

Abstract
Objective:
To determine the molecular basis of a severe neurologic disorder in a large consanguineous family with complete agenesis of the corpus callosum (ACC), pontocerebellar hypoplasia (PCH), and peripheral axonal neuropathy.
Methods:
Assessment included clinical evaluation, neuroimaging, and nerve conduction studies (NCSs). Linkage analysis used genotypes from 7 family members, and the exome of 3 affected siblings was sequenced. Molecular analyses used Sanger sequencing to perform segregation studies and cohort analysis and Western blot of patient-derived cells.
Results:
Affected family members presented with postnatal microcephaly and profound developmental delay, with early death in 3. Neuroimaging, including a fetal MRI at 30 weeks, showed complete ACC and PCH. Clinical evaluation showed areflexia, and NCSs revealed a severe axonal neuropathy in the 2 individuals available for electrophysiologic study. A novel homozygous stopgain mutation in adenosine monophosphate deaminase 2 (AMPD2) was identified within the linkage region on chromosome 1. Molecular analyses confirmed that the mutation segregated with disease and resulted in the loss of AMPD2. Subsequent screening of a cohort of 42 unrelated individuals with related imaging phenotypes did not reveal additional AMPD2 mutations.
Conclusions:
We describe a family with a novel stopgain mutation in AMPD2. We expand the phenotype recently described as PCH type 9 to include progressive postnatal microcephaly, complete ACC, and peripheral axonal neuropathy. Screening of additional individuals with related imaging phenotypes failed to identify mutations in AMPD2, suggesting that AMPD2 mutations are not a common cause of combined callosal and pontocerebellar defects.
Callosal malformations include complete or partial agenesis, hypoplasia, and dysgenesis.1,2 Clinical sequelae range from no or mild manifestations to severe neurodevelopmental disability.3,4 The latter is more common when the callosal malformation is accompanied by other brain abnormalities, is part of a multiple congenital anomaly syndrome, or is secondary to a metabolic or degenerative disorder.5 Agenesis of the corpus callosum (ACC) occurs in approximately 1:5,000 live births and >80 congenital syndromes,6,7 although the etiology remains unknown for the majority. Pontocerebellar hypoplasia (PCH) syndromes are rare, usually autosomal recessive, disorders characterized by degeneration of the cerebellum and brainstem. The term “hypoplasia” is used although the changes are thought to reflect prenatal onset of atrophy.8 Ten PCH syndromes have been described with additional features that variably include microcephaly, a movement disorder, epilepsy, optic atrophy, and axonal neuropathy.
The combination of corpus callosum (CC) hypoplasia and PCH secondary to mutations in the gene encoding adenosine monophosphate deaminase 2 (AMPD2) was recently reported (PCH type 9, Online Mendelian Inheritance in Man [OMIM] #615809).9 The authors described 8 affected individuals from 5 families with severe disabilities, hypoplasia of the CC, and a characteristic midbrain “figure of 8” appearance on axial MRI. AMPD2 encodes 1 of 3 of adenosine monophosphate (AMP) deaminase homologs, which convert AMP to inosine monophosphate. Guanine nucleotide deficiency was shown to be central to the pathogenesis of PCH9 and associated with defective protein synthesis.9 In this study, we characterize a large consanguineous Middle Eastern family and expand the phenotype of PCH9, describing the novel combination of complete ACC, PCH, and an axonal neuropathy.
METHODS
Standard protocol approvals, registrations, and patient consents.
Study approval was provided by the Royal Children's Hospital Research Ethics Committee (Human Research Ethics Committee #28097), with informed consent provided by participants or their guardians.
Clinical and molecular studies.
