

Abstract
Herpes simplex virus 1 (HSV-1) and HSV-2 remain major human pathogens despite the development of anti-HSV therapeutics as some of the first antiviral drugs. Current therapies are incompletely effective and frequently drive the evolution of drug-resistant mutants. We recently determined that certain natural troponoid compounds such as β-thujaplicinol readily suppress HSV-1 and HSV-2 replication. Here, we screened 26 synthetic α-hydroxytropolones with the goals of determining a preliminary structure-activity relationship for the α-hydroxytropolone pharmacophore and providing a starting point for future optimization studies. Twenty-five compounds inhibited HSV-1 and HSV-2 replication at 50 μM, and 10 compounds inhibited HSV-1 and HSV-2 at 5 μM, with similar inhibition patterns and potencies against both viruses being observed. The two most powerful inhibitors shared a common biphenyl side chain, were capable of inhibiting HSV-1 and HSV-2 with a 50% effective concentration (EC50) of 81 to 210 nM, and also strongly inhibited acyclovir-resistant mutants. Moderate to low cytotoxicity was observed for all compounds (50% cytotoxic concentration [CC50] of 50 to >100 μM). Therapeutic indexes ranged from >170 to >1,200. These data indicate that troponoids and specifically α-hydroxytropolones are a promising lead scaffold for development as anti-HSV drugs provided that toxicity can be further minimized. Troponoid drugs are envisioned to be employed alone or in combination with existing nucleos(t)ide analogs to suppress HSV replication far enough to prevent viral shedding and to limit the development of or treat nucleos(t)ide analog-resistant mutants.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) and HSV-2 are highly related human herpesviruses. They have colinear genomes, share 87% amino acid sequence identity, and are ubiquitous pathogens (1). HSV-1 and HSV-2 infect mucosal surfaces and abraded skin and transit within sensory nerve fibers to the ganglia, where they establish lifelong latency. Periodic reactivation of HSV-1 or HSV-2 replication results in recurrent disease, but frequent asymptomatic shedding occurs and is a source of transmissible virus (2). HSV-1 typically causes common cold sores, rare but debilitating encephalitis, and over 400,000 cases annually of sight-threatening corneal disease (3). HSV-1 also causes an increasing proportion of genital infections (4–8), although HSV-2 has historically been the source of most genital ulcerative disease. Nearly 1 in 6 Americans has been exposed to HSV-2 (10), and the estimated number of infected persons exceeds half a billion worldwide (11). Anogenital HSV ulcers increase the chance of acquiring and transmitting HIV infection (12, 13), and peripartum virus shed in the genital tract of pregnant women can result in widely disseminated severe disease in their newborns (14).
Nucleoside analog therapy with drugs such as acyclovir (ACV) is used to treat acute infections and reduces viral shedding and disease associated with HSV reactivations but is incompletely effective (9, 15, 16). In addition, viral resistance to nucleos(t)ide analog drugs has been observed in immunocompromised adults (17–22), pediatric patients (23), and immunocompetent persons with herpetic stromal keratitis or uveitis (24–26). The emergence of drug-resistant and multidrug-resistant viruses has been linked to severe complications in immunocompromised individuals (27–30). More effective suppression of HSV will require new drugs that will likely be used in combination with nucleos(t)ide analogs.
Nucleotidyltransferase superfamily (NTS) enzymes perform many functions in nucleic acid metabolism. HSV DNA replication requires several viral activities consistent with those of NTS enzymes: the ends of the linear genome are annealed upon entry, Okazaki fragments are removed during viral DNA replication, and genome concatemers are cleaved into unit length and packaged into nascent capsids. Viral NTS enzymes include the distantly related HIV integrase and RNase H and hepatitis B virus (HBV) RNase H (31, 32). We previously demonstrated that numerous compounds selected for the capacity to suppress HIV or HBV RNase H activity or close chemical relatives of these compounds also inhibit HSV replication in cell cultures (33). One of these compounds, the natural product β-thujaplicinol (34), is the most widely studied member of a class of troponoids called α-hydroxytropolones that have been identified as anticancer agents (35, 36) as well as lead therapeutic agents for a number of infections, including HIV (37–41), HBV (42), malaria (43), and many bacteria (44). This antimicrobial activity is often attributed to the capacity of the compounds to sequester divalent cations in the active sites of dinuclear metalloenzymes, a feature arising from the three contiguous oxygen atoms on the troponoid ring (45–47). Here, 26 synthetic α-hydroxytropolones were screened for their capacity to inhibit HSV-1 and HSV-2 replication to assess whether α-hydroxytropolones may be attractive candidates for development as anti-HSV drugs.
MATERIALS AND METHODS
Compound acquisition and synthesis.
The compounds employed are listed in Table 1, and their structures are shown in Fig. S1 in the supplemental material. Compound 46 was acquired from the National Cancer Institute (NCI) Developmental Therapeutics Program. α-Hydroxytropolone (compound 172) was synthesized in 3 steps from tropolone based on procedures described previously by Takeshita et al. (48). Compounds 95 to 104 were synthesized from manicol as previously described (39) and exceeded 95% purity, as determined by liquid chromatography-mass spectrometry (LCMS). Compounds 106 to 117, 119, 120, 143 to 147, and 173 were synthesized in 5 to 7 steps from kojic acid as previously described (49–51). Synthetic α-hydroxytropolones were pure as determined by 1H nuclear magnetic resonance (NMR) analysis, as reported previously, and a spectrum of compound 118 is provided in Fig. S2 in the supplemental material. Compound 118 was also synthesized from kojic acid, as described below. All compounds were dissolved in dimethyl sulfoxide (DMSO) and stored at −80°C.
