

Abstract
Many pharmaceuticals on the market today belong to a large class of natural products called nonribosomal peptides (NRPs). Originating from bacteria and fungi, these peptide-based natural products consist not only of the 20 canonical L-amino acids, but also non-proteinogenic amino acids, heterocyclic rings, sugars, and fatty acids, generating tremendous chemical diversity. As a result, these secondary metabolites exhibit a broad array of bioactivity ranging from antimicrobial to anticancer. The biosynthesis of these complex compounds is carried out by large multimodular megaenzymes called nonribosomal peptide synthetases (NRPSs). Each module is responsible for incorporation of a monomeric unit into the natural product peptide and is composed of individual domains that perform different catalytic reactions. Biochemical and bioinformatic investigations of these enzymes have uncovered the key principles of NRP synthesis, expanding the pharmaceutical potential of their enzymatic processes. Progress has been made in the manipulation of this biosynthetic machinery to develop new chemoenzymatic approaches for synthesizing novel pharmaceutical agents with increased potency. This review focuses on the recent discoveries and breakthroughs in the structural elucidation, molecular mechanism, and chemical biology underlying the discrete domains within NRPSs.
1 Introduction
The biosynthetic pathways of natural products produced by microorganisms have been investigated extensively over the past few decades due to the broad spectrum of biological activity exhibited by these compounds. Many of these structurally diverse and complex molecules display valuable medicinal activities extending from antibiotic to immunosuppressive, to anticancer properties.1 On account of their wide range of therapeutic properties, these natural products have gained substantial consideration in the field of modern medicine. Not only do they continue to be an important source of drugs used today, but they also show promise as scaffolds for the development of new more potent pharmaceutical agents in the future.2
Prominent among these microbial metabolites is the large subclass of natural products known as nonribosomal peptides (NRPs). These peptide-based natural products are synthesized by large multifunctional megaproteins called nonribosomal peptide synthetases (NRPSs).3 The structural framework of these biosynthetic enzymes are comprised of modules that are responsible for the incorporation of a single building block into the natural product. The modules can be organized on a single polypetide (type I NRPS), as seen in many fungal species,4,5 or on several discrete interacting proteins (type II NRPS), as seen in many bacterial systems.6,7 Each module is further divided into catalytic domains accountable for a single reaction in the incorporation and modification of a monomer unit in the chain elongation process. Due to the modular architecture of NRPSs, considerable attention has focused on the reprogramming of their biosynthetic machinery for the biocombinatorial synthesis of novel peptides with improved bioactivity.8,9 In the past decade, reengineered synthetases, generated by either exchanging or fusing modules10,11 or translocating domains12,13 were able to produce a library of NRP derivatives. However, the productivity of these engineered synthetases is hindered by poor substrate selectivity14,15 and improper protein interactions.16,17,18 Consequently, the extent to which these methods can be improved will depend on our understanding of the structural and mechanistic features of the protein interactions involved in the biosynthetic strategies of NRPSs.
In the past, great advances have been made in the structural and functional elucidation of NRPSs. Extensive biochemical investigations have revealed the molecular mechanisms underlying the biosynthesis of therapeutic NRPs.19 At present, the X-ray and nuclear magnetic resonance (NMR) structures of the core domains of these large biosynthetic assembly lines have been solved.20 In addition, the mechanistic function and overall modular structure of these NRPS units as well as several tailoring domains have been uncovered.21 The focus of this review will be on these investigations, which have examined the structural and functional aspects of NRPSs.
2 Structural Diversity of Nonribosomal Peptides
Nonribosomal peptides display a wide variety of structurally complex features that generate their chemical diversity (Fig. 1). These unique features often transform the peptides into their bioactive conformations, allowing them to interact specifically with select binding pockets on their particular molecular target. As a result, these secondary metabolites hold great pharmaceutical potential. Responsible for the incorporation of these characteristic components, such as D-amino acids, heterocyclic rings, and N-methylated residues are the auxiliary domains of NRPSs. In addition, other tailoring enzymes incorporate sugars and fatty acids to confer biological activity to the natural product peptide.1
Fig. 1.

A variety of nonribosomal peptides with structural features that confer their bioactivity (highlighted).
Bacitracin and vibriobactin are examples of nonribosomal peptides that contain heterocyclic rings, incorporated by the cyclization (Cy) domain (Fig. 1). Produced by the organism Bacillus licheniformis for protection against other bacteria,22 bacitracin is commonly used as an antibiotic ointment for the treatment of skin infections. It contains a thiazoline ring that maintains the structural configuration responsible for complexation with a divalent cation and the phosphate moiety of C(55)-isoprenyl pyrophosphate.23 This complexation prevents dephosphorylation of the lipid phosphate, a key step in bacterial cell wall biosynthesis, which leads to inhibition of peptidoglycan synthesis and thus killing the unwanted bacteria.24
Vibriobactin is a catecholate siderophore first isolated in 1984 from the pathogenic bacterium Vibrio cholerae.25 It contains two oxazoline rings, derived from threonine, that help position the hydroxyl groups of the catechols to chelate metal ions such as Fe(III).26,27 In doing so, it functions as a virulence factor by sequestering iron in iron-limited environments.
One of the hallmarks in nonribosomal peptides is the incorporation of D-amino acids. Cyclosporin A and tyrocidine A are cyclic peptides that have immunosuppressant28 and antibacterial properties,29 respectively. They contain D-amino acids, which provide stereochemical constraints for proper cyclization into the final product.30 In doing so, the regio- and stereoselective formation of these cyclic peptides enables compatibility with their specific biological target.
The modifications mentioned thus far occur during NRP synthesis. They are carried out by the auxiliary domains located within the synthetase (in cis). Additional chemical alterations also take place, but by discrete tailoring domains (in trans). Some of these reactions include glycosylation, halogenation, oxidative crosslinking, and lipidation.31 Typically, these reactions occur after the peptide has been synthesized, but in some cases they occur beforehand. For instance, initiation of daptomycin synthesis was recently reported to begin with condensation between a fatty acyl chain and its N-terminus amino acid, tryptophan.32 Daptomycin was originally discovered in the early 1980’s by Eli Lilly and Company and has since gained considerable attention as a novel drug for the treatment of Gram-positive infections.33 Unlike most antibacterial agents, which target the proteins in control of cell wall synthesis, daptomycin disrupts the cell membrane directly to kill bacteria. Due to the presence of a fatty acid side chain, when Ca(II) binds to the cyclic peptide, the antibiotic is rendered more amphiphilic so that it can insert itself into the membrane. In doing so, the cell membrane is perforated, causing leakage of ions and eventually cell death.34 Fengycin, produced by Bacillus subtilis strain F-29-3, also operates in a similar manner due to its lipid moiety, but specifically inhibits filamentous fungi.35,36 Balhimycin is a glycopeptide antibiotic and syringomycin E is a lipopeptide fungicide, each modified with halogens. It has been shown that the presence of chlorine increases the antibiotic activity of balhimycin 8- to 16-fold37 and the antifungal potency of syringomycin E 3-fold.38 Two biaryl ethers and a biaryl ring, formed through oxidative crosslinking by cytochrome P450-like oxygenases,39 are also present in balhimycin, giving it the tricyclic peptide backbone required for antimicrobial activity.40 The extent of structural diversity is further illustrated by luzopeptin A and echinomycin. Exhibiting antitumor activity, they consist of bicyclic chromophores, enabling them to intercalate DNA. As a result, DNA replication is inhibited in cancerous cells.41,42 On a final note, many of the chemical functionalities featured in Fig. 1 not only provide a wide scope of biological activity, but also increase the stability of these natural product peptides, preventing proteolytic degradation.43
3 Modular Nonribosomal Peptide Biosynthesis
Although NRPs are marked by structural diversity, their mode of synthesis is highly conserved. Classified into three categories, biosynthesis of NRPs can occur in a linear (Type A), iterative (Type B), or nonlinear (Type C) manner, as shown in Fig. 2.44 In the linear strategy (Type A, Fig. 2), the number and sequence of the modules in the NRPS matches the number and order of amino acids in the peptide. In type B (Fig. 2), the modules or domains of the synthetase are used more than once to synthesize the peptide, which consists of repeated sequences. Lastly, nonlinear NRPSs generate peptides in which the sequence of amino acids does not correlate to the arrangement of modules on the synthetase template (Type C, Fig. 2).
Fig. 2.

Biosynthetic strategies for assembling nonribosomal peptides (NRPS).
Regardless of the biosynthetic strategy utilized by the megasynthetases, all NRP synthesis is mediated by the peptidyl carrier protein (PCP) found in each NRPS module. The ~80 aa PCP acts as a scaffold, tethering the amino acid building blocks and peptidyl intermediates as they are modified and condensed by other domains of the NRPS.45 In addition to the PCP, the adenylation (A) and condensation (C) domains constitute the three core domains that define a typical NRPS module. The A domain recognizes and activates the amino acid building block by formation of an amino acyl adenylate intermediate through the consumption of ATP. Then the activated substrate is transferred to the thiol group of the 4′-phosphopantetheine on PCP and covalently bound through a thioester linkage.46 Derived from coenzyme A (CoA), phosphopantetheine is posttranslationally attached to a conserved serine residue of the PCP by a phosphopantetheinyl transferase (PPTase).47 The activated PCP acts as a flexible arm permitting the bound substrate to travel from one catalytic center to another. Once the amino acyl substrate has undergone all necessary modifications, the C domain catalyzes the peptide bond formation with the downstream amino acyl unit that is tethered to the PCP of the adjacent module.48 While the typical module contains these three domains, the first module of the NRP system, known as the initiation module, lacks a C domain. In addition to the core domains are the tailoring domains, which are responsible for the various modifications that create the diversity in NRPs (see Section 2). Some of these chemical alterations occur during peptide synthesis and are performed by domains such as the epimerase (E), cyclization (Cy), methyltransferase (MT), and oxidation (Ox) domains. After these modifications, the final product is released from the PCP of the termination module through either hydrolysis or macrocyclization by the thioesterase (TE) domain.49 Once released, the NRP products can be further subjected to modifications by glycosyltransferases, halogenases, and oxygenases.
4 Posttranslation Modification and Mispriming
4.1 Phosphopantetheinyl Transferases (PPTases)
The PPTases form a particular group of enzymes essential for activating NRPSs to their functional state. Prior to NRP biosynthesis, all PCPs undergo posttranslational modification by these external enzymes. The 4′-phosphopantetheine moiety of the cofactor CoA is transferred on to the conserved serine residue of the PCP by a dedicated PPTase in a Mg2+-dependent reaction, converting the inactive apo-PCP to the active holo-PCP. Although the PPTases share low primary sequence homology, they have been organized into three classes based on both size and substrate selectivity. The first class is the E. coli AcpS-type, which is ~120 aa in length and displays strict substrate selectivity, recognizing mainly carrier proteins involved in primary metabolism. As such, many of the AcpS-type PPTases are responsible for modification of the fatty acid synthase acyl carrier protein (FAS ACP). Characterized by broad substrate recognition tolerance, the Sfp-type PTTases are ~240 aa in length and form the second class. They are capable of modifying carrier proteins from both primary and secondary metabolism, which include NRPSs. They include the PPTases B. brevis Gsp,50 B. licheniformis Bli,51 Vibrio cholerae VibD,52 and S. aurantiaca MtaA.53 The third class of PPTases modify the fatty acid synthase of eukaryotes. Unlike the first two classes of PPTases, this class is directly attached to their corresponding biosynthetic system. For example, the yeast PPTase found in B. ammoniagenes was found to be an integral part of the yeast FAS2 gene and responsible for activation of the acyl carrier proteins in its type I FAS.54
In 1999, the crystal structure of the Bacillus subtilis Sfp protein complexed with CoA in its active site was resolved at 1.8 Å (Fig 3A).55 The PPTase enzyme exhibited an α/β-fold with pseudo 2-fold symmetry, dividing it into two similar halves with the active site pocket located at the interface. It was suggested that Mg2+ complexed with the α- and β-phosphates of the pyrophosphate moiety of CoA along with the carboxylate moieties of active site residues D107, E109, and E151.56 In addition, H90 was observed to participate in binding CoA to the active site through salt bridges between the ε- and δ-nitrogens of the imidazole group of H90 to the 3′-and α-phosphate moieties of CoA, respectively. This was in accordance with an earlier study demonstrating the importance of the 3′-phosphate for CoA binding and the strict pH dependence of Sfp activity.57
Fig. 3.

(A) Crystal structure of the Bacillus subtilis Sfp protein (red and yellow) complexed with CoA (purple). Residues D107, E109, and E151 coordinate with Mg2+ ion (blue sphere) and H90 binds to CoA (purple). Mutational analysis of residues K112, E117, and K120 (orange) determined the loop region (cyan) that forms the binding pocket for the PCP. (B) Coordination diagram depicting the proposed mechanism of the phosphopantetheinylation reaction.
In a more recent study, structure-based mutational analysis of the PPTase enzyme Sfp revealed the PCP binding region and the reaction mechanism for activating the PCP.58 According to the model presented in this study, invariant residue E151 of Sfp is proposed to deprotonate the hydroxyl group of the active site serine residue of PCP in order for it to attack the β-phosphate of CoA through an addition-elimination mechanism, causing the release of 3′,5′-ADP (Fig. 3B). The proton transferred on to E151’s carboxyl group is passed to D107 and then to K155, where it is finally handed off to the oxygen of the α-phosphate of CoA. Based on the cocrystal structure of Sfp•CoA,55 all residues participating in shuttling the proton are within hydrogen-bonding distance. In addition, mutations of these proton shuttling residues resulted in a 1000-fold reduction in catalytic efficiency for Sfp, further corroborating the proposed mechanism. To determine the binding pocket for the PCP, residues K112, E117, and K120 were selected for mutational analysis based on comparison to the AcpS–ACP cocrystal structure.59 These Sfp mutants displayed 15- to 24-fold reduced Km values for PCP, indicating the importance of these residues for PCP binding. Likewise, the location of these residues on the loop region between β4 and α5 suggests that the Sfp protein utilizes this flexible loop region to recognize various carrier proteins.
4.2 Type II Thioesterase Domain
NRPSs utilize a specialized thioesterase domain to ensure efficient and accurate production of the final NRP product. Unlike the type I thioesterases (TEIs), which catalyze the release of the final peptide product from the synthetase, type II thioesterases (TEIIs) function as repair enzymes, regenerating the functional 4′-phosphopantetheine arm of a misprimed PCP. Due to the large percentage (~80%) of acylated CoA in bacteria,60 along with the promiscuity of the PPTase Sfp, mispriming of PCP domains occurs quite frequently, rendering the protein inactive and incapable of producing the natural product peptide. Additionally, mispriming of the PCP can occur when the holo-PCP is loaded with an incorrect amino acid. As a result, the TEII is required to restore functionality to the PCP domain by hydrolyzing the acyl or peptidyl group off of the misprimed acyl-4′-phosphopantetheine arm. This enzyme must be able to recognize all PCPs and hydrolyze off incorrect acyl or peptidyl groups while avoiding untimely cleavage of the correct growing peptide. The functional activity of this domain in relation to NRPS was first realized in a study in which the TEII protein associated with the antibiotics surfactin (TEIIsrf) and bacitracin (TEIIbac) were investigated for hydrolysis of acetyl-PCP substrates.61 The TEII domains were determined to hydrolyze acetyl-PCPs as well as aminoacyl- and peptidyl-CPs. The overall rate of NRP production was unaffected by the activity of the TEIIs, supporting the hypothesis that the main role of TEII enzymes is to regenerate the misprimed PCP domains of NRPSs.
Shortly after the discovery of TEIIs, a mutational study of the surfactin TEII domain was conducted, revealing three catalytic residues and a residue potentially important for structural stability.62 The three catalyic residues identified form a catalytic triad, similar to that found in TEI domains, consisting of S86, D190, and H216. The alignment of these residues suggested that the TEII domains belong to the α/β-hydrolase superfamily. In addition, the conserved residue D163 was found to be structurally important since a D163A mutant maintained hydrolytic activity but was unable to remain structurally stable. Having established the TEII active site residues necessary for catalysis, efforts turned towards uncovering the specific protein interactions with the PCP domain. NMR titration experiments of SrfTEII were performed with TycC3–PCP to elucidate its structure and uncover the key factors governing the conformational changes during its interaction with the PCP along with the structural basis for substrate selectivity.63 SrfTEII exhibited a typical α/β-hydrolase fold, with a central 7-stranded β-sheet surrounded by 8 helices (Fig. 4A). The short ‘lid’ region containing the helix-turn-helix motif was observed to only partially cover the TEII active site. The key residues comprising the interaction surface of the SrfTEII enzyme were also determined in this structural analysis and they included the N-terminal loop, the loop regions between the α-helices and β-sheets, and the “lid” region. Like the TEI domain, the TEII enzyme was ascertained to also exhibit two distinct conformations based on NMR chemical shifts of certain key amino acids, with preference for one conformation when interacting with its native substrate, the acetylated holo PCP. This observation of an equilibrium between two different conformations demonstrates that a conformational exchange process is occurring in solution. Interestingly, similar exchange processes had been detected in the structural study of the EntF PCP–TEI didomain.64
Fig. 4.

