

Abstract
5-Azacytidine (5-aza-C) is a ribonucleoside analog that induces the lethal mutagenesis of human immunodeficiency virus type 1 (HIV-1) by causing predominantly G-to-C transversions during reverse transcription. 5-Aza-C could potentially act primarily as a ribonucleotide (5-aza-CTP) or as a deoxyribonucleotide (5-aza-2′-deoxycytidine triphosphate [5-aza-dCTP]) during reverse transcription. In order to determine the primary form of 5-aza-C that is active against HIV-1, Illumina sequencing was performed using proviral DNA from cells treated with 5-aza-C or 5-aza-dC. 5-Aza-C and 5-aza-dC were found to induce highly similar patterns of mutation in HIV-1 in terms of the types of mutations observed, the magnitudes of effects, and the distributions of mutations at individual sequence positions. Further, 5-aza-dCTP was detected by liquid chromatography–tandem mass spectrometry in cells treated with 5-aza-C, demonstrating that 5-aza-C was a substrate for ribonucleotide reductase. Notably, levels of 5-aza-dCTP were similar in cells treated with equivalent effective concentrations of 5-aza-C or 5-aza-dC. Lastly, HIV-1 reverse transcriptase was found to incorporate 5-aza-CTP in vitro at least 10,000-fold less efficiently than 5-aza-dCTP. Taken together, these data support the model that 5-aza-C enhances the mutagenesis of HIV-1 primarily after reduction to 5-aza-dC, which can then be incorporated during reverse transcription and lead to G-to-C hypermutation. These findings may have important implications for the design of new ribonucleoside analogs directed against retroviruses.
INTRODUCTION
RNA viruses exhibit high mutation rates and have been postulated to replicate near the error threshold—the maximal mutation rate compatible with the maintenance of genetic information (1, 2). Thus, these viruses may be particularly sensitive to small molecules that promote viral mutations, an antiviral strategy called lethal mutagenesis (3). Lethal mutagenesis has been pursued as a potential antiviral approach for many different RNA viruses (4). Most small-molecule candidates for lethal mutagenesis identified thus far have been nucleoside analogs with altered base-pairing properties. These nucleoside analogs base pair promiscuously due to ionization, structural rearrangement, tautomerization, or conformational flexibility (5). Alternatively, small molecules can be used to promote viral mutagenesis by leveraging host nucleic acid-editing enzymes that are part of the innate immune response. For example, molecules have been identified that block the degradation of APOBEC3 enzymes by the human immunodeficiency virus type 1 (HIV-1) accessory protein Vif (6–9). These molecules ultimately promote the incorporation of APOBEC3 proteins into virions, resulting in lethal G-to-A hypermutation during the next cycle of replication.
The ribonucleoside analog 5-azacytidine (5-aza-C) reduces the infectivity of HIV-1 by inducing lethal mutagenesis (10). 5-Aza-C is active during both the early and late phases of viral replication, reflecting incorporation during both reverse transcription and the transcription of viral genomic RNA, respectively. When added during the late phase of viral replication, 5-aza-C induces primarily C-to-G transversions in HIV-1. In contrast, during the early phase of replication, 5-aza-C induces primarily G-to-C transversions in the virus. These G-to-C transversions are thought to be caused by the incorporation of 5-aza-C into minus-strand viral DNA, followed by the hydrolysis of 5-aza-C and its deformylation into ring-opened remnants (10). These ring-opened remnants can then mispair with deoxycytidine during plus-strand synthesis, leading to the fixation of G-to-C transversions in proviral DNA. However, it remains possible that 5-aza-C hydrolysis products are directly incorporated by HIV-1 reverse transcriptase (RT) as well. Notably, 5-aza-C is closely related to 5-aza-2′-deoxycytidine (5-aza-dC), another nucleoside analog that has been explored for the lethal mutagenesis of HIV-1 (11, 12); the primary difference is that 5-aza-dC is much more potent than 5-aza-C and likely is incorporated only into viral DNA.
Two different mechanisms could account for the antiviral activity of 5-aza-C during the early phase of replication: First, 5-aza-C could be incorporated during reverse transcription primarily as a deoxyribonucleotide (i.e., as 5-aza-2′-deoxycytidine 5′-triphosphate [5-aza-dCTP]). For this to occur, the cellular enzyme ribonucleotide reductase (RNR) would have to first convert 5-aza-C (in its diphosphate form) to 5-aza-dC, which could then be phosphorylated to form 5-aza-dCTP. Notably, one previous study has demonstrated that ∼10 to 20% of 5-aza-C is reduced to 5-aza-dC by RNR (13), suggesting that 5-aza-dCTP would likely be available for incorporation during reverse transcription. However, the reduction of 5-aza-C to 5-aza-dC has not yet been demonstrated in cell types for which antiviral activity has been reported. In further support of this possibility, HIV-1 RT has been shown to selectively exclude ribonucleotides by using a residue (Y115) that acts as a steric gate (14–16). Alternatively, 5-aza-C might be incorporated directly as a ribonucleotide (i.e., as 5-aza-CTP) during reverse transcription. Notably, HIV-1 RT has been found to incorporate significant levels of endogenous ribonucleotides when the levels of deoxyribonucleotides are very low (resulting in high nucleoside triphosphate [NTP]/deoxynucleoside triphosphate [dNTP] ratios), as they are in macrophages (17, 18). Previous studies have found that high concentrations of 5-aza-C are required to elicit antiviral activity in cell culture (10), potentially skewing the NTP/dNTP ratio enough to allow for significant 5-aza-CTP incorporation. However, these findings could also reflect the inefficient reduction of 5-aza-C to 5-aza-dC.
In order to determine the primary form of 5-aza-C that is active during HIV-1 reverse transcription, Illumina high-throughput sequencing was performed to compare viral mutagenesis in the presence of 5-aza-C with that in the presence of 5-aza-dC. 5-Aza-C and 5-aza-dC caused similar levels of G-to-C and C-to-G transversions in HIV-1. Further, G-to-C and C-to-G transversions at individual sequence positions were distributed in highly similar patterns for 5-aza-C and 5-aza-dC. In addition, 5-aza-dCTP was detected in cells treated with 5-aza-C, indicating that 5-aza-C was reduced to 5-aza-dC by RNR. Importantly, 5-aza-dCTP levels were similar in cells treated with equivalent effective concentrations of 5-aza-C or 5-aza-dC. Lastly, 5-aza-CTP was incorporated in vitro by HIV-1 RT, but much less efficiently than 5-aza-dCTP. Overall, the data support the conclusion that 5-aza-C enhances HIV-1 mutagenesis and diminishes HIV-1 infectivity primarily by acting as 5-aza-dCTP, which may have important implications for the design of antiretroviral ribonucleoside analogs.