TABLE 1.
HSV suppression by hydroxytropolones
| Compounda | Compound name | Log10 HSV-1 suppressionb |
Log10 HSV-2 suppressionb |
CC50 (μM) | IC50 (μM)c |
|||
|---|---|---|---|---|---|---|---|---|
| 50 μM | 5 μM | 50 μM | 5 μM | Anti-HBV RNase H | Anti-human RNase H1 | |||
| Natural-product tropolones or α-hydroxylated tropolones | ||||||||
| 46 | Beta-thujaplicinol | 4.8 | 5.0 | 5.1 | 5.3 | >100 | 5.9 | 53 |
| 47 | Beta-thujaplicin | 1.9 | 0.31 | 1.4 | 0.22 | − | − | |
| 56 | Manicol | 5.8 | 5.1 | 5.8 | 6.0 | 100 | +/− | − |
| 195 | Purpurogallin | 0.20 | 0.17 | 0.11 | −0.04 | − | +++ | |
| Synthetic α-hydroxylated tropolones | ||||||||
| 95 | JKJ07-26 | 6.3 | 0.60 | + | − | − | − | |
| 96 | JKJ08-04 | 6.6 | 2.5 | + | − | − | + | |
| 99 | JKJ07-36 | − | − | − | − | − | − | |
| 102 | JKJ08-42 | + | − | + | − | − | ICA | |
| 103 | JKJ08-45 | + | 0.11 | + | 0.02 | − | − | |
| 104 | JKJ07-79 | + | 3.0 | + | 2.0 | − | − | |
| 106 | CM1012-6a | 4.1 | −0.07 | 2.9 | −0.55 | 30 | 65 | |
| 108 | CM1012-6c | + | 0.00 | + | 0.00 | + | ++ | |
| 109 | CM1012-6d | 4.0 | 0.21 | 4.2 | −0.22 | + | ++ | |
| 110 | CM1012-6e | 5.1 | −0.47 | 4.2 | −0.65 | 35 | 88 | |
| 111 | CM1012-6f | T | 4.3 | T | 3.2 | >50 | − | +++ |
| 112 | CM1012-6i | 5.3 | 0.02 | 3.2 | −0.40 | >100 | 96 | |
| 113 | RM-YM-1-0613 | 5.4 | 2.3 | 5.6 | 2.1 | >100 | >100 | 27 |
| 114 | RM-YM-2-0613 | 4.8 | 4.8 | 5.3 | 3.9 | >100 | − | >100 |
| 115 | RM-YM-3-0613 | 4.1 | 4.3 | 5.1 | 4.5 | >100 | − | >100 |
| 117 | RM-CM-2-0613 | 4.7 | 0.76 | 5.0 | −0.16 | − | − | |
| 118 | RM-MD-2-0813 | 6.3 | 5.1 | 5.6 | 4.2 | >100 | − | − |
| 119 | RM-YM-3BrPh | 5.3 | 5.2 | 4.9 | 5.0 | 72 | − | − |
| 120 | RM-MD-1-0713 | 5.5 | 4.8 | 5.1 | 5.4 | >50 | + | +++ |
| 143 | MD-1-138 | 5.9 | 0.25 | 4.8 | 0.00 | + | + | |
| 144 | DH-1-148 | 3.8 | −0.34 | 5.2 | 0.65 | − | − | |
| 145 | DH-1-163 | 4.0 | 4.0 | 4.3 | 4.1 | >100 | − | − |
| 146 | DH-2-8 | 6.2 | 5.7 | 5.3 | 5.7 | >100 | − | +++ |
| 147 | DH-2-4 | 5.8 | 5.9 | 4.1 | 4.3 | 75 | − | − |
| 172 | 7-HT | 4.1 | 3.3 | 4.6 | 1.8 | >100 | − | ++ |
| 173 | MD-1-152 | 5.8 | 5.9 | 5.9 | 5.3 | >100 | − | ++ |
| Nucleoside analog | ||||||||
| ACV | Acyclovir | 5.4 | 3.6 | 4.7 | 2.9 | >100 | − | − |
Compounds 46, 47, and 56 were previously reported (33).
For HSV screening, + indicates a modest reduction in cytopathic effect compared to the DMSO control as determined by a colorimetric assay, and − indicates no change in cytopathic effect as determined by a colorimetric assay. Numerical data from a plaque reduction assay were not obtained due to the insufficient amount of compound available. T, toxic.
For HBV and human RNase H1 screening, 50% inhibitory concentrations (IC50s) are shown either in micromolar concentrations or as +++ for inhibition at 10 μM, ++ for inhibition at 20 μM, + for inhibition at 60 μM, or − for no inhibition at 60 μM. ICA, insufficient compound available. IC50s for compounds 106 to 172 against HBV RNase H and human RNase H1 were previously reported (42).
4-([1,1′-Biphenyl]-4-carbonyl)-2,7-dihydroxy-5-methylcyclo-hepta-2,4,6-trien-1-one (compound 118) synthesis.