(A) Structure of Bacillus subtilis SrfTEII (red and yellow) in complex with TycC3–PCP (blue), a PCP from the tyrocidine system. Residues S86, D190, and H216 of SrfTEII form the catalytic triad. Key residues (purple), including the ‘lid’ region (orange) of SrfTEII that interact with TycC3–PCP are highlighted. (B) Structure of RifR. Residues S94, D200, H228 form the catalytic triad of the RifR active site and the flexible linker region (purple) as well as the ‘lid’ region (orange) are indicated.
Another distinct feature of the TEII domain that was revealed in this study was its substrate specificity. NMR titration experiments with 15N-labelled SrfTEII and unlabelled TycC3–PCP domain loaded with either a single amino acid or a tripeptide were performed. Chemical shift changes were observed for active site residues of SrfTEII when titrated with Ala-loaded TycC3–PCP domain but not with the tripeptide loaded on, indicating a preference for small substrates in the TEII active site. Consequently, the active site of the TEII was determined to be embedded in a shallow groove capable of accommodating only small acyl substituents.65
Recently, another external thioesterase domain, RifR, from the hybrid NRPS–PKS rifamycin, demonstrated broad substrate specificity, hydrolyzing various-size carboxylated and decarboxylated acyl thioesters, with a preference for misacylated carrier proteins over the natural building blocks of rifamycin.66 Interestingly, RifR also showed preferential hydrolysis of misacylated carrier proteins over misacylated CoA analogues, emphasizing the importance of protein-protein interactions in the functionality of the TEII domains.
As for the overall structure of RifR, it maintained the same α/β-hydrolase fold as seen by previously characterized NRPS TEII domains, with a three α-helix ‘lid’ governing proper substrate binding by controlling access to the substrate chamber (Fig. 4B). In addition, a flexible linker region, forming part of the substrate chamber, was identified as being responsible for the broad substrate specificity of RifR. Although RifR displayed broad substrate selectivity, the catalytic efficiency for the hydrolysis of the different acyl thioesters varied, suggesting that the lid somehow affects the linker region, dictating the extension it undergoes in order to accommodate the different length substrates.
5 Core NRPS Domains
5.1 Adenylation Domain
As the initial determinants of substrate selectivity in NRP biosynthesis, the adenylation domains (~550 aa) function as the gatekeepers of the NRPS assembly line. Based on previous studies,67,68 the A domains recognize and activate their cognate amino acids in a two-step process. First, they selectively bind the cognate amino acid and convert it into an aminoacyl adenylate intermediate at the expense of an ATP. Then the adenylated substrate undergoes a nucleophilic attack by the thiol of the 4′-phosphopantetheine arm of the PCP, forming a thioester bound aminoacyl-S-PCP (Fig. 5A).
Fig. 5.

(A) Adenylation reaction in NRPS. (B) Crystal structure of the adenylation domain, consisting of the large N-terminal domain (red and yellow) and the smaller C-terminal domain (gray), from the gramicidin S synthetase, GrsA, complexed with AMP (purple) in the presence of Mg2+ion (blue sphere). The 10 catalytic residues (orange) termed ‘codons’ are highlighted. (C) Crystal structure of DltA, consisting of the large N-terminal domain (red and yellow), the linker region (orange) and the smaller C-terminal domain (gray), bound to ATP (purple). Invariant residues K492, E298, and R397 stabilize ATP in the presence of Mg2+ ion (light blue sphere).
In 1997, the first crystal structure of an NRPS A domain, the phenylalanine-activating domain (PheA) of the gramicidin S initiation module, was determined.69 Since then, only two other A domain has been resolved, the stand-alone 2,3-dihydroxybenzoic acid-activating domain (DhbE) of the bacillibactin NRPS,70 and the A domain of the NRPS gene sidN discovered in the fungus Neotyphodium lolii, which activates N(Δ)-cis-anhydromevalonyl-N-(Δ)-hydroxy-L-ornithine.71 Based on these crystal structures, the overall structure of the A domain consists of a large N-terminal domain and a small C-terminal domain, with the active site located at the junction of these two subdomains (Fig. 5B). Within the active site, 8–10 residues were identified and confirmed to be relevant for catalysis through sequence alignments of other A domains and mutational studies.72,73 Referred to as the ‘codons’ of nonribosomal peptide synthesis, these 8–10 catalytic residues appeared to be degenerate and were further exploited for synthesizing new peptide antibiotics.74 These biochemical studies altered the specificity-conferring code of A domains to recognize noncognate amino acids with minor changes in polarity and size compared to the cognate substrate. In addition to directly modifying the specificity conferring amino acids, computational redesign has been employed to alter the substrate specificity of the A domain.75 Using the crystal structure of PheA as a starting model, computational algorithms predicted specific active-site mutations that would accommodate alternate substrates that are not usually activated by PheA, including Leu, Arg, Glu, Lys, and Asp. The predicted mutations successfully altered the substrate specificity of PheA when tested in vitro. In contrast with earlier mutational studies, residues not involved in the specificity conferring code were analyzed to further increase specificity towards non-natural substrates.
Endeavors towards a better grasp of the structural orientations affecting substrate selectivity in A domains have focused on examination of other adenylate-forming enzymes, such as the crystal structure of the acetyl-CoA synthetase (Acs) from Salmonella enterica76 and the D-alanyl carrier protein ligase (DltA) from Bacillus cereus.77 The crystal structure of the acteyl-CoA synthetase (Acs) complexed with adenosine-5′-propyl phosphate and CoA gave the first insight on the thioesterification reaction between the adenylated substrate and CoA. It was suggested that in the second half reaction, the C-terminal domain undergoes a conformational change, exposing new catalytic residues for CoA to attack the adenylated intermediate. Hence, the idea of multiple structural orientations for the two half reactions was proposed as a model for members of the adenylate-forming enzymes. In this ‘domain alternation hypothesis’,78 the K609A and G524L mutants of Acs were shown to be inactive in the adenylation half-reaction and the thioester-formation half-reaction, respectively, through kinetic analysis, demonstrating the importance of these residues in catalysis of each half reaction. Furthermore, the crystal structures of Acs in the adenylate-forming conformation displayed residue G524 25–30 Å from the active site, supporting the need for the C-terminal domain to undergo a ~140° rotation for the second half reaction.
In a more recent study, the crystal structure of another adenylate-forming enzyme, the D-alanine carrier protein ligase (DltA), complexed with ATP was reported (Fig. 5C),79 enabling the examination of the pre-adenylation state. Prior to this development, two crystal structures of DltA, one in the thioester-forming state,80 and another in the post-adenylation state81 had been elucidated, further corroborating the findings of the studies with PheA, DhbE, and Acs. The structure of the pre-adenylation state suggests that there are three distinct conformations adopted by adenylation domains during thioester formation. The crystal structure of the DltA•ATP complex described the catalytic role of invariant residues K492, E298 and R397, which could not be explained in the previous two DltA crystal structures. It was proposed that E298 and R397 were responsible for stabilizing the pyrophosphate leaving group of ATP in the adenylation reaction. K492 was observed to be in different conformations in the DltA/ATP complex, as opposed to the DltA/adenylate complex. Due to this mobility, K492 is able to facilitate the electron transfer from the carboxylate group of D-alanine to the pyrophosphate group via a pentavalent transition state. In addition to proposing a molecular mechanism of catalysis for AMP-forming enzymes, this study also revealed a 48° reconfiguration of the C-terminal domain for complexation with ATP. Hence, two conformational changes appear to be necessary for the adenylation domains to properly recognize and activate their substrates for NRP biosynthesis. Powerful bioinformatics tools have been developed to accurately predict the preferred substrate of adenylation domains.82,83 Based largely on data obtained from the crystal structure of PheA, 24 residues within 8Å of the bound phenylalanine substrate, in addition to the previously described 8–10 specificity-conferring active site residues,72 were selected for the comptuational prediction of substrate specificity. These bioinformatics tools enable quick and accurate analysis of newly identified NRPS systems, allowing for more complete characterization of novel NRPS gene clusters.
Biochemical investigations have also shed light on characteristics governing substrate specificity of the adenylation domain. A comprehensive survey defining the limits of A domain substrate specificity was recently published,84 revealing how hydrophobicity along with shape complementarity between the substrate and the binding site on the protein play a major role in substrate recognition by the A domain. The adenylation activity of TycA, which has a native affinity for Phenylalanine, was tested against a panel of 30 amino acid substrates using a discontinuous ATP/pyrophosphate-exchange assay.85 Charged substrates displayed no activity, indicating the importance of the electronic character of the side chain. It was speculated that the energy of desolvation of a charged group may be too large to overcome, preventing the substrate from binding within the A domain hydrophobic active site. In addition, the Hansch log P value was also calculated for the side chain of each amino acid substrate to measure its hydrophobicity. Based on the kinetic data gathered correlated to the log P values, a general trend was observed in which the catalytic efficiency of the enzyme increased with the hydrophobicity of the amino acid’s side chain. However, there were discrepancies from the general trend, which were rationalized by additional factors, such as shape complementarity and van der Waals interactions. For example, the natural substrate L-Phe deviated from the general trend. This discrepancy was rationalized by its high degree of shape complementarity, arising from the stacking of the phenyl ring side chain between two walls of the binding pocket, which promoted van der Waals interactions to improve catalytic efficiency. A third factor affecting substrate recognition was determined to be the size of the side chain. Larger substrates exhibited reduced catalytic efficiency, as observed for L-Tyr, which had an 800-fold lower catalytic efficiency than L-Phe. From the findings uncovered in this study, it was concluded that the A domain of TycA displayed a substantial degree of specificity, despite its tolerance for alternate substrates.
Directed evolution of the specificity conferring code has been used to exploit the alternate substrate tolerence of TycA.86 Eight active site residues were mutated using successive saturation mutagenesis. The resulting mutants were analyzed for adenylation activity against alternate substrates using a PPi/ATP exchange assay adapted to a 96-well plate form.87 Mutants that showed increased activity towards smaller, hydrophobic substrates were selected and subjected to further rounds of mutation. The resulting mutants exhibited a significantly smaller active site, which showed increased activity with smaller substrates, and severely reduced activity with the natural Phe substrate.
A class of small ~70 amino acid proteins, known as MbtH-like proteins, are sometimes encoded within NRPS gene clusters,88 and have been discovered to associate with the A domain.89 To aid in the elucidation of the exact function of these small proteins, the structures of PA2412 from Pseudomonas aeruginosa90 and MbtH from Mycobacterium tuberculosis,91 from which this class of proteins derives its name, have been solved., PA2412 has been analyzed both x-ray crystallography and NMR, while MbtH has been solved using NMR and CD (Fig. 6). These proteins exhibit a conserved three-stranded anti parallel β-sheet, which interact with an adjacent α-helix. Structural data of PA2412 indicate a second α-helix at the C-terminus of the protein, which is observed to be mobile in the NMR solution structure. Similarly, MbtH exhibits flexibility and disorder at the C-terminus. Biochemical studies have helped elucidate possible roles of MbtH-like proteins in NRP biosynthesis. In a study of the vicibactin biosynthetic pathway of the genus Rhizobium, the MbtH-like protein vbsG was found to be necessary for A domain activity of the module vbsS.92 These genes, which are adjacent to one another in the genome, were shown to copurify. This suggests a strong interaction between A domains and MbtH-like proteins. Similarly, two A-PCP modules from the gene clusters responsible for the biosynthesis of the anti-tuberculosis agents capreomycin and viomycin were studied.93 It was discovered that the A-PCP domains CmnO and VioO, which contain A domains specific for β-Lys, required the presence of their cognate MbtH-like proteins, CmnN and VioN, in stoichiometric amounts to activate β-Lys. Additionally, both CmnO and VioO copurified with their respective MbtH-like protein. In the same study, NRPS modules from the Enterobactin biosynthetic pathway were also investigated. EntF, a module containing an A domain specific for Ser, co-purified with the MbtH-like protein YmbZ. EntF in the presence of YmbZ exhibited 15-fold higher affinity for Ser. Conversely, a module from the same biosynthetic pathway containing an A domain specific for 2,3 dihydroxybenzoate, EntE, did not show increased activity in the presence YmbZ, nor did the two proteins copurify. This suggests that MbtH-like proteins are not necessary for all A domains. Interestingly, in another recent study, the module GlbF from Glidobactin synthesis in Burkholderia sp. K481-B101 was found to only express solubly when coexpressed with the MbtH-like protein GlbF, which is adjacent to GlbE in the genome.94 In this case, the MbtH-like protein apparently serves as a chaparone for its cognate NRPS module. While the exact function is still unclear, these studies have greatly increased our understanding of the biological role MbtH-like proteins.
Fig. 6.
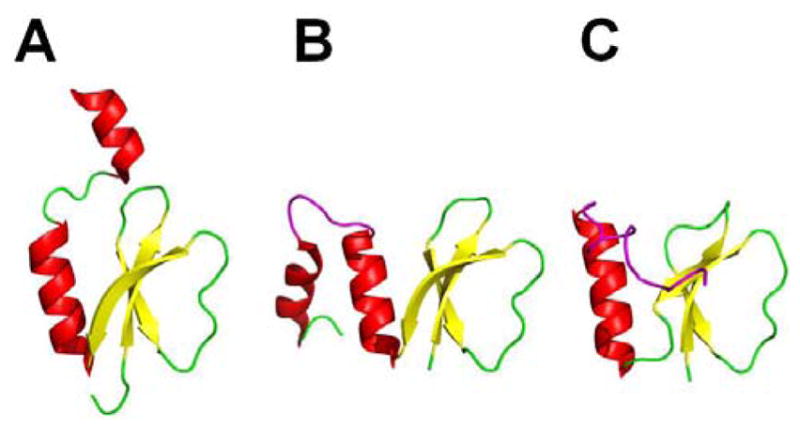
Structures of MbtH-like proteins. (A) Crystal structure of PA2412. (B) NMR structure of PA2412. Disordered residues at the N-terminus have been omitted for clarity. Flexible region between the two α-helices is shown in purple. (C) NMR solution structure of MbtH. Disordered residues at the N-terminus have been omitted for clarity. Flexible C-terminus is shown in purple.
5.2 Peptidyl Carrier Protein Domain
Central in mediating nonribosomal peptide biosynthesis is the small ~80–100 residue peptidyl carrier protein (PCP) domain. The PCP carries the growing natural product chain throughout the catalytic steps of NRP biosynthesis. As mentioned in section 3, the PCP is post-translationally modified by a PPTase, which covalently transfers the 4′-phosphopantetheine arm of a CoA molecule onto a conserved serine residue located at the PCP active site. In doing so, the PCP is converted from its inactive apo form to its active holo form, which can then covalently bind its aminoacyl substrate via a thioester linkage with the terminal thiol of the 4′-phosphopantetheine arm. Once bound, the substrates are shuttled between the PCPs of each module throughout the cycles of peptide elongation. The initial structure of a PCP was determined to be the typical four-helix bundle observed in many carrier proteins, as demonstrated by the NMR solution structure of TycC3–PCP, the carrier protein from the third module of the tyrocidine synthetase of Bacillus subtilis.95 However, in a more recent evaluation of the TycC3–PCP solution structure,96 it was reported that the apo and holo forms of the carrier protein maintained two different stable configurations, the A-state and H-state, respectively. In addition, a third common structural state shared by both the apo and holo forms, the A/H state, was revealed in which the configuration resembled the canonical four-helix carrier protein structure. In the A-state (Fig. 7A), the PCP is in its most flexible and extended conformation. Helices αI and αII are primarily uncoiled with loop III stretched out and lodged in between helices αII and αIV. Based on a molecular dynamics simulation,97,98 it was proposed that the embedment of loop III within the core of the protein permitted the formation of a hydrogen bond between the carbonyl of active site residue S45 and the side chain of R47, preventing modification with phosphopantetheine. In the A/H-state (Fig. 7B), the helices are orientated similarly as in the A state but are longer and more stable. The unstructured loop III is tightened into helix αIII, and is situated outside of the protein core. When phosphopantetheine is finally attached in the H-state (Fig. 7C), helix αIII unfolds, causing αIV to align parallel with αI. Helix αII is also relocated along with the active site serine residue, resulting in a large movement of the phosphopantetheine arm across the PCP face. This observation provided the first structural evidence at the molecular level for the “swinging arm” model.95
Fig. 7.
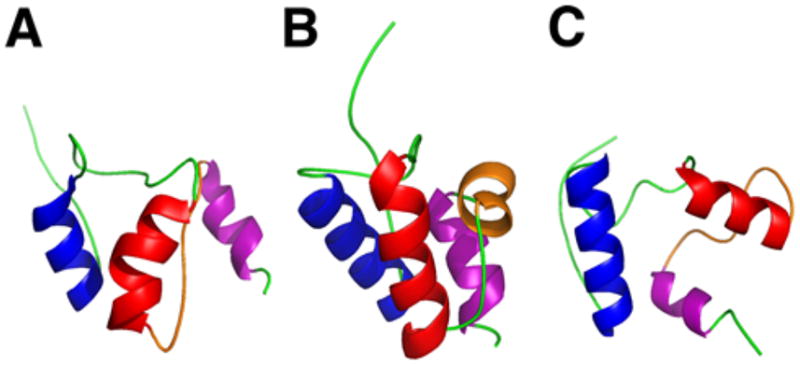
Ribbon diagrams of the NMR solution structures of TycC3–PCP in three different conformations (A) the A-state, (B) the A/H-state and (C) the H-state. Helices αI (blue), αII (red), αIII/loop III(orange), and αIV (purple) undergo conformational changes in each state.
With the elucidation of the dynamic conformational changes exhibited by the PCP in its three different states, examination of the interactions and conformational changes of the PCP during its communication with other domains in the NRPS system followed. NMR studies were carried out in which 15N-labeled TycC3–PCP was titrated with the Bacillus subtilis phosphopantetheinyl transferase, Sfp, and the surfactin type II thioesterase domain, SrfTEII.96 The results of the titration experiment with Sfp showed that the PCP residues with chemical shift deviations constituted a surface that interacts with the PPTase in the A-state conformer rather than the A/H-state. Based on this data and the mutational analysis of the PCP–Sfp interaction,58 a model was proposed suggesting that the selectivity of the A-state conformer by Sfp was due to the unfolding of helix αIII, causing less steric hindrance for interaction with the PPTase. Likewise, titration of holo-TycC3–PCP with SrfTEII uncovered a protein interaction surface, composed of the loop III region, the loop containing the active site S45, and the N-terminal part of helix αII in the H-state. Further investigations of the enterobactin system corroborated the importance of helix III of PCP in recognizing its cognate partner domains in trans. However, two mutagenesis studies on the aryl carrier protein of the enterobactin synthetase, EntB–ArCP, identified two distinct recognition sites for PPTases EntD and Sfp,99 and the serine-incorporating NRPS module, EntF.100 Key residues G242 and D244 constituted an interaction surface on EntB–ArCP for EntD and Sfp, while the residues F264 and A268 on helix III were identified as the recognition surface for EntF. Recently, it was shown that chimeric constructs of EntB-PCP were unable to participate in a bond-forming reaction with EntE, an adenylation domain.101 All EntB-PCP constructs other than the wild type showed no evidence of adenylation, which further highlights the importance of specific protein-protein interactions between PCP and catalytic domains.
Complementing these findings were the results from another study in which aryl carrier proteins (ArCP) from VibB of Vibrio cholerae vibriobactin and HMWP2 of Yersinia pestis yersiniabactin NRPSs were evolved by random mutagenesis to reconstitute enterobactin production activity.102 Three specific surfaces on the VibB-ArCP, which corresponded to those found previously on EntB–ArCP, were identified to be responsible for recognizing catalytic domains from the enterobactin system. Altogether, these results indicate that the PCP undergoes various conformational changes in order to interact in trans with its many catalytic partner domains.
The peptidyl carrier protein also interacts with other core domains in cis for proper production of natural product peptides. In the enterobactin system, the interdomain interaction between EntF–PCP and the thioesterase domain was examined by combinatorial mutagenesis.103 Residues G1027 and M1030, located in the helix III region of EntF-PCP, were identified to be key factors in recognizing the downstream TE I domain. This data and the results from the studies with EntB–ArCP and TycC3–PCP imply that mediation of domain interactions downstream of PCP are controlled by helix III, acting as a conformational switch. More recently, the solution structure of the EntF PCP–TE didomain elucidated the intra- and interdomain motions of the PCP during its interaction with other catalytic domains in NRP biosynthesis (Fig. 8).64
Fig. 8.
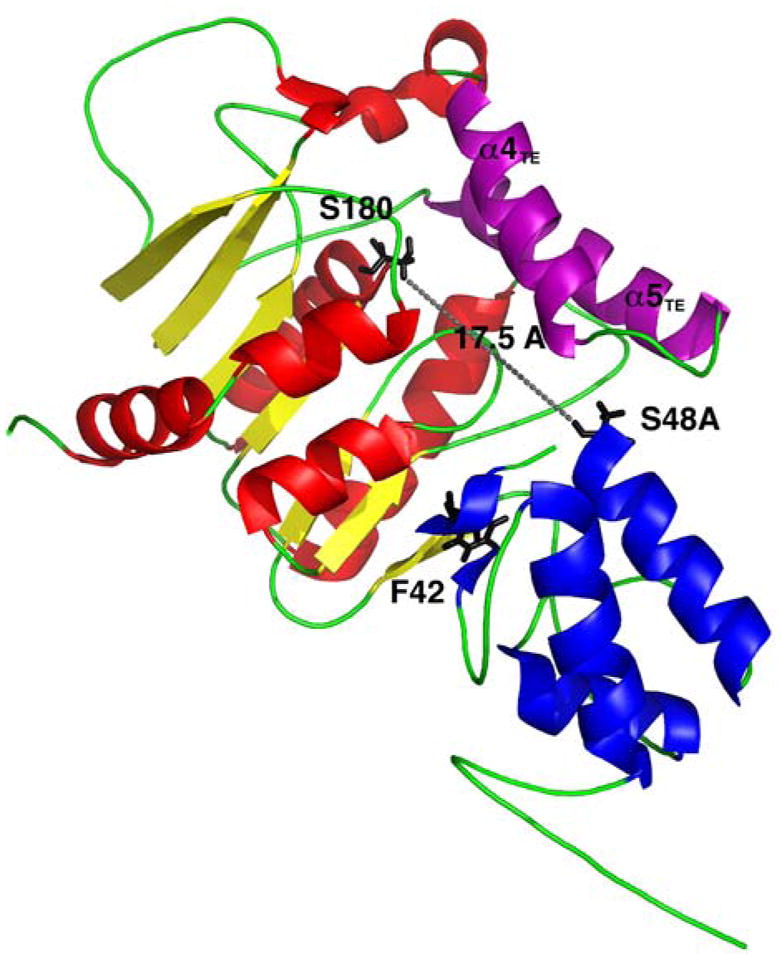
Structure of the PCP–TE didomain of the Escherichia coli enterobactin synthetase, EntF. Active site residues S180 of the TE domain (red and yellow) and S48A of the PCP (blue) are 17.5 Å apart. F42 stabilizes the interactions between the PCP and TE and helices α4TE-α5TE form the ‘lid’ region (purple).
Based on NMR analysis, the PCP domain exhibited a three-helical bundle wedged between the core of the TE and the two α-helices (α4TE-α5TE) protruding from this core. From the dynamics data in this study, it was established that these two α-helices formed a lid covering the active sites of both domains, which opens to allow access by the 4′-phosphopantetheine arm. The active sites of the PCP and TE domains were also found to be within 17 Å in this structure, enabling the 20 Å prosthetic arm to reach the active site of the TE domain. The PCP domain demonstrated internal mobility, leading to low intensity NMR signals for loop L3T of the PCP and fast NH exchange. Residue F42 was also shown, through mutagenesis, to play a key role in stabilizing the PCP fold to maintain the protein interactions with the TE domain. By utilizing other domains which interact in cis, such as the EntF C domain, and in trans, such as Sfp, with the EntF PCP domain, the dynamic interdomain motions were observed between the PCP and TE domains.
When Sfp was titrated with the PCP–TE didomain, new signals and chemical shifts were observed for the open form of the PCP–TE complex, indicating that the PPTase drives the dynamic equilibrium towards the dislodged form of the didomain. In addition, the EntF C domain was also titrated with the didomain and specific chemical shift changes were observed for the C-domain binding face of the PCP.
To gain further insight into the interdomain interactions that occur in cis between the PCP and other NRPS domains, crystal structures of the PCP were resolved within a bidomain in the tyrocidine system, TycC5–6 PCP–C,104 and an entire termination module of the surfactin NRPS, SrfA–C (Fig. 9).105 In both structures, the PCP was in the A/H state without the presence of phosphopantetheine. The structural arrangement of the PCP in the bidomain structure, however, was concluded to not be the peptide bond-forming conformation due to the large distance (47 Å) between the active site residues of the two domains, making it impossible for the phosphopantetheine arm (20 Å) to span this distance. Additionally, the critical residues necessary for productive interaction with the downstream C domain confirmed by the EntB–ArCP study,99 did not interact with the surface of the TycC6 condensation domain. The PCP conformation in the termination module, however, did bring the active site residues of the two domains within 16 Å, which would place phosphopantetheine at the acceptor site entry of the C domain. Likewise, the corresponding amino acids of the PCP domain, M1007 and F1027, which were critical for condensation activity in the EntB–ArCP study, formed hydrophobic interactions with F24, L28, and Y337 of the SrfA–C condensation domain, validating this structural orientation to be the peptide bond-forming conformation of the PCP.
Fig. 9.

