

Abstract
When a behavior is repeated performance often improves, i.e., repetition priming occurs. Although repetition priming is ubiquitous, mediating mechanisms are poorly understood. We address this issue in the feeding network of Aplysia. Similar to the priming observed elsewhere, priming in Aplysia is stimulus specific, i.e., it can be either “ingestive” or “egestive.” Previous studies demonstrated that priming alters motor and premotor activity. Here we sought to determine whether sensorimotor transmission is also modified. We report that changes in sensorimotor transmission do occur. We ask how they are mediated and obtain data that strongly suggest a presynaptic mechanism that involves changes in the “background” intracellular Ca2+ concentration ([Ca2+]i) in primary afferents themselves. This form of plasticity has previously been described and generated interest due to its potentially graded nature. Manipulations that alter the magnitude of the [Ca2+]i impact the efficacy of synaptic transmission. It is, however, unclear how graded control is exerted under physiologically relevant conditions. In the feeding system changes in the background [Ca2+]i are mediated by the induction of a nifedipine-sensitive current. We demonstrate that the extent to which this current is induced is altered by peptides (i.e., increased by a peptide released during the repetition priming of ingestive activity and decreased by a peptide released during the repetition priming of egestive activity). We suggest that this constitutes a behaviorally relevant mechanism for the graded control of synaptic transmission via the regulation of the [Ca2+]i in a neuron.
Keywords: mollusc, invertebrate, small cardioactive peptide, cerebral peptide 2, Aplysia
when a behavior is repeated performance often improves, i.e., repetition priming occurs (e.g., Fowler et al. 1985; Kristjansson and Campana 2010; Salimpoor et al. 2010). Although neural correlates of repetition priming have been described (e.g., Engell and McCarthy 2014; Heusser et al. 2013), few studies have characterized underlying mechanisms at the network or cellular level. This is true even in model systems such as the one we study (the feeding system of Aplysia). Studies of repetition priming in the context of feeding have focused on its “motor” side, i.e., motor neurons and elements of the central pattern generator (CPG) that directly control motor activity (Dacks et al. 2012; Dacks and Weiss 2013; Friedman and Weiss 2010; Friedman et al. 2015; Proekt et al. 2004). However, the feeding CPG, like many other CPGs, receives sensory input from the periphery that plays an important role in generating motor programs (Cropper et al. 2004b; Jahan-Parwar et al. 1983). An issue not previously addressed is whether repetition priming modifies this afferent transmission.
Previous studies of repetition priming in the feeding system have demonstrated that, similar to the priming observed in many other systems, it is stimulus specific (Huettel et al. 2002; Proekt et al. 2004). It is ingestive when one type of CPG input is activated, and egestive with the activation of a second type (Proekt et al. 2004). Input stimulation releases “ingestive” and “egestive” modulatory peptides (e.g., Koh et al. 2003; Wu et al. 2010), which modify properties of the motor circuit (Dacks and Weiss 2013; Friedman and Weiss 2010; Friedman et al. 2015). Here, we ask whether sensorimotor transmission is similarly altered by peptides and input activation. We demonstrate that changes in sensorimotor transmission are observed and ask how they are mediated.
Our experiments focus on an identified sensory neuron, the mechanoafferent input (B21). This neuron makes a direct excitatory synaptic connection with motor neurons that close the food-grasping organ in Aplysia (Fig. 1A) (Rosen et al. 2000b). We focused on this circuitry because radula opening/closing movements are reconfigured during repetition priming, whereas other movements are not (Cropper et al. 2004a). We have shown that subthreshold depolarization of B21 results in an increase in the “background” [Ca2+]i, which modifies subsequent spike-mediated synaptic transmission (Ludwar et al. 2009). A previous study demonstrated that this process is nifedipine sensitive (Ludwar et al. 2009), suggesting that depolarization activates a dihydropyridine (DHP)-sensitive current in B21. We now show that this is the case. Furthermore, we demonstrate that peptides released during repetition priming modify the magnitude of DHP-sensitive currents. Currents are increased by an ingestive peptide [cerebral peptide 2 (CP2)] and decreased by an egestive peptide [small cardioactive peptide b (SCPb)].
Fig. 1.

A: experimental setup. Experiments were conducted with one or two sharp electrodes in both B21 and B8. Ingestive priming was induced by stimulating cerebral buccal interneuron 2 (CBI-2) with a sharp electrode. Egestive repetition priming was induced via extracellular stimulation of the esophageal nerve (EN). B: activation of physiologically relevant inputs to the feeding central pattern generator (CPG) modifies the efficacy of B21–B8 synaptic transmission. In all experiments, B21 was depolarized to −40 mV, and spikes were triggered via the injection of brief current pulses. Induced excitatory postsynaptic current (EPSCs) were recorded from B8 neurons held at −60 mV using two-electrode voltage clamp mode (TEVC). EPSCs were recorded in high-divalent (HiDi) saline before (control) and after input activation. B1: CBI-2 stimulation potentiates B21–B8 synaptic transmission (n = 4). B2: group data for the experiment shown in A1. C1: EN stimulation depresses B21–B8 synaptic transmission (n = 4). C2: group data for the experiment shown in C1. *P < 0.05 and **P ≤ 0.01.
Plasticity mediated via an alteration in the background intracellular Ca2+ concentration ([Ca2+]i) has been reported in other systems (Awatramani et al. 2005; Christie et al. 2011; Fekete et al. 2014; Ivanov and Calabrese 2003; Shu et al. 2006). An interesting feature of this type of control is its potentially graded nature. Manipulations that alter the magnitude of the [Ca2+]i impact postsynaptic potential (PSP) amplitude. This potentially adds an analog component to what is sometimes considered a digital form of communication (spike-mediated synaptic transmission) (see Alle and Geiger 2008; Debanne et al. 2013; Marder 2006 for discussion of this issue). Graded control will, however, only be of biological importance if it is invoked under behaviorally relevant conditions. Our research identifies a situation where this is likely to occur.
MATERIALS AND METHODS
Animals/preparation used.
