalo A, Jones WD, García JJ. Organometallics. 2007;26:1712.
- (12).Cremin DJ, Hergarty AF, Begley MJ. J. Chem. Soc., Perkin Trans. 1980;2:412. [Google Scholar]
- (13).The equilibrium constant for exchange of 3 and 2a with 1 and 4 is 0.27 at 23 °C in deuterated benzene. See SI for further details.
- (14).For a recent example of the use of boronic acids as Lewis acids see: Zheng H, Ghanbari S, Nakamura S, Hall DG. Angew. Chem., Int. Ed. 2012;51:6187. doi: 10.1002/anie.201201620. references therein.
- (15).For examples wherein a boronic acid has been proposed as both activator and coupling partner in a Suzuki reaction see: Trost BM, Spagnol MD. J. Chem. Soc., Perkin Trans. 1995;1:2083. Tsukamoto H, Sato M, Kondo Y. Chem. Commun. 2004:1200. Tsukamoto H, Uchiyama T, Suzuki T, Kondo Y. Org. Biomol. Chem. 2008;6:3005. doi: 10.1039/b804991b. Yu D-A, Shi Z-J. Angew. Chem., Int. Ed. 2011;50:7097. doi: 10.1002/anie.201101461. Li M-B, Wang Y, Tian S-K. Angew. Chem., Int. Ed. 2012;51:2968. doi: 10.1002/anie.201109171.
- (16).In competition experiments among the same three boroxines with 5-triflate, p-CF3 was favored over p-H and p-OMe by 1.8:1.1:1.0 respectively. Since this ratio is considerably lower than that observed for the competition experiment starting from 3, we believe that the data with 3 reflect the electronic bias in oxidative addition rather than transmetalation or reductive elimination.
- (17).See SI for full experimental procedures and data.
- (18).For recent examples, see: Matsubara R, Jamison TF. J. Am. Chem. Soc. 2010;132:6880. doi: 10.1021/ja101186p. Nishikata T, Lipshutz BH. J. Am. Chem. Soc. 2009;131:12103. doi: 10.1021/ja905082c.
- (19).When complex 3 was decomposed in air, recovered EEDQ showed complete conservation of enantioenrichment by chiral HPLC.
- (20).Granberg KL, Bäckvall J-E. J. Am. Chem. Soc. 1992;114:6858. [Google Scholar]
- (21).Streitwieser A, Klein HS. J. Am. Chem. Soc. 1964;86:5170. [Google Scholar]
- (22).(a) Stang PJ, Schiaveill MD, Chenault HK, Breidegam JL. Organometallics. 1984;3:1133. [Google Scholar]; (b) Griffin TR, Cook DB, Haynes A, Pearson JM, Monti D, Morris GE. J. Am. Chem. Soc. 1996;118:3029. [Google Scholar]; (c) Habibzadeh S, Rashidi M, Nabavizadeh SM, Mahmoodi L, Hosseini FN, Puddephatt RJ. Organometallics. 2010;29:82. [Google Scholar]
- (23).Nabavizadeh SM, Habibzadeh S, Rashidi M, Puddephatt RJ. Organometallics. 2010;29:6359. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.