MATERIALS AND METHODS
Plasmids, cell lines, and reagents.
For single-cycle infections, viral stocks were produced by cotransfecting the HIV-1 envelope-deficient vector pNL4-3 MIG (19), which expresses mCherry and enhanced green fluorescent protein (EGFP), and pHCMV-G, a kind gift from J. Burns (University of California, San Diego). Viral stocks were produced in human embryonic kidney (HEK 293T) cells from the American Type Culture Collection (Manassas, VA). Viral infections were performed in U373-MAGI-CXCR4CEM cells, obtained from Michael Emerman through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH (20). 5-Aza-C was purchased from Sigma-Aldrich (St. Louis, MO), while 5-aza-dC divalerate (referred to below simply as 5-aza-dC), a stabler prodrug form of 5-aza-dC, was synthesized by the Center for Drug Design at the University of Minnesota as described previously (21). 5-Aza-CTP and 5-aza-dCTP were purchased from American Advanced Scientific (College Station, TX) and Jena Bioscience (Jena, Germany), respectively. All drugs were dissolved in the appropriate solvent and were stored in aliquots at −20°C.
Production and titration of viral stocks for Illumina sequencing.
HIV-1 stocks for Illumina sequencing were produced by cotransfecting 10 μg of pNL4-3 MIG and 1 μg of pHCMV-G per 10-cm plate of 293T cells. Transfections were carried out using the polyethyleneimine (PEI) method, as described previously (19). Viral supernatants were collected 48 h posttransfection, treated with 10 U/ml of DNase I for 2 h at 37°C to reduce plasmid carryover from transfections, and frozen in aliquots at −80°C. The titers of viral stocks were determined by infecting U373-MAGI cells (31,250 cells/well in 24-well plates) with volumes of virus ranging from 1.25 to 40 μl (2-fold series). Viral infectivity was determined by performing flow cytometry on cells collected 72 h postinfection. Viral titers were determined by plotting the volume of virus against the percentage of infected cells (i.e., cells expressing mCherry and/or EGFP).
Determination of viral mutation frequencies by Illumina sequencing.
Samples were prepared for Illumina amplicon sequencing as described previously (22), but with several minor modifications. Briefly, dimethyl sulfoxide (DMSO) (for the no-drug control), 5-aza-C (75% effective concentration [EC75], 260 μM), or 5-aza-dC (EC75, 3.75 μM) was added to U373-MAGI cells (1 million cells/sample) 2 h before infection. The cells were then infected at a multiplicity of infection (MOI) of 1.0 by adding the appropriate volume of NL4-3 MIG-VSVG. The medium was replaced 24 h postinfection, and cells were collected 72 h postinfection. Genomic DNA was purified from cell pellets, and plasmid carryover (from transfections) was then assessed by quantitative PCR (qPCR). Levels of plasmid carryover were determined by dividing the starting quantity of the ampicillin resistance gene by the starting quantity of HIV-1 vif, using previously published primers (22). The average level of plasmid carryover was found to be ∼0.4% (range, 0.1 to 0.7%). Five small (∼160- to 170-bp) amplicons (Gag, Pol, Vif, Env, Nef) were then prepared from each sample by PCR, using the primers listed in Table 1. PCRs were also performed with the purified plasmid to determine the level of background error due to PCR and Illumina sequencing. For each sample, all amplicons were gel purified, quantified, and pooled in an equimolar fashion to normalize coverage between amplicons. Sequencing libraries were prepared from each sample using the TruSeq Nano DNA kit, pooled in an equimolar fashion, and subjected to 2 × 250 paired-end sequencing on the Illumina MiSeq system. Sequencing reads were analyzed as described previously (22) but were demultiplexed based on library indices instead of internal bar codes. As before, background error hot spots (mostly G-to-T and C-to-A transversions) were identified using the plasmid controls and were masked prior to mutational analysis of the biological samples. The final sequencing data were also used in separate analyses that were focused on drug interactions between 5-aza-C and RNR inhibitors (J. M. O. Rawson, M. E. Roth, J. Xie, M. B. Daly, C. L. Clouser, S. R. Landman, C. S. Reilly, L. Bonnac, B. Kim, S. E. Patterson, L. M. Mansky, submitted for publication).
TABLE 1.
Primer sequences for Illumina amplicon sequencing of HIV-1a
| Amplicon | Forward primer | Reverse primer |
|---|---|---|
| Gag | NNAAGCAGCAGCTGACACAGGAA | NNTTCTGGGCTGAAAGCCTTCTCT |
| Pol | NNAGCAGTTCATGTAGCCAGTGGA | NNACAGGCGGCCTTAACTGTAGTA |
| Vif | NNACCCTGACCTAGCAGACCAACTAA | NNGCTGCTAGTGCCAAGTACTGTAGA |
| Env | NNACAGCTGAACACATCTGTAG | NNAGTGGCATTCCATTTTGCTC |
| Nef | NNGCAGCTGTAGATCTTAGCCA | NNTCAGTGGATATCTGACCC |
Illumina primers were designed to prepare five different amplicons (∼160 to 170 bp) from different viral genes. Primers were designed with two 5′-terminal degenerate bases (NN) to promote similar efficiencies of ligation to Illumina adapters. The Gag and Vif primers are the same as those utilized in a previous study (22) but lack bar codes, since samples were demultiplexed based on the library index rather than the internal bar code for this study.
Determination of 5-aza-CTP and 5-aza-dCTP levels by LC–MS-MS.
Samples were prepared for liquid chromatography-tandem mass spectrometry (LC–MS-MS) by splitting U373-MAGI cells onto 10-cm plates (1.3 million cells/plate), using two plates per treatment group. The medium was replaced 24 h later, and 5-aza-C, 5-aza-dC, or DMSO (for the no-drug control) was added. 5-Aza-C and 5-aza-dC were added at equivalent effective concentrations (i.e., EC25, EC50, or EC75). Cells were collected 4 h after 5-aza-C or 5-aza-dC addition, corresponding to the time of early reverse transcription (data not shown). Cell pellets were then resuspended in 750 μl of 60% methanol (stored at −20°C) and were incubated at −20°C for 18 h. The samples were vortexed, heated at 95°C for 3 min, and centrifuged at 16,000 × g for 5 min. The supernatants were transferred to new microcentrifuge tubes, dried using a Savant SPD1010 SpeedVac concentrator (Thermo Fisher Scientific, Inc.) at maximum pressure, and frozen at −80°C until the time of analysis.