N,N-Diisopropylaniline (81 μl; 0.414 mmol; 1.2 eq) was added to a suspension of 5-hydroxy-4-methoxy-2-methylpyrylium trifluoromethanesulfonate (100 mg; 0.345 mmol) and 1-([1,1′-biphenyl]-4-yl)prop-2-yn-1-one (712 mg; 3.45 mmol; 10 eq) in CH2Cl2 (5 ml). After microwave irradiation at 100°C for 1 h, the reaction mixture was concentrated and purified by chromatography (silica [10 g] with a gradient from 0% ethyl acetate-hexane to 35% ethyl acetate-hexane over 20 column volumes), yielding bicycle 6-([1,1′-biphenyl]-4-carbonyl)-3-methoxy-5-methyl-8-oxabicyclo[3.2.1]octa-3,6-dien-2-one as an orange solid (92.8 mg; 77% yield). Melting point = 156 to 159°C. Retention factor = 0.22 in 20% ethyl acetate in hexanes. Infrared (thin film, potassium bromide) 3,063 (w), 2,979 (w), 2,935 (w), 2,837 (w), 1,711 (s), 1,641 (m), 1,603 (s), 1,449 (w), 1,323 (m), 1,127 (m), 1,043 (w), 989 (w), 844 (m), 744 (s), 698 (m) cm−1. 1H NMR (400 MHz, CDCl3) δ 7.93 (d, J = 8.3 Hz, 2H), 7.71 (d, J = 8.3 Hz, 2H), 7.65 to 7.61 (m, 2H), 7.51 to 7.38 (m, 3H), 6.83 (d, J = 2.4 Hz, 1H), 6.30 (s, 1H), 5.20 (d, J = 2.5 Hz, 1H), 3.60 (s, 3H), 1.77 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 190.37, 188.72, 155.32, 146.66, 145.31, 139.84, 138.77, 135.73, 129.91, 129.29, 128.71, 127.63, 127.54, 120.62, 87.37, 87.01, 54.98, 21.07. HRMS (ESI+) m/z calculated for C22H19O4+, 347.1278; found, 347.1280.
Trifluoromethanesulfonic acid (47.3 μl; 0.536 mmol; 4 eq) was added to the above-described bicycle 6-([1,1′-biphenyl]-4-carbonyl)-3-methoxy-5-methyl-8-oxabicyclo[3.2.1]octa-3,6-dien-2-one (46.4 mg; 0.134 mmol) in CH2Cl2 (1.5 ml). The reaction mixture was stirred for 30 min, at which time it was quenched with sodium acetate (110 mg; 1.34 mmol; 10 eq), stirred for 20 min, and concentrated under reduced pressure to generate the methoxytropolone. The methoxytropolone was then dissolved in 25% hydrobromic acid in acetic acid (2 ml) and heated to 90°C for 4 h. The reaction mixture was cooled to room temperature, quenched with pH 7 phosphate buffer (10 ml), and extracted with CH2Cl2 (5 ml). The organic layer was washed thrice with 10 ml phosphate buffer, dried over Na2SO4, filtered, and concentrated to yield compound 118 as a brown oil (26.2 mg; 59% yield). Infrared (thin film, potassium bromide) 3,262 (br), 3,060 (w), 2,961 (w), 1,669 (s), 1,601 (s), 1,534 (s), 1,398 (m), 1,284 (s), 1,232 (s), 1,191 (s), 1,083 (s), 906 (m), 859 (m), 750 (s), 696 (m) cm−1. 1H NMR (400 MHz, CDCl3) δ 7.88 (d, J = 8.2 Hz, 2H), 7.71 (d, J = 8.2 Hz, 2H), 7.63 (d, J = 7.4 Hz, 2H), 7.54 (s, 1H), 7.52 to 7.39 (m, 3H), 7.36 (s, 1H), 2.36 (s, 3H) (see Fig. S1 in the supplemental material). 13C NMR (100 MHz, CDCl3) δ 197.02, 168.70, 159.03, 157.33, 147.49, 140.15, 139.85, 138.42, 134.57, 131.05, 129.41, 128.96, 128.01, 127.70, 124.67, 119.18, 24.86. HRMS (ESI+) m/z calculated for C21H17O4+, 333.1121; found, 333.1124.
Cells and viruses.
Vero cells were maintained in growth medium (Dulbecco's modified Eagle's medium [DMEM] containing 3% newborn calf serum, 3% bovine growth serum, 100 IU/ml penicillin–0.1 mg/ml streptomycin, and 2 mM l-glutamine). The HSV-1 and HSV-2 strains used for screening were deidentified clinical isolates from the Saint Louis University Hospital passaged once in culture. Wild-type HSV-1 and HSV-2 used in the experiments in Fig. 4 were laboratory strains 17 and 333, respectively. ΔTK is a thymidine kinase (TK)-deficient mutant of HSV-1 strain 17 (52). ΔTK− is a TK-deficient mutant of HSV-2 strain 333 (53). HSV-1 strain McKrae6β (David Leib, unpublished data) was made in a fashion identical to that for KOS6β (54). McKrae6β and HSV-2 strain 333vhsB (55) express β-galactosidase under the control of the ICP6 promoter from the UL49/UL50 intergenic region and the vhs locus, respectively. Virus stocks were grown and titers were determined on Vero cells (56, 57).
FIG 4.