(A) Crystal structure of the TycC5–6 PCP–C bidomain from the tyrocidine NRPS. The active site residues H224 of the C domain (red and yellow) and S43 from the PCP (blue) are positioned 47 Å apart. Residues (orange) responsible for proper interaction between the PCP and C domain are highlighted. (B) Crystal structure of the termination module from the Bacillus subtilis surfactin NRPS, SrfA–C. Linker regions (green) connecting the C domain (orange), A domain (gray and purple), PCP (blue), and TE domain (red) are indicated. Active site residues H147 of the C domain and S1003 from the PCP are 16.4 Å apart. Residues (black) determined to be responsible for proper interaction between the PCP and C domain are highlighted.
Recently, biochemical studies have characterized the interaction between the PCP and the adenylation domain.106 The study employed the use of a truncated A-PCP construct of the gramacidin initiation module GrsA. Both the apo- and holo-A-PCP constructs were subjected to an in-gel trypsin digest, a method previously used to study TycA.107 The digest of A-PCP with no ligand was compared with the digest when either its native substrates, ATP and L-Phe, or a phenylalanyl adenylate mimetic were added to the proteolysis reaction. A significant decrease in cleavage between the A domain and the PCP was observed when either ATP and L-Phe or the phenylalanyl adenylate mimetic were present, suggesting conformational changes upon substrate binding that increase domain-domain interaction. These findings were complemented by native PAGE and gel filtration analysis of the A-PCP. Using Texas Red bromoacetamide to detect the free sulfhydryl group of the phosphopantethine arm, it was discovered that the Ppant arm of A-PCP was less accessable in the presence of the phenylalanyl adenylate mimetic. These data suggested that both the phosphopantetheine modification and the binding of the substrates necessary for adenylation lead to conformational changes of the A and PCP domains required for adenylation activity.
5.3 Condensation Domain
Monomeric precursors in NRP synthesis are joined together by the condensation domain, a large ~450 aa monomeric enzyme situated at the N-terminus of each elongation module that catalyzes the peptide bond formation of two aminoacyl substrates bound to PCPs of adjacent modules.108 The thioester group of the upstream donor substrate undergoes a nucleophilic attack by the α-amino group of the downstream acceptor substrate, forming an amide bond and transferring the peptide intermediate from one module to the next (Fig. 10A). Based on the crystal structures of the stand-alone condensation domain, VibH,109 and the PCP–C bidomain, TycC5–6 PCP–C,104 the C domain is composed of an N- and C-terminal subdomain, arranged in a V-shape manner with the active site located at the junction of these subdomains (Fig. 10B). Initially, the second histidine of the ‘His-motif’ found in the C domain active site was thought to deprotonate the α-ammonium group of the acceptor substrate in order to attack the electrophilic carboxyl-thioester group of the donor substrate.110 However, based on mutational studies109 and analysis of pKa values for the catalytic residues,111 it was suggested that electrostatic interactions rather than general acid/base catalysis governs peptide bond formation in C domains. The crystal structure of VibH also revealed a solvent channel running through the active site, providing access from the N- and C-faces. These two faces act as the binding sites for the adjacent upstream and downstream PCPs, facilitating extension of the pantetheinyl arms into the solvent channel to present their corresponding substrates for catalysis. Biochemical investigations of different C domains from the tyrocidine synthetase revealed strict substrate stereoselectivity for the acceptor site (N-face) of the C domain.112,113 In contrast, the donor (C-face) site did not discriminate against noncognate amino acids. Thus, the C domain also acts as a filter for selectivity in NRP biosynthesis.
Fig. 10.

(A) Peptide bond formation catalyzed by the C domain. (B) X-ray crystal structure of the stand-alone C domain, VibH, from the Vibrio cholerae vibrioactin synthetase. The N-terminal (red) and C-terminal (blue) subdomains are connected by a linker region (purple), forming a V-shaped canyon. The ‘His’ motif (black), consisting of the catalytic residue H126, marks the active site, which is located at the junction of these two subdomains.
In addition to aminoacyl and peptidyl substrates, the C domain is capable of condensing polyketide (PK) intermediates in hybrid polyketide-nonribosomal peptide (PK/NRP) systems, as well as non-proteinogenic amino acids. These hybrid systems employ the C domain to catalyze the nucleophilic substitution between the acyl group of the polyketide intermediate bound to the ACP of the upstream module and the amino group of its aminoacyl substrate bound to the PCP of the downstream module (Fig. 11A). Some key examples include the systems for bleomycin,114 myxothiazol,115 rapamycin,116 and nostopeptolide.117 Furthermore, some of these hybrid enzymes also incorporate non-proteinogenic amino acids. For instance, rapamycin, FK506, and FK520 are hybrid peptide-polyketide immunosuppressant macrolides that contain a pipecolate moiety, derived from L-pipecolic acid, a nonproteinogenic, six-membered proline analogue (Fig. 11B).118,119,120 This amino acid is incorporated into the growing polyketide chain by a pipecolate-incorporating enzyme (PIE).118 In the biosynthetic pathways of rapamycin and FK520, the PIEs were determined to be RapP and FkbP, respectively. The first condensation domain of FkbP, the four-domain (C–A–PCP–C) NRPS found in the FK520 biosynthetic pathway, was proven to condense the pipecolate moiety with the acyclic polyketide chain of the natural product.121 Even though the condensation domain of hybrid PK/NRP systems can accept both nonproteinogenic amino acids and polyketide intermediates, it does not display any distinct characteristics compared to the conventional NRPS C domain. This is not surprising considering that the C domain from the tyrocidine system was previously reported to exhibit relaxed substrate specificity for its donor substrate.112 Fatty acids are incorporated at the N-terminus of several NRPs, including daptomycin122 and surfactin.123 Recent investigation of the surfactin biosynthetic pathway has shed light on the mechanism of fatty acid incorporation for these lipopeptides.124 In contrast to hybrid PK/NRP systems, the fatty acid is added by the initiation module, which has a domain organization of C-A-PCP. The C domain facilitates amide bond formation between the amino acid loaded on the PCP domain and a CoA activated fatty acid.
Fig. 11.

