

Abstract
T-cell prolymphocytic leukemia (T-PLL) is a rare, mature T-cell neoplasm with distinct features and an aggressive clinical course. Early relapse and short overall survival are commonplace. Use of the monoclonal anti-CD52 antibody alemtuzumab has improved the rate of complete remission and duration of response to over 50% and between 6–12 months, respectively. Despite this advance, without an allogeneic transplant, resistant relapse is inevitable. We report complete (7) and partial (1) remissions in eight patients receiving alemtuzumab and cladribine with or without an HDAC inhibitor. These data show that administration of epigenetic agents overcomes alemtuzumab resistance. We report epigenetically induced expression of the surface receptor protein CD30 in T-PLL. Subsequent treatment with the anti-CD30 antibody drug conjugate brentuximab vedotin overcame organ specific (skin) resistance to alemtuzumab. Our findings demonstrate activity of combination epigenetic and immunotherapy in the incurable illness T-PLL, particularly in the setting of prior alemtuzumab therapy.
Introduction
Prolymphocytic leukemia is a rare, aggressive disease compromising 2% of mature lymphoid neoplasms. T-cell variant (T-PLL) is responsible for about 20% of cases.(1) Median age of onset is between 65 and 70 years, and there is a male predilection.(2) Common presenting signs include splenomegaly (73%), lymphadenopathy (53%), hepatomegaly (40%), skin manifestations (27%), pleural effusions (12%) and high leukocyte count (> 100 × 109 cells/L in 75%). T-PLL cells usually express CD2, CD5, CD7 and are TdT−. The majority of cases also have a CD4+/CD8− (65%) phenotype, though CD4−/CD8+ (13%) and CD4+/CD8+ (21%) variants exist. Analysis of the peripheral blood shows characteristic prolymphocyte morphology with basophilic cytoplasm, a single nucleolus and surface protrusions. (2, 3) Human T-lymphotropic virus 1 (HTLV-1) must be negative by serology and PCR as well.(4)
T-PLL is considered incurable, and treatment is difficult.(5) CHOP (cyclophosphamide, vincristine, doxorubicin, prednisone) and single agent 2′-deoxycoformycin (DCF), cladribine and fludarabine have shown little success. (3, 6, 7) CD52 is highly expressed on all normal lymphocytes, as well as T-PLL cells providing the rationale for use of alemtuzumab, an anti-CD52 monoclonal antibody, in T-PLL.(8) Although approved for B-cell chronic lymphocytic leukemia (B-CLL), single agent alemtuzumab has become first line therapy for T-PLL, with higher response rates than previous regimens.(9) The mechanism of action of alemtuzumab and other monoclonal antibodies remains poorly characterized. Antibody-dependent cell-mediated cytotoxicity (ADCC), complement-mediated cytotoxicity (CMC) and direct antitumor effects have been proposed. However, alemtuzumab alone is not a curative approach for T-PLL due to resistance.(5)
Aberrant activation and deactivation of transcription due to epigenetic changes are associated with tumorigenesis. (10, 11) Two changes instrumental in gene silencing are methylation of DNA and acetylation of histone tail lysine residues. The purine analog cladribine has mechanisms of action that make it useful as an epigenetic agent. It inhibits SAH hydrolyase through inhibition of donation of methyl groups by S-adenosyl methionine (SAM).(7, 12, 13) Vorinostat and romidepsin are both inhibitors of pan-histone deacetylase (HDAC) enzymes and are both approved for treatment of cutaneous T-cell lymphoma (CTCL) and PTCL. There are many other HDAC inhibitor (HDACi) compounds in development as well.(14) Thus, the combination of HDAC inhibitors with hypomethylating agents, such as cladribine, is potentially synergistic. Administration of HDACi subsequent to DNA methyltransferase inhibitors synergistically increases expression of silenced tumor suppressors and promotes cell death.(15) The ability of cladribine to inhibit both DNA and histone methylation may be critical to the success of this combination therapy.
CD30 (TNFRSF8) is a 130kD membrane glycoprotein receptor, originally identified on Reed-Sternberg cells of Hodgkin’s Lymphoma (HL). As a member of the TNF receptor superfamily, it is preferentially expressed in developing and activated lymphocytes (16–20) as well as in many cancers, including HL, CTCL, anaplastic large cell lymphoma (ALCL) and mastocytosis (21–23). Members of the TNFR superfamily are classically associated with transducing death signals, but CD30 does not contain the death domain of other TNF receptors.(24) Regardless of its normal function, CD30 is a target for delivery of potent chemotherapeutics such as brentuximab vedotin.
Brentuximab vedotin is an antibody drug conjugate (ADC) consisting of three components: a chimeric immunoglobulin G1 specific for CD30, a microtubule disrupting agent (auristatin E) and a protease cleavable covalent linker.(25, 26) It binds CD30 on the cell surface. When internalized, the linker is cleaved in the lysosome, releasing auristatin E into the cell. As a targeted therapy, it is currently approved for third line and second line treatment of HL and ALCL, respectively.(27)
We present a case series of eight T-PLL patients who were treated with cladribine, vorinostat/romidepsin/valproic acid and alemtuzumab. We report overcoming alemtuzumab resistance with these epigenetic drugs in T-PLL. Proteomic and transcriptomic analysis showed such therapy can activate expression of CD30 in T-PLL cells. This increase in CD30 allows for successful treatment with brentuximab vedotin.
Results
Patient response and outcomes
Patients were predominantly male and aged between 57 and 77 years. They all met the diagnostic criteria of T-PLL. Clinicopathologic features at presentation are summarized in Table S1. Patient responses to combined epigenetic and immunotherapy are summarized in Table 1. Full case descriptions of each patient are in the supplemental. We treated eight patients with both cladribine and alemtuzumab in combination, seven achieved CR. The rationale for adding cladribine was based on published data and a recently completed phase II trial demonstrating synergistic effects between cladribine, vorinostat and the monoclonal antibody rituximab in the aggressive\B cell malignancy mantle cell lymphoma (MCL).(28–30) Additionally, in vivo studies of B-CLL and breast cancer and in vitro study of mantle cell lymphoma (MCL) cell lines support the ability of cladribine to be both a DNA and histone methylation inhibitor (Fig. S1).(7, 31) A full description and diagram of the treatment schedule is presented in Figure 1. Patient 1 presented with high white blood cell count, anemia and thrombocytopenia and was first treated with IV alemtuzumab alone. White blood cell count dropped briefly but then continued to rise while on treatment. Cladribine was added and she achieved CR. She remained in CR for more than one year, relapsed and again achieved CR with cladribine and alemtuzumab. In contrast to the primary refractory pattern of patient 1, patient 2 was representative of the relapse, retreatment pattern. He presented with alemtuzumab resistant relapse but went into remission after the addition of cladribine and vorinostat. Although he relapsed several times, his disease remained susceptible to treatment with alemtuzumab, cladribine and vorinostat (Fig. 2). An attempt to identify the cell death mechanism utilized by combination therapy showed a lack of apoptotic cells despite the rapid decrease in cell count in patients 2 and 3 (Fig. S2). Patients 3, 4, 5, 6 and 8 were treated with combination cladribine and alemtuzumab with or without vorinostat as well. With the exception of patient 3, who achieved PR, these patients also achieved CRs; subsequent relapses remained susceptible to treatment (Fig. S3). Patient 7 was treated with cladribine and alemtuzumab but only achieved CR when valproic acid was added. Like vorinostat, valproic acid has HDACi properties.(32) It was used, because vorinostat and romidepsin were not available due to insurance issues. These results show that addition of epigenetic agents, such as cladribine and HDACis, to alemtuzumab treatment overcomes resistance to alemtuzumab in T-PLL. Resistance and subsequent re-sensitization to alemtuzumab was not due to silencing of and then re-expression of CD52 after treatment, respectively (Fig. S4). Major toxicities were hematologic and immune suppression. One patient experienced a fatal CNS hemorrhage, a potential rare complication of T-PLL and alemtuzumab therapy (33) (Table S2). Of the eight patients, five died with persistent disease; one remains in remission after allogeneic transplant; and two remain alive in early relapse (Table 1).