For LC–MS-MS, dried extracts were dissolved in 200 μl of water containing 10 μM 5-iodo-dCTP, an internal standard. The samples were centrifuged at 14,000 rpm for 5 min at 4°C, and the supernatants were analyzed by LC–MS-MS. 5-Aza-C and 5-aza-dC samples were reconstituted immediately prior to LC–MS-MS injection in order to minimize their degradation during the procedure. LC–MS-MS was used to determine the levels of 5-aza-CTP, 5-aza-dCTP, riboguanylurea 5′-triphosphate (RGU-TP; the final hydrolysis product of 5-aza-CTP), and 2′-deoxyriboguanylurea 5′-triphosphate (dRGU-TP; the final hydrolysis product of 5-aza-dCTP) by a method published previously (23) with minor modifications. Purified 5-aza-CTP and 5-aza-dCTP were included as standards to verify the procedure. The LC–MS-MS system consisted of an AB Sciex QTrap 5500 mass spectrometer and an Agilent 1260 Infinity high-performance liquid chromatography (HPLC) system. Chromatographic separation of analytes was achieved using a Thermo Scientific Hypercarb column (length, 100 mm; inside diameter, 3 mm; particle size, 5 μm). The two eluents were 0.5% diethylamine in water, with the pH adjusted to 10 with acetic acid (A), and 50% acetonitrile in water (B). The mobile phase was delivered at a flow rate of 0.5 ml/min using stepwise gradients of A and B as follows: from 0 to 20 min, 0 to 25% (vol/vol) B; from 20 to 28 min, 25 to 50% (vol/vol) B; from 28 to 28.5 min, 50 to 95% (vol/vol) B; from 28.5 to 30.5 min, 95% (vol/vol) B; from 30.5 to 31 min, 95 to 0% (vol/vol) B; from 31 to 39 min, 0% (vol/vol) B. Only the eluate from 10 to 30 min was diverted into the mass spectrometer for analysis. The analytes were detected by MS-MS using an electrospray ionization (ESI) ion source with multiple reaction monitoring (MRM) detection in negative mode, with the curtain gas set to 20 lb/in2. The ion spray voltage was set at −4,500 V and the temperature at 650°C. The nebulizer gas (GS1) and turbo gas (GS2) were both set at 45 lb/in2.
HIV-1 RT in vitro incorporation assay.
The HIV-1 RT single nucleotide extension assay was performed as in a previous study (24) but with minor modifications. Briefly, a 32P 5′-end-labeled DNA primer of 18 nucleotides (nt) (5′-GTCCCTCTTCGGGCGCCA-3′) was annealed to a DNA template of 19 nt (3′-CAGGGAGAAGCCCGCGGTG-5′) at a 1:2 ratio. Single nucleotide extension indicates that the compound of interest (5-aza-CTP, 5-aza-dCTP, CTP, or dCTP) was successfully incorporated. The reaction mixtures contained 200 fmol template-primer, 2 μl of the test compound at the indicated concentrations (or 2 μl of dNTPs, for a final concentration of 50 μM for the positive control), 4 μl of purified RT (HIV-1 NL4-3), 25 mM Tris-HCl (pH 8.0), 2 mM dithiothreitol, 100 mM KCl, 5 mM MgCl2, and 10 μM oligo(dT), for a final volume of 20 μl/reaction mixture. Reaction mixtures were incubated at 37°C for 5 min, and the reactions were then quenched with 10 μl of 40 mM EDTA and 99% (vol/vol) formamide at 95°C for 2 min. The reactions were resolved on a 14% urea-PAGE gel (AmericanBio, Inc., Natick, MA), and the results were analyzed using a PharosFX molecular imager (Bio-Rad) and Image Lab software (Bio-Rad).
Statistical analyses.
All figures were created in Microsoft Office for Mac 2011, version 14.5.2 (Redmond, WA), or GraphPad Prism, version 5.0 (GraphPad Software, Inc., La Jolla, CA). To determine the EC25, EC50, and EC75 of 5-aza-C and 5-aza-dC, infectivity data were normalized to the data for the no-drug control, plotted against log-transformed drug concentrations, and subjected to nonlinear regression in GraphPad Prism. In order to determine whether differences in 5-aza-dCTP levels between cells treated with 5-aza-C and those treated with 5-aza-dC were statistically significant, normalized data were analyzed by one-way repeated-measures analysis of variance (ANOVA) (without assuming equal variability of differences) in GraphPad Prism. Sidak's posttest was used to compare the relative quantity of 5-aza-dCTP at each effective concentration (i.e., EC25, EC50, or EC75). To test for variables that may influence mutation frequencies, generalized linear mixed-effects models were applied to processed Illumina sequencing data. The raw counts for each type of mutation (e.g., transitions) were modeled as overdispersed Poisson random variables, with an offset given by the total number of reference bases. The type of sample, the type of amplicon, and their interactions were treated as fixed effects, while the replicate was treated as a random effect. The logarithmic link was used, as is standard for Poisson outcomes, and penalized quasi-likelihood was used to estimate the model parameters (25). These computations were conducted using R, version 3.1.0, and the MASS package.
Nucleotide sequence accession number.
All Illumina sequencing data supporting the results of the manuscript have been deposited into the NCBI Sequence Read Archive (SRA) under accession no. SRP068916.
RESULTS
5-Azacytidine and 5-aza-2′-deoxycytidine induce similar levels of G-to-C and C-to-G transversions in HIV-1.