Different condensation reactions catalyzed by the C domain. (A) The C domain in the bleomycin NRPS subunit, BlmVII, condenses an aminoacyl substrate (blue) with a ketide unit (gold). (B) The C domain in the FK520 NRPS subunit, FkbP, catalyzes condensation between pipecolate (green) and a ketide unit (yellow). (C) The free-standing C domain, SgcC5, from the C-1027 NRPS catalyzes ester bond formation.
Besides forming peptide bonds between NRP precursors, the C domain also catalyzes ester bond formation, as first observed in the biosynthesis of fumonisin, a polyketide-derived mycotoxin produced by Fusarium verticillioides.125 In this study, FUM14 was predicted to encode a PCP and C domain. As a result, FUM14 deletion mutants were generated and analyzed. Extracts of the FUM14 mutants were found to produce only pre-fumonisin compounds, lacking the tricarballylic esters, suggesting Fum14p to be responsible for esterification of fumonisin. This result was further corroborated by expressing FUM14 in E. coli and performing in vitro assays with Fum14p to convert pre-fumonisin to fumonisin using tricarballylic thioesters. As expected, Fum14p catalyzed the esterification of the tricarballylic thioesters to C-14 and C-15 of the fumonisin backbone.
In the biosynthesis of C-1027, a stand-along C domain, SgcC5, catalyzes ester bond formation (Fig. 11C).126 Isolated from Streptomyces globisporus, the antitumor antibiotic C-1027 belongs to the enediyne family of antibiotics. Like all other enediynes, its mode of action begins with a cycloaromatization process via a Myers-Saito or Bergman-type rearrangement.127 This generates a benzenoid biradical, which can abstract hydrogens from DNA, resulting in oxygen-mediated DNA double-strand breaks.128 In previous studies, (S)-3-chloro-5-hydroxy-β-tyrosyl-(S)-SgcC2 was identifed as the PCP-bound donor substrate for SgcC5.129,130,131 Thus, it was employed with SgcC5 to test for ester bond formation with the enediyne core mimic, (R)-1-phenyl-1,2-ethanediol. Based on these findings, SgcC5 was reported to catalyze the regiospecific esterification of the C-2 hydroxyl group (corresponding to the C-14 of the enediyne core) of the acceptor substrate, substantiating its role in C-1027 biosynthesis. Additionally, using 3-chloro-5-hydroxy-β-tyrosine as a donor substrate instead of the PCP-bound substrate produced no esterified product, verifying that SgcC5 requires its donor substrate to be presented by a carrier protein. Specificity of SgcC5 toward acceptor substrates was also examined by checking for product formation with enediyne core mimics (R)-2-amino-1-phenyl-1-ethanol and (R)-2-phenylglycinol. By accepting the other enediyne core mimics, SgcC5 exhibited relaxed selectivity for its acceptor substrate, a quality differing from previously reported C domains, which displayed strict specificity for the acceptor substrate. Consequently, SgcC5 is the first known C domain to be able to catalyze both ester and amide bond formation in NRP biosynthesis, providing possible insight into how a canonical amide-forming C domain can be engineered to catalyze esterification.
6 Tailoring Domains
6.1 Epimerization Domain
One of the striking characteristics of nonribosomal peptides is the presence of D-amino acids, which give these natural products their unique conformations for biological activity.132 D-amino acids also serve functional roles, orientating the configurations of growing peptides for further modification by tailoring domains, as seen for penicillin and vancomycin. Incorporation of these unnatural amino acids can occur by either direct activation of a D-amino acid produced by an external racemase, via an A domain, such as the sixth A domain in the fusaricidin NRPS,133 or by in situ epimerization of the Cα center of the PCP-bound L-amino acid during peptide elongation. The latter is the more common route performed by the ~450 aa epimerization (E) domain, believed to be similar in structure as the C domain based on sequence alignments and secondary structure predictions.109 It was also discovered that dual function C and E domains incorporate D-amino acids into the biosurfactant arthrofactin along with other lipopeptides syringomycin, syringopeptin, and ramoplanin.134
Biochemical studies have been performed elucidating key catalytic residues, substrate specificity, and timing of epimerization. In a mutational analysis of the E domain in the initiation module of gramicidin S synthetase, the second histidine, H753, of the His-motif along with residues D757 and Y976 were deduced to be essential for proton transfer at the Cα center of the PCP-bound L-Phe to form D-Phe.135 In another study, noncognate substrates were found to be epimerized by the E domain, but with lower efficiency.136 In addition, it was uncovered that the E domain only catalyzes epimerization of the amino acid while it is tethered to the PCP and not in its free form, signifying the critical role played by the PCP for efficient catalysis. Prior to this study, it was demonstrated that the aminoacyl substrate in an elongation module is first condensed with the upstream peptidyl intermediate before being epimerized, whereas the E domain of an initiation module epimerizes its substrate before condensing with the downstream amino acid.137 Although the E domain found in initiation modules typically epimerizes aminoacyl substrates, it was reported to be able to also epimerize peptidyl intermediates. In contrast, the E domain in an elongation module was not as tolerant and preferred peptidyl intermediates over aminoacyl substrates.138 Apparently, there was no correlation between the substrate specificity of E domains and the specificity of the module they arose from. However, in a follow-up study of the two different E domains,139 it was determined that the E domain does impact the intermodular transfer of correct intermediates, making certain that misinitation of certain D-isomers does not occur.
Recently, the structure of an epimerization domain of the initiation module of tyrocidine biosynthesis was solved (Fig. 12).140 This initiation module is responsible for the loading of Phe and subsequent epimerization to the D isomer for further downstream biosynthetic steps. It is important to note that this is the first structural data of an epimerization domain. Along with previous biochemical studies, this structure will aid in better understanding the mechanism, selectivity, and intermodular interactions of the E domain.
Fig. 12.
Crystal structure of the Epimerization domain from Tyrocidine synthetase A (TycA).
6.2 Heterocyclization and Oxidation/Reduction Domains
Another structurally distinctive feature in nonribosomal peptides, such as vibriobactin and anguibactin, is the presence of heterocyclic rings, namely oxazolines and thiazolines. These 5-membered rings, derived from either cysteine (thiazoline) or serine/threonine (oxazoline), are incorporated into the natural product by a cyclization (Cy) domain through three chemical steps (Fig. 13A). First, the amino acid containing the β-nucleophile side chain condenses with the upstream activated acyl donor group. Then, the newly formed amide bond undergoes an intramolecular nucleophilic attack by the cysteine thiol or serine/threonine hydroxyl group, forming the 5-membered ring. Finally, dehydration gives the final thiazoline/oxazoline ring observed in antibiotic bacitracin and siderophore mycobactin, respectively.
Fig. 13.

Enzymatic reactions of the tailoring domains. (A) The cyclization (Cy) domain from the Vibrio cholerae vibriobactin NRPS subunit, VibF, catalyze cyclization of threonine to form the oxazoline ring in three steps. (B) The oxidation (Ox) domain from the epothilone synthetase B, EpoB, oxidizes the thiazoline ring to the thiazole in the presence of the cofactor flavin mononucleotide (FMN). (C) The reduction (R) domain from the pyochelin synthetase, PchF, reduces the thiazoline ring to the thiazolidine in the presence of NADPH.
Upon closer examination of the substrate selectivity of the Cy domain, it was discovered that the Cy domain exhibits strong specificity for its donor substrate in regards to its condensation activity as illustrated by the tandem Cy domains found in VibF.141 In addition, the specificity for its acceptor substrate for the heterocyclization reaction showed tolerance only for β-functionalized amino acids serine and cysteine. Interestingly, the catalytic efficiency of both reactions increased when the substrates were loaded on to a PCP, verifying yet again the vital role played by the PCP in NRP biosynthesis.
Although the Cy and C domains share significant structural and functional homology, heterocyclization and condensation reactions are performed independently within the Cy domain. One of the early indications for this separation of condensation and heterocyclization processes was demonstrated by the mutational study of the two different Cy domains found in the VibF NRPS of the vibriobactin system.142 Given the conserved DXXXXDXXS motif of Cy domains,143 mutation of the catalytic aspartic acid residues in the first Cy domain of VibF resulted in lower heterocyclic product formation while maintaining condensation activity, whereas the same mutation in the second Cy domain had the opposite effect. As a result, the second Cy domain was designated the role of the condensation reaction and the first for heterocyclization. Reaffirmation of this distinction between the Cy and C domains was demonstrated in the mutational analysis of the Cy domain found in the bacitracin system.144 Mutants N900A and S984A of the model system consisting of the first two modules of bacitracin synthetase A fused to the TE domain of tyrocidine synthetase, BacA1–2–TE, produced linear dipeptides, indicating that these residues were critical for heterocyclization but not condensation activity. From secondary structure predictions and comparison with the crystal structure of the C domain, VibH, N900 and S984 were determined to be involved in the formation of the Cy solvent channel, indicating a structural role for these residues in the heterocyclization reaction.
Discussion of the Cy domain so far has been limited to those found associated with other NRPS domains. However, a recent study of the NRPS responsible for the biosynthesis of anguibactin, a siderophore produced by Vibrio anguillarum,145 reported the first system to comprise of two stand-alone Cy domains. Based on mutational analysis of the conserved aspartic residues in the Cy domain core motif, both Cy domains were established to be responsible for siderophore production. Sequence alignment of the second Cy domain with the second Cy domain of VibF revealed 32% homology and the residues identified to be important for cyclization but not condensation were not conserved in either of the second Cy domains. However, designation of heterocyclization and condensation reactions for each Cy domain could not be confirmed based on the in vivo studies alone. Only with a detailed structural study tied in with mutational analysis of the catalytic residues of the Cy domain will the elucidation of the mechanistic features involved in heterocyclization and condensation be resolved.
Following heterocyclization, the thiazoline/oxazoline can either be oxidized to the thiazole/oxazole, as observed in thiostrepton and epothilone, by the oxidation (Ox) domain146 (Fig. 13B) or reduced by the reduction (R) domain to form the thiazolidine/oxazolidine, as seen in pyochelin (Fig. 12C).147 In the former process, flavin mononucleotide (FMN) is used as a cofactor for the two electron oxidation reaction, whereas the latter path is dependent on NADPH. At present, there are no crystal structures for either the Ox or R domains, but biochemical studies have revealed the Ox domain to be located either downstream of the PCP or incorporated into the C-terminal portion of the A domain.148 Moreover, the R domain of the MxcG synthetase was found to be responsible for the release of the final peptide product, myxochelin A, through a four-electron reduction of the PCP-bound thioester.149
6.3 N- and C-Methyltransferase Domains
Many of the nonribosomal peptides seen in Figure 2 consist of N-and C-methylated amino acids, which are responsible for the natural product’s bioactivity and provide the structural conformation for subsequent reactions in the biosynthetic pathway. Accountable for methylation of these amino acids is the methyltransferase (MT) domain, which transfers the methyl group from its cosubstrate (S)-adenosyl methionine (SAM). Typically, N-methylation of the amino acid occurs while it is tethered to the 4′-phosphopantetheine arm of the PCP by the ~420 aa N-MT located at the C-terminal of the associated A domain, as observed in the cyclosporin150 and pyochelin synthetases (Fig. 14A).151 However, N-methylation of external substrates, which are not bound to the PCP of the associated module, takes place as well. For instance, the N-MT of the enniatin synthetase from Fusarium scirpi152 was able to N-methylate aminoacyl-N-acetylcysteamine thioesters (aminoacyl-SNACs) of L-Val, L-Ile, and L-Leu. It also exhibited enantioselectivity for the L-isomers of the amino acids and mutational analysis established the functional boundaries of the domain.153
Fig. 14.

N- and C-methylation of amionacyl substrates in NRP biosynthesis. (A) The N-methyltransferase (NMT) domain from the pyochelin NRPS, PchF, transfers a methyl group from S-adenosyl methionine (SAM) to the amine group of the substrate tethered on the PCP. (B) Crystal structure of NMT, MtfA, from the chloroeremomycin synthetase complexed with SAM (purple stick model) with one monomer designated in gray. (C) The C-methyltransferase (CMT), GlmT, from the CDA producer Streptomyces coelicolor transfers the methyl group from SAM to the β-carbon of α-ketoglutarate in a stereospecific manner.
In addition to in cis-acting N-MTs that are linked to their associated module, discrete 234–280 aa N-MTs, such as MtfA from the chloroeremomycin synthetase, interact in trans to methylate aminoacyl and peptidyl substrates.154 The recently solved crystal structure of MtfA depicted the protein as a dimer with two wing-like structures extending from the main body of the protein (Fig. 14B).155 With 9 α-helices and 11 βsheets, the bulk of the protein folded into an extended Rossman fold domain with the cofactor, SAM, bound near the β2/α3 loop, in which the characteristic SAM-binding motif ExAxGxG was located. Mutational analysis determined H228 to be the base residue required for catalysis and molecular modeling illustrated that the antibiotic substrate binds in a cleft located at the dimer interface.
Despite its similar role in nonribosomal peptide synthesis as N-MTs, C-MTs do not directly methylate PCP-tethered amino acids. Instead, it has been demonstrated that these enzymes methylate precursors leading to the final nonproteogenic amino acid.156 For instance, the acidic lipopeptides, daptomcyin and calcium-dependent antibiotics (CDA), represent a class of antibiotics which contain βmethylated glutamate, produced by these C-MTs. The methyltransferase, GlmT, found in the CDA producer S. coelicolor catalyzes the SAM-dependent stereospecific methylation of α-ketoglutarate to (3R)-3-methyl-2-oxoglutarate (Fig.14C).157 Then, in the presence of an excess of valine, acting as the amino group donor, the branched chain aminotransferase, IlvE, transforms 3-methyl-2-oxoglutarate to 3-MeGlu, the nonproteogenic amino acid that is incorporated into the final lipopeptide. With the mechanism of the methylation performed by GlmT uncovered, methyltransferases DptI from the daptomycin producer S. roseosporus and LptI from the A5145 producer S. fradiae were examined due to their high homology with GlmT. The two homologues were assayed and determined to catalyze the same methylation reaction as GlmT.
6.4 Formylation Domain
Commonly observed in the initation of ribosomal peptide biosynthesis in prokaryotes,158 N-formylation also occurs in the biosynthesis of linear gramicidins from Bacillus brevis ATCC 8185,159 as well as the anabaenopeptilides from Anabaena strain 90.160 Responsible for catalyzing this reaction is the formylation (F) domain, one of the least explored tailoring domains in the NRPS system. In 2006, the small ~24 kDa F domain of module LgrA1 was determined to formylate the first residue of linear gramicidin, valine, in the presence of the cofactor N10-formyltetrahydrofolate (fH4F) (Fig. 15).161 Utilizing a formylation assay with LgrA1, it was shown that the F domain exhibited strict substrate selectivity for its PCP-bound substrate valine. In addition, an elongation assay with the truncated dimodular system of LgrA F–A1– PCP–C–A2–PCP demonstrated that only formyl-valine and not valine condensed with glycine, the second amino acid of the linear gramicidin, signifying the importance of the formylation reaction on the initation of linear gramicidin biosynthesis. The inability of the A domain of LgrA1 to accept formyl-valine as a substrate further supported the necessity of the F domain for initiation as well as demonstrating the critical role played by the 4′-phosphopantetheine arm of the PCP domain in positioning the substrate for formylation.
Fig. 15.
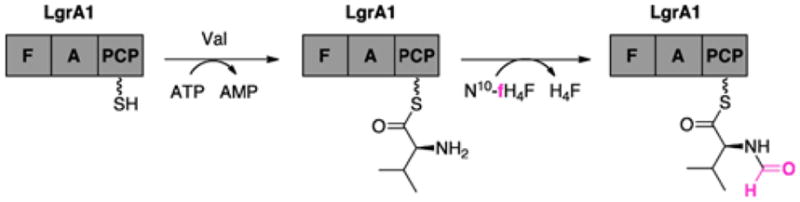
The formylation (F) domain of the Bacillus brevis linear gramicidin NRPS subunit, LgrA1, catalyzes formylation of valine in the presence of N10-formyltetrahydrofolate.
6.5 Halogenase Domain
With over 4,000 halogenated natural products reported to date,162 our understanding of the structural and mechanistic features of halogenating enzymes has grown considerably in the past decade. At present, five different classes of halogenating enzymes have been uncovered, with two of them being responsible for the incorporation of halogens into nonribosomal peptides. They are the flavin-dependent halogenases and non-heme iron-dependent halogenases. Halogenation of nonribosomal peptides within their electron-rich aromatic or heteroaromatic ring systems, as seen in the antibiotics balhimycin and pyoluteorin, are executed by the flavin-dependent halogenases.163 The non-heme iron-dependent halogenases are responsible for halogenating nonribosomal peptides at unactivated aliphatic carbon centers, as observed in the phytotoxin syringomycin E and the molluscicidal compound barbamide.164 As a result of the halogen’s presence, the therapeutic potency of the natural products is noticeably greater. For example, balhimycin exhibits an 8- to 16-fold increase in antibiotic activity towards certain pathogenic bacteria and syringomycin E shows a 3-fold increase in antifungal activity due to the presence of a halogen.
Based on enzymatic and structural studies, the flavin-dependent halogenases have been categorized into two groups: those that halogenate free small-molecule substrates (PrnA and RebH)165 and those that modify substrates bound to the PCP in the NRPS system (CndH and SgcC3).166 Until recently, only two crystal structures of the flavin-dependent halogenases involved in NRPS systems had been resolved, PrnA from Pseudomonas fluorescens167 and RebH from Lechevalieria aerocolonigenes.168 Both of these halogenases belong to the first group and have aided in the elucidation of the reaction mechanism and structural features for flavin-dependent halogenation. In general, these ~500 aa flavin-dependent halogenases consist of two conserved regions, a flavin-binding site (GxGxxG) near the N-terminus and a second conserved motif (WxWxIP) situated in the middle of the enzyme.169 They require reduced flavin (FADH2), which is provided by an NADH-dependent partner reductase, molecular oxygen, and a chloride ion.170 Currently, the proposed mechanism for flavin-dependent halogenation begins with reduced flavin reacting with O2 to form a FAD–C4a–OOH intermediate (Fig. 16A). The intermediate reacts with Cl−, oxidizing it to Cl+, to produce hypochlorous acid (HOCl).171 Based on the most recent finding, HOCl then reacts with conserved active site residue K79 to form lysine chloramine K79–εNH2–Cl,172 which acts as the chlorinating agent for the substrate. This is based on the observation that K79 is located between the flavin and tryptophan binding site, and the fact that HOCl is known to react with the εNH2 of lysine.173 Upon chlorination of the aromatic substrate, a Wheland complex is formed, which is then deprotonated by a conserved basic E residue to form the halogenated aromatic product.
Fig. 16.