Table 1. Summary of Patient Treatment Response.
Disease status, previous treatments, treatment administered, clinical and molecular remission, duration of response, overall survival and expected survival were tabulated. Survival times are calculated from first treatment using alemtuzumab. CR, complete remission; PR, partial response (decrease in white blood cell counts); allo, allogeneic; Mean time between relapses was 6 months.
| Patient | Disease status prior to epigenetic therapy |
Previous Treatment | Treatment Administered |
Clinical Remission |
Molecular Remission |
Duration of Response | Overall survival time |
* Expected survival |
|---|---|---|---|---|---|---|---|---|
| 1 | First Relapse | CVP (cyclophosphamide, vincristine, prednisone) | alemtuzumab, cladribine | CR | Yes | 16 months | 34.3 months | 4 months |
| Second Relapse | alemtuzumab, cladribine | alemtuzumab, cladribine | PR | N/A | 11.6 months | |||
| Third Relapse | alemtuzumab, cladribine | alemtzumab, brentuximab vedotin | PR | N/A | 6.7 months - death, persistent disease | |||
| 2 | First Relapse | alemtuzumab (6 mo. CR) | alemtuzumab, cladribine, vorinostat | CR | Yes | 5.3 months | 17 months | 7.5–10 months |
| Second Relapse | alemtuzumab, cladribine, vorinostat | alemtuzumab, cladribine, vorinostat | PR | N/A | 2 months | |||
| Third Relapse | alemtuzumab, cladribine, vorinostat | alemtuzumab, cladribine, vorinostat | PR | N/A | 3.7 month - death, persistent disease | |||
| 3 | Initial Presentation | N/A | alemtuzumab, cladribine | PR | N/A | 5.7 months | 5.7 months | 20 months |
| 4 | Initial Presentation | N/A | alemtuzumab, cladribine | CR | Yes | 13.3 months | 14.8 months | 20 months |
| First Relapse | alemtuzumab, cladribine | alemtuzumab, cladribine | PR | N/A | 1.5 months - death, persistent disease CNS hemorrhage | |||
| 5 | Initial Presentation | N/A | alemtuzumab, cladribine, vorinostat | CR | Yes | 6.3 months | 15.3 months | 20 months |
| First Relapse w/skin involvement | alemtuzumab, cladribine, vorinostat | alemtuzumab, cladribine, romidepsin | PR | N/A | 4 months-blood | |||
| Second Relapse w/skin involvement | alemtuzumab, cladribine, romidepsin | brentuximab vedotin | PR | N/A | 5 months - Allo transplant death, persistent disease | |||
| 6 | Initial Presentation | N/A | alemtuzumab, cladribine | CR | Yes | 0.8 months - Allo transplant | 0.8 months+ | N/A |
| 7 | First Relapse | alemtuzumab (0.3 months) | alemtuzumab, cladribine, valproic acid | CR | Yes | 12 months - Allo transplant referral | 12 months+ | 4 months |
| 8 | First Relapse | Alemtuzumab (16 months) | alemtuzumab, cladribine | CR | Yes | 6.7 months | 23.7 months+ | 7.5–10 months |
| Second Relapse | alemtuzumab, cladribine | alemtuzumab, cladribine | CR | Yes | 1 month + |
Expected survival without transplant.(5)
Figure 1. Patient treatment protocol.
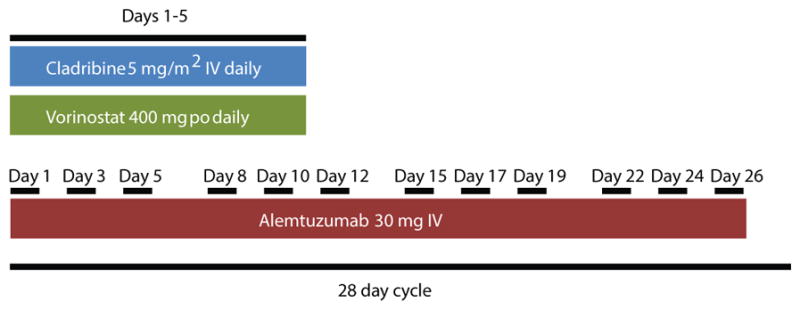
Patients received cladribine at 5 mg/m2 intravenously each day on days 1–5. Those treated with vorinostat were given 400 mg orally each day on days 1–5 as well. Patients received alemtuzumab 30 mg intravenously once a day on days 1, 3, 5, 8, 12, 15, 17, 19, 22, 24 and 26. One treatment cycle was 28 days. All patients were treated with alemtuzumab and cladribine. Those whose white blood cell counts did not respond well after relapse were also treated with vorinostat. Status of clinical and molecular remissions was evaluated after 2–3 cycles.
Figure 2. Epigenetic therapy decreased leukemic white blood cell counts despite multiple relapses.

Patient 2’s white blood cell counts were monitored during treatment. Day 0 was the initial white blood cell count at each respective presentation. The bars beneath the graph represent treatment received by the patient over the respective time periods. Graphs for all treated patients are available in Fig. S3.
Induction of CD30 gene expression and changes in CD30 promoter chromatin markers after epigenetic therapy in T-PLL patients
The addition of cladribine induced the expression of CD30 in three patients after initial treatment (Fig. 3) and one more after first relapse (Fig. 4). Two patients (1 and 5) were then successfully treated with brentuximab vedotin. We did not observe increases in CD30 in 4 leukemic MCL patients after treatment with epigenetic drugs indicating T-PLL specific CD30 induction (Fig. S5). We demonstrate CD30 induction with epigenetic drugs in vivo in patients with a T-cell malignancy.
Figure 3. Epigenetic therapy induced CD30 gene expression and chromatin reorganization.

(A) Patient mRNA samples were assayed for CD30 gene expression using qRT-PCR before and 5 days after treatment with epigenetic therapy. Fold change represents the fold increase or decrease of expression after treatment relative to before treatment. Numbers in parentheses indicate actual fold change. Genomic DNA from patients 1 (B) and 5 (C) was assessed for changes in DNA and chromatin methylation as well as RNA polymerase II binding before and after treatment using ChIP assays. For patient 5, samples were taken before and 5 days post-treatment of relapse using romidepsin instead of vorinostat. For patient 1, samples were taken before and 5 days post-treatment of initial epigenetic treatment. Fold change indicates fold increase or decrease in binding after treatment as measured by qRT-PCR. Three regions of the CD30 promoter were considered in this assay spanning roughly 400 bp on each side of the transcription start site. Prox refers to a 200 bp region around the transcription start site. RNA polymerase II (Pol 2), 5-methylcytosine (5meC), histone 3 lysine 9 trimethylation (H3Ly9Me3), histone 3 lysine 27 trimethylation (H3Ly27Me3), * p<0.05, ** p<0.005, ***p<0.0005. Bars represent mean fold change +/− SEM. N=3.
Figure 4. CD30 positivity correlates with response to brentuximab vedotin in patient 5.

For time points at which blood samples of patient 5 were available, CD30 status was assessed by qRT-PCR. This is represented by the bar graph with days on the x-axis. Values are reported as fold change compared to initial treatment naïve samples collected prior to study intervention. White blood cell counts (in red) from these same time points were also collected and graphed above their corresponding dates (black line graph). Antibody treatment received during each period is labeled above the graph. The inset to the right shows flow cytometry data of CD30 negative (top), CD52 positive (bottom) cell populations of the last time point at which CD30 expression was observed to disappear by qRT-PCR and subsequently treatment was switched back to alemtuzumab. ***p<0.0005. Bars represent mean fold change +/− SEM. N=3.
To further explore the transcriptional effect of epigenetic drugs on TNFRSF8 (CD30), chromatin immunoprecipitation (ChIP) assays were carried out. The −100bp to −1bp regions upstream of the transcription start site from patients 1 and 5 were analyzed. These two patients were chosen, because they showed CD30 induction and were successfully treated with brentuximab vedotin. There were increases in RNA polymerase II (PolII) binding as well as decreased amounts of methylated cytosine residues (5meC), histone 3 lysine 9 trimethylation (H3K9Me3) and histone 3 lysine 27 trimethylation (H3K27Me3) after treatment (Fig. 3B,C).