Aplysia californica obtained from Marinus (Long Beach, CA) were anesthetized by injection of isotonic MgCl2. In most experiments buccal ganglia were desheathed and pinned in a cooled recording chamber (15–17°C) that was continuously superfused (0.3–0.5 ml/min) with either artificial seawater (ASW) containing (in mM) 460 NaCl, 10 KCl, 55 MgCl2, 11 CaCl2, and 10 Na-HEPES, adjusted to pH 7.6, or a high-divalent saline containing (in mM) 398 NaCl, 8 KCl, 110 MgCl2, 13.6 CaCl2, and 10 Na-HEPES, also adjusted to a pH of 7.6 (Trudeau and Castellucci 1992).
Recording techniques.
Ca2+ currents were recorded from B21 neurons using single-electrode voltage clamp (SEVC). B21 was impaled with electrodes filled with 3 M CsCl with resistances of ∼2 MΩ. Ca2+ currents were isolated by bath applying a Na+-free solution, and using Ba2+ as the charge carrier, which increases the amplitude of the evoked current, and avoids Ca2+-dependent inhibition of Ca2+ channels (e.g., Dunn and Sossin 2013; Yue et al. 1990). The seawater used in these experiments also included drugs to block other currents induced by depolarization. Its ionic composition was (in mM): 450 tetraethylammonium chloride (TEA), 11 BaCl2, 55 MgCl2, 10 K-HEPES, 10 4-aminopyridine (4-AP), and 10 CsCl, with the pH adjusted to 7.6 (modified from Edmonds et al. 1990). Ca2+ currents were evoked by depolarizing voltage steps that had a duration of 150 ms. The holding potential was −50 mV, and the step increment was 10 mV (Dunn and Sossin 2013; Edmonds et al. 1990). Control conditions were established by doing multiple runs before peptide application to ensure current stability.
For experiments analyzing the effect of substances on synaptic transmission from B21 to B8, B21 was impaled with two sharp electrodes filled with 0.6 mM K2SO4 and 60 mM KCl (9–11 MΩ) (e.g., Fig. 1A), or with one electrode filled with 3 M potassium acetate and 0.1 M KCl (3–4 MΩ). Results were similar with the two protocols. The advantage of the first procedure was that separate electrodes were used for current passing and recording. The advantage of the second procedure was the single impalement of B21. In one set of experiments K2SO was used as a substitute for potassium acetate since potassium acetate can impact chemical transmission (Coleman et al. 1995). B21 was depolarized to a subthreshold voltage of approximately −40 mV to ensure spike propagation to its lateral process (which makes synaptic contact with B8) (Evans et al. 2003). Depolarizations of a similar magnitude are observed during the retraction phase of motor programs (Koh et al. 2003; Ludwar et al. 2009). Action potentials were triggered in B21 by injecting brief depolarizing current pulses. Excitatory postsynaptic currents (EPSCs) induced in B8 were monitored in SEVC, or two-electrode voltage clamp mode (TEVC) (Fig. 1A). B8 was either impaled with one electrode with a resistance of 6–7 MΩ, and one electrode with a resistance of 9–10 MΩ, or with one electrode with a lower resistance (3–4 MΩ). Unless otherwise indicated B8 was held at its normal resting potential (−60 to −70 mV). Neuropeptides were bath applied for 10 min (for SCPb) and 5 min (for CP2) before B21 was depolarized and stimulated. EPSCs were measured from the baseline to the peak amplitude and normalized (i.e., EPSC amplitude is expressed as a percent of the amplitude of the last EPSC recorded before peptide application).
In all experiments in which effects of exogenous peptides were studied, peptides were bath applied at 1 μM for at least 5 min. Effects of endogenous peptides were studied in experiments in which we stimulated either cerebral buccal interneuron 2 (CBI-2) or the esophageal nerve (EN) (Fig. 1A). CBI-2 was stimulated by injecting brief current pulses at 10 Hz for 30 s (Koh and Weiss 2005), which is a physiologically relevant frequency (Jing and Weiss 2005). The EN was stimulated extracellularly using a suction electrode for ∼2 min so that approximately five cycles of motor activity were induced (Friedman et al. 2015; Friedman et al. 2009; Proekt et al. 2004).
Signals were acquired and amplified using standard equipment, i.e., a DigiData 1322A and AxoClamp 2B (Molecular Devices, Sunnyvale, CA) in bridge, discontinuous current clamp, SEVC, or TEVC, and a PC equipped with Clampex 9 or Axoscope software (Molecular Devices).
Reagents.
TEA, 4-AP, and nifedipine were purchased from Sigma-Aldrich (St. Louis, MO). SCPb was obtained from Anaspec (Fremont, CA), and CP2 was obtained from SynPep (Dublin, CA).
Statistics.
Statistical significance was evaluated using an appropriate t-test (for 2-group comparisons) or ANOVA (for multigroup analyses) with subsequent comparisons performed using a Bonferroni correction. Data are presented as means ± SE. Statistics were performed using Prism (GraphPad Software, San Diego, CA). Effects were considered significant at P < 0.05.
RESULTS
Input activation and modulation of B21–B8 synaptic transmission.
A goal of this study was to determine whether repetition priming alters sensorimotor transmission in the feeding network. In particular, we focused on an identified radula mechanoafferent input (B21), which makes a direct excitatory synaptic connection with the B8 radula closer motor neurons (Fig. 1A) (Rosen et al. 2000b). We focused on this circuitry because radula opening/closing movements are reconfigured during repetition priming (whereas other movements are not) (Cropper et al. 2004a). Repetition priming in the feeding network can be induced by stimulating inputs to the feeding CPG (Dacks et al. 2012; Dacks and Weiss 2013; Friedman and Weiss 2010; Friedman et al. 2015; Proekt et al. 2004). Ingestive priming is induced by stimulating the command-like neuron CBI-2 (Fig. 1A) (Dacks et al. 2012; Dacks and Weiss 2013; Friedman and Weiss 2010; Proekt et al. 2004).