Clinical information was obtained by review of medical records and examination of affected individuals. Neuroimaging was reviewed and included cranial ultrasound, head CT, and brain MRI. Nerve conduction studies (NCSs) were performed using a 4-channel Keypoint Electromyography machine (Natus Medical Incorporated, Pleasanton, CA), as previously described.10 Genomic DNA was isolated from peripheral blood. Single nucleotide polymorphism (SNP) genotype data were generated with the Human610-Quad BeadChip Kit (Illumina, San Diego, CA) for 7 family members (I-1, I-2, II-2, II-3, II-5, II-6, and II-8) and extracted from the whole-exome sequence (WES) data of individual II-9. Genotypes were processed using LINKDATAGEN. Parametric multipoint linkage analysis was subsequently performed using MERLIN,11 specifying a rare recessive disease model. Genomic DNA derived from 3 affected siblings (II-2, II-5, and II-9) was enriched using an Illumina TruSeq capture. Sequencing was performed by Otogenetics Corporation (Norcross, GA) with 100-base paired-end reads on an Illumina HiSeq 2000 platform. Reads were aligned to the reference human genome (GRch37/hg19) with ambiguous nucleotide codes at dbSNP132 locations using Novoalign (www.novocraft.com). Variants were called using SAMtools 0.1.17 and annotated using ANNOVAR. The candidate list was filtered to retain rare variants (minor allele frequency ≤0.01) within the linkage regions using public databases and our own in-house database of 132 unrelated exomes. Sanger sequencing confirmed segregation. A cohort of 42 additional individuals with imaging features of complete or partial ACC and brainstem and/or cerebellar hypoplasia was ascertained. Potentially causative copy number variation had previously been excluded by SNP array (data not shown). Sanger sequence analysis of AMPD2 was performed by PCR amplification using M13-tagged primers (primer sequences available on request). Western blot analysis was performed as previously described12 using monoclonal anti-AMPD2 (1:1000 HPA036471, Sigma-Aldrich, St. Louis, MO) and anti-β-actin antibody (1:10000 A5441, Sigma-Aldrich). The reference sequences used for AMPD2 were NG_034075.1, NM_001257360.1, and NP_001244289.1.
RESULTS
We describe a Middle Eastern family with 5 affected offspring from a first cousin marriage (figure 1A). Clinical and imaging details are summarized in table 1. Affected individuals had severe neurodevelopmental disability of early onset, with early death in 3 of 5. All had a normal head circumference at birth but developed microcephaly within the first year and were hypereflexic in the first decade. Dysmorphic features in the 3 patients examined (II-2, II-5, and II-9) included bitemporal narrowing, hypotonic facies, midface hypoplasia, short or downslanting palpebral fissures, a high or broad nasal bridge with a prominent columella, a short upper lip, macroglossia, abnormal ears with a thickened antihelix and a grooved or folded lobe, tapering fingers with dimples over the knuckles, camptodactyly of the distal interphalangeal joints, a few large pigmented nevi, and trophic skin changes of the feet. The 2 surviving individuals (II-2 and II-5) were later noted to have absent lower limb reflexes and distal lower extremity wasting. NCSs performed on upper and lower extremities in these individuals (ages 10 and 20 years) showed low-amplitude or absent motor responses with a mild reduction in motor nerve conduction velocity. Sensory responses were normal in both. EMG showed positive sharp waves and markedly reduced recruitment of high-amplitude polyphasic motor unit potentials, consistent with either a motor axonal neuropathy or neuronopathy. Neuroimaging comprised brain MRI in 2, head CT in 2, and cranial ultrasound in 1. Complete ACC and hypoplasia of the cerebellum and/or brainstem were confirmed in all. Fetal MRI in one showed that all of the brain abnormalities were present at 30 weeks' gestation (figure 2), suggesting prenatal onset of PCH.
Figure 1. Novel stopgain mutation in adenosine monophosphate deaminase 2 segregates with disease in family ACC1 and causes complete loss of protein.