Alemtuzumab resistant skin lesions treated with brentuximab vedotin and epigenetic drugs
Patient 5 relapsed with both leukemic and dermatologic infiltration by prolymphocytes. He initially underwent treatment with alemtuzumab, cladribine and vorinostat. After two months of treatment, he achieved CR (Fig. S3). After 6.3 months of remission, he relapsed and did not respond to treatment. At this time, vorinostat was replaced with romidepsin, and the patient achieved a second remission lasting 4 months.
At the time of his second relapse, he showed a resurgence of leukemic cells as well as infiltration of prolymphocytes into the dermis of the skin. He was treated again with alemtuzumab, cladribine and romidepsin. His peripheral blood counts responded, but his skin did not. Lymphocytes in both compartments remained CD52 positive (Fig. S6). The patient’s skin disease was progressing and was refractory to treatment with alemtuzumab, vorinostat, romidepsin. Pralatrexate was utilized as it is approved for use in T-cell malignancies but also had no effect. A biopsy showed dermis infiltrating lymphocytes testing weakly positive for CD30 surface expression (Fig. 5A). His peripheral disease tested positive for CD30 throughout his second relapse as well (Fig. 4). He was subsequently started on brentuximab vedotin. His skin scabbed and cleared over the next month (Fig. 5B). Repeat biopsy of a healing skin lesion showed lymphocyte depletion. He remained in remission for 5 months. Shortly thereafter, his lymphocyte count began to increase again, from 1,180 to 5,500 (Fig. 4). They were found to be CD30−, CD52+ (Fig. 4 inset). Alemtuzumab was initiated again. In the interim, the patient was matched and received an allogeneic stem cell transplant complicated by graft vs. host disease with persistent lymphadenopathy and died shortly thereafter.
Figure 5. Brentuximab vedotin cleared CD30+ skin lesions in patient 5.

(A) Immunohistochemistry of biopsies of these lesions showed CD30+ infiltrate (left) in the dermis before treatment with brentuximab vedotin and absent CD30+ T-PLL cells after 1 month of treatment (right). Nucleus (blue), CD30 (brown), scale bars are 20 μM (B) Images of the upper left shoulder (top) and lateral left chest wall (bottom) depict plaques infiltrated with CD30+ T-PLL cells before (left) and after (right) treatment with brentuximab vedotin.
mRNA microarray and qRT-PCR analyses of lymphocyte proliferations after epigenetic treatment
Microarray analysis before and after epigenetic therapy was performed using four patients from whom there was sufficient amount of quality RNA (Fig. S7). Genes were filtered using a 2-fold increase in expression after treatment as cutoff. Targets that showed increases in 2 or more patients after therapy were confirmed by quantitative RT-PCR (qRT-PCR). The genes with the most notably increased expression after epigenetic therapy were interferon inducible proteins and mediators of MAPK activity, mainly the HIN-200 and CCAAT ester binding protein (CEBP) families. We studied the HIN-200 family because of its known tumor biology and sequential location on chromosome 1q21-23.(34) All four members of the HIN-200 family, MNDA, IFI16, PYHIN and AIM2, were assayed for expression changes by qRT-PCR after treatment. Not all patient samples upregulated the same family member, but they all upregulated at least one of the four (Fig. 6). The CEBP family contains six members, CEBPα, β, δ, ε, γ and ζ. CEBP α, β, and δ have unique functions. (35) As with the HIN-200 family, not all post-treatment samples upregulated the same CEBP gene, but they all upregulated at least one family member after treatment (Fig. 6). After analysis of microarray mRNA expression of patients after therapy, TRIB1, which codes for the tribbles1 protein, was the only gene that showed at least 2 fold increase in expression in all four tested patients. qRT-PCR confirmed four of six T-PLL patients upregulated TRIB1 expression (Fig. 7A). ChIP assays on the TRIB1 promoter of samples from patient 1 and 5 showed significant increases in RNA Polymerase II (Pol II) and pan-H4 acetylation and corresponding decreases in H3K9Me3. Patient 5 showed more pronounced increases in Pol II and pan-H4 acetylation than patient 1 and large decreases in 5meC, H3K9Me3 and H3K27Me3 (Fig. 7B). These changes are consistent with decreased repressive chromatin marks.(36) Microarray data also suggested globin genes, HBA and HBB, were upregulated (Fig. S7). qRT-PCR of these genes confirmed microarray results. All patient samples except for patients 3 and 4 showed increases in globin mRNA expression after treatment (Fig. 6).
Figure 6. Epigenetic therapy induces HIN-200, CEBP and globin gene expression.

Changes in expression of AIM2, MNDA, IFI16, PYHIN, CEBPA, CEBPB, CEBPD, HBA and HBB were assessed by qRT-PCR before and 5 days after treatment of six patients with epigenetic therapy. Sufficient samples for mRNA analysis were only available for patients 1–6. Only the three unique CEBP family members were tested. Values are shown as fold changes as compared to treatment naïve levels of expression in each patient. Numbers in parentheses indicate actual fold change. * p<0.05, ** p<0.005, ***p<0.0005. Bars represent mean fold change +/− SEM. N=3.
Figure 7. Epigenetic therapy induces TRIB1 expression and results in increased active chromatin marks.

(A) Changes in expression of TRIB1 were assessed by qRT-PCR before and after treatment of six patients with epigenetic therapy. Values are shown as fold changes as compared to treatment naïve levels of expression in each patient. Numbers in parentheses indicate actual fold change. (B) Genomic DNA from patients 1 (grey) and 5 (black) was assessed for changes in DNA and chromatin methylation as well as RNA polymerase II binding before and 5 days after treatment using ChIP, the same time point as fig. 3B and C. Fold change indicates fold increase or decrease in binding after treatment as measured by qRT-PCR. The region represented is 400bp around the transcriptional start of the TRIB1 gene. RNA polymerase II (Pol II), 5-methylcytosine (5meC), histone 3 lysine 9 trimethylation (H3K9Me3), histone 3 lysine 27 trimethylation (H3K27Me3) and Acytl (pan-histone acetylation), * p<0.05, ** p<0.005, ***p<0.0005. Bars represent mean fold change +/− SEM. N=3.
We also assessed other candidate genes involved in the biology of T-PLL after treatment using qRT-PCR. BAX, ATM, CDKN1B (p27), TCL1, MTCP1 and DUSP16 are known to have aberrant expression in cases of T-PLL in general.(37–41) We found mRNA levels of TCL1 and CDKN1B to be abnormal in our patients. Patients 1, 2, 3 and 5 showed overexpression of TCL1 (Fig. S8), which is overexpressed in 70% of T-PLL.(2) All patients showed less than half the levels of CDKN1B mRNA as normal controls, in line with published reports that p27 haploinsufficiency plays a critical role in T-PLL pathogenesis (Fig. S8).(37) Aberrations in expression of these genes did not change after treatment with epigenetic drugs, indicating that though important, these elements of T-PLL tumor biology are not epigenetically regulated in our patients. However, the presence of these dysregulated genes confirms the T-PLL signature in the microarray data.
STAT5B mutational status of T-PLL patients
All patients were tested for the N642H STAT5B mutation by PCR amplification and DNA sequencing. Additionally, DNA banked from two previous T-PLL patients (402, 429) not on our study were sequenced for this mutation as well. 6/10 (60%) were wild type STAT5B. 3/10 (30%) were heterozygous for the N642H mutation, and 1/10 (10%) was homozygous for the mutation (Fig. S9).
Discussion
Here we found that eight patients with T-PLL responded to combination therapy with alemtuzumab and epigenetic agents. The median survival post-first relapse, censored for living patients (patients 6–8) was 13 months. The median overall survival was 15.3 months in such patients. Toxicities experienced were mainly infectious in nature and managed with conventional monitoring and antibiotic therapy. One patient did experience a fatal CNS hemorrhage possibly related to therapy. Although the patient numbers are small, it is interesting to look at overall survival in our patients stratified by previous therapy. Two patients initially resistant to alemtuzumab had overall survival of 34.3 and 12+ months (patients 1 and 7, respectively), compared to an expected survival of 4 months in such patients.(5, 42) Patient 2 who had relapsed after initial alemtuzumab therapy had an overall survival of 17 months, compared to expected survival of between 7.5 to 10 months. Patient 8 was also treated after initial alemtuzumab relapse and has survived 23.7+ months.(5, 9, 43) Four of our patients were treated initially with the combination epigenetic therapeutic approach. One remains in remission following allogeneic transplant. The survival of the others was 5.7, 14.8 and 15.3 months compared to overall survival of 20 months for patients treated with initial alemtuzumab alone.(5) These data suggest that addition of epigenetic therapy might be best suited for patients with alemtuzumab refractory or relapsed disease.