In order to determine the primary form of 5-aza-C that is active against HIV-1, the effects of 5-aza-C and 5-aza-dC on HIV-1 mutagenesis were compared using Illumina amplicon sequencing. The direct incorporation of 5-aza-C as a ribonucleotide (5-aza-CTP) was predicted to lead to a mutational pattern distinct from that of 5-aza-dCTP, because incorporated ribonucleotides can (i) be replaced (correctly or incorrectly) by RNase H2-mediated repair, (ii) cause mutations if left unrepaired, particularly short (2- to 5-bp) deletions, and (iii) elevate the HIV-1 RT mismatch extension frequency on the opposite strand (26–28). To prepare samples for sequencing, U373-MAGI cells were treated with DMSO (i.e., no drug), 5-aza-C (EC75, ∼260 μM) or 5-aza-dC (EC75, ∼3.8 μM). Cells were then infected 2 h after drug addition at an MOI of 1.0 with NL4-3 MIG-VSVG. Genomic DNA was purified from cells collected 72 h postinfection, and PCR was performed to prepare multiple amplicons (Gag, Pol, Vif, Env, Nef) from proviral DNA. Plasmid control amplifications were included to measure the level of background error from PCR and sequencing. The amplicons were pooled, used to prepare libraries, and subjected to 2 × 250 paired-end sequencing on the Illumina MiSeq system. Illumina sequencing generated ∼5.5 million read pairs after all bioinformatics processing steps, containing ∼570,000 mutations and ∼630 million reference bases. 5-Aza-C and 5-aza-dC were found to significantly increase the frequencies of G-to-C and C-to-G transversions compared to the no-drug control in all amplicons examined (P, <0.0001 for all comparisons), although C-to-G transversions were ∼5- to 6-fold less prevalent than G-to-C transversions (Fig. 1). In the presence of 5-aza-C, ∼67% of all mutations were G-to-C transversions and ∼12% were C-to-G transversions (compared with 64% and 12% for 5-aza-dC). C-to-G transversions were not observed previously when 5-aza-C was added during the early phase of viral replication (10), likely due to the lower sequencing depth of earlier studies (see Discussion). Notably, 5-aza-C and 5-aza-dC increased the levels of G-to-C transversions (Fig. 1A) and C-to-G transversions (Fig. 1B) to similar extents in all five amplicons, and the resulting mutation frequencies did not differ significantly (P, >0.05 for all comparisons). Depending on the amplicon, 5-aza-C and 5-aza-dC raised G-to-C transversion frequencies by ∼45- to 75-fold and C-to-G frequencies by ∼25- to 60-fold. Further, we did not observe short (2- to 5-bp) deletions that are characteristic of ribonucleoside incorporation in the 5-aza-C-treated sample (data not shown). These data demonstrate that 5-aza-C and 5-aza-dC cause very similar changes in viral mutation frequencies and spectra, arguing that 5-aza-C acts primarily as 5-aza-dCTP during reverse transcription.
FIG 1.
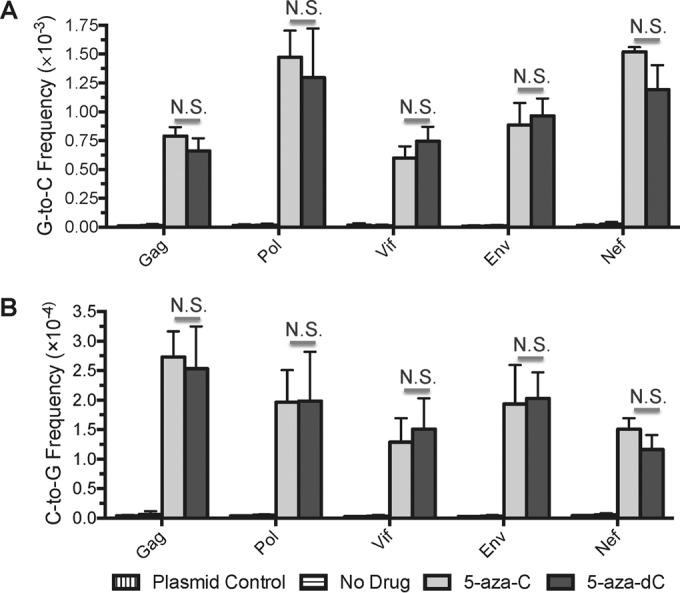
5-Azacytidine and 5-aza-2′-deoxycytidine induce similar levels of G-to-C and C-to-G transversion mutations during HIV-1 replication. In order to determine whether 5-azacytidine (5-aza-C) and 5-aza-2′-deoxycytidine (5-aza-dC) induce similar changes in HIV-1 mutation frequencies and spectra, U373-MAGI cells were treated with DMSO (no-drug control), 5-aza-C, or 5-aza-dC. 5-Aza-C and 5-aza-dC were added 2 h before infection at the EC75 (∼260 or 3.8 μM, respectively). Cells were infected at an MOI of 1.0 with NL4-3 MIG-VSVG and were collected 72 h postinfection for the purification of genomic DNA. PCR was performed to prepare multiple amplicons (Gag, Pol, Vif, Env, Nef) from proviral DNA; these were then pooled, used to prepare libraries, and analyzed by 2 × 250 paired-end sequencing on the Illumina MiSeq system. Plasmid control amplifications were performed to determine the levels of background errors resulting from PCR and sequencing. Mutation frequencies for each amplicon, expressed as the number of mutations per base pair, were calculated by dividing the number of mutations by the number of reference bases (mutations + wild-type bases). Data represent means ± standard deviations for three independent biological replicates; N.S., not significant (P > 0.05).
5-Azacytidine and 5-aza-2′-deoxycytidine cause highly similar HIV-1 mutational patterns.
5-Aza-dC has been demonstrated previously to cause highly variable levels of G-to-C and C-to-G transversions depending on the specific sequence position (12). If 5-aza-C acts against HIV-1 after reduction to 5-aza-dC, the susceptibilities of individual bases to 5-aza-C and 5-aza-dC should closely resemble one another. To address this, mutation frequencies were determined at individual sequence positions in the five amplicons. G-to-C mutation frequencies were determined at every guanine position (124 in total), while C-to-G mutation frequencies were determined at every cytosine position (116 in total). The extent of correlation between 5-aza-C- and 5-aza-dC-induced mutation frequencies was then examined. For both 5-aza-C and 5-aza-dC, significant variability (as much as ∼20-fold) was observed between G-to-C and C-to-G frequencies at individual sequence positions (Fig. 2). 5-Aza-C- and 5-aza-dC-mediated mutation frequencies were found to be strongly positively correlated, both for G-to-C transversions (Pearson's r, 0.93 [95% confidence interval {CI}, 0.90 to 0.95]; P < 0.0001) and for C-to-G transversions (Pearson's r, 0.96 [95% CI, 0.94 to 0.97]; P < 0.0001). This correlation was able to explain most of the variability in the observed data (R2, 0.87 [G-to-C transversions] or 0.91 [C-to-G transversions]). The slopes of the best-fit linear regression lines were also determined, since they would equal 1.0 if the 5-aza-C- and 5-aza-dC-induced mutation frequencies matched perfectly. The regression lines exhibited slopes of 0.71 for G-to-C transversions (95% CI, 0.66 to 0.76) and 0.88 for C-to-G transversions (95% CI, 0.83 to 0.93), indicating that the frequencies of 5-aza-C-mediated mutations were slightly higher than those of 5-aza-dC-mediated mutations. These findings indicate that the patterns of viral mutagenesis in the presence of 5-aza-C and 5-aza-dC closely resembled one another, further indicating that 5-aza-C likely reduces HIV-1 infectivity after conversion to 5-aza-dC.