To determine whether stimulation of CBI-2 impacts B21–B8 transmission we triggered spikes in B21 and recorded EPSCs from B8. B21 was centrally depolarized so that spikes propagated to the lateral process, which contacts B8 (Evans et al. 2003). B8 was voltage clamped at its normal resting membrane potential (−60 mV). Action potentials were triggered by injecting brief current pulses, and CBI-2 was stimulated using a protocol that induces peptide release (10 Hz for 30 s) (Koh and Weiss 2005). We measured EPSC amplitude shortly after stimulation of CBI-2 (i.e., at ∼30 s) and after effects of CBI-2 stimulation are expected to dissipate (i.e., after at least 240 s) (Koh and Weiss 2005). CBI-2 stimulation had a significant effect on EPSC amplitude [1-way ANOVA: F(2,6) = 13.25; P ≤ 0.01; n = 4] (Fig. 1, B1 and B2). EPSCs were potentiated so that they were 146.2 ± 5.4% of control values (t = 5.108, P < 0.05) and recovered to control values (t = 2.004, P > 0.05 for the control vs. recovery comparison).
Egestive repetition priming of the feeding network is induced by stimulating the EN (Fig. 1A) (e.g., Friedman et al. 2015; Friedman et al. 2009; Proekt et al. 2004). To determine whether egestive priming impacts B21–B8 synaptic transmission, we stimulated the EN using an established protocol (Friedman et al. 2015; Friedman et al. 2009; Proekt et al. 2004). We measured EPSC amplitude when effects of egestive priming are apparent (i.e., 1 min after EN stimulation) (Friedman et al. 2009) and after effects of egestive priming have dissipated [i.e., 6 or more min after EN stimulation (Friedman et al. 2009)]. EN stimulation had a significant effect on EPSC amplitude [1-way ANOVA: F(2,6) = 9.597, P < 0.05, n = 4] (Fig. 1, C1 and C2). EPSCs were depressed at 1 min (i.e., were 53.2 ± 7.5% of control values: t = 4.263, P < 0.05) but had recovered at the later time point (for the before vs. recovery comparison: t = 1.258, P > 0.05, n = 4).
Peptidergic modulation of synaptic transmission at the B21–B8 synapse.
When inputs to the feeding CPG are activated, peptides are released, e.g., CP2 is released from CBI-2 (Koh et al. 2003; Koh and Weiss 2005). CP2 plays an important role in the induction of the repetition priming of motor activity (Friedman and Weiss 2010; Friedman et al. 2015). We asked whether exogenous CP2 mimics effects of CBI-2 stimulation on B21–B8 synaptic transmission and used the protocol described above (i.e., we induced spiking in B21 by injecting brief current pulses and monitored evoked EPSCs in B8). Like CBI-2 stimulation, CP2 increased EPSC amplitude. After CP2, EPSCs were 206.9 ± 21.1% of control values (t-test: t = −4.80, n = 5, P < 0.01) (Fig. 2, A1 and A3).
Fig. 2.

Exogenous peptides modify the efficacy of B21–B8 synaptic transmission. In all experiments B21 was depolarized to −40 mV, and spikes were triggered via the injection of brief current pulses. Resulting EPSCs were recorded from B8 neurons held at −60 mV using TEVC. EPSCs were recorded in HiDi saline (control) and in the presence of 1 μM peptide. A: effect of cerebral peptide 2 (CP2) in the presence (A1) and absence (A2) of 10 μM nifedipine (n = 5 in both cases). Note that CP2 produced a larger increase in peak EPSC amplitude in the absence of nifedipine. A3: group data for the experiments shown in A1 and A2. Plotted is normalized amplitude (i.e., EPSC amplitude is expressed as a percent of the amplitude of the last EPSC recorded before peptide application). B: effect of small cardioactive peptide b (SCPb) in the presence (B1) (n = 7) and absence (B2) (n = 5) of 10 μM nifedipine. Note that SCPb decreased the peak EPSC amplitude in the absence of nifedipine but had no significant effect it its presence. B3: group data for the experiments shown in B1 and B2. *P < 0.05, **P ≤ 0.01, and ***P ≤ 0.001.
When afferents contained within the EN are stimulated, the peptide SCPb is released (Wu et al. 2010). Previous work has demonstrated that SCPb primes motor activity (Friedman et al. 2015). B21 itself is also SCP containing (Miller et al. 1994), but it does not usually spike during either CBI-2- or EN-induced motor programs (Evans et al. 2003; Ludwar et al. 2009). A second set of experiments sought to assess whether EN stimulation is mimicked by exogenous SCPb. Like EN stimulation, SCPb decreased EPSC amplitude. After SCPb, EPSCs were 57.4 ± 6.9% of control values (t-test: t = 5.957, P < 0.01, n = 7) (Fig. 2, B1 and B3). These data suggest that effects of input activation on B21–B8 synaptic transmission are likely to be at least partially mediated by effects of modulatory peptides.
Characterization of Ca2+ currents in B21.
Previous studies of the regulation of B21–B8 synaptic transmission have implicated a presynaptic form of control that has been described in other systems and generated considerable general interest. It is mediated by alterations in background [Ca2+]i that impact subsequent spike-mediated synaptic transmission (i.e., determine the amplitude of the induced PSP) (Awatramani et al. 2005; Fekete et al. 2014; Ivanov and Calabrese 2003; Ludwar et al. 2009; Shapiro et al. 1980; Shu et al. 2006). In previous experiments we demonstrated that B21–B8 synaptic transmission is potentiated by subthreshold depolarizations. With imaging techniques we demonstrated that these depolarizations produce increases in background [Ca2+]i that are not observed in the presence of nifedipine (Ludwar et al. 2009). Taken together previous data suggest that there is a nifedipine-sensitive Ca2+ current in B21 that plays an important role in regulating the efficacy of synaptic transmission to B8.