Treatment with these drugs showed altered mRNA expression profile on microarray analysis, implicating the CCAAT ester binding proteins, HIN-200 family and tribbles (TRIB1) proteins in epigenetic dysregulation during malignancy. This treatment regimen activated expression of CD30 in four patients; two then received successful treatment with brentuximab vedotin. We demonstrate epigenetic induction of CD30 and subsequent successful treatment with brentuximab vedotin. These findings represent a potential effective treatment paradigm for T-PLL.
We hypothesize that cladribine and HDACis may activate effector cells such as NK- and T-cells or granulocytes to enhance the antitumor effects of alemtuzumab through ADCC and re-express silenced genes. The absence of apoptotic changes in T-PLL cells after treatment supports enhanced ADCC. The increases in CD30 expression and decreases in CD30 promoter histone and DNA methylation observed after treatment point to epigenetic silencing in T-PLL. Despite these findings, the full mechanism by which epigenetic agents induce the expression of CD30 has not been fully elucidated. The two patients in whom CD30 induction was not observed were started on alemtuzumab, cladribine and vorinostat. The presence of vorinostat most likely silenced the expression of CD30. This observation is consistent with in vitro findings from our laboratory that vorinostat can transiently ablate CD30 expression.(44) This is further supported by the additional observation that patient 5 showed no CD30 upregulation until he was switched from vorinostat to romidepsin. These data support the ability of epigenetic therapy to modulate gene expression in T-PLL.
Induction of CD30 provides an additional method to circumvent resistance in T-PLL. Patients 1 and 5 were successfully treated with brentuximab vedotin after epigenetic induction of CD30. Treatment with brentuximab vedotin was only considered in patients who showed CD30 upregulation and had exhausted epigenetic treatment options due to treatment failure or toxicity (patients 1 and 5). Their response to treatment indicates that epigenetic therapy not only overcomes resistance to alemtuzumab but also generates sensitivity to another drug class. This finding proved important to treatment of patient 5’s skin lesions. His blood responded to treatment with alemtuzumab but his skin did not. Certain aspects of the dermal environment were likely not conducive to alemtuzumab function despite CD52 positivity. Clark et al. showed that leukemia cells that do not re-enter blood circulation in CTCL are resistant to alemtuzumab despite CD52 positivity and alemtuzumab penetration into the dermis.(45) Regardless of the mechanism, epigenetic therapy enabled the use of brentuximab vedotin to circumvent alemtuzumab resistance in skin lesions in T-PLL.
Correlative experiments found three upregulated gene families that potentially link dysregulation of the MAP kinase and interferon pathways with the extreme proliferative drive of T-PLL. These two pathways can be connected by CEBP family transcription factors, particularly CEBPB. CEBPB is induced by interferon gamma and is required to activate expression of DAPK1, a protein that regulates the cell cycle and apoptosis.(46) DAPK1 induction by CEBPB requires ERK signaling.(46, 47) The tribbles family of proteins, mainly TRIB1, regulates MAP kinase signaling through scaffold-like interactions of MEK and ERK and interact with CEBPB.(48–50) Increased expression of these two gene families after epigenetic therapy could help to reinitiate apoptosis and slow cell growth. The HIN-200 family of genes also links growth suppressing interferon pathways to T-PLL by acting as regulators of cell cycle progression and differentiation and therefore tumor suppressor proteins.(51, 52) Reversal of HIN-200 silencing through epigenetic therapy could re-sensitize cells to death signals. Overall, re-expression of the HIN-200, CEBP and tribbles families may contribute to the dramatic fall in circulating leukemia cells after epigenetic treatment through regaining control of the cell cycle.
The N642H STAT5B mutation has been identified in several types of aggressive T-cell lymphomas, including T-cell large granular lymphocyte leukemia (T-LGL) where it was discovered(53), pediatric acute lymphoblastic leukemia(54) and in aggressive NK-cell lymphomas(55). Because T-PLL presents as an aggressive disease (high white blood cell counts, organomegaly, cytopenias, skin manifestations, etc.), we hypothesized that STAT5B mutations may be present in T-PLL as well. The T to G point mutation replaces an asparagine with a histidine at position 642 in the src-like homology 2 domain. This change leads to hyperactivated STAT5B.(53) We noted that 4/10 (40%) of our patients carried the mutation in their leukemic clone. The presence of the mutation did not seem to have an effect on the efficacy of epigenetic therapy. A recent study from Kiel et al. showed STAT5B was hyperactive in T-PLL in general and that targeted inhibition of STAT5B led to apoptosis.(56) Similarly to the gene families found upregulated after epigenetic therapy, hyperactivated STAT5B with the N642H mutation dysregulates interferon signaling and the JAK/STAT pathway. This mutation further implicates the interferon pathway in the aggressiveness of T-PLL. Our findings suggest that targeting the interferon pathway, potentially through development of anti-N642H STAT5B therapeutics, may have merit in the treatment of T-PLL.
Our study has several limitations, particularly regarding the relatively small sample size. We were only able to obtain blood samples before and after epigenetic treatment on 6 of the 8 patients. All patients received some form of epigenetic therapy, but the epigenetic drugs varied somewhat. Although correlative studies identified gene families upregulated with epigenetic therapy, we were unable to definitively determine the mechanism that governed the susceptibility of T-PLL to such therapy. The promising results of this pilot study need validation in a larger prospective trial.
This study presents an effective epigenetic combination drug therapy for T-PLL, an aggressive T-cell leukemia for which there are few treatment options and a poor prognosis. Such therapy appears to overcome drug resistance. Although patients eventually relapsed, leukemia cells at relapse remained sensitive to this regimen. The re-induction of complete remission in resistant disease creates multiple opportunities to pursue curative therapy with allogeneic stem cell transplant. The most clinically relevant epigenetic change noted was the induction of drug target CD30. This observation formed the basis for successful treatment with brentuximab vedotin. We suggest that the combination of cladribine with or without an HDACi and an appropriate tumor specific monoclonal antibody may be an important therapeutic platform for the treatment of cancer.
Materials and Methods
Study Design
This was a pilot study performed under the supervision of the two senior authors at primarily one institution. Due to the rarity of T-PLL, 8 patients were studied over the course of 7 years. Diagnosis of T-PLL was made based on immunophenotype, peripheral blood smear, clinical presentation and laboratory values as outlined previously.(5) Complete remission was defined as the absence of disease detectable by morphology and resolution of splenomegaly/lymphadenopathy confirmed by physical examination and/or CT scanning. Repeat marrows were not obtained in all patients. Molecular remission was defined as both negative flow cytometry for minimal residual disease and no detectable clone by TCR rearrangement using PCR. Partial remissions were defined by 50% or more decrease in circulating tumor cell counts but lack of normalization of other complete remission criteria.(5, 9, 42, 43) Patients with lymphadenopathy or splenomegaly were followed by CT scans (Table S1). Patients were treated until they achieved remission (1–3 cycles), opted to end therapy, or received an allogeneic bone marrow transplant. The end point of this study was allogeneic transplant or death. The objective of this study was to determine if epigenetic based therapies could overcome treatment resistance in T-PLL. One cycle of therapy is 28 days and defined as follows (Figure 1): Alemtuzumab given at 30 mg intravenously on days 1, 3, 5, 8, 10, 12, 15, 17, 19, 22, 24 and 26. Cladribine was given at 5 mg/m2 on days 1–5. Vorinostat was given orally at 400mg a day for days 1–5. Romidepsin was dosed at 14 mg/m2 on days 1, 8 and 15 when used. In the case of CD30 induction and treatment with brentuximab vedotin, 1.8 mg/kg once a week for 3 weeks was used. Pralatrexate dose was 30 mg/m2 IV. Patients were treated with 2–3 cycles of therapy with only alemtuzumab and cladribine initially. If white blood cell counts did not respond, vorinostat was added. After noting that the addition of vorinostat led to better responses, vorinostat was added routinely with alemtuzumab and cladribine given that hematologic and infectious toxicities were manageable with supportive care. Alemtuzumab was continued three times a week for two weeks after patients achieved CR. No maintenance alemtuzumab was used because of risk of opportunistic infections and lack of added benefit.(5) Patients were prophylaxed for infectious complications with sulfamethoxazole/trimethoprim, levofloxacin if neutropenic and daily acyclovir and monitored for infections, especially CMV. Clinical data collected were white blood cell counts, molecular and flow cytometric analyses of the blood and bone marrow, skin biopsies and toxicities. 6 of the 8 patients were consented for collection of blood under a Penn State Hershey IRB approved protocol (No. 2000-186). Blood was collected prior to initiating therapy, at day 3 post-therapy and at day 5 post-therapy. The remaining two patients were treated at other institutions and were not consented for blood collection during their initial treatments. Blood samples from the six consented patients were used for laboratory correlative studies.