(A) Proposed mechanism of flavin-dependent halogenases. (B) Crystal structure of the chondrochloren halogenase, CndH (red and yellow), from the myxobacterium Chondromyces crocatus Cm c5 complexed with FAD (purple) and in the presence of Cl− ion (blue sphere). Active site residue K76 reacts with HOCl to form the chlorinating reagent and E387 (black) acts as the base to complete halogenation.
This mechanism also holds true for the second group of flavin-dependent halogenases, which utilize a carrier protein bound substrate. However, there are some slight differences between the two groups, both in structure and mechanism. Based on the crystal structure of the chondrochloren halogenase (CndH) from the myxobacterium Chondromyces crocatus Cm c5, the 512 amino acid enzyme differs from the halogenases PrnA and RebH in that it lacks the 45 residue segment near position 100, which is near the active site center, and deviates in the C-terminal domain (Fig. 16B).174 Instead of covering the active center of CndH, as is the case for PrnA, the C-terminal domain is disordered in the CndH crystal structure, leaving the active site in the open conformation. As a result, this creates a large non-polar surface patch, which is believed to interact with the carrier protein domain. In looking at the active site of CndH, the conserved lysine (K76) is present, but the glutamine residue that deprotonates the Wheland complex is missing. It was suggested that the base needed to complete the halogenation reaction may be provided by a residue at its C-terminal domain (E387) or by a more likely candidate, a residue from the carrier protein.
Halogenation of unactivated aliphatic carbon centers in nonribosomal peptides are catalyzed by the recently discovered class of non-heme Fe(II) and α-ketoglutarate (αKG)-dependent halogenases.175 Thus far, four of these halogenases have been characterized, with the crystal structures for two of these halogenases being resolved: SyrB2 from the syringomycin E biosynthetic pathway176 (Fig. 17A) and more recently, CytC3 from the NRPS of the Streptomyces antibiotic, γ,γ-dichloroaminobutyrate (Fig. 17B).177 Non-heme Fe(II)/αKG-dependent halogenases exhibit a common cupin fold, comprised of antiparallel β–strands in a jelly roll motif.178 At the center of the jelly roll is the iron cofactor, which is coordinated by two histidine residues. Molecular oxygen, αKG, and the chloride coordinate with the iron to carry out the halogenation reaction.179 First, molecular oxygen binds to the iron center causing oxidative decarboxylation of αKG, forming an Fe(IV)-oxo intermediate, which is proposed to be responsible for hydrogen abstraction of the substrate. Then the substrate radical reacts with Cl• to form the chlorinated product (Fig. 17C).180 Comparison of the open conformation (chloride is not bound to iron) observed in the crystal structure of CytC3 with the closed conformation (chloride bound to iron) of SyrB2 revealed two important factors affecting chloride binding in this class of halogenases: the hydrogen-bonding network between the chloride and the surrounding enzyme residues and the hydrophobic pocket of the chloride binding site. In SyrB2, residues N123, T143, and R254 form hydrogen bonds with the chloride through water molecules, stablizing the halogen within the active site (Fig. 17A). In contrast, the corresponding residues in CytC3 are too far from the chloride binding site to form this same hydrogen-bonding (Fig. 17B). Furthermore, residues A118, F121, and the β-carbon of S231 of SyrB2 form a large hydrophobic pocket (absent in CytC3) in which the chloride resides and is believed to be important for binding.
Fig. 17.

Structure and mechanism of αKG-dependent halogenases. (A) Crystal structure of the halogenase domain, SyrB2, from the syringomycin E synthetase complexed with αKG (purple) in the presence of Fe(II) ion (blue sphere). Residues N123, T143, and R254 (orange) form hydrogen bonds with the Cl− ion (green sphere) and residues A118, F121, and S231 (gray) form the hydrophobic pocket. (B) Crystal structure of the halogenase domain, CytC3, from the γ,γ-dichloroaminobutyrate synthetase complexed with αKG (purple) in the presence of Fe2+ ion (blue sphere). Corresponding residues from SyrB2 are indicated as well. (C) Proposed mechanism of non-heme Fe(II)/αKG-dependent halogenases.
7 Thioesterase Domain
Termination of nonribosomal peptide biosynthesis is carried out by the thioesterase (TE) domain with the release of the full-length peptidyl chain from the final PCP of the termination module.181 This 250 amino acid long enzyme, which is only found in termination modules of NRPSs,182 utilizes an active site serine residue as a nucleophilic catalyst in the two-step process liberating its peptide cargo. To initiate catalysis, the active site serine carries out a nucleophilic attack of the PCP-bound peptide thioester to form an acyl-O-TE intermediate.183 Immediately following, this attack, the peptide chain can undergo either hydrolysis to produce a linear peptide, such as vancomycin, or macrocyclization by attack of an internal nucleophile to form a macrocyclic peptide, such as surfactin or daptomycin (Fig. 18). Based on the structures of typical nonribosomal peptides,184 it is clear that the latter pathway is the most commonly observed mechanism for peptide release, given that the structural constraint of the cyclic peptide provides resistance to proteolytic degradation and enhanced bioactivity. Although the TE domain performs a single process in nonribosomal synthesis, it provides a diversity of different size macrocycles, suggesting a high degree of specialization for catalyzing cyclization and substrate specificity.
Fig. 18.

(A) The thioesterase (TE) domain from the vancomycin NRPS catalyzes release of the linear peptide through hydrolysis after three crosslinking reactions. (B) The TE domain from the daptomycin NRPS catalyzes the release of the peptidic product through an intramolecular macrocyclization.
The first look into the structural conformation of the TE domain was provided by the crystal structure of the surfactin TEI domain (Fig. 19).185 With the exception of an insertion “lid” region consisting of three α-helices covering the active site, the SrfTEI domain exhibits similar structural features as observed for members of the α/β-hydrolase family. Located centrally at the bottom of a bowl-shaped hydrophobic cavity, the catalytic triad of residues S80, H207, and A107 marks the active site of the SrfTEI domain. The backbone amides of A81 and V27 form the oxyanion hole which stabilizes the tetrahedral intermediate during acyl-O-TE formation. Based on sequence alignments, the nonconserved “lid” region differed greatly from other TE domains, suggesting that this region was responsible for substrate recognition of the SrfTEI domain. Moreover, two monomers were observed for the crystal structure of SrfTEI, each with the lid in different conformations. In the ‘O’ (open) monomer of the SrfTEI domain, the lid is flipped back allowing access to the active site, whereas in the ‘C’ (closed) monomer, the lid is closed excluding water and permitting only small molecules entry. Active site residues K111, R120, and P26 were found essential in preventing hydrolysis of the lipoheptapeptide.186 It was also determined that the side chains of C-terminal residues D-Leu6 and Leu7 of the lipopeptide surfactin bound specifically within well-defined hydrophobic pockets of the active site in order for macrocyclization.
Fig. 19.
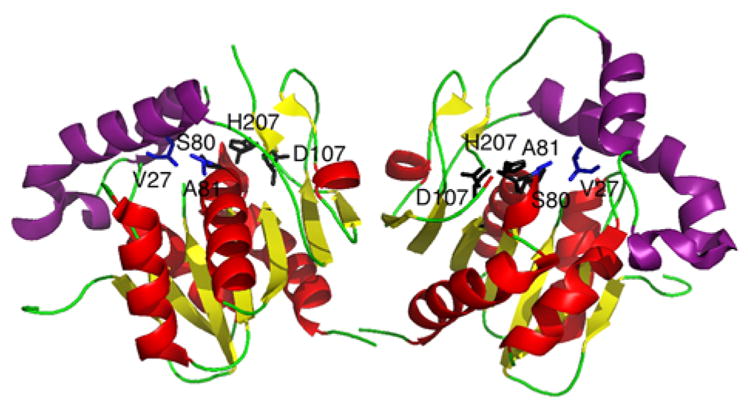
Crystal structure of the surfactin thioesterase domain, SrfTEI, depicted as an asymmetric dimer. The monomer on the left is in the closed ‘C’ conformation with the ‘lid’ region (purple) covering the active site, while the other is in the open ‘O’ conformation with the ‘lid’ flipped back. Residues S80, D107, and H207 (black) form the catalytic triad and A81 and V27 (blue) form the oxyanion hole.
Similar in structure to SrfTEI as well as other members of the α/β-hydrolase family, the fengycin TE domain (FenTEI) also bears a catalytic triad for substrate binding and catalysis of macrocyclization, composed of residues S84, D111, and H201 (Fig. 20).187 These catalytic residues are located at the bottom of the central region of the crevice-like active site. Likewise, the backbone amide groups of A85 and I30 formed the oxyanion hole which stabilizes the tetrahedral intermediate in substrate binding. Depicted as an α/βsandwich with a central β-sheet, the FenTEI structure differs from the SrfTEI domain structure in the lid region. Twelve residues shorter than the SrfTEI lid region, the corresponding region in FenTEI domain lacks an α-helical element, αL1, which is responsible for blocking parts of the active site in the SrfTEI. As a result, the FenTEI domain was observed only in the open state conformation, defined earlier in the SrfTEI study, suggesting that this lid region may not have any effect in substrate recognition and only participate in preventing access of unwanted macromolecular substrates. However, the idea that the FenTEI may exist in a closed state in solution cannot be excluded due to the crystallization conditions utilized in this study. With the lid region being excluded as a crucial player in substrate recognition for the FenTEI, modeling, molecular dynamics and enzymatic assays were utilized to examine the orientation of the substrate and to determine the factors affecting its binding. Based on these investigations, the cyclic portion of the fengycin peptide was observed sitting “edge-on” the active site canyon with residues D-Orn3, L-Gln9, D-Tyr10, and L-Ile11 contributing to substrate binding and recognition during cyclization, in accordance with earlier findings on the substrate specificity of NRPS TE domains.185 In addition, the molecular dynamics of FenTEI placed the C-terminus of the lipopeptide in contact with the western side of the active site canyon and the nucleophilic residue interacting with the eastern and central regions of the active site.
Fig. 20.
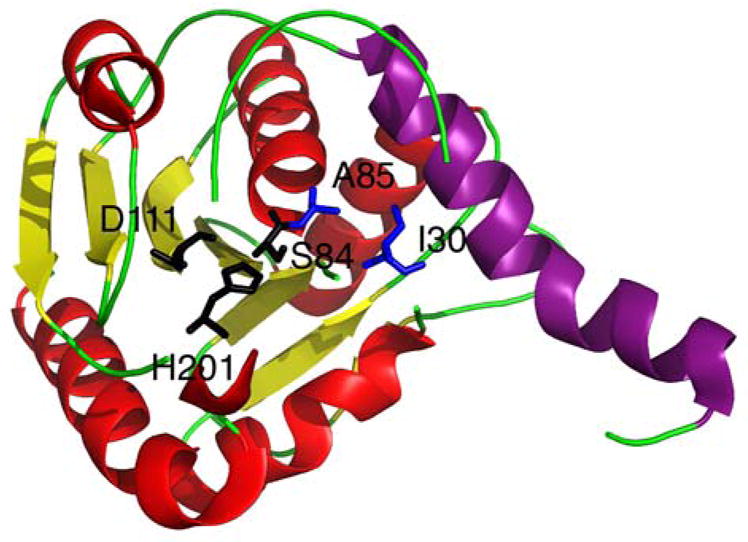
Crystal structure of the fengycin thioesterase domain, FenTEI. The ‘lid’ region (purple) is flipped back designating FenTEI in the open conformation. Residues S84, D111, and H201 (black) form the catalytic triad and A85 and I30 (blue) form the oxyanion hole.
In contrast to most TEI enzymes, such as SrfTEI and FenTEI, which are located at the C-terminus of the termination module, the TEI enzyme involved in the biosynthesis of phosphinothricin tripeptide (PTT), a peptide antibiotic produced by S. viridochromogenes Tü494, is located at the N-terminus of the first synthetase, PhsA, of the PTT system.188 To determine if this TE domain was required for PTT production, a phsA deletion mutant (MphsA) was complemented with two different phsA constructs carrying mutations in the thioesterase motif. In one construct, the conserved active site serine residue of the TE GXSXG motif was changed to an alanine, which would abolish TE activity. Complementation of MphsA with this construct did not restore PTT production, suggesting that this TE domain is required for release of the peptide antibiotic. Consequently, the PCP domains of the other two synthetases, PhsB and PhsC, in the PTT biosynthetic pathway must be arranged in a manner so that they can interact with this TE domain. This is the first example of an NRPS organized in a cyclic manner, in which the synthesis begins and ends with the same module.
Recently, a prototypical TE was discovered to catalyze both macrolactonization and macrothiolactonization (Fig. 21).189 The TioS PCP–TE bidomain, excised from the thiocoraline NRPS of Micromonospora ML1,190 was examined for substrate selectivity and utilized for in vitro generation of thiocoraline analogs. At first, linear tetrapeptide thioester substrates were employed to establish the characteristics of the individual amino acids within the substrate that regulate the cyclization mechanism of the TE domain. The steric demand of the C-terminal amino acid was proven to suppress hydrolysis by protecting the acyl-O-TE oxoester intermediate from the nucleophilic attack of water. The cysteine residue at the third position of the tetrapeptide also exhibited a strong influence on the ligation and cyclization reaction of the TE by forming a disulfide crossbridge prior to cyclization, orientating the substrate in a fold to facilitate macrocyclization. This finding was in accord with the backward mechanism proposed for iteratively working thioesterases, where the PCP acts as a docking port for dimerized products.191 In addition, the D-configuration of the N-terminal amino acid was found to be crucial for cyclization because it creates the Burgi-Dunitz trajectory for an intramolecular nucleophilic attack of the acyl-O-TE oxoester intermediate.192 Interestingly, the cyclization reaction was determined to be temperature-dependent, suggesting that the production of thiocoraline is thermally regulated. It was speculated that at lower temperatures the enzyme becomes more compact, reducing premature hydrolysis and increasing the stability of the PCP bound substrate intermediate for intramolecular macrocyclization.
Fig. 21.
The thioesterase domain from the thiocoraline synthetase, TioS, catalyzes macrothiolactonization.
8 Intermodular Protein Interactions
Throughout this review, investigations of the different catalytic domains have emphasized the importance of intramodular protein-protein interactions in regulating NRP biosynthesis. However, intermodular protein interactions also play a vital part in governing proper processing of peptide intermediates. The following two sections will discuss the discovery of the domains responsible for the intermodular protein interactions which have advanced our understanding and ability to manipulate the NRP synthetases in order to produce more potent bioactive peptidic compounds.
8.1 COM Domains
Initially, it was believed that condensation between peptide intermediates of consecutive NRPS enzymes was mediated by protein-protein interactions between the C-terminal domain of the first synthetase and the N-terminal domain of the second synthetase. However, small 15–25 aa recognition regions called communication-mediating (COM) domains were determined to be important for regulating the intermodular protein interactions between NRPSs.17 Located at the C-terminus of the donor NRPS, the donor COM domain (COMD) interacts specifically with its cognate partner, the acceptor COM domain (COMA), which is situated at the N-terminus of the acceptor NRPS (Fig. 22A). The two COM domains form a compatible set, ensuring selective protein interactions in order to condense the appropriate peptide intermediates in NRP biosynthesis. Based on COM domain-swapping studies,13,18 it was shown that these domains could be manipulated both in vitro and in vivo to generate new peptide intermediates, demonstrating great potential for further biocombinatorial methods.
Fig. 22.