To determine whether this is the case, we performed SEVC experiments, using a step protocol, in ASW, and in the presence of 10 μM nifedipine (Fig. 3, A1, A2, and 3B). Nifedipine had a significant effect on the magnitude of induced currents [2-way ANOVA: F(1,60) = 7.427, P < 0.01, n = 7]. The reduction in total current did not depend on membrane potential [F(9, 60) = 0.96, P > 0.05, n = 7]. The amount of current remaining in the presence of nifedipine was, however, greater at steps to more depolarized potentials. For example, the mean current observed in the presence of nifedipine with the step to 0 mV was only −0.03 ± 0.02 nA, whereas the mean current remaining with the step to 30 mV was −1.46 ± 0.21 nA (t = 6.54, P < 0.001, n = 7). To confirm the identity of induced currents, we added Cd2+ at the conclusion of experiments (a nonspecific Ca2+ channel blocker in Aplysia). Cd2+ completely blocked all inward currents (n = 5; Fig. 3, A3 and 3B). Thus, our data indicate that there are both nifedipine-sensitive and nifedipine-insensitive currents in B21. The current or currents that are nifedipine insensitive are larger with steps to more depolarized potentials.
Fig. 3.
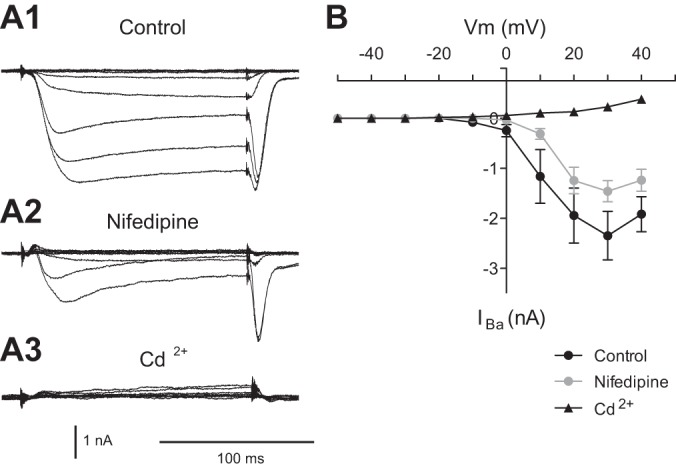
Nifedipine reduces barium currents in B21. A1: barium currents in the absence of nifedipine (Control). B21 was held at −50 mV, and inward currents were induced by 150-ms steps to 40 mV (with 10-mV increments). A2: currents induced as in A1 but in the presence of 10 μM nifedipine. A3: at the conclusion of experiments, currents were induced in 2 mM Cd2+, a nonspecific Ca2+ channel blocker in Aplysia. B: group data for the experiments shown in A1–A3 (n = 7).
Peptidergic modulation of the nifedipine-sensitive Ca2+ current in B21.
We conducted experiments in the presence of CP2 to examine whether the nifedipine-sensitive current can be modulated. Because we predicted a potentiating effect, CP2 was tested under conditions where total currents are small, and the nifedipine-sensitive current predominates. CP2 had a significant effect on the magnitude of induced currents [2-way ANOVA: F(2, 36) = 4.27, P < 0.05, n = 4] (Fig. 4, A1 and A2). More specifically, CP2 did not induce currents at membrane potentials where currents were not present to begin with (i.e., with steps in the −50 to −20 mV range) (Fig. 4A2). At membrane potentials where currents were observed in the absence of peptide, however, CP2 had a significant effect, i.e., with steps to −10 and 0 mV, currents were larger in the presence of CP2 (for the control vs. CP2 comparison: t = 3.08, P < 0.05 in both cases) (Fig. 4, A1 and A2). Effects of CP2 were reversible (for the control vs. wash comparison: t = 0.02, P > 0.05 for the step to −10 mV; and t = 0.03, P > 0.05 for the step to 0 mV) (Fig. 4, A1 and A2).
Fig. 4.
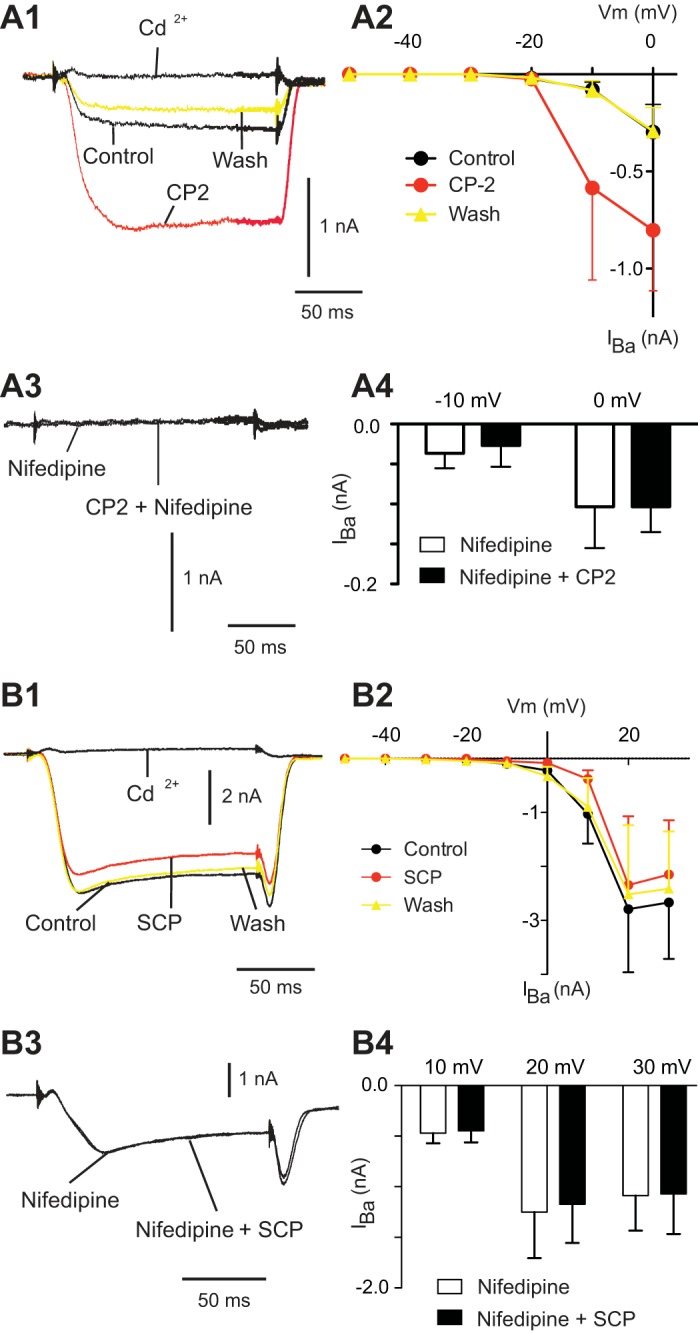
Exogenous CP2 and SCPb have significant effects on barium currents in B21. A1: B21 was held at −50 mV, and inward currents were induced by a 150-ms step to 0 mV. Currents were induced under control conditions (no peptide), in the presence of 1 μM CP2, after peptide washout, and in the presence of 2 mM Cd2+. Note the potentiating effect of the CP2. A2: group data for experiments in which currents were induced before, during, and after exogenous CP2 (n = 4). A3: currents were induced as described for A1 after preincubation in 10 μM nifedipine. Note the reduction in the magnitude of the current induced. A4: group data for experiments using the protocol shown in A3 (n = 3). B1: B21 was held at −50 mV, and inward currents were induced by a 150-ms step to 30 mV. Currents were induced under control conditions (no peptide), in the presence of 1 μM SCPb, after peptide washout, and in the presence of 2 mM Cd2+. Note that SCPb reduced the magnitude of the induced current. B2: group data for experiments in which currents were induced before, during, and after exogenous SCPb (n = 5). B3: currents were induced as described for B1 after preincubation in 10 μM nifedipine. Note that the magnitude of the induced current is no longer altered by SCPb application. B4: group data for experiments using the protocol shown in B3 (n = 3).