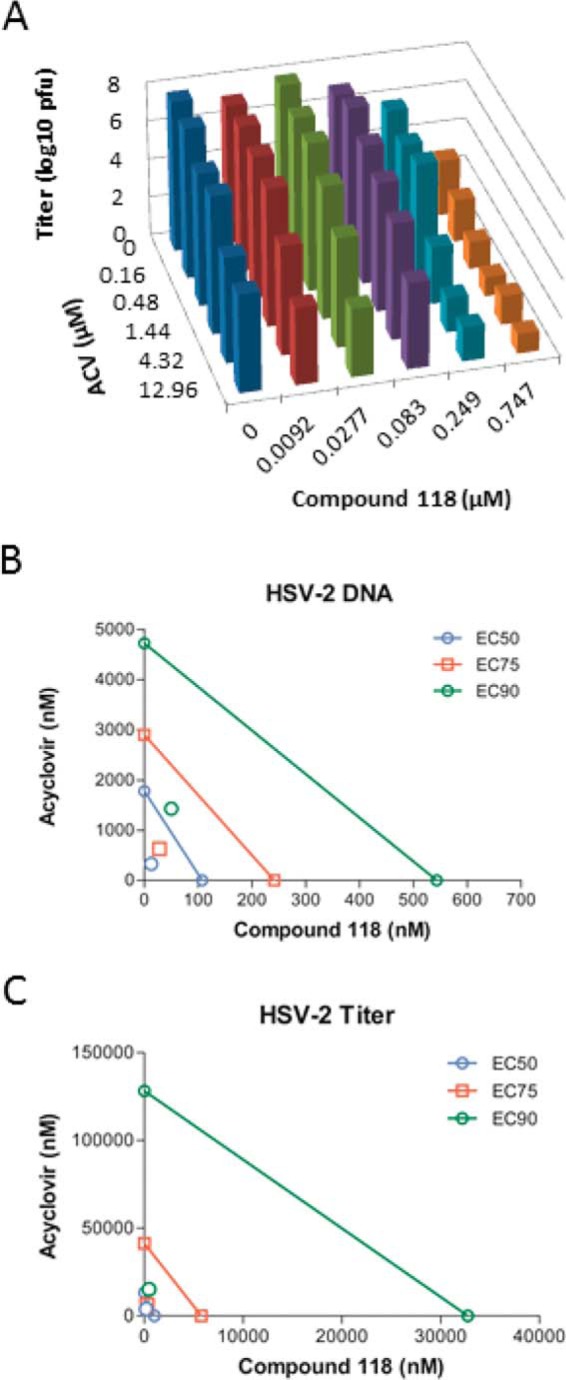
ACV and compound 118 synergistically inhibit HSV-2 replication and DNA accumulation. ACV and compound 118 were diluted in 3-fold steps and added alone or together in constant ratios to Vero cell monolayers infected with HSV-2 at an MOI of 0.1. After 24 h, the cultures were collected and divided into two aliquots. Shown are a representative volume plot of viral titers for combinations of ACV and/or compound 118 against HSV-2, as determined by a plaque assay (A), and isobolograms for qPCR for HSV-2 genomic DNA (B) and viral titer (C) data, as analyzed by the Chou-Talalay method. In panels B and C, the x axis indicates the effective concentration in this experiment for compound 118 alone, and the y axis indicates the effective concentration for ACV alone. The colored lines represent efficacy expected from mixing compound 118 and ACV at various proportions if the effects of the compounds are additive. The area below the lines indicates synergism, and the area above indicates antagonism. EC90, EC75, and EC50 values were calculated in this experiment from the combinations of compound 118 and ACV.
Colorimetric screening assay.
Vero cells (1 × 104 cells per well) were seeded into 96-well plates and incubated in growth medium as indicated above. After 24 h, the medium was removed, and cells were infected at a multiplicity of infection (MOI) of 0.2 with HSV-1 McKrae6β or HSV-2 333vhsB. After 1 h of infection, virus-containing medium was removed; wells were rinsed with phosphate-buffered saline (PBS); and compounds which had been diluted to 50 μM, 5 μM, and 0.5 μM in DMEM containing 2% newborn calf serum, 100 IU/ml penicillin–0.1 mg/ml streptomycin, and 2 mM l-glutamine were added to duplicate wells. Equivalent concentrations of DMSO were added to additional wells as a diluent control. Twenty-four hours after compound addition (HSV-1) or 20 h after compound addition (HSV-2), the plates were inspected by phase-contrast microscopy for toxicity. Subsequently, the medium was removed, cells were lysed, and chlorophenol red-β-d-galactopyranoside (CPRG) substrate (5 mg/ml) was added. After 45 min of incubation at 37°C, the absorbance at 570 nm was determined by using a plate reader. A compound was considered to have suppressed β-galactosidase accumulation at a given concentration if the optical density reading was <50% of that of the DMSO control.
HSV-1 and HSV-2 replication inhibition assay.
Compounds were diluted in PBS containing 2% newborn calf serum and 2 mM l-glutamine and were added to confluent cell monolayers in 24-well plates. Equivalent dilutions of DMSO were used as vehicle controls. HSV-1 and HSV-2 were diluted in supplemented PBS medium and added so that the final compound concentrations were 50 μM or 5 μM and the HSV MOI was 0.1. The cells were incubated at 37°C for 1 h, the virus-containing inoculum was removed, the wells were washed once in PBS, and the compound (50 μM or 5 μM) in supplemented DMEM was added. Cells were incubated at 37°C for an additional 23 h, and the plates were then inspected by phase-contrast microscopy for cytopathic effect (CPE) or toxicity. Cells in wells showing less CPE than in DMSO-treated control wells were harvested by scraping, along with cells from additional wells with significant CPE for comparison. Samples were frozen at −80°C, thawed, and sonicated, and virus titers were then determined by a plaque assay on Vero cells. Each experiment was repeated at least once. The 50% effective concentrations (EC50s) were determined as described above except that serial dilutions of the compounds were employed. Values were calculated with GraphPad Prism using the three-parameter log(inhibitor)-versus-response algorithm with the bottom value set to zero.