White blood cell separation
White blood cells were separated from blood on ficoll®-paque PLUS (GE Healthcare) gradients and either lysed for protein, DNA or RNA. Remaining cells were stored in highly concentrated aliquots (>107 cells) in FBS with 10% DMSO in locked liquid N2 dewars (−195°C).
Patient Photographs
After obtaining written consent, patient photographs were taken using a Canon T2i camera.
Flow Analyses and Immunohistochemistry (IHC)
All clinical flow analyses and IHC analyses of skin biopsies were carried out by the Penn State Hershey Department of Pathology. IHC was done on a Ventana Benchmark XT automated immunoperoxidase stainer. Briefly, slides were cut from paraffin embedded tissue blocks and deparaffinized. Cells were conditioned in pH 8.0 buffer for 30 minutes prior to addition of CD30 antibody at 1:100 dilution for 24 minutes. Slides were then blocked in avidin/biotin blocker and counter stained with hematoxylin eosin stain.
mRNA microarray analysis
mRNA microarray analysis was carried out using a microarray chip (Illumina) and the facilities of the Penn State Genomics Core. cDNA was made from extracted mRNA from four patients before and after therapy using High Capacity cDNA RT kits (AB&I). Only four of the six patients had sufficient quality mRNA for microarray experimentation. Results were analyzed using MS excel and Gene Set Enrichment Analysis (GSEA) from the Broad institute.
Quantitative RT-PCR (qRT-PCR)
White blood cells were isolated, washed and lysed in TRIzol® (Ambion) as per the manufacturer’s instructions. All RNA was then subjected to RQ1 RNase-Free DNase (Promega) in order to eliminate residual DNA. After DNase treatment, samples were reverse transcribed and qRT-PCR was then carried out using SYBR® green (Qiagen) and BLAST designed primers (IDT). Primer sequences can be found in Table S3.
Chromatin Immunoprecipitation (ChIP) Assays
Frozen (90%FBS, 10%DMSO) patient cells were thawed and suspended in 10mL PBS and fixed in 0.4% formaldehyde for 10 min rocking at room temperature. Fixation was stopped by 500μl of 2.5M glycine for 5 min, followed by cold PBS wash. Cells were suspended in 500μl cold lysis buffer (50mM Tris, 10mM EDTA, 1%SDS) with 1:100 protease inhibitor cocktail (Sigma P8340) and incubated on ice for 30 min. Lysates were sonicated using a Misonix Microson XL at 25% power for 6 cycles of 30s on, 30s off, incubated on ice. Insoluble material was cleared with a 10 min, 17,000xg spin. 50μl of input chromatin was diluted to 500μl in dilution buffer (20mM Tris, 2mM EDTA, 150mM NaCl, 1% Triton X-100). 15μl of Magna ChIP™ Protein A and Protein G beads (Millipore) were added, as well as 10ug of specific antibody, H3 Lysine 27 Trimethyl (Millipore 05-1951), H3 Lysine 9 trimethyl (Millipore 07-523), normal rabbit antibody (Santa Cruz sc-2345) or 10μl of histone H3 positive control antibody (Cell Signaling #9715). Mixtures were precipitated overnight at 4°C with rotation, then washed with 750μl of the following buffers, each with 5 min incubation at 4°C with rotation: low salt buffer (50mM NaCl, 0.1% SDS, 1% Triton X-100, 10mM Tris-HCL, 1mM EDTA), high salt buffer (150mM NaCl, 0.1% SDS, 1% Triton X-100, 10mM Tris-HCL, 1mM EDTA), LiCl buffer (250mM LiCl, 1% Deoxycholic Acid, 1%NP40, 10mM Tris-HCL, 1mM EDTA), and twice with TE (10mM Tris-HCL, 1mM EDTA). Following washes, chromatin was eluted from beads in 100μl elution buffer (1% SDS, 100mM NaHCO3) plus 1μl Proteinase K (Qiagen) at 64°C overnight. Eluted DNA was purified using a PCR purification kit (Qiagen). DNA was detected by qRT-PCR using a Quantitect SYBR Green PCR kit (Qiagen) on a Bio-Rad C-1000 real time thermocycler. All ChIP assay PCRs were done in triplicate and independently repeated at least twice.
STAT5B N642H sequencing
Sequencing for the N642H STAT5B mutation was carried out using genomic DNA from patients as template. STAT5B mutation forward and reverse primers (Table S3), spanning an ~250 bp region, were used to amplify out the mutation site using Takara Hot Start Taq Polymerase (Clontech). The reaction was PCR purified (Qiagen) and sent for sequencing (fisher/operon) using the STAT5B mutation reverse primer. Paired normal germ-line controls were unavailable.
DNA methylation analysis by HELP
Genomic DNA was isolated using a standard high-salt procedure. HELP assay, a comparative isoschizomer profiling method interrogating cytosine methylation status on a genomic scale, was carried out as previously described. (57–59) Briefly, genomic DNA from the samples was digested by a methylcytosine-sensitive enzyme, HpaII, and with MspI, a methylcytosine-insensitive enzyme. The HpaII and MspI digested products were amplified by ligation-mediated PCR optimized to amplify fragments between 200 and 2000 base pairs with the preferential amplification of cytosine-phosphate-guanosine (CpG) dinucleotide-dense regions. Each fraction is then labeled with a specific dye and cohybridized onto a human HG17 custom-designed oligonucleotide array (50-mers) covering 25626 HpaII amplifiable fragments (HAFs) located at gene promoters and imprinted regions across the genome.(58) HAFs are defined as genomic sequences between two flanking HpaII sites found within 200 to 2000 bp from each other. Each HAF on the array is represented by 15 individual probes. All samples for microarray hybridization were processed at the Roche-NimbleGen Service Laboratory. PCR fragment length bias was corrected by quantile normalization. Further quality control and data analysis of HELP microarrays were performed as described in Thompson et al.(60)
TUNEL Assay
T-PLL cells were attached to glass slides using a cytospin at 500 rpm for 5 minutes prior to processing and staining with the click-iT TUNEL Alexa Fluor Imaging Assay (Invitrogen: C10245) and accompanying protocol prior to imaging on an Olympus BX 60 upright fluorescent microscope.
Statistical Analyses
All significant values were derived from two tailed student’s T-tests with a cut off of p<0.05 and can be found in Table S4. All experiments were repeated twice. Survival times were calculated from the start of therapy.