To determine whether the current modulated by CP2 was nifedipine sensitive, we preincubated preparations with 10 μM nifedipine and then stepped to −10 and 0 mV in the presence and absence of CP2. There was no difference in the magnitude of the induced inward current [2-way ANOVA: F(1,4) = 0.12, P > 0.05, n = 3] (Fig. 4, A3 and A4). These data indicate that CP2 potentiates a nifedipine-sensitive current that is induced in B21 when it is depolarized.
In a second set of experiments, we tested SCPb. Because a depressing effect was predicted, we additionally tested it with steps to more depolarized potentials (i.e., under conditions where total currents are larger) (Fig. 4B2). Overall SCPb had a significant effect on the magnitude of induced currents [2-way ANOVA: F(2,72) = 10.75, P < 0.001, n = 5]. More specifically, currents were significantly reduced with steps to 10, 20, and 30 mV (for the control vs. SCPb comparison: t = 4.806, P < 0.001 for the step to 10 mV; t = 3.31, P < 0.05 for the step to 20 mV; and t = 3.82, P < 0.01 for the step to 30 mV) (Fig. 4, B1 and B2). Effects of SCPb were reversible (for the control vs. wash comparison: t = 1.0, P > 0.05 for the step to 10 mV; t = 2.0, P > 0.05 for the step to 20 mV; and t = 1.9, P > 0.05 for the step to 30 mV) (Fig. 4, B1 and B2). We next preincubated preparations in 10 μM nifedipine and then stepped to different membrane potentials before and after SCPb to demonstrate that the currents modulated by SCPb were nifedipine sensitive. SCPb no longer produced a decrease in the magnitude of the induced current [2-way ANOVA: F(1,6) = 1.41, P > 0.05, n = 3] (Fig. 4, B3 and B4). These data indicate that SCPb reduces the magnitude of the nifedipine-sensitive inward current induced in B21.
Peptide application in the presence of nifedipine.
To determine whether effects of peptides on B21–B8 synaptic transmission are mediated, at least in part, via modulation of the nifedipine-sensitive Ca2+ current, we performed a final set of experiments in which preparations were preincubated with nifedipine before peptide application. Under these conditions, we found that CP2 increased EPSC amplitude (EPSCs were 126.9 ± 6.5% of control values; t-test: t = −3.05, P ≤ 0.05, n = 5), but peptide-induced increases were significantly less in the presence of nifedipine (t-test; t = 3.631, P < 0.01, n = 5 for the CP2 vs. CP2 + nifedipine comparison) (Fig. 2, A1 vs. A2 and A3). When SCPb was applied in the presence of nifedipine, it had no significant effect on EPSC amplitude (t-test: t = −0.64, P > 0.05, n = 5) (Fig. 2, B2 and B3). These data indicate that modulatory effects of peptides on the nifedipine-sensitive current in B21 are sufficient to significantly alter B21–B8 synaptic transmission.
DISCUSSION
An important limitation of previous studies of repetition priming in the feeding network is that they focused on changes in motor and premotor activity. However, the feeding CPG is similar to many other CPGs in that under physiological conditions its activity is modified by afferent feedback from the periphery (Cropper et al. 2004b; Jahan-Parwar et al. 1983). Potential effects of repetition priming on this feedback were not previously studied. One goal of the current research was, therefore, to determine whether repetition priming modifies afferent input to the feeding CPG.
Furthermore, it was of interest to characterize mechanisms mediating any observed alterations of sensorimotor transmission. Mechanistic studies of the priming of motor activity have demonstrated a prominent role for changes in the excitability of motor neurons and interneurons (Dacks et al. 2012; Dacks and Weiss 2013; Friedman and Weiss 2010; Friedman et al. 2015; Proekt et al. 2004). Thus, when priming increases the strength of movements, this occurs to a large extent as a result of an increase in the number of action potentials in the relevant motor neuron and/or the interneuron driving this motor neuron. When priming decreases the strength of movements, excitability decreases are observed. Previous work has therefore demonstrated that alterations in the strength in movements necessary for effective repetition priming are very effectively mediated via the regulation of neural excitability. In the context of sensorimotor transmission, however, changes in excitability might be less desirable. For example, centrally mediated increases in excitability could lead to a situation where spikes are triggered in response to central inputs (e.g., primary afferent depolarization) in the absence of a peripheral stimulus. Thus, it was important to determine whether repetition priming could modify sensorimotor transmission via a mechanism that would not necessarily alter peripheral encoding.