Cytotoxicity assays.
Vero cells (1 × 104 cells per well) were seeded into 96-well plates and incubated in DMEM as indicated above. The compounds were diluted in medium to the indicated concentrations with 1% DMSO and added to the cells 24 h after plating, with each concentration being tested in triplicate. Twenty-four hours after compound addition, thiazolyl blue tetrazolium bromide (MTT) (Sigma-Aldrich) was added to 0.25 mg/ml, the cultures were incubated for 60 min, the metabolites were solubilized in acidic isopropanol, and the absorbance was read at 570 nm. The most inhibitory compounds were also incubated with cells for 72 h prior to the addition of a tetrazolium compound [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS)] and phenazine ethosulfate (PES) (Promega). The 50% cytotoxic concentration (CC50) values were calculated with GraphPad Prism by using the four-parameter variable-response log(inhibitor)-versus-response algorithm with the bottom value set to zero.
Quantitative PCR.
Samples for quantitative PCR (qPCR) were thawed, and cells and cellular debris containing virus were pelleted by centrifugation for 1 h at 21,100 × g. Total DNA was isolated by using a QIAamp DNA minikit (Qiagen) according to the manufacturer's instructions. Primers and probes for qPCR amplified a region of HSV-2 DNA harboring part of the latency-associated transcript. The forward primer was 5′-GAGCTAACACTCGGCTTGCT-3′, the reverse primer was 5′-TCTCCTCCCCGTCTTTCC-3′ (IDT), and the probe was Universal Probe Library 10 (5′–6-carboxyfluorescein [FAM]–GGAGGTG–dark quencher–3′) (Roche Diagnostics Corporation). PCR mixtures consisted of FastStart Universal Probe master mix containing Rox (Roche Diagnostics Corporation), 900 nM each primer, 250 nM probe 10, and 100 ng of the DNA template. Reactions were performed in triplicate with 25-μl volumes in a 7500 Fast real-time PCR system (Applied Biosystems). The PCR parameters were 1 cycle at 90°C for 10 min followed by 50 cycles at 90°C for 20 s and 60°C for 1 min. A standard curve was generated by using purified HSV-2 strain 333 DNA diluted from 108 to 102 copies/μl. Data were analyzed by using ABI 7500 sequence detection system software and expressed as HSV-2 genome equivalents per nanogram of total DNA.
Synergy assay.
Vero cells were seeded into 24-well plates and incubated overnight in growth medium as indicated above. HSV-2 was diluted in PBS supplemented with 1% newborn calf serum and 1% glucose and added to wells at an MOI of 0.1. The plates were incubated at 37°C for 1 h, the virus-containing inoculum was then removed, and the wells were washed once in PBS. Acyclovir and compound 118 were each serially diluted in 3-fold steps from 9× EC50 to 1/9× EC50, using a diluent containing a consistent concentration of DMSO. The compound dilutions were added to infected Vero cell monolayers alone or in constant combination ratios according to the Chou-Talalay method (58). Plates were incubated at 37°C for an additional 23 h, and cells were then harvested by scraping, divided into two aliquots, and stored at −80°C. Infectious virus was quantified by a plaque assay as described above, and viral DNA content was determined by qPCR. For viral titer determinations, samples were thawed and sonicated, and viral titers were determined by a plaque assay on Vero cells. Dose and effect data were evaluated with CompuSyn (http://combosyn.com/) to generate an isobologram and combination index (CI) values, which quantitatively depict antagonism (CI > 1), additive effects (CI = 1), or synergism (CI < 1), according to the Chou-Talalay method (58).
RESULTS
Inhibition of HSV-1 and HSV-2 replication.
Twenty-six synthetic α-hydroxytropolones plus purpurogallin (see Fig. S2 in the supplemental material) were initially screened for activity against HSV-1 and HSV-2 in a semiquantitative colorimetric assay at 50, 5, and 0.5 μM. Only 2 of the 27 compounds were inactive at 50 μM (Table 1). Compounds that had obvious inhibitory activity at 5 μM, plus some noninhibitory compounds for comparison, were then tested for their capacity to reduce virus replication in cell culture. For the replication inhibition assay, Vero cells were infected at an MOI of 0.1, and compounds were added at final concentrations of 50 and 5 μM (certain compounds were tested in the replication inhibition assay at 5 μM only due to limited supply). Screening was performed against single-passage clinical isolates of HSV-1 and HSV-2 to ensure that screening hits were specific for clinically relevant virus.
Thirteen compounds inhibited the replication of HSV-1 at 50 μM but not at 5 μM (<1.0 log10 inhibition), and 14 had activity against HSV-2 at 50 μM but not at 5 μM (Table 1). Three additional compounds reduced HSV-1 replication by 1 to 3 log10 units at 5 μM, and 3 compounds inhibited HSV-2 to a similar extent. Eleven of 27 compounds (41%) inhibited HSV-1 replication by >3 log10 units at 5 μM, and 10 of 27 compounds (37%) similarly inhibited HSV-2 by >3 log10 units at 5 μM. The inhibition patterns and potencies against HSV-1 and HSV-2 were very similar. In general, compounds that were most effective against HSV-1 and HSV-2 did not inhibit the HBV RNase H enzyme, as tested by an oligonucleotide-directed RNA cleavage assay for anti-HBV RNase H activity (42), demonstrating that inhibition was largely HSV specific for this set of α-hydroxytropolones.