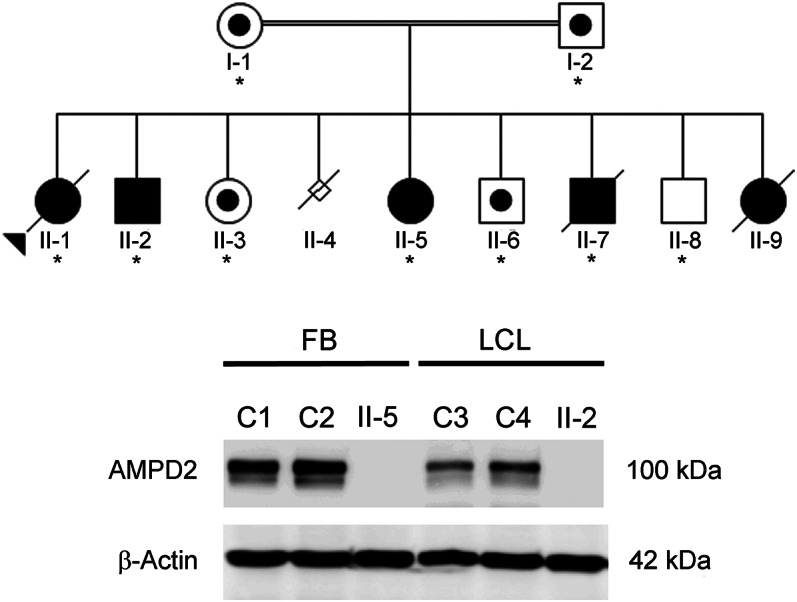
(A) The ACC1 family pedigree is shown with affected individuals shaded in black and unaffected individuals in white. A dot in the pedigree symbol indicates an unaffected carrier. The proband is indicated by an arrowhead. Family members whose DNA was tested are indicated by an asterisk. Homozygosity for the adenosine monophosphate deaminase 2 (AMPD2) mutation segregated with disease in all family members tested. (B) Immunoblot analysis with an AMPD2-specific antibody identified a ∼100-kDa protein in control fibroblasts (C1, C2) and control lymphoblasts (C3, C4) that was absent in extracts derived from affected individuals II-5 (fibroblasts) and II-2 (lymphoblasts). An antibody directed against β-actin confirmed equivalent protein loading.
Table 1.
Clinical phenotype and investigation of family ACC1
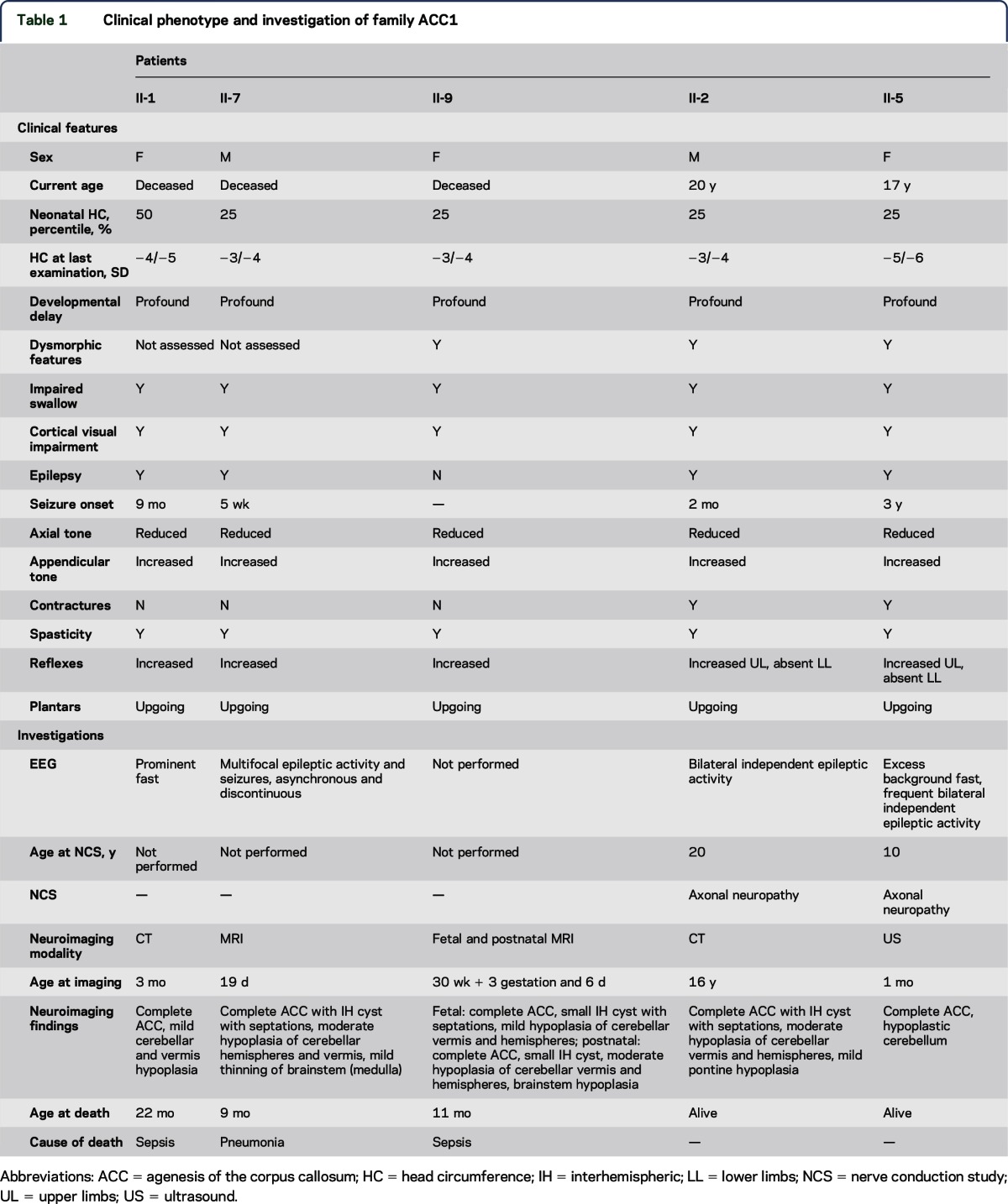
Figure 2. Loss of adenosine monophosphate deaminase 2 results in progressive loss of brainstem and cerebellar parenchyma with prenatal onset.

Axial (top row), coronal (middle row), and midline sagittal (bottom row) T1- and T2-weighted images. All images show a characteristic “figure of 8” appearance to the midbrain (circles), hypoplasia/atrophy of the brainstem (arrows) and cerebellar hemispheres > vermis, and complete agenesis of the corpus callosum. Comparison of the fetal and postnatal MRIs of patient II-9 suggests progressive loss of volume of the pontine belly in the last trimester.
The clinical features suggested a potentially novel genetic cause; therefore, a linkage-gene discovery strategy was used. Two regions on chromosome 1 achieved the maximal autosomal logarithm of the odds score of 2.73 (figure e-1 at Neurology.org/ng). WES and in silico filtering identified 1 variant within the linkage regions, homozygous in all 3 affected siblings, that was predicted to be damaging to the protein sequence. The novel stopgain mutation (NM_001257360.1:c.2256C>G, p.Tyr752*) in AMPD2 (figure e-1) was subsequently shown to segregate with disease in the extended family. Western blot analysis of control cells using a monoclonal antibody directed against AMPD2 identified a single band of the expected size. However, this protein was absent in fibroblast and lymphoblast cell extracts derived from affected individuals (figure 1B). Sequence analysis of AMPD2 was performed in a cohort of 42 unrelated individuals with a neuroimaging phenotype of combined abnormalities of the CC and cerebellum. A number of known polymorphisms were observed (table e-1), but no causative mutations were identified.