Several sources of afferent input to the feeding CPG have been described. However, not all aspects of feeding are altered when repetition priming occurs, i.e., radula protraction precedes radula retraction during all characterized feeding movements (Cropper et al. 2004a). Thus, it is activity of the radula opener/closer circuitry that is reconfigured. Consequently, we focused on the most well characterized source of afferent input to these neurons. We studied a radula mechanoafferent input (B21) that makes an excitatory connection to the radula closer motor neurons (i.e., the B8 neurons) (Rosen et al. 2000b). We demonstrate that the efficacy of B21–B8 synaptic transmission is increased after ingestive priming (Fig. 5). This makes sense given our current understanding of feeding behavior. B21–B8 synaptic transmission will occur during the radula retraction phase of motor programs. Increased excitatory input to B8 is likely to increase the amplitude of radula closing movements (Friedman et al. 2009). Enhanced closing during retraction will presumably aid in pulling food into the buccal cavity (Kupfermann 1974). Therefore, it will make a positive contribution to ingestive priming. In contrast, the efficacy of B21–B8 synaptic transmission is decreased after egestive priming (Fig. 5). This suppression is likely to prevent sensorimotor transmission that would be counterproductive during egestive priming. Egestion is most effective when the radula is closed during protraction and not retraction (Kupfermann 1974). We therefore demonstrate that the efficacy of B21–B8 synaptic transmission is modified by both ingestive and egestive repetition priming in a manner that is likely to facilitate behavior.
Fig. 5.
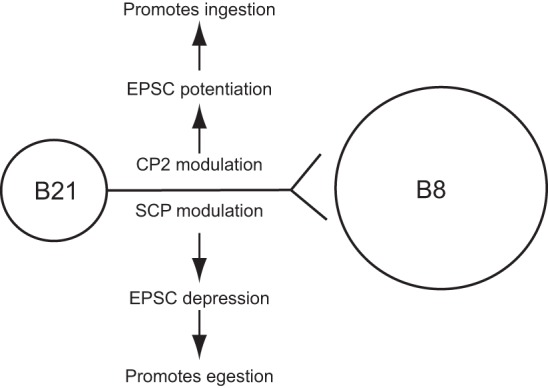
A schematic showing the effects of CP2 and SCPb at the B21–B8 synapse. The sensory neuron B21 makes a direct (monosynaptic) excitatory connection with the radula closer motor neuron B8. Top, CP2 potentiates B21–B8 transmission, which is likely to promote ingestive behavior. Bottom, in contrast, SCPb depresses B21–B8 transmission, which is likely to promote egestive behavior.
Other experiments in this study characterized a mechanism used to alter B21–B8 transmission. They focused on a form of plasticity identified in the B21–B8 circuit in previous work (Ludwar et al. 2009). This form of plasticity consists of increases in the background [Ca2+]i that are induced by subthreshold depolarizations. Increases in the Ca2+ concentration potentiate subsequent spike-mediated transmission. This form of plasticity was originally described >30 years ago in experiments conducted in Aplysia (e.g., Edmonds et al. 1990; Shapiro et al. 1980). It has, however, received renewed attention as a result of its identification in mammals (e.g., Awatramani et al. 2005; Christie et al. 2011; Fekete et al. 2014; Shu et al. 2006). A goal of ongoing research in this field is to determine how this form of plasticity is invoked under physiologically relevant conditions.
In some contexts Ca2+ currents that contribute to increases in background [Ca2+]i are induced by repeated (phasic) subthreshold depolarizations. These depolarizations can consist of oscillatory changes in membrane potential in the brain (e.g., Shu et al. 2006) or depolarizations that occur during rhythmic motor programs (e.g., Ivanov and Calabrese 2003). Previous studies have shown that B21 receives subthreshold depolarizing input during one of the two antagonistic phases of feeding motor programs, i.e., the radula retraction phase (Evans et al. 2003; Rosen et al. 2000a). Furthermore, we have demonstrated that motor program-induced depolarizations are sufficient to increase the background [Ca2+]i in B21 (Ludwar et al. 2009). A question that has not been addressed in previous work, however, is whether the magnitude of these subthreshold depolarizations is regulated to take advantage of the potentially graded nature of this form of control. We address this issue here.
Previous experiments showed that changes in background [Ca2+]i in B21 are sensitive to application of a DHP (nifedipine) (Ludwar et al. 2009). We hypothesized that depolarization activates a DHP-sensitive Ca2+ current in B21. Here, we confirm the existence of this current and demonstrate that the magnitude of its induction is modulated by peptides that are released during repetition priming. Namely, we demonstrate that current induction is increased by CP2, which is released from the ingestive command neuron CBI-2 (Koh et al. 2003; Koh and Weiss 2005). We also demonstrate that SCPb, which is released from the EN (Wu et al. 2010), decreases current induction. These data taken together with previous findings suggest that the extent to which motor program-generated depolarizations induce the nifedipine-sensitive current is influenced by the release of physiologically relevant peptides.
We suggest that effects of modulatory peptides may be manifested in a graded manner in that they will become progressively more pronounced during the course of repetition priming. That this may be the case is suggested by previous studies of repetition priming in the context of motor activity (Dacks et al. 2012; Dacks and Weiss 2013; Friedman and Weiss 2010; Friedman et al. 2015; Proekt et al. 2004). In these experiments there is no progressive change in the firing frequency of the CPG inputs. Nevertheless, there are progressive changes in patterns of motor activity. We have suggested that, at least in part, this is a consequence of the fact that the modulators that mediate repetition priming exert second messenger-mediated effects that persist long enough for summation to occur when cycles of activity are repeatedly generated (Cropper et al. 2014). It is likely that effects of peptides on Ca2+ currents in B21 will also summate. In all Aplysia neurons where cellular/molecular effects of CP2 and SCPb have been studied, second messengers have been identified (e.g., Abrams et al. 1984; Bernier et al. 1982; Buttner and Siegelbaum 2003; Friedman and Weiss 2010; Friedman et al. 2015; Jarrard et al. 1993; Ocorr and Byrne 1986). Thus, we suggest that, as motor programs become ingestive, progressive potentiation of B21 afferent transmission to B8 will occur. In a similar vein, as motor programs become more egestive, afferent transmission will undergo progressive depression. If so, the mechanism that we describe may constitute a behaviorally relevant mechanism for the “graded” modulation of the background [Ca2+]i in a neuron. In other systems, the potential for graded control has been demonstrated experimentally. Other work has, however, not demonstrated how this control occurs under behaviorally relevant conditions.