To help select compounds for which EC50s would be determined, the most potent inhibitors of HSV-1 at 5 μM underwent a preliminary screen at lower concentrations in the replication inhibition assay to rank their efficacy (Fig. 1). Compounds 115, 118, and 146 maintained some capacity to suppress HSV-1 replication even at 0.17 μM. EC50s against both HSV-1 and HSV-2 were determined for these three compounds plus compound 114 for comparison. Compounds 114, 146, and 115 had EC50s of ≤0.58 μM against both HSV-1 and HSV-2 (Table 2 and Fig. 2). The most active compound, compound 118, had EC50s of 120 nM and 80 nM against HSV-1 and HSV-2, respectively. These α-hydroxytropolones were comparable in potency to the commonly used drug ACV against HSV-1 (EC50 = 0.16 μM) and significantly exceed ACV's activity against HSV-2 (EC50 = 1.4 μM) (Table 1 and Fig. 2). Compounds 115 and 118 also suppressed three primary clinical isolates of HSV-2 by >5 log10 units at 5 μM (data not shown), indicating that they have broad activity against clinically relevant strains.
FIG 1.
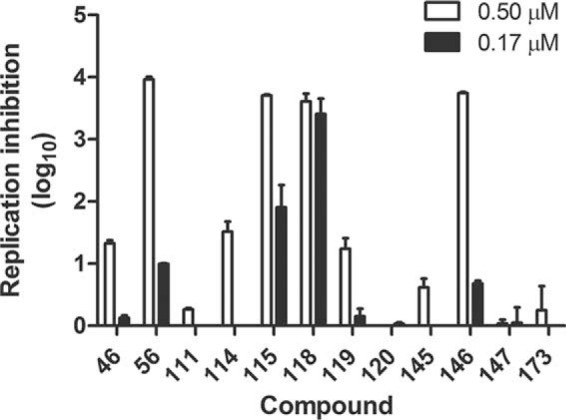
Screen of selected α-hydroxytropolones at low concentrations. Vero cells were infected with a primary clinical isolate of HSV-1 or HSV-2 at an MOI of 0.1 in the presence of each compound or the diluent control and incubated for 24 h. Cultures were then collected, and the virus titer was determined by a plaque assay. The log10 reduction in plaque counts for each compound at 0.5 μM or 0.17 μM compared to the diluent control is shown. Values are the averages of data from duplicate samples ± ranges from one experiment.
TABLE 2.
CC50, EC50, and TI values for the most potent α-hydroxytropolones
| Compound | CC50 (μM) | Anti-HSV-1 |
Anti-HSV-2 |
||
|---|---|---|---|---|---|
| Mean EC50 (μM) ± SD | TI | Mean EC50 (μM) ± SD | TI | ||
| 114 | >100 | 0.58 ± 0.27 | >170 | 0.38 ± 0.20 | >260 |
| 146 | >100 | 0.47 ± 0.03 | >210 | 0.27 ± 0.10 | >390 |
| 115 | >100 | 0.18 ± 0.07 | >560 | 0.21 ± 0.12 | >480 |
| 118 | >100 | 0.12 ± 0.04 | >830 | 0.081 ± 0.000 | >1,200 |
| ACV | >100 | 0.16 ± 0.17 | >630 | 1.40 ± 0.04 | >71 |
FIG 2.

EC50s for α-hydroxytropolone inhibitors of HSV-2. EC50s were determined over a range of 12 to 14 concentrations. EC50 curves are shown for compounds 114, 115, 118, and 146 against HSV-2. The EC50 curves are from representative assays, and the EC50s values in Table 1 are the averages ± 1 standard deviation from two or three independent assays.
Cytotoxicity.
The cytotoxicity of 12 compounds that suppressed the replication of both HSV-1 and HSV-2 at 5 μM by >2 log10 units was assessed in Vero cells by using an MTT assay under the culture conditions employed for the replication inhibition assays. Moderate cytotoxicity was observed for four compounds, with 50% cytotoxic concentration (CC50) values ranging from >50 to 75 μM (Table 1). The other eight compounds showed no discernible cytotoxicity in this assay (CC50 values of >100 μM). This resulted in apparent therapeutic index (TI) (CC50/EC50) values for compounds 114, 146, 115, and 118 ranging from 170 to 830 against HSV-1 and ranging from >260 to >1200 against HSV-2 (Table 2). In contrast, the apparent therapeutic index values for ACV were >630 and >71 against HSV-1 and HSV-2, respectively. Compounds 114, 146, 115, and 118 were also incubated with Vero cells for 72 h, with no toxicity being noted in an MTS assay (CC50 values of >100 μM).
Counterscreening against human RNase H1.
All but 1 of the 27 compounds were counterscreened against recombinant human RNase H1 in an initial effort to further evaluate the specificity of inhibition. Compound 102 was not tested because an insufficient amount of this compound was available. The inhibition patterns differed between the HSVs and human RNase H1, with only 3 compounds having strong efficacy against both the human enzyme and the HSVs at 5 to 10 μM (Table 1). Four compounds were active against the human enzyme at 10 μM, and 8 had activity at concentrations of between 20 μM and 65 μM. Notably, the two most active compounds against HSV-1 and HSV-2, compounds 115 and 118, did not inhibit human RNase H1. These data confirmed the largely different patterns of inhibition between HSV and the human enzyme.