(A) COM domains from the tyrocidine system regulate the protein interactions between the first two modules, TycA and TycB1, for proper processing of the aminoacyl substrates, L-Phe and L-Pro, to form the dipeptidyl intermediate. (B) Pantetheine azide and difluorocyclooctyne pantetheine modified the PCPs of TycA and TycB1, respectively, and displayed sensitivity to the protein interactions governed by the COM domains of these cognate partner NRPSs through crosslinking.
Examination of the crystal structure of the surfactin termination module, SrfA-C, provided tentative structural analysis of the COM domains.105 An unexpected interaction was observed between the C-terminal myc-his6 tag of the SrfA–C module and the N-terminal condensation domain of a neighboring SrfA–C module. A sequence alignment comparison between the C-terminus of the upstream module SrfA–B and the C-terminus myc-his6 tag of the SrfA–C showed high homology, suggesting the C-terminal myc-his6 tag accurately represents the COMD domain of SrfA–B. Interestingly, a three-stranded β-sheet, in conjunction with the previously defined COMA domain of SrfA-C,193 formed a ‘COM hand’ that provides a docking site for the myc-his6 tag (Fig. 23), in contrast to the four-helix bundle of docking domains found in PKSs.194 The ‘COM hand’ motif suggests that COM domain docking is more complex than previously thought.
Fig. 23.
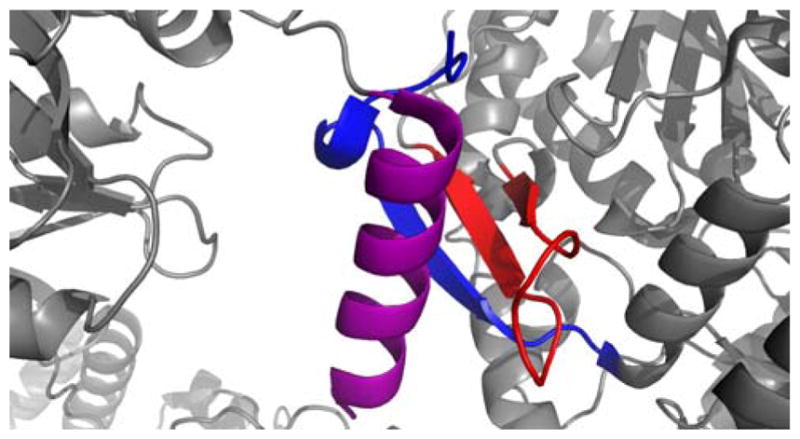
Ribbon diagram depicting the COM hand interaction between two SrfA-C monomers. The myc-His6 tag (purple) interacts not only with the putative COMA sequence (blue) of SrfA-C, but also two additional βsheets (red) located on the C domain.
More recently, crosslinking probes were developed to investigate the protein-protein interactions mediated by the COM domains in the tyrocidine NRPS system.195 Utilizing a one-pot chemoenzymatic carrier protein modification method,196 synthesized pantetheine azides and alkynes were loaded on to the PCP of the first two modules of the tyrocidine system, TycA and TycB1. Then through a crosslinking assay, these bioorthogonally-tagged modules were examined for crosslinking through gel-shift SDS-PAGE analysis. Based on the results, the protein interactions between the COM domains of TycA and TycB1 were too transient to catalyze the in situ [3+2] cycloaddition of the PCP-modified azides and alkynes. However, a ring strain-activated cyclooctyne pantetheine analogue exhibited selective protein-protein interaction-dependent crosslinking of cognate NRPS modules (Fig. 22B). This technique provides a method for covalently linking two interacting NPRS modules, which allows for structural analysis and elucidation of important module-module interactions.
8.2 Docking Domains in Hybrid PKS/NRPS Systems
With the discovery of hybrid NRP–PK natural products, exploration of the docking domains governing the protein-protein interactions between NRPS and PKS enzymes in hybrid systems became a necessity in order to comprehend the interplay between these two types of modular synthases for combinatorial biosynthesis. Earlier studies of the mixed PKS–NRPS epothilone system illustrated that manipulation of the docking domains between its synthases was plausible; however, this came at the expense of decreased chain elongation efficiency.197,198 Despite the conserved architecture and catalytic function shared by these synthases, it is surprising to find that the docking system used in hybrid systems are quite different from those in purely PKS or NRPS systems.
In a recent structural study of the tubulysin system of Angiococcus disciformis An d48, a new family of N-terminal docking domains was identified.199 Based on multiple sequence alignments of mixed PKS–NRPS systems, TubCdd, the N-terminal docking domain of TubC, was established to be a representative of the N-terminal docking domains found between NRPS–NRPS and PKS–NRPS interfaces in these hybrid systems. Structural analysis of TubCdd by NMR revealed this domain to be homodimeric, suggesting that NRPS enzymes in hybrid systems self-associate in order to recognize and interact with C-terminal PKS partner domains. Likewise, a new αββαα fold was elucidated for this domain with an exposed β-hairpin, comprised of charged residues that make up a docking code responsible for proper protein-protein interaction with its cognate partner docking domain (Fig. 24).
Fig. 24.
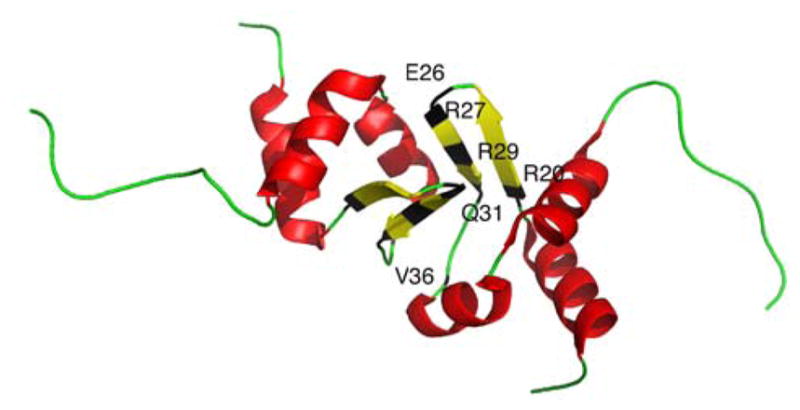
Ribbon diagram of the tubulysin docking domain, TubCdd (red and yellow). A new αββαα-fold is displayed with residues R20, E26, R27, R29, Q31 and V36 (black) making up the docking code.
9 Summary and Outlook
This review summarizes the key investigations and remarkable breakthroughs in the structural and mechanistic elucidation of the catalytic domains engaged in NRP biosynthesis. The knowledge we have attained from these studies have significantly broadened our understanding of the distinct structural elements as well as the dynamic interactions mediating the enzymatic processes of these large multidomain assembly lines. Given the modular organization of NRPSs, it is clear that defining the intra- and intermodular protein interactions governing proper processing of NRP precursors is vital to the comprehension of such intricately designed molecular machines. As a result, many of the endeavors discussed in this review have focused on resolving the issues regarding the selective communication between NRPS enzymes.
In particular, mutational, NMR, and X-ray crystallographic studies have identified key residues, facilitating proper protein-protein interactions, at the recognition interfaces of several NRPS subunits. Given the central role of the PCP, many of these endeavors focused on defining the architectural determinants regulating the interdomain communication between the PCP and its partner proteins. From the mutational study of the EntB–ArCP, specific recognition features on helix 3 of the ArCP were determined to regulate proper interactions in trans with the C domain of its cognate partner module, EntF, for peptide bond formation. Interestingly, the corresponding surface residues on helix 3 of the PCP from the SrfA–C termination module were also resolved to govern proper interactions in cis with the C domain in this termination module. Investigations of the interactions that occur in trans between TycC3–PCP and Sfp, as well as Srf–TEII revealed that each of these external enzymes interact with different regions of helix 3 of the PCP. NMR solution structures of TycC3–PCP demonstrated that helix 3 undergoes structural transformations as the PCP goes from its apo form to its holo form as it interacts with these different enzymes. On account of this structural flexibility and the critical role it plays in governing the interactions between the PCP and its partner enzymes, investigating the dynamic conformational changes of helix 3 of the PCP will be essential for elucidating other interdomain interactions with the PCP.
Another critical issue regarding NRP synthesis highlighted in this review is the identification of the active site residues governing substrate specificity of the domains. For instance, we now recognize the physico-chemical and thermodynamic forces discriminating against unwanted substrates of the A domain as well as the TEI domain from structural and mutagenesis studies of PheA, DhbE, DltA, FenTEI, and SrfTEI. Using this data, tools that enable the alteration of A domain specificity are currently being developed and optimized that could lead to tailor-made enzyme systems.
In addition, the strict substrate stereoselectivity of the C domain at its acceptor site along with the binding pocket residues of the halogenases essential for accomodating its substrate and cofactors have also been established. However, there still remains much to be learned about the dynamic interactions of these halogenases with the PCP. Moreover, with the discovery of SgcC5, further investigations exploring the mechanistic and structural aspects of the this C domain holds great promise into gaining a better understanding of the selectivity for a hydroxy or amine nucleophile of the acceptor substrate in the condensation reaction of NRP synthesis. Although the characterization of the SrfTEI and FenTEI domains elucidated certain fundamental features involved in macrocyclization, further investigations into other NRPS TE domains is required to fully understand the determinants of intramolecular cyclization versus intermolecular hydrolysis as well.
Finally, the recent discovery of the COM domains in NRPSs as well as the docking domains situated at the NRPS–PKS interface found in hybrid systems revealed another level of regulation in these multienzyme complexes. Development of chemical tools, such as crosslinking probes, will aid in distinguishing the structural characteristics controlling the conformational arrangement of the individual domains within the synthetase for the appropriate intermodular communication.
It is without a doubt that future endeavors towards deciphering the protein interactions at both the domain and modular level, by means of biochemical and structural techniques, will bring forth new findings that will help us gain a better grasp of the subtle intricacies influencing the enzymatic reactions involved in NRP synthesis. Furthermore, explorations into establishing the substrate selectivity and identifying the surface recognition elements of the tailoring enzymes mentioned in this review, as well as uncovering the dynamic conformational changes accompanying their interactions with the PCP, will lead to a comprehensive understanding of the architectural design of these multimodular enzyme complexes. Only then, will we be able to successfully manipulate these complex, natural product-producing assembly lines for the combinatorial biosynthesis of new valuable non-ribosomal peptide compounds.
Acknowledgments
We gratefully acknowledge the US National Institutes of Health grants R01GM094924 and R01GM095970 for funding.
References
- 1.Finking R, Marahiel MA. Annu Rev Microbiol. 2004;58:453–488. doi: 10.1146/annurev.micro.58.030603.123615. [DOI] [PubMed] [Google Scholar]
- 2.Cane DE, Walsh CT, Khosla C. Science. 1998;282:63–68. doi: 10.1126/science.282.5386.63. [DOI] [PubMed] [Google Scholar]
- 3.Stein T, Vater J, Kruft V, Otto A, Wittmann-Liebold B, Franke P, Panico M, McDowell R, Morris HR. J Biol Chem. 1996;271:15428–15435. doi: 10.1074/jbc.271.26.15428. [DOI] [PubMed] [Google Scholar]
- 4.Weber G, Schorgendorfer K, Schneider-Scherzer E, Leitner E. Curr Genet. 1994;26:120–125. doi: 10.1007/BF00313798. [DOI] [PubMed] [Google Scholar]
- 5.Jirakkakul J, Punya J, Pongpattanakitshote S, Paungmoung P, Vorapreeda N, Tachaleat A, Klomnara C, Tanticharoen M, Cheevadhanarak S. Microbiology. 2008;154:995–1006. doi: 10.1099/mic.0.2007/013995-0. [DOI] [PubMed] [Google Scholar]
- 6.Guenzi E, Galli G, Grguirina I, Gross DC, Grandi G. J Biol Chem. 1998;273:32857–32863. doi: 10.1074/jbc.273.49.32857. [DOI] [PubMed] [Google Scholar]
- 7.Keating TA, Marshall CG, Walsh CT. Biochemistry. 2000;39:15522–15530. doi: 10.1021/bi0016523. [DOI] [PubMed] [Google Scholar]
- 8.Keller U, Schauwecker F. Comb Chem High Throughput Screen. 2003;6:527–540. doi: 10.2174/138620703106298707. [DOI] [PubMed] [Google Scholar]
- 9.Menzella HG, Reeves CD. Curr Opin Microbiol. 2007;10:238–245. doi: 10.1016/j.mib.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 10.Schneider A, Stachelhaus T, Marahiel MA. Mol Gen Genet. 1998;257:308–318. doi: 10.1007/s004380050652. [DOI] [PubMed] [Google Scholar]
- 11.Mootz HD, Schwarzer D, Marahiel MA. Proc Natl Acad Sci USA. 2000;97:5848–5853. doi: 10.1073/pnas.100075897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Ferra F, Rodriguez F, Tortora O, Tosi C, Grandi G. J Biol Chem. 1997;272:25304–25309. doi: 10.1074/jbc.272.40.25304. [DOI] [PubMed] [Google Scholar]
- 13.Chiocchini C, Linne U, Stachelhaus T. Chem Biol. 2006;13:899–908. doi: 10.1016/j.chembiol.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 14.Suo Z. Biochemistry. 2005;44:4926–4938. doi: 10.1021/bi047538s. [DOI] [PubMed] [Google Scholar]
- 15.Duerfahrt T, Eppelmann K, Müller R, Marahiel MA. Chem Biol. 2004;11:261–271. doi: 10.1016/j.chembiol.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 16.Linne U, Stein DB, Mootz HD, Marahiel MA. Biochemistry. 2003;42:5114–5124. doi: 10.1021/bi034223o. [DOI] [PubMed] [Google Scholar]
- 17.Hahn M, Stachelhaus T. Proc Natl Acad Sci USA. 2004;101:15585–15590. doi: 10.1073/pnas.0404932101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hahn M, Stachelhaus T. Proc Natl Acad Sci USA. 2006;103:275–280. doi: 10.1073/pnas.0508409103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sieber SA, Marahiel MA. Chem Rev. 2005;105:715–738. doi: 10.1021/cr0301191. [DOI] [PubMed] [Google Scholar]
- 20.Challis GL, Naismith JH. Curr Opin Struct Biol. 2004;14:748–756. doi: 10.1016/j.sbi.2004.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grünewald J, Marahiel MA. Microbiol Mol Biol Rev. 2006;70:121–146. doi: 10.1128/MMBR.70.1.121-146.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Froyshov O. FEBS Lett. 1974;44:75–78. doi: 10.1016/0014-5793(74)80309-1. [DOI] [PubMed] [Google Scholar]