FIG 2.

5-Azacytidine and 5-aza-2′-deoxycytidine induce similar patterns of mutation during HIV-1 replication. Using the Illumina sequencing data, G-to-C and C-to-G transversion frequencies were determined at every individual guanine (124 in total) or cytosine (116 in total) position within the sequences of the five amplicons. Mutation frequencies for each amplicon were calculated by dividing the number of mutations by the number of reference bases (mutations + wild-type bases) and are represented as mutations per base pair (m/bp). 5-Aza-C- and 5-aza-dC-induced mutation frequencies were then plotted against each other for each sequence position, and the resulting data were subjected to linear regression and correlation analyses. Data represent averages for three independent biological replicates. R2 denotes the extent to which the best-fit regression line explains the observed variability in the data; P indicates the significance of the correlation; and m indicates the slope of the best-fit regression line.
Levels of 5-aza-dCTP are similar in cells treated with 5-azacytidine or 5-aza-2′-deoxycytidine.
While a previous report indicated that ∼10 to 20% of 5-aza-C is reduced to 5-aza-dC in one particular cell line (13), the conversion of 5-aza-C to 5-aza-dC has not yet been assessed in cell lines for which antiviral activity has been demonstrated. To address this, cells were treated with multiple concentrations of either 5-aza-C or 5-aza-dC for 4 h (corresponding to a time point during which reverse transcription should be actively occurring); nucleotides were then extracted, and LC–MS-MS was performed to determine the relative quantities of 5-aza-CTP and 5-aza-dCTP (see Materials and Methods). The relative levels of RGU-TP and dRGU-TP, which are the final hydrolysis products of 5-aza-CTP and 5-aza-dCTP, respectively, and may be relevant to antiviral activity, were also determined. Cells were treated with equivalent effective concentrations (EC25, EC50, or EC75) of 5-aza-C or 5-aza-dC. If 5-aza-C acts primarily as 5-aza-dCTP against HIV-1, cells treated with 5-aza-C or 5-aza-dC should contain approximately equal levels of 5-aza-dCTP at equivalent effective concentrations. 5-Aza-CTP and RGU-TP were detected only in cells treated with 5-aza-C (data not shown), as expected considering the lack of cellular pathways for the conversion of deoxyribonucleotides to ribonucleotides. In contrast, 5-aza-dCTP was detected in cells treated with either 5-aza-C or 5-aza-dC (Fig. 3A), indicating that 5-aza-C (in its diphosphate form) was reduced to 5-aza-dC by RNR. Further, the levels of 5-aza-dCTP either did not differ significantly between cells treated with 5-aza-C and cells treated with 5-aza-dC (EC25 and EC75) or were higher in cells treated with 5-aza-C (at the EC50) (P < 0.05). Similar trends were observed for dRGU-TP, the final hydrolysis product of 5-aza-dCTP (Fig. 3B). These findings further indicate that 5-aza-C reduces the infectivity of HIV-1 primarily after reduction to 5-aza-dC, since HIV-1 RT should incorporate 5-aza-dCTP much more readily than 5-aza-CTP.
FIG 3.

5-Aza-dCTP levels are comparable in cells treated with 5-aza-C or 5-aza-dC. In order to determine the extent to which 5-aza-C is reduced to 5-aza-dC intracellularly, U373-MAGI cells were incubated with varying concentrations (EC25, EC50, or EC75) of 5-aza-C or 5-aza-dC. Cells were collected for analysis 4 h after drug addition, a time corresponding to the expected time of early reverse transcription. LC–MS-MS was then used to determine the relative levels of 5-aza-dCTP and 2′-deoxyriboguanylurea 5′-triphosphate (dRGU-TP). dRGU-TP is the final hydrolysis product of 5-aza-dCTP and is potentially relevant to antiviral activity. Data represent means ± standard deviations for three independent experiments, normalized to the EC75 of 5-aza-dC. N.S., not significant; *, P < 0.05.
HIV-1 reverse transcriptase incorporates 5-aza-CTP in vitro much less efficiently than 5-aza-dCTP.
Although similar levels of 5-aza-dCTP were observed in cells treated with 5-aza-C or 5-aza-dC, the extents to which 5-aza-CTP and 5-aza-dCTP are incorporated by HIV-1 RT had not yet been determined. 5-Aza-CTP was hypothesized to be incorporated only weakly, while 5-aza-dCTP was hypothesized to be incorporated relatively efficiently, since 5-aza-dCTP can be efficiently incorporated by cellular DNA polymerases (29, 30). To address this, the incorporation of 5-aza-CTP and that of 5-aza-dCTP were compared using an HIV-1 RT in vitro single nucleotide extension assay, with dCTP and CTP included for comparative purposes. Additional control reactions were performed by incubation with all four standard dNTPs (50 μM) (positive control) or by the omission of RT (negative control). It was found that HIV-1 RT incorporated 5-aza-dCTP relatively efficiently (Fig. 4), resulting in total primer extension at 5 μM (lane 10) and some detectable extension at 5-aza-dCTP concentrations as low as 50 nM (lane 8). However, 5-aza-dCTP was incorporated somewhat less effectively than dCTP (Fig. 4, compare lanes 7 to 9 with lanes 19 to 21). In contrast, HIV-1 RT incorporated 5-aza-CTP and CTP only at the highest concentration tested (500 μM) (Fig. 4, lanes 6 and 18). These data demonstrate that 5-aza-CTP can be directly incorporated by HIV-1 RT, but only at very high concentrations and in the absence of competing deoxyribonucleotides. 5-Aza-dCTP was incorporated at least 10,000-fold more efficiently than 5-aza-CTP (Fig. 4, compare lanes 6 and 8). These observations are consistent with previous findings that HIV-1 RT exhibits high selectivity for deoxyribonucleotides, largely due to a steric gate (Y115) that blocks incoming ribonucleotides (14–16). Considering that similar levels of 5-aza-dCTP were observed in cells treated with equivalent effective concentrations of 5-aza-C or 5-aza-dC (Fig. 3), these findings strongly indicate that 5-aza-C exerts activity against HIV-1 primarily through its reduction to 5-aza-dC.