DISCUSSION
We used linkage mapping and WES to identify loss of function of AMPD2 as the molecular basis of a severe neurologic disorder in a consanguineous family presenting with progressive postnatal microcephaly, complete ACC, PCH, and a motor axonal neuropathy/neuronopathy. AMPD2 mutations have recently been shown to cause PCH9, a disorder with clinical features that overlap with those of our family (table 1).9 Fetal and postnatal MRI support a prenatal onset to the neurodegenerative process in PCH9, as is postulated for other PCH forms.8 Like all reported individuals with PCH9, the mutation in our family is within the conserved AMP deaminase domain (figure 3). It is currently unclear why our patients display a severe phenotype with both central and peripheral neurologic manifestations. The 8 previously reported patients are described as having hypoplasia of the CC, whereas our patients have complete ACC. Although there is no specific mention of neurophysiologic testing of the previously reported patients, all were noted to have brisk or hyperactive reflexes. The areflexia seen in our family was only noted after the second decade, suggesting age-dependent onset. In the absence of autopsy findings, it is not possible to determine whether the lower motor neuron involvement identified in our family reflects a pure motor axonal neuropathy or an anterior horn cell disorder similar to that described in PCH1 (OMIM #607596).
Figure 3. Schematic representation of the structure of the 5 predicted adenosine monophosphate deaminase 2 transcripts and exonic distribution of all mutations reported to date.

The protein-coding sequence is colored yellow, the conserved adenosine monophosphate (AMP) deaminase domain (also protein coding) purple, and untranslated regions red. All pontocerebellar hypoplasia type 9 mutations reside within the catalytic AMP deaminase domain and therefore affect all 5 protein isoforms, while the one spastic paraplegia type 63 mutation falls outside this region and disrupts the coding region of only 3 of 5 adenosine monophosphate deaminase 2 transcripts. aNovarino et al.13 bAkizu et al.9 cACC1.
A frameshift mutation in AMPD2 was recently reported to cause hereditary spastic paraplegia type 63 (OMIM #615686) in a single family.13 The clinical presentation was relatively mild, with normal cognition, normal NCSs, and no cerebellar signs. MRI of 1 affected individual identified thinning of the CC and periventricular deep white matter changes but no brainstem or cerebellar abnormalities. The identified truncation [p.(Cys107Alafs*80)] disrupts only 3 of 5 predicted AMPD2 isoforms (figure 3), whereas disruption of the deaminase domain affects all, suggesting functional redundancy of isoforms as a molecular explanation for variability in disease severity. Functional redundancy has been observed in mutant mice. Targeted knockout of Ampd2 resulted in proteinuria but no overt brain phenotype,13 whereas a severely shortened lifespan and a neurodegenerative phenotype were observed in mice when 2 AMP deaminase homologs (Ampd2 and Ampd3) were knocked out.9
This is the second report of mutations in AMPD2 as a cause of a callosal abnormality associated with PCH and is the largest multiplex pedigree reported with this disorder. The clinical phenotype associated with loss of AMPD2 function extends from spastic paraplegia to PCH with hypoplasia of the CC, PCH with complete ACC, and motor neuropathy/neuronopathy. Our patients expand the spectrum of emerging PCH disorders with combined callosal, brainstem, and peripheral nervous system manifestations.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the family for participating in this study. They are grateful for the generous support of the Lefroy and Handbury families. They thank Greta Gillies, Hayley Mountford, and Elizabeth Fitzpatrick (MCRI) for assistance with patient recruitment and sample preparation.