Inhibition of acyclovir-resistant viruses.
Thymidine kinase (TK)-deficient mutants of HSV are insensitive to ACV because ACV is a nucleoside analog prodrug that must be phosphorylated by the viral TK to become a substrate for the viral DNA polymerase (59). Because resistance to nucleoside analogs is relatively common (25, 60, 61), we determined whether the two α-hydroxytropolones that were most effective in reducing HSV-1 and HSV-2 replication could also suppress the replication of TK-deficient mutants. Vero cells were infected with a laboratory strain of HSV-2 and an engineered TK-deficient mutant of the same strain. The cells were treated with 50 μM or 5 μM ACV or compound 115 or 118, and viral yields at 24 h postinfection were measured by a plaque assay. ACV inhibited wild-type HSV-2 replication but could not suppress replication of the TK-deficient mutant (Fig. 3). In marked contrast, compounds 115 and 118 efficiently inhibited the wild-type and TK-deficient mutant strains of HSV-2 (Fig. 3) and also HSV-1 (data not shown). Therefore, these α-hydroxytropolones do not require phosphorylation by the viral TK enzyme to be active, and they suppress HSV-1 and HSV-2 replication in a different manner than ACV.
FIG 3.
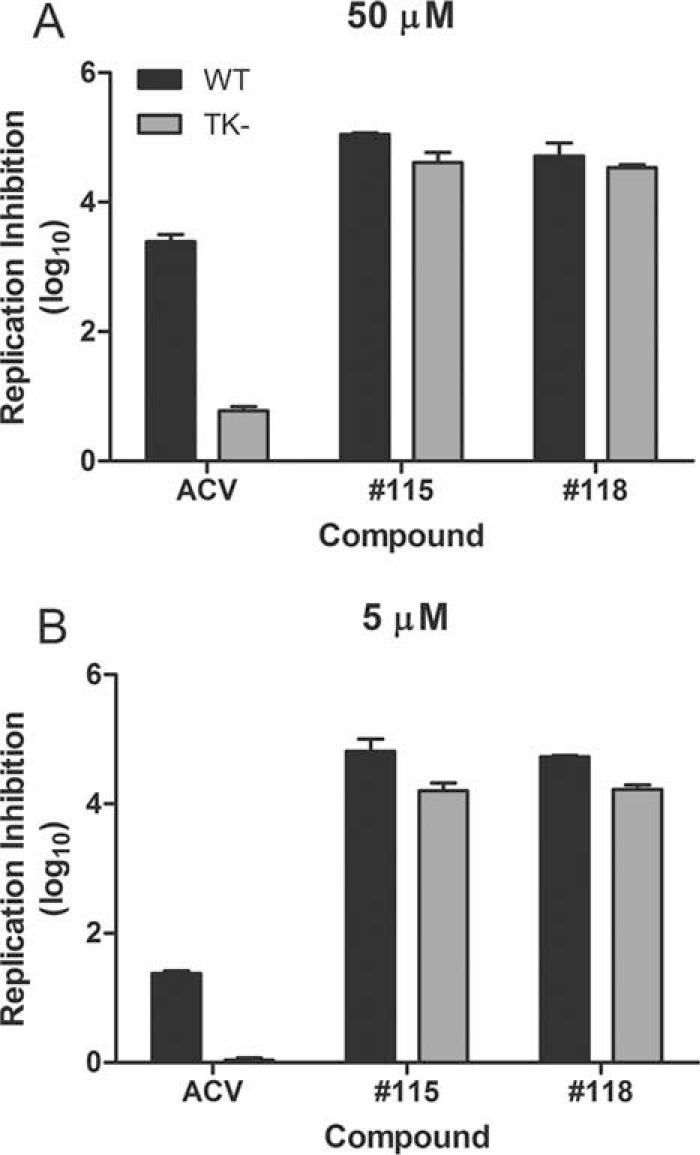
α-Hydroxytropolones inhibit an ACV-resistant mutant of HSV-2. ACV or the indicated α-hydroxytropolone compound was added at 50 μM (A) or 5 μM (B) to cultures infected with wild-type HSV-2 or a TK-deficient mutant of the same strain. Log10 suppression was determined relative to the diluent control. Data are the averages of data from duplicate samples ± ranges from one of two independent experiments at each concentration.
Synergy between ACV and compound 118.
ACV and compound 118 were used in a matrix assay according to the Chou-Talalay method (58) to assess their combined effect against HSV-2 DNA and infectious virus accumulation. The compounds were added to infected Vero cells alone or combined in ratios resulting in a concentration gradient ranging from 1/9× EC50 to 9× EC50 of each compound. qPCR and plaque reduction assays were performed to determine the extent of viral inhibition across the concentration ranges. The combined effect of ACV and compound 118 was greater than the effect of either compound alone, as exemplified by their impact on HSV-2 replication (Fig. 4A). As expected due to its known mechanism of action, ACV suppressed HSV DNA accumulation in a dose-dependent manner (Fig. 4B). Compound 118 also suppressed viral DNA replication at the highest concentration of 9× EC50, but it had a more potent effect when combined with ACV. ACV and compound 118 more dramatically suppressed viral titers (Fig. 4C), and the combination of the two achieved nearly complete suppression of viral replication when both compounds were added at 3× EC50. Analyses of suppressive activity yielded CI values, with a CI of <1 indicating synergism, a CI of ∼1 indicating additive interactions, and a CI of >1 revealing antagonism. At 95% effective doses (ED95s), CI values of 0.44 in the qPCR assay and 0.12 in the viral titer reduction assay were found. Weighted CI values that emphasize data from the highest efficacy levels were 0.40 and 0.17 in the qPCR and viral titer assays, respectively. These CI values of ≪1 indicate that ACV and compound 118 are highly synergistic.