FIG 4.
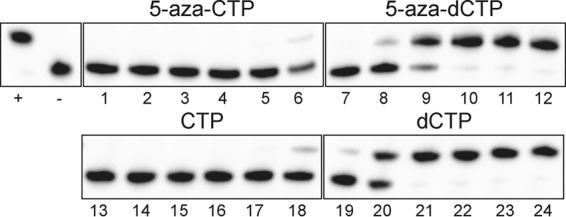
HIV-1 RT incorporates 5-aza-CTP much less efficiently than 5-aza-dCTP in vitro. The relative abilities of HIV-1 RT to incorporate 5-aza-CTP, 5-aza-dCTP, CTP, and dCTP were determined using an in vitro single nucleotide extension assay. The HIV-1 RT was incubated with a radiolabeled primer (18 nt) annealed to a DNA template (19 nt) in the presence of each compound at 5 nM to 500 μM (a 10-fold series, from left to right). Additional control reactions were performed by incubating with all four standard dNTPs (50 μM) (+) or by omitting RT (−). All of the reactions were analyzed on the same gel.
DISCUSSION
The ribonucleoside analog 5-aza-C could potentially act as a ribonucleotide or as a deoxyribonucleotide to reduce HIV-1 infectivity during the early phase of viral replication. To address this, Illumina sequencing was performed in order to compare the enhancement of HIV-1 mutagenesis by 5-aza-C and 5-aza-dC. 5-Aza-C and 5-aza-dC were found to cause similar changes in viral mutation frequencies regardless of the amplicon examined (Fig. 1). Both drugs elicited primarily G-to-C transversions in HIV-1, but also significant levels of C-to-G transversions, which were not observed in a previous study of 5-aza-C (10). 5-Aza-C was not found to induce other types of mutations that are considered indicative of ribonucleotide incorporation, such as short (2- to 5-bp) deletions (27). While the mechanism of C-to-G transversions remains unclear, they may result from the incorporation of 5-aza-dCTP into plus-strand viral DNA or, alternatively, from the direct incorporation of 5-aza-dCTP hydrolysis products (e.g., dRGU-TP) into minus-strand viral DNA. 5-Aza-C-mediated C-to-G transversions were not observed in a previous study (10), possibly due to the reduced depth of Sanger sequencing, the lower concentration of 5-aza-C used, or other differences in the mutational assay. Additionally, 5-aza-C- and 5-aza-dC-mediated mutational patterns correlated strongly with one another even at the level of individual sequence positions in HIV-1 (Fig. 2), further indicating that 5-aza-C exerts antiviral activity after reduction to 5-aza-dC.
5-Aza-dCTP was detected in cells treated with 5-aza-C, indicating that 5-aza-C was reduced to 5-aza-dC (Fig. 3), and, importantly, similar levels of 5-aza-dCTP were observed in cells treated with equivalent effective concentrations of 5-aza-C and 5-aza-dC. Notably, the absolute concentrations of 5-aza-C were ∼70- to 300-fold higher than those of 5-aza-dC in these experiments, indicating that intracellular conversion of 5-aza-C to 5-aza-dCTP was much less efficient than the conversion of 5-aza-dC to 5-aza-dCTP. By use of an in vitro incorporation assay, very high concentrations of 5-aza-CTP were found to be successfully incorporated by HIV-1 RT (Fig. 4). However, 5-aza-dCTP was incorporated at least 10,000-fold more efficiently than 5-aza-CTP, further supporting the notion that 5-aza-dCTP is the principal form of 5-aza-C that is active against HIV-1.
Taken together, these findings clearly indicate that 5-aza-dCTP is the primary form of 5-aza-C that is active during the early phase of HIV-1 replication. Further, 5-aza-C has been shown to exert much more potent antiviral activity during the early phase of replication than during the late phase of replication (10), indicating that 5-aza-C likely acts primarily as 5-aza-dCTP during spreading infections as well. These observations lead to the following model for the antiviral activities of 5-aza-C and 5-aza-dC (Fig. 5). 5-Aza-C and 5-aza-dC enter the cell by facilitated diffusion through a transporter protein and are then phosphorylated by the appropriate cellular kinases. 5-Aza-CTP is incorporated into viral RNA during the late phase of replication, leading to the fixation of C-to-G transversions during the following round of reverse transcription. 5-Aza-C (in its diphosphate form) is also converted to 5-aza-dC by RNR, and the resulting 5-aza-CDP can then be phosphorylated to form 5-aza-dCTP. Lastly, 5-aza-dCTP is incorporated into viral DNA during reverse transcription, resulting in the fixation of G-to-C and (to a lesser extent) C-to-G transversions in HIV-1.
FIG 5.
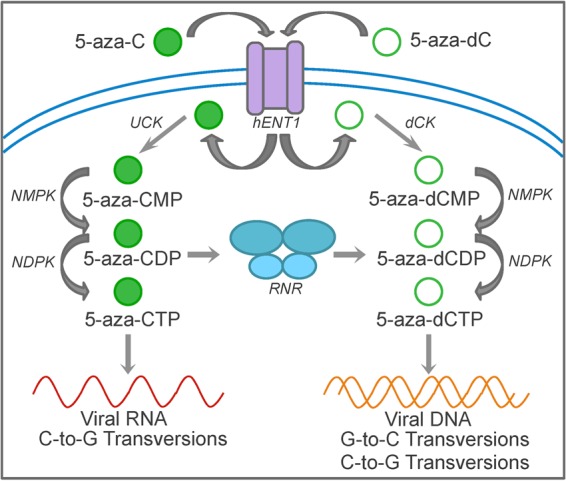
Model of 5-azacytidine- and 5-aza-2′-deoxycytidine-mediated HIV-1 mutagenesis. 5-Aza-C and 5-aza-dC are first transported into the cell by facilitated diffusion through human equilibrative nucleoside transporter 1 (hENT1). 5-Aza-C and 5-aza-dC are then phosphorylated by uridine-cytidine kinase (UCK) and deoxycytidine kinase (dCK), respectively, to the monophosphate forms. Nucleoside monophosphate and diphosphate kinases (NMPK and NDPK) phosphorylate the monophosphate and diphosphate forms, respectively, resulting in the formation of 5-aza-CTP and 5-aza-dCTP. 5-Aza-CTP can be incorporated during the transcription of viral genomic RNA, resulting in C-to-G transversions. 5-Aza-CDP is also reduced to 5-aza-dCDP, ultimately forming 5-aza-dCTP. 5-Aza-dCTP is incorporated during reverse transcription, resulting primarily in G-to-C transversions (reflecting minus-strand incorporation) but also in low levels of C-to-G transversions. The mechanism by which C-to-G transversions are formed is still unclear, but they may be due to plus-strand incorporation of 5-aza-dCTP or minus-strand incorporation of 5-aza-dCTP hydrolysis products.