Supplementary Material
Figure S1: Cladribine inhibits DNA methylation in vivo and histone methylation in vitro
Figure S2: Epigenetic therapy does not induce apoptosis in T-PLL
Figure S3: Epigenetic therapy lowered leukemic white blood cell counts despite multiple relapses
Figure S4: CD52 levels were unchanged by epigenetic therapy
Figure S5: CD30 induction was not observed in circulating mantle cell lymphoma cells
Figure S6: Skin biopsy from patient 5 shows CD52 positive dermis infiltrating lymphocytes
Figure S7: Epigenetic treatment of T-PLL causes mRNA expression changes in the HIN-200, CEBP and tribbles families
Figure S8: All tested patients showed CDKN1B haploinsufficiency and 4/6 were TCL1 positive
Figure S9: 40% of patients were positive for the N642H STAT5B Mutation
Table S1: Patient presentation at diagnosis
Table S2: Patient characteristics and adverse events
Table S3: Primer sequences
Table S4: Significant p-values for all statistically significant comparisons in figures
Acknowledgments
We thank the Epigenomics and Genomics Cores of the Albert Einstein College of Medicine for expert technical assistance with HELP data. We thank Rob Brucklacher and the Penn State Genomics Core along with Tom Olson and Xin Liu for microarray experimentation and analysis.
Funding: Supported by the Chemotherapy Foundation to S.P., Gabrielle’s Angel Foundation to S.P., Leukemia and Lymphoma Society Translational Research Project Grant to S.P., Paul Calabresi Career Development Award K12-CA132783-01 to S.P, grants to E.E. from the Lymphoma Research Foundation and Tobacco Settlement funds of PA and RO1- CA098472 from the National Cancer Institute to T.P.L.
Footnotes
Author Contributions: Z.S.H and E.E. designed experiments, collected and interpreted data, prepared the manuscript and clinical case studies; K.S. and B.S helped with manuscript and clinical case study preparation; T.P.L and S.P. were involved with data interpretation and manuscript preparation; A.S., S.S., M.S., V.L. and S.P. were involved with data collection and data interpretation; J.E. performed and interpreted tissue IHC.
Competing Interests: E.E. and K.S. Speakers Bureau Celgene; E.E. Speakers Bureau Seattle Genetics
Data and materials availability: The microarray data for this study has been deposited in the GEO database (GSE67368).
References
- 1.Robak T, Robak P. Current treatment options in prolymphocytic leukemia. Med Sci Monit. 2007;13:RA69–80. [PubMed] [Google Scholar]
- 2.Dearden CE. T-cell prolymphocytic leukemia. Med Oncol. 2006;23:17–22. doi: 10.1385/MO:23:1:17. [DOI] [PubMed] [Google Scholar]
- 3.Matutes E, Brito-Babapulle V, Swansbury J, Ellis J, Morilla R, Dearden C, Sempere A, Catovsky D. Clinical and laboratory features of 78 cases of T-prolymphocytic leukemia. Blood. 1991;78:3269–3274. [PubMed] [Google Scholar]
- 4.Pawson R, Schulz TF, Matutes E, Catovsky D. The human T-cell lymphotropic viruses types I/II are not involved in T prolymphocytic leukemia and large granular lymphocytic leukemia. Leukemia. 1997;11:1305–1311. doi: 10.1038/sj.leu.2400725. [DOI] [PubMed] [Google Scholar]
- 5.Dearden C. How I treat prolymphocytic leukemia. Blood. 2012;120:538–551. doi: 10.1182/blood-2012-01-380139. [DOI] [PubMed] [Google Scholar]
- 6.Mercieca J, Matutes E, Dearden C, MacLennan K, Catovsky D. The role of pentostatin in the treatment of T-cell malignancies: analysis of response rate in 145 patients according to disease subtype. J Clin Oncol. 1994;12:2588–2593. doi: 10.1200/JCO.1994.12.12.2588. [DOI] [PubMed] [Google Scholar]
- 7.Spurgeon S, Yu M, Phillips JD, Epner EM. Cladribine: not just another purine analogue? Expert Opin Investig Drugs. 2009;18:1169–1181. doi: 10.1517/13543780903071038. [DOI] [PubMed] [Google Scholar]
- 8.Ginaldi L, De Martinis M, Matutes E, Farahat N, Morilla R, Dyer MJ, Catovsky D. Levels of expression of CD52 in normal and leukemic B and T cells: correlation with in vivo therapeutic responses to Campath-1H. Leuk Res. 1998;22:185–191. doi: 10.1016/s0145-2126(97)00158-6. [DOI] [PubMed] [Google Scholar]
- 9.Keating MJ, Cazin B, Coutre S, Birhiray R, Kovacsovics T, Langer W, Leber B, Maughan T, Rai K, Tjonnfjord G, Bekradda M, Itzhaki M, Herait P. Campath-1H treatment of T-cell prolymphocytic leukemia in patients for whom at least one prior chemotherapy regimen has failed. J Clin Oncol. 2002;20:205–213. doi: 10.1200/JCO.2002.20.1.205. [DOI] [PubMed] [Google Scholar]
- 10.Lin HY, Chen CS, Lin SP, Weng JR. Targeting histone deacetylase in cancer therapy. Med Res Rev. 2006;26:397–413. doi: 10.1002/med.20056. [DOI] [PubMed] [Google Scholar]
- 11.Fukuda H, Sano N, Muto S, Horikoshi M. Simple histone acetylation plays a complex role in the regulation of gene expression. Brief Funct Genomic Proteomic. 2006;5:190–208. doi: 10.1093/bfgp/ell032. [DOI] [PubMed] [Google Scholar]
- 12.Wyczechowska D, Fabianowska-Majewska K. The effects of cladribine and fludarabine on DNA methylation in K562 cells. Biochem Pharmacol. 2003;65:219–225. doi: 10.1016/s0006-2952(02)01486-7. [DOI] [PubMed] [Google Scholar]
- 13.Gowher H, Jeltsch A. Mechanism of inhibition of DNA methyltransferases by cytidine analogs in cancer therapy. Cancer Biol Ther. 2004;3:1062–1068. doi: 10.4161/cbt.3.11.1308. [DOI] [PubMed] [Google Scholar]
- 14.Khan O, La Thangue NB. HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol Cell Biol. 2012;90:85–94. doi: 10.1038/icb.2011.100. [DOI] [PubMed] [Google Scholar]
- 15.Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet. 1999;21:103–107. doi: 10.1038/5047. [DOI] [PubMed] [Google Scholar]
- 16.Lee SY, Park CG, Choi Y. T cell receptor-dependent cell death of T cell hybridomas mediated by the CD30 cytoplasmic domain in association with tumor necrosis factor receptor-associated factors. J Exp Med. 1996;183:669–674. doi: 10.1084/jem.183.2.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han S, Koo J, Bae J, Kim S, Baik S, Kim MY. Modulation of TNFSF expression in lymphoid tissue inducer cells by dendritic cells activated with Toll-like receptor ligands. BMB Rep. 2011;44:129–134. doi: 10.5483/BMBRep.2011.44.2.129. [DOI] [PubMed] [Google Scholar]
- 18.Biswas P, Galli A, Capiluppi B, Ciuffreda D, Lazzarin A, Tambussi G. Selective enrichment of CD30-expressing cells within the blast region of lymphocytes from patients with primary HIV infection (PHI) J Biol Regul Homeost Agents. 2002;16:33–36. [PubMed] [Google Scholar]
- 19.Saini D, Ramachandran S, Nataraju A, Benshoff N, Liu W, Desai N, Chapman W, Mohanakumar T. Activated effector and memory T cells contribute to circulating sCD30: potential marker for islet allograft rejection. Am J Transplant. 2008;8:1798–1808. doi: 10.1111/j.1600-6143.2008.02329.x. [DOI] [PubMed] [Google Scholar]
- 20.Halim MA, Al-Otaibi T, Al-Muzairai I, Mansour M, Tawab KA, Awadain WH, Balaha MA, Said T, Nair P, Nampoory MR. Serial soluble CD30 measurements as a predictor of kidney graft outcome. Transplant Proc. 2010;42:801–803. doi: 10.1016/j.transproceed.2010.03.033. [DOI] [PubMed] [Google Scholar]