In general, our work characterizes a mechanism for the induction of repetition priming that is likely to be broadly relevant. Many networks that undergo use-dependent forms of plasticity contain modulatory neurotransmitters. In terms of the forms of priming observed in humans, the type of mechanism we describe is likely to be most relevant to the related phenomenon “automaticity” that clearly occurs without a change in “higher-order” function. Automaticity is defined as the ability to do things without occupying the mind with low-level details (e.g., Logan 1990). It is thought to be essential for the execution of vital activities such as locomotion and speaking. Without automaticity, it is unlikely that humans would be able to multitask in the manner that is required to cope with their complex environment.
GRANTS
The work was supported by the National Institute of Mental Health Grant MH-051393.
DISCLOSURES
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
Author contributions: E.S. and C.G.E. performed experiments; E.S., C.G.E., and E.C.C. analyzed data; E.S., C.G.E., and E.C.C. interpreted results of experiments; E.S., C.G.E., and E.C.C. prepared figures; E.S., C.G.E., and E.C.C. edited and revised manuscript; E.S., C.G.E., and E.C.C. approved final version of manuscript; E.C.C. conception and design of research; E.C.C. drafted manuscript.
ACKNOWLEDGMENTS
We thank Dr. David Parker and Dr. Klaudiusz R. Weiss for discussions and valuable comments on this manuscript.
REFERENCES
- Abrams TW, Castellucci VF, Camardo JS, Kandel ER, Lloyd PE. Two endogenous neuropeptides modulate the gill and siphon withdrawal reflex in Aplysia by presynaptic facilitation involving cAMP-dependent closure of a serotonin-sensitive potassium channel. Proc Natl Acad Sci USA 81: 7956–7960, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alle H, Geiger JR. Analog signalling in mammalian cortical axons. Curr Opin Neurobiol 18: 314–320, 2008. [DOI] [PubMed] [Google Scholar]
- Awatramani GB, Price GD, Trussell LO. Modulation of transmitter release by presynaptic resting potential and background calcium levels. Neuron 48: 109–121, 2005. [DOI] [PubMed] [Google Scholar]
- Bernier L, Castellucci VF, Kandel ER, Schwartz JH. Facilitatory transmitter causes a selective and prolonged increase in adenosine 3′:5′-monophosphate in sensory neurons mediating the gill and siphon withdrawal reflex in Aplysia. J Neurosci 2: 1682–1691, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttner N, Siegelbaum SA. Antagonistic modulation of a hyperpolarization-activated Cl(-) current in Aplysia sensory neurons by SCP(B) and FMRFamide. J Neurophysiol 90: 586–598, 2003. [DOI] [PubMed] [Google Scholar]
- Christie JM, Chiu DN, Jahr CE. Ca(2+)-dependent enhancement of release by subthreshold somatic depolarization. Nat Neurosci 14: 62–68, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman MJ, Meyrand P, Nusbaum MP. A switch between two modes of synaptic transmission mediated by presynaptic inhibition. Nature 378: 502–505, 1995. [DOI] [PubMed] [Google Scholar]
- Cropper EC, Evans CG, Hurwitz I, Jing J, Proekt A, Romero A, Rosen SC. Feeding neural networks in the mollusc Aplysia. Neurosignals 13: 70–86, 2004a. [DOI] [PubMed] [Google Scholar]
- Cropper EC, Evans CG, Jing J, Klein A, Proekt A, Romero A, Rosen SC. Regulation of afferent transmission in the feeding circuitry of Aplysia. Acta Biol Hung 55: 211–220, 2004b. [DOI] [PubMed] [Google Scholar]
- Cropper EC, Friedman AK, Jing J, Perkins MH, Weiss KR. Neuromodulation as a mechanism for the induction of repetition priming. Curr Opin Neurobiol 29: 33–38, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dacks AM, Siniscalchi MJ, Weiss KR. Removal of default state-associated inhibition during repetition priming improves response articulation. J Neurosci 32: 17740–17752, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dacks AM, Weiss KR. Latent modulation: a basis for non-disruptive promotion of two incompatible behaviors by a single network state. J Neurosci 33: 3786–3798, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debanne D, Bialowas A, Rama S. What are the mechanisms for analogue and digital signalling in the brain? Nat Rev Neurosci 14: 63–69, 2013. [DOI] [PubMed] [Google Scholar]
- Dunn TW, Sossin WS. Inhibition of the Aplysia sensory neuron calcium current with dopamine and serotonin. J Neurophysiol 110: 2071–2081, 2013. [DOI] [PubMed] [Google Scholar]
- Edmonds B, Klein M, Dale N, Kandel ER. Contributions of two types of calcium channels to synaptic transmission and plasticity. Science 250: 1142–1147, 1990. [DOI] [PubMed] [Google Scholar]
- Engell AD, McCarthy G. Repetition suppression of face-selective evoked and induced EEG recorded from human cortex. Hum Brain Mapp 35: 4155–4162, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans CG, Jing J, Rosen SC, Cropper EC. Regulation of spike initiation and propagation in an Aplysia sensory neuron: gating-in via central depolarization. J Neurosci 23: 2920–2931, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fekete A, Johnston J, Delaney KR. Presynaptic T-type Ca2+ channels modulate dendrodendritic mitral-mitral and mitral-periglomerular connections in mouse olfactory bulb. J Neurosci 34: 14032–14045, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler CA, Napps SE, Feldman L. Relations among regular and irregular morphologically related words in the lexicon as revealed by repetition priming. Mem Cognit 13: 241–255, 1985. [DOI] [PubMed] [Google Scholar]