In the future, it would be of interest to investigate the antiviral activity of 5-aza-C in other cell types, such as primary CD4+ T cells and macrophages. 5-Aza-C would likely act primarily as 5-aza-dCTP in activated CD4+ T cells, since they divide rapidly and thus have high levels of RNR and dNTPs (17, 31). In contrast, 5-aza-CTP could potentially be directly incorporated at significant levels in macrophages, which have low levels of RNR, low dNTP pools, and much higher ratios of NTPs to dNTPs (17, 31). Indeed, previous work has shown that HIV-1 RT can incorporate significant levels of endogenous ribonucleotides in macrophages and in vitro under macrophage-like conditions (17, 18). Further, the ribonucleoside analog 3′-deoxyadenosine, which likely cannot be reduced, exhibits antiviral activity in macrophages but not in CD4+ T cells, indicating that ribonucleoside analog triphosphates can be directly incorporated in macrophages (17).
Notably, the findings in this study have important implications for the design of ribonucleoside analogs directed against retroviruses. Ribonucleoside analogs are of interest as antiretroviral agents because their synthesis is often less complicated, less time-intensive, and more affordable than that of their deoxyribonucleoside counterparts. Ribonucleoside analogs may also offer other advantages in certain instances, such as better activation by cellular enzymes or improved antiviral activity by the targeting of both viral RNA and DNA. However, the findings in this study suggest that it is important to consider the efficiency at which ribonucleoside analogs are reduced—at least for the targeting of viral replication in cell types with high dNTP pools, such as CD4+ T cells. In contrast, efficient reduction may not be necessary or even desired for the targeting of viral replication in cell types with low dNTP pools, such as macrophages. Ribonucleoside analogs that cannot be reduced (such as those lacking a 3′ hydroxyl group, e.g., 3′-deoxyadenosine) could be used to specifically target viral replication in macrophages, an important viral reservoir (17). In sum, these findings inform efforts to identify additional nucleoside analogs that target HIV-1 RT, which continue due to significant concerns regarding the development of drug resistance and off-target effects.
ACKNOWLEDGMENTS
We thank the staff of the University of Minnesota Genomics Center for helpful advice on the design, performance, and analysis of Illumina sequencing experiments. We also acknowledge the support of the Minnesota Supercomputing Institute in providing computing, software, and data storage support for this project.
Funding Statement
This research was funded by fellowships (to J.M.O.R. and S.R.L.) from the University of Minnesota Graduate School. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Domingo E, Sheldon J, Perales C. 2012. Viral quasispecies evolution. Microbiol Mol Biol Rev 76:159–216. doi: 10.1128/MMBR.05023-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eigen M. 2002. Error catastrophe and antiviral strategy. Proc Natl Acad Sci U S A 99:13374–13376. doi: 10.1073/pnas.212514799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Loeb LA, Essigmann JM, Kazazi F, Zhang J, Rose KD, Mullins JI. 1999. Lethal mutagenesis of HIV with mutagenic nucleoside analogs. Proc Natl Acad Sci U S A 96:1492–1497. doi: 10.1073/pnas.96.4.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perales C, Domingo E. 21 August 2015. Antiviral strategies based on lethal mutagenesis and error threshold. Curr Top Microbiol Immunol doi: 10.1007/82_2015_459. [DOI] [PubMed] [Google Scholar]
- 5.Bonnac LF, Mansky LM, Patterson SE. 2013. Structure-activity relationships and design of viral mutagens and application to lethal mutagenesis. J Med Chem 56:9403–9414. doi: 10.1021/jm400653j. [DOI] [PubMed] [Google Scholar]
- 6.Matsui M, Shindo K, Izumi T, Io K, Shinohara M, Komano J, Kobayashi M, Kadowaki N, Harris RS, Takaori-Kondo A. 2014. Small molecules that inhibit Vif-induced degradation of APOBEC3G. Virol J 11:122. doi: 10.1186/1743-422X-11-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nathans R, Cao H, Sharova N, Ali A, Sharkey M, Stranska R, Stevenson M, Rana TM. 2008. Small-molecule inhibition of HIV-1 Vif. Nat Biotechnol 26:1187–1192. doi: 10.1038/nbt.1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pery E, Sheehy A, Nebane NM, Brazier AJ, Misra V, Rajendran KS, Buhrlage SJ, Mankowski MK, Rasmussen L, White EL, Ptak RG, Gabuzda D. 2015. Identification of a novel HIV-1 inhibitor targeting Vif-dependent degradation of human APOBEC3G protein. J Biol Chem 290:10504–10517. doi: 10.1074/jbc.M114.626903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zuo T, Liu D, Lv W, Wang X, Wang J, Lv M, Huang W, Wu J, Zhang H, Jin H, Zhang L, Kong W, Yu X. 2012. Small-molecule inhibition of human immunodeficiency virus type 1 replication by targeting the interaction between Vif and ElonginC. J Virol 86:5497–5507. doi: 10.1128/JVI.06957-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dapp MJ, Clouser CL, Patterson S, Mansky LM. 2009. 5-Azacytidine can induce lethal mutagenesis in human immunodeficiency virus type 1. J Virol 83:11950–11958. doi: 10.1128/JVI.01406-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clouser CL, Patterson SE, Mansky LM. 2010. Exploiting drug repositioning for discovery of a novel HIV combination therapy. J Virol 84:9301–9309. doi: 10.1128/JVI.01006-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rawson JM, Landman SR, Reilly CS, Bonnac L, Patterson SE, Mansky LM. 17 August 2015. Lack of mutational hotspots during decitabine-mediated HIV-1 mutagenesis. Antimicrob Agents Chemother doi: 10.1128/AAC.01644-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li LH, Olin EJ, Buskirk HH, Reineke LM. 1970. Cytotoxicity and mode of action of 5-azacytidine on L1210 leukemia. Cancer Res 30:2760–2769. [PubMed] [Google Scholar]