- 21.Kocabas E, Turel Ermertcan A, Akinci S, Temiz P, Gunduz K. Primary cutaneous CD30-positive anaplastic large cell lymphoma in a 16-year-old girl. Int J Dermatol. 2011 doi: 10.1111/j.1365-4632.2011.05157.x. [DOI] [PubMed] [Google Scholar]
- 22.Hussain R, Bajoghli A. Primary Cutaneous CD30-Positive Large T-Cell Lymphoma in an 80-Year-Old Man: A Case Report. ISRN Dermatol. 2011;2011:634042. doi: 10.5402/2011/634042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maric I, Calvo KR. Mastocytosis: the new differential diagnosis of CD30-positive neoplasms. Leuk Lymphoma. 2011;52:732–733. doi: 10.3109/10428194.2011.561389. [DOI] [PubMed] [Google Scholar]
- 24.Tarkowski M. Expression and a role of CD30 in regulation of T-cell activity. Curr Opin Hematol. 2003;10:267–271. doi: 10.1097/00062752-200307000-00003. [DOI] [PubMed] [Google Scholar]
- 25.Francisco JA, Cerveny CG, Meyer DL, Mixan BJ, Klussman K, Chace DF, Rejniak SX, Gordon KA, DeBlanc R, Toki BE, Law CL, Doronina SO, Siegall CB, Senter PD, Wahl AF. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood. 2003;102:1458–1465. doi: 10.1182/blood-2003-01-0039. [DOI] [PubMed] [Google Scholar]
- 26.Doronina SO, Toki BE, Torgov MY, Mendelsohn BA, Cerveny CG, Chace DF, DeBlanc RL, Gearing RP, Bovee TD, Siegall CB, Francisco JA, Wahl AF, Meyer DL, Senter PD. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat Biotechnol. 2003;21:778–784. doi: 10.1038/nbt832. [DOI] [PubMed] [Google Scholar]
- 27.Younes A, Yasothan U, Kirkpatrick P. Brentuximab vedotin. Nat Rev Drug Discov. 2012;11:19–20. doi: 10.1038/nrd3629. [DOI] [PubMed] [Google Scholar]
- 28.Leshchenko VV, Hasanali Z, Stuart A, Shimko S, Spurgeon SE, Parekh S, Epner EM. Combined Epigenetic and Immunotherapy For Newly Diagnosed Mantle Cell Lymphoma: Correlative Studies Suggest The Importance Of Enhanced ADCC, Mechanisms of Resistance and Cyclin D1 Nuclear Localization Genotype. 2013 Annual Meeting Abstracts, American Society of Hematology; New Orleans, LA. Dec. 7–10 2013; p. abstract 3063. [Google Scholar]
- 29.Sharma Kamal, Hasanali Zainul, Spurgeon Stephen, Okada Craig, Stuart August, Shimko Sara, Leshchenko Violetta, Parekh Samir, Chen Yiyi, Kirschbaum Mark, Epner Elliot M. Proceedings: AACR 104th Annual Meeting; 2013; Washington, D.C: American Association for Cancer Research; Apr 6–10, 2013. p. abstract LBA140. [Google Scholar]
- 30.Sun X, Hasanali ZS, Chen A, Zhang D, Liu X, Wang HG, Feith DJ, Loughran TP, Jr, Xu K. Suberoylanilide hydroxamic acid (SAHA) and cladribine synergistically induce apoptosis in NK-LGL leukaemia. Br J Haematol. 2014 doi: 10.1111/bjh.13143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krawczyk B, Rudnicka K, Fabianowska-Majewska K. The effects of nucleoside analogues on promoter methylation of selected tumor suppressor genes in MCF-7 and MDA-MB-231 breast cancer cell lines. Nucleosides Nucleotides Nucleic Acids. 2007;26:1043–1046. doi: 10.1080/15257770701509594. [DOI] [PubMed] [Google Scholar]
- 32.Gottlicher M, Minucci S, Zhu P, Kramer OH, Schimpf A, Giavara S, Sleeman JP, Lo F, Nervi C, Pelicci PG, Heinzel T. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001;20:6969–6978. doi: 10.1093/emboj/20.24.6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cuker A, Coles AJ, Sullivan H, Fox E, Goldberg M, Oyuela P, Purvis A, Beardsley DS, Margolin DH. A distinctive form of immune thrombocytopenia in a phase 2 study of alemtuzumab for the treatment of relapsing-remitting multiple sclerosis. Blood. 2011;118:6299–6305. doi: 10.1182/blood-2011-08-371138. [DOI] [PubMed] [Google Scholar]
- 34.Ludlow LE, Johnstone RW, Clarke CJ. The HIN-200 family: more than interferon-inducible genes? Exp Cell Res. 2005;308:1–17. doi: 10.1016/j.yexcr.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 35.Tsukada J, Yoshida Y, Kominato Y, Auron PE. The CCAAT/enhancer (C/EBP) family of basic-leucine zipper (bZIP) transcription factors is a multifaceted highly-regulated system for gene regulation. Cytokine. 2011;54:6–19. doi: 10.1016/j.cyto.2010.12.019. [DOI] [PubMed] [Google Scholar]
- 36.Kim J, Kim H. Recruitment and biological consequences of histone modification of H3K27me3 and H3K9me3. ILAR journal/National Research Council, Institute of Laboratory Animal Resources. 2012;53:232–239. doi: 10.1093/ilar.53.3-4.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Le Toriellec E, Despouy G, Pierron G, Gaye N, Joiner M, Bellanger D, Vincent-Salomon A, Stern MH. Haploinsufficiency of CDKN1B contributes to leukemogenesis in T-cell prolymphocytic leukemia. Blood. 2008;111:2321–2328. doi: 10.1182/blood-2007-06-095570. [DOI] [PubMed] [Google Scholar]
- 38.Yokohama A, Saitoh A, Nakahashi H, Mitsui T, Koiso H, Kim Y, Uchiumi H, Saitoh T, Handa H, Jimbo T, Murayama K, Sakura T, Murakami H, Karasawa M, Nojima Y, Tsukamoto N. TCL1A gene involvement in T-cell prolymphocytic leukemia in Japanese patients. Int J Hematol. 2012;95:77–85. doi: 10.1007/s12185-011-0986-5. [DOI] [PubMed] [Google Scholar]
- 39.De Schouwer PJ, Dyer MJ, Brito-Babapulle VB, Matutes E, Catovsky D, Yuille MR. T-cell prolymphocytic leukaemia: antigen receptor gene rearrangement and a novel mode of MTCP1 B1 activation. Br J Haematol. 2000;110:831–838. doi: 10.1046/j.1365-2141.2000.02256.x. [DOI] [PubMed] [Google Scholar]
- 40.Roos J, Hennig I, Schwaller J, Zbaren J, Dummer R, Burg G, Tobler A, Virgilio L, Croce CM, Fey MF, Borisch B. Expression of TCL1 in hematologic disorders. Pathobiology: journal of immunopathology, molecular and cellular biology. 2001;69:59–66. doi: 10.1159/000048758. [DOI] [PubMed] [Google Scholar]
- 41.Pekarsky Y, Hallas C, Croce CM. Targeting mature T cell leukemia: new understanding of molecular pathways. American journal of pharmacogenomics: genomics-related research in drug development and clinical practice. 2003;3:31–36. doi: 10.2165/00129785-200303010-00005. [DOI] [PubMed] [Google Scholar]
- 42.Dearden CE, Khot A, Else M, Hamblin M, Grand E, Roy A, Hewamana S, Matutes E, Catovsky D. Alemtuzumab therapy in T-cell prolymphocytic leukemia: comparing efficacy in a series treated intravenously and a study piloting the subcutaneous route. Blood. 2011;118:5799–5802. doi: 10.1182/blood-2011-08-372854. [DOI] [PubMed] [Google Scholar]
- 43.Dearden CE, Matutes E, Cazin B, Tjonnfjord GE, Parreira A, Nomdedeu B, Leoni P, Clark FJ, Radia D, Rassam SM, Roques T, Ketterer N, Brito-Babapulle V, Dyer MJ, Catovsky D. High remission rate in T-cell prolymphocytic leukemia with CAMPATH-1H. Blood. 2001;98:1721–1726. doi: 10.1182/blood.v98.6.1721. [DOI] [PubMed] [Google Scholar]