- 23.Stone KJ, Strominger JL. Proc Natl Acad Sci USA. 1971;68:3223–3227. doi: 10.1073/pnas.68.12.3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Storm DR, Stone KJ. Fed Proc. 1972;31:910. [Google Scholar]
- 25.Griffiths GL, Sigel SP, Payne SM, Neilands JB. J Biol Chem. 1984;259:383–385. [PubMed] [Google Scholar]
- 26.Keating TA, Marshall CG, Walsh CT. Biochemistry. 2000;39:15522–15530. doi: 10.1021/bi0016523. [DOI] [PubMed] [Google Scholar]
- 27.Quadri LE. Mol Microbiol. 2000;37:1–12. doi: 10.1046/j.1365-2958.2000.01941.x. [DOI] [PubMed] [Google Scholar]
- 28.Cardenas ME, Zhu D, Heitman J. Curr Opin Nephrol Hypertens. 1995;4:472–477. doi: 10.1097/00041552-199511000-00002. [DOI] [PubMed] [Google Scholar]
- 29.Marques MA, Citron DM, Wang CC. Bioorg Med Chem. 2007;15:6667–6677. doi: 10.1016/j.bmc.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trauger JW, Kohli RM, Mootz HD, Marahiel MA, Walsh CT. Nature. 2000;407:215–218. doi: 10.1038/35025116. [DOI] [PubMed] [Google Scholar]
- 31.Walsh CT, Chen H, Keating TA, Hubbard BK, Losey HC, Luo L, Marshall CG, Miller DA, Patel HM. Curr Opin Chem Biol. 2001;5:525–534. doi: 10.1016/s1367-5931(00)00235-0. [DOI] [PubMed] [Google Scholar]
- 32.Wittmann M, Linne U, Pohlmann V, Marahiel MA. FEBS J. 2008;275:5343–5354. doi: 10.1111/j.1742-4658.2008.06664.x. [DOI] [PubMed] [Google Scholar]
- 33.Steenbergen JN, Alder J, Thorne GM, Tally FP. J Antimicrob Chemother. 2005;55:283–288. doi: 10.1093/jac/dkh546. [DOI] [PubMed] [Google Scholar]
- 34.Straus SK, Hancock RE. Biochim Biophys Acta. 2006;1758:1215–1223. doi: 10.1016/j.bbamem.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 35.Vanittanakom N, Loeffler W, Koch U, Jung G. J Antibiot (Tokyo) 1986;39:888–901. doi: 10.7164/antibiotics.39.888. [DOI] [PubMed] [Google Scholar]
- 36.Deleu M, Paquot M, Nylander T. Biophys J. 2008;94:2667–2679. doi: 10.1529/biophysj.107.114090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bister B, Bischoff D, Nicholson GJ, Stockert S, Wink J, Brunati C, Donadio S, Pelzer S, Wohlleben W, Süssmuth RD. Chembiochem. 2003;4:658–662. doi: 10.1002/cbic.200300619. [DOI] [PubMed] [Google Scholar]
- 38.Vaillancourt FH, Yin J, Walsh CT. Proc Natl Acad Sci USA. 2005;102:10111–10116. doi: 10.1073/pnas.0504412102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stegmann E, Pelzer S, Bischoff D, Puk O, Stockert S, Butz D, Zerbe K, Robinson J, Süssmuth RD, Wohlleben W. J Biotechnol. 2006;124:640–653. doi: 10.1016/j.jbiotec.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 40.Walsh CT, Fisher SL, Park IS, Prahalad M, Wu Z. Chem Biol. 1996;3:21–28. doi: 10.1016/s1074-5521(96)90079-4. [DOI] [PubMed] [Google Scholar]
- 41.Waring MJ, Wakelin LP. Nature. 1974;252:653–657. doi: 10.1038/252653a0. [DOI] [PubMed] [Google Scholar]
- 42.Huang CH, Mong S, Crooke ST. Biochemistry. 1980;19:5537–5542. doi: 10.1021/bi00565a012. [DOI] [PubMed] [Google Scholar]
- 43.Blout ER. Biopolymers. 1981;20:1901–1912. [Google Scholar]
- 44.Mootz HD, Schwarzer D, Marahiel MA. Chembiochem. 2002;3:490–504. doi: 10.1002/1439-7633(20020603)3:6<490::AID-CBIC490>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 45.Lai JR, Koglin A, Walsh CT. Biochemistry. 2006;45:14869–14879. doi: 10.1021/bi061979p. [DOI] [PubMed] [Google Scholar]
- 46.Stachelhaus T, Marahiel MA. J Biol Chem. 1995;270:6163–6169. doi: 10.1074/jbc.270.11.6163. [DOI] [PubMed] [Google Scholar]
- 47.Lambalot RH, Gehring AM, Flugel RS, Zuber P, LaCelle M, Marahiel MA, Reid R, Khosla C, Walsh CT. Chem Biol. 1996;3:923–936. doi: 10.1016/s1074-5521(96)90181-7. [DOI] [PubMed] [Google Scholar]
- 48.Stachelhaus T, Mootz HD, Bergendahl V, Marahiel MA. J Biol Chem. 1998;273:22773–22781. doi: 10.1074/jbc.273.35.22773. [DOI] [PubMed] [Google Scholar]
- 49.Schneider A, Marahiel MA. Arch Microbiol. 1998;169:404–410. doi: 10.1007/s002030050590. [DOI] [PubMed] [Google Scholar]
- 50.Borchert S, Stachelhaus T, Marahiel MA. J Bacteriol. 1994;176:2458–2462. doi: 10.1128/jb.176.8.2458-2462.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gaidenko TA, Belitsky BR, Haykinson MJ. Biotechnologia. 1992:13–19. [Google Scholar]
- 52.Wyckoff EE, Smith SL, Payne SM. J Bacteriol. 2001;183:1830–1834. doi: 10.1128/JB.183.5.1830-1834.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gaitatzis N, Hans A, Müller R, Beyer S. J Biochem. 2001;129:119–124. doi: 10.1093/oxfordjournals.jbchem.a002821. [DOI] [PubMed] [Google Scholar]
- 54.Stuible HP, Meier S, Schweizer E. Eur J Biochem. 1997;248:481–487. doi: 10.1111/j.1432-1033.1997.00481.x. [DOI] [PubMed] [Google Scholar]
- 55.Reuter K, Mofid MR, Marahiel MA, Ficner R. EMBO J. 1999;18:6823–6831. doi: 10.1093/emboj/18.23.6823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lambalot RH, Walsh CT. Methods Enzymol. 1997;279:254–262. doi: 10.1016/s0076-6879(97)79029-3. [DOI] [PubMed] [Google Scholar]
- 57.Quadri LE, Weinreb PH, Lei M, Nakano MM, Zuber P, Walsh CT. Biochemistry. 1998;37:1585–1595. doi: 10.1021/bi9719861. [DOI] [PubMed] [Google Scholar]
- 58.Mofid MR, Finking R, Essen LO, Marahiel MA. Biochemistry. 2004;43:4128–4136. doi: 10.1021/bi036013h. [DOI] [PubMed] [Google Scholar]
- 59.Parris KD, Lin L, Tam A, Mathew R, Hixon J, Stahl M, Fritz CC, Seehra J, Somers WS. Structure. 2000;8:883–895. doi: 10.1016/s0969-2126(00)00178-7. [DOI] [PubMed] [Google Scholar]
- 60.Strieker M, Tanović A, Marahiel MA. Curr Opin Struct Biol. 2010;20:234–240. doi: 10.1016/j.sbi.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 61.Schwarzer D, Mootz HD, Linne U, Marahiel MA. Proc Natl Acad Sci USA. 2002;99:14083–14088. doi: 10.1073/pnas.212382199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Linne U, Schwarzer D, Schroeder GN, Marahiel MA. Eur J Biochem. 2004;271:1536–1545. doi: 10.1111/j.1432-1033.2004.04063.x. [DOI] [PubMed] [Google Scholar]
- 63.Koglin A, Löhr F, Bernhard F, Rogov VV, Frueh DP, Strieter ER, Mofid MR, Güntert P, Wagner G, Walsh CT, Marahiel MA, Dötsch V. Nature. 2008;454:907–911. doi: 10.1038/nature07161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Frueh DP, Arthanari H, Koglin A, Vosburg DA, Bennett AE, Walsh CT, Wagner G. Nature. 2008;454:903–906. doi: 10.1038/nature07162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Koglin A, Walsh CT. Nat Prod Rep. 2009;26:987–1000. doi: 10.1039/b904543k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Claxton HB, Akey DL, Silver MK, Admiraal SJ, Smith JL. J Biol Chem. 2009;284:5021–5029. doi: 10.1074/jbc.M808604200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kleinkauf H, Gevers W, Lipmann F. Proc Natl Acad Sci USA. 1969;62:226–233. doi: 10.1073/pnas.62.1.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee SG, Lipmann F. Proc Natl Acad Sci USA. 1977;74:2343–2347. doi: 10.1073/pnas.74.6.2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Conti E, Stachelhaus T, Marahiel MA, Brick P. EMBO J. 1997;16:4174–4183. doi: 10.1093/emboj/16.14.4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.May JJ, Kessler N, Marahiel MA, Stubbs MT. Proc Natl Acad Sci USA. 2002;99:12120–12125. doi: 10.1073/pnas.182156699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee TV, Johnson LJ, Johnson RD, Koulman A, Lane GA, Lott JS, Arcus VL. J Biol Chem. 2010;285:2415–2427. doi: 10.1074/jbc.M109.071324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stachelhaus T, Mootz HD, Marahiel MA. Chem Biol. 1999;6:493–505. doi: 10.1016/S1074-5521(99)80082-9. [DOI] [PubMed] [Google Scholar]
- 73.Challis GL, Ravel J, Townsend CA. Chem Biol. 2000;7:211–224. doi: 10.1016/s1074-5521(00)00091-0. [DOI] [PubMed] [Google Scholar]
- 74.Eppelmann K, Stachelhaus T, Marahiel MA. Biochemistry. 2002;41:9718–9726. doi: 10.1021/bi0259406. [DOI] [PubMed] [Google Scholar]
- 75.Chen CY, Georgiev I, Anderson AC, Donald BR. Proc Nat Acad Sci USA. 2009;106:3764–3769. doi: 10.1073/pnas.0900266106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gulick AM, Starai VJ, Horswill AR, Homick KM, Escalante-Semerena JC. Biochemistry. 2003;42:2866–2873. doi: 10.1021/bi0271603. [DOI] [PubMed] [Google Scholar]
- 77.Turgay K, Krause M, Marahiel MA. Mol Microbiol. 1992;6:529–546. doi: 10.1111/j.1365-2958.1992.tb01498.x. [DOI] [PubMed] [Google Scholar]
- 78.Reger AS, Carney JM, Gulick AM. Biochemistry. 2007;46:6536–6546. doi: 10.1021/bi6026506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Osman KT, Du L, He Y, Luo Y. J Mol Biol. 2009;388:345–355. doi: 10.1016/j.jmb.2009.03.040. [DOI] [PubMed] [Google Scholar]
- 80.Yonus H, Neumann P, Zimmermann S, May JJ, Marahiel MA, Stubbs MT. J Biol Chem. 2008;283:32484–32491. doi: 10.1074/jbc.M800557200. [DOI] [PubMed] [Google Scholar]
- 81.Du L, He Y, Luo Y. Biochemistry. 2008;47:11473–11480. doi: 10.1021/bi801363b. [DOI] [PubMed] [Google Scholar]
- 82.Rausch C, Weber T, Kohlbacher O, Wohlleben W, Huson DH. Nucleic Acids Res. 2005;33:5799–5808. doi: 10.1093/nar/gki885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Röttig M, Medema MH, Blin K, Weber T, Rausch C, Kohlbacher O. Nucleic Acids Res. 2011;39:W362–W367. doi: 10.1093/nar/gkr323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Villiers BRM, Hollfelder F. Chembiochem. 2009;10:671–682. doi: 10.1002/cbic.200800553. [DOI] [PubMed] [Google Scholar]
- 85.Linne U, Marahiel MA. Methods Enzymol. 2004;388:293–315. doi: 10.1016/S0076-6879(04)88024-8. [DOI] [PubMed] [Google Scholar]
- 86.Villiers B, Hollfelder F. Chem Biol. 2011;18:1290–1299. doi: 10.1016/j.chembiol.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 87.Otten LG, Schaffer ML, Villiers BRM, Stachelhaus T, Hollfelder F. Biotechnol J. 2007;2:232–240. doi: 10.1002/biot.200600220. [DOI] [PubMed] [Google Scholar]
- 88.Quadri LEN, Sello J, Keating TA, Weinreb PH, Walsh CT. Chem Biol. 1998;5:631–645. doi: 10.1016/s1074-5521(98)90291-5. [DOI] [PubMed] [Google Scholar]
- 89.Baltz RH. J Ind Microbiol Biotechnol. 2011;38:1747–1760. doi: 10.1007/s10295-011-1022-8. [DOI] [PubMed] [Google Scholar]
- 90.Drake EJ, Cao J, Qu J, Shah MB, Straubinger RM, Gulick AM. J Biol Chem. 2007;282:20425–20434. doi: 10.1074/jbc.M611833200. [DOI] [PubMed] [Google Scholar]
- 91.Buchko GW, Kim CY, Terwilliger TC, Myler PJ. Tuberculosis (Edinb) 2010;90:245–251. doi: 10.1016/j.tube.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Heemstra JR, Jr, Walsh CT, Sattely ES. J Am Chem Soc. 2009;131:15317–15329. doi: 10.1021/ja9056008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Felnagle EA, Barkei JJ, Park H, Podevels AM, McMahon MD, Drott DW, Thomas MG. Biochemistry. 2010;49:8815–8817. doi: 10.1021/bi1012854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Imker HJ, Krahn D, Clerc J, Kaiser M, Walsh CT. Chem Biol. 2010;17:1077–1083. doi: 10.1016/j.chembiol.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Weber T, Baumgartner R, Renner C, Marahiel MA, Holak TA. Structure. 2000;8:407–418. doi: 10.1016/s0969-2126(00)00120-9. [DOI] [PubMed] [Google Scholar]
- 96.Koglin A, Mofid MR, Löhr F, Schäfer B, Rogov VV, Blum MM, Mittag T, Marahiel MA, Bernhard F, Dötsch V. Science. 2006;312:273–276. doi: 10.1126/science.1122928. [DOI] [PubMed] [Google Scholar]
- 97.Van Gunsteren WF, Hünenberger PH, Mark AE, Smith PE, Tironi IG. Comput Phys Commun. 1995;91:305–319. [Google Scholar]
- 98.Berendsen HJC, van der Spoel D, van Drunen R. Comput Phys Commun. 1995;91:43–56. [Google Scholar]
- 99.Lai JR, Fischbach MA, Liu DR, Walsh CT. J Am Chem Soc. 2006;128:11002–11003. doi: 10.1021/ja063238h. [DOI] [PubMed] [Google Scholar]
- 100.Lai JR, Fischbach MA, Liu DR, Walsh CT. Proc Natl Acad Sci USA. 2006;103:5314–5319. doi: 10.1073/pnas.0601038103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Worthington AS, Hur GH, Burkart MD. Mol Biosyst. 2011;7:365–370. doi: 10.1039/c0mb00251h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhou Z, Lai JR, Walsh CT. Proc Natl Acad Sci USA. 2007;104:11621–11626. doi: 10.1073/pnas.0705122104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhou Z, Lai JR, Walsh CT. Chem Biol. 2006;13:869–879. doi: 10.1016/j.chembiol.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 104.Samel SA, Schoenafinger G, Knappe TA, Marahiel MA, Essen LO. Structure. 2007;15:781–792. doi: 10.1016/j.str.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 105.Tanovic A, Samel SA, Essen LO, Marahiel MA. Science. 2008;321:659–663. doi: 10.1126/science.1159850. [DOI] [PubMed] [Google Scholar]
- 106.Zettler J, Mootz HD. FEBS J. 2010;277:1159–1171. doi: 10.1111/j.1742-4658.2009.07551.x. [DOI] [PubMed] [Google Scholar]
- 107.Dieckmann R, Pavela-Vrancic M, Von Döhren H, Kleinkauf H. J Mol Biol. 1999;288:129–140. doi: 10.1006/jmbi.1999.2671. [DOI] [PubMed] [Google Scholar]
- 108.Stachelhaus T, Mootz HD, Bergendahl V, Marahiel MA. J Biol Chem. 1998;273:22773–22781. doi: 10.1074/jbc.273.35.22773. [DOI] [PubMed] [Google Scholar]
- 109.Keating TA, Marshall CG, Walsh CT, Keating AE. Nat Struct Biol. 2002;9:522–526. doi: 10.1038/nsb810. [DOI] [PubMed] [Google Scholar]
- 110.Bergendahl V, Linne U, Marahiel MA. Eur J Biochem. 2002;269:620–629. doi: 10.1046/j.0014-2956.2001.02691.x. [DOI] [PubMed] [Google Scholar]
- 111.Gordon JC, Myers JB, Folta T, Shoja V, Heath LS, Onufriev A. Nucleic Acids Res. 2005;33:W368–W371. doi: 10.1093/nar/gki464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Belshaw PJ, Walsh CT, Stachelhaus T. Science. 1999;284:486–489. doi: 10.1126/science.284.5413.486. [DOI] [PubMed] [Google Scholar]