- Friedman AK, Weiss KR. Repetition priming of motoneuronal activity in a small motor network: intercellular and intracellular signaling. J Neurosci 30: 8906–8919, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman AK, Weiss KR, Cropper EC. Specificity of repetition priming: the role of chemical coding. J Neurosci 35: 6326–6334, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman AK, Zhurov Y, Ludwar B, Weiss KR. Motor outputs in a multitasking network: relative contributions of inputs and experience-dependent network states. J Neurophysiol 102: 3711–3727, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heusser AC, Awipi T, Davachi L. The ups and downs of repetition: modulation of the perirhinal cortex by conceptual repetition predicts priming and long-term memory. Neuropsychologia 51: 2333–2343, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huettel SA, Mack PB, McCarthy G. Perceiving patterns in random series: dynamic processing of sequence in prefrontal cortex. Nat Neurosci 5: 485–490, 2002. [DOI] [PubMed] [Google Scholar]
- Ivanov AI, Calabrese RL. Modulation of spike-mediated synaptic transmission by presynaptic background Ca2+ in leech heart interneurons. J Neurosci 23: 1206–1218, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahan-Parwar B, Wilson AH Jr, Fredman SM. Role of proprioceptive reflexes in control of feeding muscles of Aplysia. J Neurophysiol 49: 1469–1480, 1983. [DOI] [PubMed] [Google Scholar]
- Jarrard HE, Goldsmith BA, Abrams TW. In Aplysia sensory neurons, the neuropeptide SCPB and serotonin differ in efficacy both in modulating cellular properties and in activating adenylyl cyclase: implications for mechanisms underlying presynaptic facilitation. Brain Res 616: 188–199, 1993. [DOI] [PubMed] [Google Scholar]
- Jing J, Weiss KR. Generation of variants of a motor act in a modular and hierarchical motor network. Curr Biol 15: 1712–1721, 2005. [DOI] [PubMed] [Google Scholar]
- Koh HY, Vilim FS, Jing J, Weiss KR. Two neuropeptides colocalized in a command-like neuron use distinct mechanisms to enhance its fast synaptic connection. J Neurophysiol 90: 2074–2079, 2003. [DOI] [PubMed] [Google Scholar]
- Koh HY, Weiss KR. Peptidergic contribution to posttetanic potentiation at a central synapse of aplysia. J Neurophysiol 94: 1281–1286, 2005. [DOI] [PubMed] [Google Scholar]
- Kristjansson A, Campana G. Where perception meets memory: a review of repetition priming in visual search tasks. Atten Percept Psychophys 72: 5–18, 2010. [DOI] [PubMed] [Google Scholar]
- Kupfermann I. Feeding behavior in Aplysia: a simple system for the study of motivation. Behav Biol 10: 1–26, 1974. [DOI] [PubMed] [Google Scholar]
- Logan GD. Repetition priming and automaticity: common underlying mechanisms. Cognitive Psychol 22: 1–35, 1990. [Google Scholar]
- Ludwar B, Evans CG, Jing J, Cropper EC. Two distinct mechanisms mediate potentiating effects of depolarization on synaptic transmission. J Neurophysiol 102: 1976–1983, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder E. Neurobiology: extending influence. Nature 441: 702–703, 2006. [DOI] [PubMed] [Google Scholar]
- Miller MW, Rosen SC, Schissel SL, Cropper EC, Kupfermann I, Weiss KR. A population of SCP-containing neurons in the buccal ganglion of Aplysia are radula mechanoafferents and receive excitation of central origin. J Neurosci 14: 7008–7023, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocorr KA, Byrne JH. Evidence for separate receptors that mediate parallel effects of serotonin and small cardioactive peptide-B (Scpb) on adenylate-cyclase in Aplysia-Californica. Neurosci Lett 70: 283–288, 1986. [DOI] [PubMed] [Google Scholar]
- Proekt A, Brezina V, Weiss KR. Dynamical basis of intentions and expectations in a simple neuronal network. Proc Natl Acad Sci USA 101: 9447–9452, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen SC, Miller MW, Cropper EC, Kupfermann I. Outputs of radula mechanoafferent neurons in Aplysia are modulated by motor neurons, interneurons, and sensory neurons. J Neurophysiol 83: 1621–1636, 2000a. [DOI] [PubMed] [Google Scholar]
- Rosen SC, Miller MW, Evans CG, Cropper EC, Kupfermann I. Diverse synaptic connections between peptidergic radula mechanoafferent neurons and neurons in the feeding system of Aplysia. J Neurophysiol 83: 1605–1620, 2000b. [DOI] [PubMed] [Google Scholar]
- Salimpoor VN, Chang C, Menon V. Neural basis of repetition priming during mathematical cognition: repetition suppression or repetition enhancement? J Cogn Neurosci 22: 790–805, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro E, Castellucci VF, Kandel ER. Presynaptic membrane potential affects transmitter release in an identified neuron in Aplysia by modulating the Ca2+ and K+ currents. Proc Natl Acad Sci USA 77: 629–633, 1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y, Hasenstaub A, Duque A, Yu Y, McCormick DA. Modulation of intracortical synaptic potentials by presynaptic somatic membrane potential. Nature 441: 761–765, 2006. [DOI] [PubMed] [Google Scholar]
- Trudeau LE, Castellucci VF. Contribution of polysynaptic pathways in the mediation and plasticity of Aplysia gill and siphon withdrawal reflex: evidence for differential modulation. J Neurosci 12: 3838–3848, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JS, Vilim FS, Hatcher NG, Due MR, Sweedler JV, Weiss KR, Jing J. Composite modulatory feedforward loop contributes to the establishment of a network state. J Neurophysiol 103: 2174–2184, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue DT, Backx PH, Imredy JP. Calcium-sensitive inactivation in the gating of single calcium channels. Science 250: 1735–1738, 1990. [DOI] [PubMed] [Google Scholar]