- 44.Hasanali ZS, Epner EM, Feith DJ, Loughran TP, Jr, Sample CE. Vorinostat Downregulates CD30 and Decreases Brentuximab Vedotin Efficacy in Human Lymphocytes. Mol Cancer Ther. 2014 doi: 10.1158/1535-7163.MCT-14-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clark RA, Watanabe R, Teague JE, Schlapbach C, Tawa MC, Adams N, Dorosario AA, Chaney KS, Cutler CS, Leboeuf NR, Carter JB, Fisher DC, Kupper TS. Skin effector memory T cells do not recirculate and provide immune protection in alemtuzumab-treated CTCL patients. Sci Transl Med. 2012;4:117ra117. doi: 10.1126/scitranslmed.3003008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gade P, Roy SK, Li H, Nallar SC, Kalvakolanu DV. Critical role for transcription factor C/EBP-beta in regulating the expression of death-associated protein kinase 1. Mol Cell Biol. 2008;28:2528–2548. doi: 10.1128/MCB.00784-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li H, Gade P, Xiao W, Kalvakolanu DV. The interferon signaling network and transcription factor C/EBP-beta. Cellular & molecular immunology. 2007;4:407–418. [PMC free article] [PubMed] [Google Scholar]
- 48.Kiss-Toth E, Bagstaff SM, Sung HY, Jozsa V, Dempsey C, Caunt JC, Oxley KM, Wyllie DH, Polgar T, Harte M, O’Neill LA, Qwarnstrom EE, Dower SK. Human tribbles, a protein family controlling mitogen-activated protein kinase cascades. J Biol Chem. 2004;279:42703–42708. doi: 10.1074/jbc.M407732200. [DOI] [PubMed] [Google Scholar]
- 49.Sung HY, Francis SE, Crossman DC, Kiss-Toth E. Regulation of expression and signalling modulator function of mammalian tribbles is cell-type specific. Immunol Lett. 2006;104:171–177. doi: 10.1016/j.imlet.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 50.Yamamoto M, Uematsu S, Okamoto T, Matsuura Y, Sato S, Kumar H, Satoh T, Saitoh T, Takeda K, Ishii KJ, Takeuchi O, Kawai T, Akira S. Enhanced TLR-mediated NF-IL6 dependent gene expression by Trib1 deficiency. J Exp Med. 2007;204:2233–2239. doi: 10.1084/jem.20070183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xaus J, Cardo M, Valledor AF, Soler C, Lloberas J, Celada A. Interferon gamma induces the expression of p21waf-1 and arrests macrophage cell cycle, preventing induction of apoptosis. Immunity. 1999;11:103–113. doi: 10.1016/s1074-7613(00)80085-0. [DOI] [PubMed] [Google Scholar]
- 52.Mogensen KE, Bandu MT. Kinetic evidence for an activation step following binding of human interferon alpha 2 to the membrane receptors of Daudi cells. Eur J Biochem. 1983;134:355–364. doi: 10.1111/j.1432-1033.1983.tb07575.x. [DOI] [PubMed] [Google Scholar]
- 53.Rajala HL, Eldfors S, Kuusanmaki H, van Adrichem AJ, Olson T, Lagstrom S, Andersson EI, Jerez A, Clemente MJ, Yan Y, Zhang D, Awwad A, Ellonen P, Kallioniemi O, Wennerberg K, Porkka K, Maciejewski JP, Loughran TP, Jr, Heckman C, Mustjoki S. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood. 2013;121:4541–4550. doi: 10.1182/blood-2012-12-474577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bandapalli OR, Schuessele S, Kunz JB, Rausch T, Stutz AM, Tal N, Geron I, Gershman N, Izraeli S, Eilers J, Vaezipour N, Kirschner-Schwabe R, Hof J, von Stackelberg A, Schrappe M, Stanulla M, Zimmermann M, Koehler R, Avigad S, Handgretinger R, Frismantas V, Bourquin JP, Bornhauser B, Korbel JO, Muckenthaler MU, Kulozik AE. The activating STAT5B N642H mutation is a common abnormality in pediatric T-cell acute lymphoblastic leukemia and confers a higher risk of relapse. Haematologica. 2014;99:e188–192. doi: 10.3324/haematol.2014.104992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kucuk C, Jiang B, Hu X, Zhang W, Chan JK, Xiao W, Lack N, Alkan C, Williams JC, Avery KN, Kavak P, Scuto A, Sen E, Gaulard P, Staudt L, Iqbal J, Cornish A, Gong Q, Yang Q, Sun H, d’Amore F, Leppa S, Liu W, Fu K, de Leval L, McKeithan T, Chan WC. Activating mutations of STAT5B and STAT3 in lymphomas derived from gammadelta-T or NK cells. Nature communications. 2015;6:6025. doi: 10.1038/ncomms7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kiel MJ, Velusamy T, Rolland D, Sahasrabuddhe AA, Chung F, Bailey NG, Schrader A, Li B, Li JZ, Ozel AB, Betz BL, Miranda RN, Medeiros LJ, Zhao L, Herling M, Lim MS, Elenitoba-Johnson KS. Integrated genomic sequencing reveals mutational landscape of T-cell prolymphocytic leukemia. Blood. 2014 doi: 10.1182/blood-2014-03-559542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Khulan B, Thompson RF, Ye K, Fazzari MJ, Suzuki M, Stasiek E, Figueroa ME, Glass JL, Chen Q, Montagna C, Hatchwell E, Selzer RR, Richmond TA, Green RD, Melnick A, Greally JM. Comparative isoschizomer profiling of cytosine methylation: the HELP assay. Genome research. 2006;16:1046–1055. doi: 10.1101/gr.5273806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shaknovich R, Figueroa ME, Melnick A. HELP (HpaII tiny fragment enrichment by ligation-mediated PCR) assay for DNA methylation profiling of primary normal and malignant B lymphocytes. Methods Mol Biol. 2010;632:191–201. doi: 10.1007/978-1-60761-663-4_12. [DOI] [PubMed] [Google Scholar]
- 59.Leshchenko VV, Kuo PY, Shaknovich R, Yang DT, Gellen T, Petrich A, Yu Y, Remache Y, Weniger MA, Rafiq S, Suh KS, Goy A, Wilson W, Verma A, Braunschweig I, Muthusamy N, Kahl BS, Byrd JC, Wiestner A, Melnick A, Parekh S. Genomewide DNA methylation analysis reveals novel targets for drug development in mantle cell lymphoma. Blood. 2010;116:1025–1034. doi: 10.1182/blood-2009-12-257485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thompson RF, Suzuki M, Lau KW, Greally JM. A pipeline for the quantitative analysis of CG dinucleotide methylation using mass spectrometry. Bioinformatics. 2009;25:2164–2170. doi: 10.1093/bioinformatics/btp382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tuset E, Matutes E, Brito-Babapulle V, Morilla R, Catovsky D. Immunophenotype changes and loss of CD52 expression in two patients with relapsed T-cell prolymphocytic leukaemia. Leuk Lymphoma. 2001;42:1379–1383. doi: 10.3109/10428190109097766. [DOI] [PubMed] [Google Scholar]
- 62.Gocmen S, Kutlay M, Erikci A, Atabey C, Sayan O, Haholu A. Central nervous system involvement of T-cell prolymphocytic leukemia diagnosed with stereotactic brain biopsy: case report. Turkish journal of haematology: official journal of Turkish Society of Haematology. 2014;31:75–78. doi: 10.4274/Tjh.2012.0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Cladribine inhibits DNA methylation in vivo and histone methylation in vitro
Figure S2: Epigenetic therapy does not induce apoptosis in T-PLL
Figure S3: Epigenetic therapy lowered leukemic white blood cell counts despite multiple relapses
Figure S4: CD52 levels were unchanged by epigenetic therapy
Figure S5: CD30 induction was not observed in circulating mantle cell lymphoma cells
Figure S6: Skin biopsy from patient 5 shows CD52 positive dermis infiltrating lymphocytes
Figure S7: Epigenetic treatment of T-PLL causes mRNA expression changes in the HIN-200, CEBP and tribbles families
Figure S8: All tested patients showed CDKN1B haploinsufficiency and 4/6 were TCL1 positive
Figure S9: 40% of patients were positive for the N642H STAT5B Mutation
Table S1: Patient presentation at diagnosis
Table S2: Patient characteristics and adverse events
Table S3: Primer sequences
Table S4: Significant p-values for all statistically significant comparisons in figures