- 113.Clugston SL, Sieber SA, Marahiel MA, Walsh CT. Biochemistry. 2003;42:12095–12104. doi: 10.1021/bi035090+. [DOI] [PubMed] [Google Scholar]
- 114.Du L, Sanchez C, Chen M, Edwards DJ, Shen B. Chem Biol. 2000;7:623–642. doi: 10.1016/s1074-5521(00)00011-9. [DOI] [PubMed] [Google Scholar]
- 115.Silakowski B, Schairer HU, Ehret H, Kunze B, Weinig S, Nordsiek G, Brandt P, Blöcker H, Höfle G, Beyer S, Müller R. J Biol Chem. 1999;274:37391–37399. doi: 10.1074/jbc.274.52.37391. [DOI] [PubMed] [Google Scholar]
- 116.König A, Schwecke T, Molnár I, Böhm GA, Lowden PAS, Staunton J, Leadlay PF. Eur J Biochem. 1997;247:526–534. doi: 10.1111/j.1432-1033.1997.00526.x. [DOI] [PubMed] [Google Scholar]
- 117.Hoffmann D, Hevel JM, Moore RE, Moore BS. Gene. 2003;311:171–180. doi: 10.1016/s0378-1119(03)00587-0. [DOI] [PubMed] [Google Scholar]
- 118.Motamedi H, Shafiee A. Eur J Biochem. 1998;256:528–534. doi: 10.1046/j.1432-1327.1998.2560528.x. [DOI] [PubMed] [Google Scholar]
- 119.Wu K, Chung L, Revill WP, Katz L, Reeves CD. Gene. 2000;251:81–90. doi: 10.1016/s0378-1119(00)00171-2. [DOI] [PubMed] [Google Scholar]
- 120.Schwecke T, Aparicio JF, Molnár I, König A, Khaw LE, Haydock SF, Oliynyk M, Caffrey P, Cortés J, Lester JB, Böhm GA, Staunton J, Leadlay PF. Proc Natl Acad Sci USA. 1995;92:7839–7843. doi: 10.1073/pnas.92.17.7839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gatto GJ, McLoughlin SM, Kelleher NL, Walsh CT. Biochemistry. 2005;44:5993–6002. doi: 10.1021/bi050230w. [DOI] [PubMed] [Google Scholar]
- 122.Miao V, Coëffet-LeGal M, Brian P, Brost R, Penn J, Whiting A, Martin S, Ford R, Parr I, Bouchard M, Silva CJ, Wrigley SK, Baltz RH. Microbiology. 2005;151:1507–1523. doi: 10.1099/mic.0.27757-0. [DOI] [PubMed] [Google Scholar]
- 123.Roongsawang N, Washio K, Morikawa M. Int J Mol Sci. 2011;12:141–172. doi: 10.3390/ijms12010141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kraas FI, Helmetag V, Wittmann M, Strieker M, Marahiel MA. Chem Biol. 2010;17:872–880. doi: 10.1016/j.chembiol.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 125.Zaleta-Rivera K, Xu C, Yu F, Butchko RA, Proctor RH, Hidalgo-Lara ME, Raza A, Dussault PH, Du L. Biochemistry. 2006;45:2561–2569. doi: 10.1021/bi052085s. [DOI] [PubMed] [Google Scholar]
- 126.Lin S, Van Lanen SG, Shen B. Proc Natl Acad Sci USA. 2009;106:4183–4188. doi: 10.1073/pnas.0808880106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Xu YJ, Zhen YS, Goldberg IH. Biochemistry. 1994;33:5947–5954. doi: 10.1021/bi00185a036. [DOI] [PubMed] [Google Scholar]
- 128.Batchelder RM, Wilson WR, Hay MP, Denny WA. Br J Cancer Suppl. 1996;27:S52–S56. [PMC free article] [PubMed] [Google Scholar]
- 129.Christenson SD, Wu W, Spies MA, Shen B, Toney MD. Biochemistry. 2003;42:12708–12718. doi: 10.1021/bi035223r. [DOI] [PubMed] [Google Scholar]
- 130.Van Lanen SG, Lin S, Dorrestein PC, Kelleher NL, Shen B. J Biol Chem. 2006;281:29633–29640. doi: 10.1074/jbc.M605887200. [DOI] [PubMed] [Google Scholar]
- 131.Lin S, Van Lanen SG, Shen B. J Am Chem Soc. 2007;129:12432–12438. doi: 10.1021/ja072311g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Peypoux F, Bonmatin JM, Wallach J. J Appl Microbiol Biotechnol. 1999;51:553–563. doi: 10.1007/s002530051432. [DOI] [PubMed] [Google Scholar]
- 133.Li J, Jensen SE. Chem Biol. 2008;15:118–127. doi: 10.1016/j.chembiol.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 134.Balibar CJ, Vaillancourt FH, Walsh CT. Chem Biol. 2005;12:1189–1200. doi: 10.1016/j.chembiol.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 135.Stachelhaus T, Walsh CT. Biochemistry. 2000;39:5775–5787. doi: 10.1021/bi9929002. [DOI] [PubMed] [Google Scholar]
- 136.Luo L, Burkart MD, Stachelhaus T, Walsh CT. J Am Chem Soc. 2001;123:11208–11218. doi: 10.1021/ja0166646. [DOI] [PubMed] [Google Scholar]
- 137.Linne U, Marahiel MA. Biochemistry. 2000;39:10439–10447. doi: 10.1021/bi000768w. [DOI] [PubMed] [Google Scholar]
- 138.Stein DB, Linne U, Marahiel MA. FEBS J. 2005;272:4506–4520. doi: 10.1111/j.1742-4658.2005.04871.x. [DOI] [PubMed] [Google Scholar]
- 139.Stein DB, Linne U, Hahn M, Marahiel MA. ChemBioChem. 2006;7:1807–1814. doi: 10.1002/cbic.200600192. [DOI] [PubMed] [Google Scholar]
- 140.Samel SA, Heine A, Marahiel MA, Essen LO. To be published, PDB 2XHG. [Google Scholar]
- 141.Marshall CG, Burkart MD, Keating TA, Walsh CT. Biochemistry. 2001;40:10655–10663. doi: 10.1021/bi010937s. [DOI] [PubMed] [Google Scholar]
- 142.Marshall CG, Hillson NJ, Walsh CT. Bichemistry. 2002;41:244–250. doi: 10.1021/bi011852u. [DOI] [PubMed] [Google Scholar]
- 143.Keating TA, Miller DA, Walsh CT. Biochemistry. 2000;39:4729–4739. doi: 10.1021/bi992923g. [DOI] [PubMed] [Google Scholar]
- 144.Duerfahrt T, Eppelmann K, Müller R, Marahiel MA. Chem Biol. 2004;11:261–271. doi: 10.1016/j.chembiol.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 145.Di Lorenzo M, Stork M, Naka H, Tolmasky ME, Crosa JH. Biometals. 2008;21:635–648. doi: 10.1007/s10534-008-9149-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Schneider TL, Shen B, Walsh CT. Biochemistry. 2003;42:9722–9730. doi: 10.1021/bi034792w. [DOI] [PubMed] [Google Scholar]
- 147.Reimmann C, Patel HM, Serino L, Barone M, Walsh CT, Haas D. J Bacteriol. 2001;183:813–820. doi: 10.1128/JB.183.3.813-820.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Du L, Chen M, Sánchez C, Shen B. FEMS Microbiol Lett. 2000;189:171–175. doi: 10.1111/j.1574-6968.2000.tb09225.x. [DOI] [PubMed] [Google Scholar]
- 149.Li Y, Weissman KJ, Müller R. J Am Chem Soc. 2008;130:7554–7555. doi: 10.1021/ja8025278. [DOI] [PubMed] [Google Scholar]
- 150.Weber G, Schörgendorfer K, Schneider-Scherzer E, Leitner E. Curr Genet. 1994;26:120–125. doi: 10.1007/BF00313798. [DOI] [PubMed] [Google Scholar]
- 151.Patel HM, Walsh CT. Biochemistry. 2001;40:9023–9031. doi: 10.1021/bi010519n. [DOI] [PubMed] [Google Scholar]
- 152.Hornbogen T, Riechers SP, Prinz B, Schultchen J, Lang C, Schmidt S, Mügge C, Turkanovic S, Süssmuth RD, Tauberger E, Zocher R. Chembiochem. 2007;8:1048–1054. doi: 10.1002/cbic.200700076. [DOI] [PubMed] [Google Scholar]
- 153.Hacker C, Glinski M, Hornbogen T, Doller A, Zocher R. J Biol Chem. 2000;275:30826–30832. doi: 10.1074/jbc.M002614200. [DOI] [PubMed] [Google Scholar]
- 154.O’Brien DP, Kirkpatrick PN, O’Brien SW, Staroske T, Richardson TI, Evans DA, Hopkinson A, Spencer JB, Williams DH. Chem Commun. 2000;1:103–104. [Google Scholar]
- 155.Shi R, Lamb SS, Zakeri B, Proteau A, Cui Q, Sulea T, Matte A, Wright GD, Cygler M. Chem Biol. 2009;16:401–410. doi: 10.1016/j.chembiol.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 156.Milne C, Powell A, Jim J, Al Nakeeb M, Smith CP, Micklefield J. J Am Chem Soc. 2006;128:11250–11259. doi: 10.1021/ja062960c. [DOI] [PubMed] [Google Scholar]
- 157.Mahlert C, Kopp F, Thirlway J, Micklefield J, Marahiel MA. J Am Chem Soc. 2007;129:12011–12018. doi: 10.1021/ja074427i. [DOI] [PubMed] [Google Scholar]
- 158.Laursen BS, Sørensen HP, Mortensen KK, Sperling-Petersen HU. Microbiol Mol Biol Rev. 2005;69:101–123. doi: 10.1128/MMBR.69.1.101-123.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kessler N, Schuhmann H, Morneweg S, Linne U, Marahiel MA. J Biol Chem. 2004;279:7413–7419. doi: 10.1074/jbc.M309658200. [DOI] [PubMed] [Google Scholar]
- 160.Rouhiainen L, paulin L, Suomalainen S, Hyytiäinen H, Buikema W, Haselkorn R, Sivonen K. Mol Microbiol. 2000;37:156–167. doi: 10.1046/j.1365-2958.2000.01982.x. [DOI] [PubMed] [Google Scholar]
- 161.Schoenafinger G, Schracke N, Linne U, Marahiel MA. J Am Chem Soc. 2006;128:7406–7407. doi: 10.1021/ja0611240. [DOI] [PubMed] [Google Scholar]
- 162.Gribble GW. J Chem Educ. 2004;81:1441–1449. [Google Scholar]
- 163.Dorrestein PC, Yeh E, Garneau-Tsodikova S, Kelleher NL, Walsh CT. Proc Natl Acad Sci USA. 2005;102:13843–13848. doi: 10.1073/pnas.0506964102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Galonić DP, Vaillancourt FH, Walsh CT. J Am Chem Soc. 2006;128:3900–3901. doi: 10.1021/ja060151n. [DOI] [PubMed] [Google Scholar]
- 165.Yeh E, Cole LJ, Barr EW, Bollinger JM, Jr, Ballou DP, Walsh CT. Biochemistry. 2006;45:7904–7912. doi: 10.1021/bi060607d. [DOI] [PubMed] [Google Scholar]
- 166.Lin S, Van Lanen SG, Shen B. J Am Chem Soc. 2007;129:12432–12438. doi: 10.1021/ja072311g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Dong C, Flecks S, Unversucht S, Haupt C, van Pée KH, Naismith JH. Science. 2005;309:2216–2219. doi: 10.1126/science.1116510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Bitto E, Huang Y, Bingman CA, Singh S, Thorson JS, Phillips GN., Jr Proteins. 2008;70:289–293. doi: 10.1002/prot.21627. [DOI] [PubMed] [Google Scholar]
- 169.van Pée KH, Zehner S. 2003:171–199. [Google Scholar]
- 170.van Pée KH, Patallo EP. Appl Microbiol Biotechnol. 2006;70:631–641. doi: 10.1007/s00253-005-0232-2. [DOI] [PubMed] [Google Scholar]
- 171.Chen X, van Pée KH. Acta Biochim Biophys Sin (Shanghai) 2008;40:183–193. doi: 10.1111/j.1745-7270.2008.00390.x. [DOI] [PubMed] [Google Scholar]
- 172.Yeh E, Blasiak LC, Koglin A, Drennan CL, Walsh CT. Biochemsitry. 2007;46:1284–1292. doi: 10.1021/bi0621213. [DOI] [PubMed] [Google Scholar]
- 173.Nightingale ZD, Lancha AH, Jr, Handelman SK, Dolnikowski GG, Busse SC, Dratz EA, Blumberg JB, Handelman GJ. Free Radical Biol Med. 2000;29:425–433. doi: 10.1016/s0891-5849(00)00262-8. [DOI] [PubMed] [Google Scholar]
- 174.Buedenbender S, Rachid S, Müller R, Schulz GE. J Mol Biol. 2009;385:520–530. doi: 10.1016/j.jmb.2008.10.057. [DOI] [PubMed] [Google Scholar]
- 175.Vaillancourt FH, Yeh E, Vosburg DA, O’Connor SE, Walsh CT. Nature. 2005;436:1191–1194. doi: 10.1038/nature03797. [DOI] [PubMed] [Google Scholar]
- 176.Blasiak LC, Vaillancourt FH, Walsh CT, Drennan CL. Nature. 2006;440:368–371. doi: 10.1038/nature04544. [DOI] [PubMed] [Google Scholar]
- 177.Wong C, Fujimori DG, Walsh CT, Drennan CL. J Am Chem Soc. 2009;131:4872–4879. doi: 10.1021/ja8097355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Dunwell JM, Purvis A, Khuri S. Phytochemistry. 2004;65:7–17. doi: 10.1016/j.phytochem.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 179.Clifton IJ, McDonough MA, Ehrismann D, Kershaw NJ, Granatino N, Schofield CJ. J Inorg Biochem. 2006;100:644–669. doi: 10.1016/j.jinorgbio.2006.01.024. [DOI] [PubMed] [Google Scholar]
- 180.Galonic DP, Barr EW, Walsh CT, Bollinger JM, Jr, Krebs C. Nat Chem Biol. 2007;3:113–116. doi: 10.1038/nchembio856. [DOI] [PubMed] [Google Scholar]
- 181.Schneider A, Marahiel MA. Arch Microbiol. 1998;169:404–410. doi: 10.1007/s002030050590. [DOI] [PubMed] [Google Scholar]
- 182.Marahiel MA. J Pept Sci. 2009;15:799–807. doi: 10.1002/psc.1183. [DOI] [PubMed] [Google Scholar]
- 183.Kohli RM, Walsh CT. Chem Commun. 2003;7:297–307. doi: 10.1039/b208333g. [DOI] [PubMed] [Google Scholar]
- 184.Grünewald J, Marahiel MA. Microbiol Mol Biol Rev. 2006;70:121–146. doi: 10.1128/MMBR.70.1.121-146.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Bruner SD, Weber T, Kohli RM, Schwarzer D, Marahiel MA, Walsh CT, Stubbs MT. Structure. 2002;10:301–310. doi: 10.1016/s0969-2126(02)00716-5. [DOI] [PubMed] [Google Scholar]
- 186.Tseng CC, Bruner SD, Kohli RM, Marahiel MA, Walsh CT, Sieber SA. Biochemistry. 2002;41:13350–13359. doi: 10.1021/bi026592a. [DOI] [PubMed] [Google Scholar]
- 187.Samel SA, Wagner B, Marahiel MA, Essen LO. J Mol Biol. 2006;359:876–889. doi: 10.1016/j.jmb.2006.03.062. [DOI] [PubMed] [Google Scholar]
- 188.Eys S, Schwartz D, Wohlleben W, Schinko E. Antimicrob Agents Chemother. 2008;52:1686–1696. doi: 10.1128/AAC.01053-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Robbel L, Hoyer KM, Marahiel MA. FEBS J. 2009;276:1641–1653. doi: 10.1111/j.1742-4658.2009.06897.x. [DOI] [PubMed] [Google Scholar]
- 190.Lombó F, Velasco A, Castro A, de la Calle F, Braña AF, Sánchez-Puelles JM, Méndez C, Salas JA. Chembiochem. 2006;7:366–376. doi: 10.1002/cbic.200500325. [DOI] [PubMed] [Google Scholar]
- 191.Hoyer KM, Mahlert C, Marahiel MA. Chem Biol. 2007;14:13–22. doi: 10.1016/j.chembiol.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Bürgi HB, Dunitz JD, Shefter E. J Am Chem Soc. 1973;95:5065–5067. [Google Scholar]
- 193.Hahn M, Stachelhaus T. Proc Natl Acad Sci USA. 2006;103:275–280. doi: 10.1073/pnas.0508409103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Broadhurst RW, Nietlispach D, Wheatcroft MP, Leadlay PF, Weissman KJ. Chem Biol. 2003;10:723–731. doi: 10.1016/s1074-5521(03)00156-x. [DOI] [PubMed] [Google Scholar]
- 195.Hur GH, Meier JL, Baskin J, Codelli JA, Bertozzi CR, Marahiel MA, Burkart MD. Chem Biol. 2009;16:372–381. doi: 10.1016/j.chembiol.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Worthington AS, Burkart MD. Org Biomol Chem. 2006;4:44–46. doi: 10.1039/b512735a. [DOI] [PubMed] [Google Scholar]
- 197.O’Connor SE, Walsh CT, Liu F. Angew Chem. 2003;115:4047–4051. [Google Scholar]
- 198.Liu F, Garneau S, Walsh CT. Chem Biol. 2004;11:1533–1542. doi: 10.1016/j.chembiol.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 199.Richter CD, Nietlispach D, Broadhurst RW, Weissman KJ. Nat Chem Biol. 2008;4:75–81. doi: 10.1038/nchembio.2007.61. [DOI] [PubMed] [Google Scholar]