GLOSSARY
- ACC
agenesis of the corpus callosum
- AMP
adenosine monophosphate
- AMPD2
adenosine monophosphate deaminase 2
- CC
corpus callosum
- NCS
nerve conduction study
- OMIM
Online Mendelian Inheritance in Man
- PCH
pontocerebellar hypoplasia
- SNP
single nucleotide polymorphism
- WES
whole-exome sequence
Footnotes
Supplemental data at Neurology.org/ng
AUTHOR CONTRIBUTIONS
A.P.L.M. formulated the study, performed molecular analysis, and cowrote the manuscript. V.L. performed linkage analysis, analyzed exome sequence data, and read/contributed to the manuscript. K.P. performed patient recruitment and read/contributed to the manuscript. C.B. and R.T. performed linkage analysis, analyzed exome sequence data, and read/contributed to the manuscript. M.M.R. and E.M.Y. provided and interpreted the clinical data and read/contributed to the manuscript. J.C.H.S. analyzed the molecular data and read/contributed to the manuscript. M.B.D., D.J.A., and G.M. provided and interpreted the clinical data and read/contributed to the manuscript. E.H.S. performed patient recruitment, provided and interpreted the clinical data, and read/contributed to the manuscript. M.B. advised on linkage and exome sequencing study design, analyzed data, and revised the manuscript. R.J.L. performed patient recruitment, provided and interpreted the clinical data, formulated the study, and cowrote the manuscript. P.J.L. formulated the study, interpreted the data, and wrote the manuscript.
STUDY FUNDING
This work was funded in part by National Health and Medical Research Council (NHMRC) Australia Program Grant 490037 to D.J.A. and M.B. and NHMRC Project Grant APP1059666 to P.J.L., R.J.L., D.J.A., and M.B.D. A.P.L.M. and R.T. were supported by an Australian Postgraduate Award. P.J.L. was supported by an NHMRC Career Development Fellowship (APP1032364). E.M.Y. was supported by an NHMRC Early Career Fellowship (APP1073323). M.M.R. was supported by NHMRC CRE grant APP1031893. M.B. was supported by an Australian Research Council Future Fellowship (FT100100764). This work was made possible through Victorian State Government Operational Infrastructure Support and Australian Government NHMRC IRIISS.
DISCLOSURE
A.P.L.M., V.L., C.B., and R.T. report no disclosures. K.P. has received research support from NHMRC and HREC. J.C.H.S. reports no disclosures. E.H.S. has served on the scientific advisory board of InVitae; holds a patent for a blood-based diagnostic for autism; has consulted for Personalis; has received research support from NINDS, the Simons Foundation, the CURE Foundation, and the John and Marsha Goldman Foundation; has stock/stock options and/or has received Board of Directors compensation with Chemocentryx; has stock/stock options/medical equipment and materials with Chemocentryx; has been involved in legal proceedings for Sheuerman, Martini & Tabari; Galloway and Lucchese; Zuger, Kirmis & Smith; Donahoe & Kearney; LaFollette, Johnson; Huff, Powell & Bailey; and Bell, Roper and Kohlmyer; and has served on the board of directors for the National Organization of Disorders of the Corpus Callosum. M.M.R. has served on the scientific advisory boards of Therapeutic Advisory Committee, Treat-NMD, and Eli-Lilly Pharmaceuticals; has served on the editorial boards of the Journal of Pediatric Neurology and the Journal of Clinical Neuroscience; has received royalties from Academic Press; and has received research support from PTC Therapeutics, Isis Pharmaceuticals, and the National Health and Medical Research Council Centre for Research Excellence. E.M.Y. has received research support from the National Health and Medical Research Council of Australia Early Career Fellowship, the Murdoch Children's Research Institute, and the Charcot-Marie-Tooth Association of Australia. M.B.D. has served on the scientific advisory board of Consultant Healthscope Pathology; has served on the editorial board of BMC Neurology; has consulted for Healthscope Pathology; and has received research support from NHMRC, FARA USA, and FARA Australasia. D.J.A. has received research support from Cancer Council Australia, NHMRC, and Cancer Australia & Prostate Cancer Foundation of Australia. G.M. reports no disclosures. M.B. has served on advisory boards for Macular Telangiectasia (MacTel) Consortium and Department of Health (Australian Government); has received