- 14.Boyer PL, Sarafianos SG, Arnold E, Hughes SH. 2000. Analysis of mutations at positions 115 and 116 in the dNTP binding site of HIV-1 reverse transcriptase. Proc Natl Acad Sci U S A 97:3056–3061. doi: 10.1073/pnas.97.7.3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cases-Gonzalez CE, Gutierrez-Rivas M, Menendez-Arias L. 2000. Coupling ribose selection to fidelity of DNA synthesis. The role of Tyr-115 of human immunodeficiency virus type 1 reverse transcriptase. J Biol Chem 275:19759–19767. doi: 10.1074/jbc.M910361199. [DOI] [PubMed] [Google Scholar]
- 16.Nguyen LA, Domaoal RA, Kennedy EM, Kim DH, Schinazi RF, Kim B. 2015. Pre-steady state kinetic analysis of HIV-1 reverse transcriptase for non-canonical ribonucleoside triphosphate incorporation and DNA synthesis from ribonucleoside-containing DNA template. Antiviral Res 115:75–82. doi: 10.1016/j.antiviral.2014.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kennedy EM, Gavegnano C, Nguyen L, Slater R, Lucas A, Fromentin E, Schinazi RF, Kim B. 2010. Ribonucleoside triphosphates as substrate of human immunodeficiency virus type 1 reverse transcriptase in human macrophages. J Biol Chem 285:39380–39391. doi: 10.1074/jbc.M110.178582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kennedy EM, Amie SM, Bambara RA, Kim B. 2012. Frequent incorporation of ribonucleotides during HIV-1 reverse transcription and their attenuated repair in macrophages. J Biol Chem 287:14280–14288. doi: 10.1074/jbc.M112.348482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rawson JM, Heineman RH, Beach LB, Martin JL, Schnettler EK, Dapp MJ, Patterson SE, Mansky LM. 2013. 5,6-Dihydro-5-aza-2′-deoxycytidine potentiates the anti-HIV-1 activity of ribonucleotide reductase inhibitors. Bioorg Med Chem 21:7222–7228. doi: 10.1016/j.bmc.2013.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vodicka MA, Goh WC, Wu LI, Rogel ME, Bartz SR, Schweickart VL, Raport CJ, Emerman M. 1997. Indicator cell lines for detection of primary strains of human and simian immunodeficiency viruses. Virology 233:193–198. doi: 10.1006/viro.1997.8606. [DOI] [PubMed] [Google Scholar]
- 21.Clouser CL, Bonnac L, Mansky LM, Patterson SE. 2014. Characterization of permeability, stability and anti-HIV-1 activity of decitabine and gemcitabine divalerate prodrugs. Antivir Chem Chemother 23:223–230. doi: 10.3851/IMP2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rawson JM, Landman SR, Reilly CS, Mansky LM. 2015. HIV-1 and HIV-2 exhibit similar mutation frequencies and spectra in the absence of G-to-A hypermutation. Retrovirology 12:60. doi: 10.1186/s12977-015-0180-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cohen S, Megherbi M, Jordheim LP, Lefebvre I, Perigaud C, Dumontet C, Guitton J. 2009. Simultaneous analysis of eight nucleoside triphosphates in cell lines by liquid chromatography coupled with tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 877:3831–3840. doi: 10.1016/j.jchromb.2009.09.030. [DOI] [PubMed] [Google Scholar]
- 24.Weiss KK, Chen R, Skasko M, Reynolds HM, Lee K, Bambara RA, Mansky LM, Kim B. 2004. A role for dNTP binding of human immunodeficiency virus type 1 reverse transcriptase in viral mutagenesis. Biochemistry 43:4490–4500. doi: 10.1021/bi035258r. [DOI] [PubMed] [Google Scholar]
- 25.Wolfinger R, O'Connell M. 1993. Generalized linear mixed models: a pseudo-likelihood approach. J Stat Comput Sim 48:233–243. doi: 10.1080/00949659308811554. [DOI] [Google Scholar]
- 26.Daddacha W, Noble E, Nguyen LA, Kennedy EM, Kim B. 2013. Effect of ribonucleotides embedded in a DNA template on HIV-1 reverse transcription kinetics and fidelity. J Biol Chem 288:12522–12532. doi: 10.1074/jbc.M113.458398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nick McElhinny SA, Kumar D, Clark AB, Watt DL, Watts BE, Lundstrom EB, Johansson E, Chabes A, Kunkel TA. 2010. Genome instability due to ribonucleotide incorporation into DNA. Nat Chem Biol 6:774–781. doi: 10.1038/nchembio.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nick McElhinny SA, Watts BE, Kumar D, Watt DL, Lundstrom EB, Burgers PM, Johansson E, Chabes A, Kunkel TA. 2010. Abundant ribonucleotide incorporation into DNA by yeast replicative polymerases. Proc Natl Acad Sci U S A 107:4949–4954. doi: 10.1073/pnas.0914857107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bouchard J, Momparler RL. 1983. Incorporation of 5-aza-2′-deoxycytidine-5′-triphosphate into DNA. Interactions with mammalian DNA polymerase alpha and DNA methylase. Mol Pharmacol 24:109–114. [PubMed] [Google Scholar]
- 30.Covey JM, D'Incalci M, Tilchen EJ, Zaharko DS, Kohn KW. 1986. Differences in DNA damage produced by incorporation of 5-aza-2′-deoxycytidine or 5,6-dihydro-5-azacytidine into DNA of mammalian cells. Cancer Res 46:5511–5517. [PubMed] [Google Scholar]
- 31.Diamond TL, Roshal M, Jamburuthugoda VK, Reynolds HM, Merriam AR, Lee KY, Balakrishnan M, Bambara RA, Planelles V, Dewhurst S, Kim B. 2004. Macrophage tropism of HIV-1 depends on efficient cellular dNTP utilization by reverse transcriptase. J Biol Chem 279:51545–51553. doi: 10.1074/jbc.M408573200. [DOI] [PMC free article] [PubMed] [Google Scholar]