funding for travel and/or speaker honoraria from The 7th International Conference on Genomics & Bio-IT APAC; International Mathematics Institute; Australian Statistical Society, International Stroke Genetics, Human Genome, and MacTel Consortium; has served on the editorial boards of Statistical Applications in Genetics and Molecular Biology and Medical Genetics; holds patents regarding Method of determining response to treatment with immunomodulatory composition Details genetic markers that predict response to Hepatitis C treatment; has consulted for MacTel, Human Genetics Society of Australia, Australian Statistical Society-Institute of Mathematical Statistics, International Conference on Systems Biology, Sixth Barossa Meeting, and the 8th International Conference on Genomics & Bio-IT APAC; has received research support from NHMRC, National Heart Foundation, Australian Research Council (ARC), Victorian Life Sciences Computing Initiative (VLSCI), Royal Melbourne Hospital Home Lottery Research Awards, Leukemia Foundation, ARC, Cancer Australia, DEST International Science Linkages, Human Genetics Society of Australia, Royal Statistical Society—Institute of Mathematical Statistics, Biometrics Society of Australasia, Australian Statistics Society, American Society of Human Genetics, and International Genetic Epidemiology Society. R.J.L. has received honoraria related to the 2015 Asia Oceania Congress of Child Neurology and has received research support from National Health and Medical Research Council and Campbell Edwards Trust Research Support. P.J.L. has served on the editorial boards of Genetics Research International and Open Access (OA) Genetics and has received research support from National Health and Medical Research Council (Australia). Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Luders E, Thompson PM, Toga AW. The development of the corpus callosum in the healthy human brain. J Neurosci 2010;30:10985–10990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Giedd JN, Rumsey JM, Castellanos FX, et al. A quantitative MRI study of the corpus callosum in children and adolescents. Brain Res Dev Brain Res 1996;91:274–280. [DOI] [PubMed] [Google Scholar]
- 3.Siffredi V, Anderson V, Leventer RJ, Spencer-Smith MM. Neuropsychological profile of agenesis of the corpus callosum: a systematic review. Dev Neuropsychol 2013;38:36–57. [DOI] [PubMed] [Google Scholar]
- 4.Paul LK, Brown WS, Adolphs R, et al. Agenesis of the corpus callosum: genetic, developmental and functional aspects of connectivity. Nat Rev Neurosci 2007;8:287–299. [DOI] [PubMed] [Google Scholar]
- 5.Paul LK. Developmental malformation of the corpus callosum: a review of typical callosal development and examples of developmental disorders with callosal involvement. J Neurodev Disord 2011;3:3–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glass HC, Shaw GM, Ma C, Sherr EH. Agenesis of the corpus callosum in California 1983-2003: a population-based study. Am J Med Genet A 2008;146A:2495–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edwards TJ, Sherr EH, Barkovich AJ, Richards LJ. Clinical, genetic and imaging findings identify new causes for corpus callosum development syndromes. Brain 2014;137:1579–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Namavar Y, Barth PG, Poll-The BT, Baas F. Classification, diagnosis and potential mechanisms in pontocerebellar hypoplasia. Orphanet J Rare Dis 2011;6:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akizu N, Cantagrel V, Schroth J, et al. AMPD2 regulates GTP synthesis and is mutated in a potentially treatable neurodegenerative brainstem disorder. Cell 2013;154:505–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yiu EM, Burns J, Ryan MM, Ouvrier RA. Neurophysiologic abnormalities in children with Charcot-Marie-Tooth disease type 1A. J Peripher Nerv Syst 2008;13:236–241. [DOI] [PubMed] [Google Scholar]
- 11.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 2002;30:97–101. [DOI] [PubMed] [Google Scholar]
- 12.Wilson GR, Sunley J, Smith KR, et al. Mutations in SH3PXD2B cause Borrone dermato-cardio-skeletal syndrome. Eur J Hum Genet 2014;22:741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Novarino G, Fenstermaker AG, Zaki MS, et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science 2014;343:506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.