DISCUSSION
We previously identified the α-hydroxytropolone natural product β-thujaplicinol (compound 46) as an inhibitor of HSV-1 and HSV-2 replication (33). β-Thujaplicinol had been tested against HSV because the structure and/or function of several enzymes involved in HSV DNA replication is consistent with that of NTS enzymes. Other viral enzymes in the nucleotidyltransferase superfamily include the HBV and HIV RNase H enzymes (31, 32), and β-thujaplicinol has efficacy against HIV RNase H (37–41), HBV RNase H (42), and foamy virus (62). Here, 27 additional tropolones were screened against HSV-1 and HSV-2. We identified 10 synthetic α-hydroxytropolones with efficacy against both viruses, as defined by the capacity to reduce replication >3 log10 units at 5 μM. Interestingly, only 1 of these 10 α-hydroxytropolones that strongly inhibit HSVs also inhibits HBV RNase H and then only weakly at 60 μM (Table 1). Thus, the specificity of these compounds for HSVs permits us to derive an initial structure-activity relationship (SAR) that could help guide their chemical optimization, even in the absence of target identification.
Fourteen of the 26 compounds that were counterscreened against human RNase H1 inhibited the enzyme to some extent, including 5 of the 11 compounds that inhibited HSV-1 and/or HSV-2 by >3 log10 units at 5 μM. Therefore, the potential exists for cross-inhibition of the human enzyme that should not be overlooked during drug development. However, differences were observed in the sensitivity profiles for the HSVs and human RNaseH1, implying that significant selectivity for the HSVs has already been achieved. For example, the two most active compounds against HSV-1 and HSV-2, compounds 115 and 118, inhibited HSV replication by >5 log10 units but had no activity against the human enzyme, even at a concentration of 60 μM (Table 1).
The cytotoxicity of most of the compounds was negligible by MTT assays conducted for the same length of time as the replication inhibition and EC50 assays and was also negligible for compounds 114, 146, 115, and 118 after 72 h by an MTS assay. EC50s for the 4 most potent α-hydroxytropolone compounds were ≤0.58 μM. These four compounds had similar EC50s against HSV-1 and HSV-2, and all four had significantly lower EC50s against HSV-2 than acyclovir. Thus, TI values for the most active α-hydroxytropolones could be as much as an order of magnitude greater than that for acyclovir. However, because definitive CC50 values have not been identified, the TI values for all compounds represent a lower limit, and the TIs for the α-hydroxytropolones relative to ACV have not yet been precisely determined. A broad range of EC50s for ACV-sensitive HSV-2 clinical isolates has been reported (63), even within a single individual over time (64), which likely contributes to the 9-fold difference in apparent EC50s that we observed for ACV against an HSV-1 versus an HSV-2 clinical isolate. A small amount of cytotoxicity (measured as a loss of mitochondrial function in MTT assays) caused by two compounds, compounds 111 and 120, may be due to inhibition of human RNase H1 because RNase H contributes to mitochondrial DNA replication and pre-rRNA processing (65–67). However, for two other compounds, compounds 119 and 147, cytotoxicity did not correlate with inhibition of RNase H1 (Table 1). This discord implies that inhibition of human RNase H1 is not the sole source of toxicity in our assays; this possibility is consistent with reports of direct toxicity against isolated mitochondria for some troponoids (68). In-depth toxicity studies will be a high priority as antiviral drug development proceeds.
The compounds were screened against primary clinical isolates of HSV-1 and HSV-2. All compounds active against HSV-1 at ≤50 μM also had a similar level of activity against HSV-2 (Table 1), indicating that the enzymatic target(s) of the α-hydroxytropolones is highly likely to be conserved between these two related viruses. Five of these compounds (compounds 96, 104, 111, 147, and 172) had efficacies that differed by >1 log10 units against the two viruses in primary screening at 5 μM, suggesting that HSV type can subtly affect compound efficacy. However, the small magnitude of these differences implies that any potential genotype specificity is unlikely to present an insurmountable hurdle during drug development for HSVs. It will be interesting to determine whether the compounds with greatest efficacy against HSV also suppress the replication of related herpesviruses such as varicella-zoster virus, human cytomegalovirus, and various veterinary herpesviruses.
Structure-function analyses from our previous studies employing natural-product troponoids (33) provided some preliminary structural insight regarding their activity against HSVs. For example, the stronger activity of the natural-product α-hydroxytropolone β-thujaplicinol than of its close tropolone analogs β- and γ-thujaplicin indicated advantages to the added oxygenation of the α-hydroxytropolones. Furthermore, the larger and more lipophilic natural-product α-hydroxytropolone manicol was 6-fold more active than β-thujaplicinol, demonstrating that the target(s) of the compounds can accommodate larger molecules and can be enhanced by substitution. Based on initial observations, a series of over two dozen α-hydroxytropolones were synthesized and tested to both confirm the initial SAR and provide leads for further optimization pursuits. In line with those previous studies, all but one of the synthetic α-hydroxytropolones tested inhibited HSV-1 and HSV-2 replication at 50 μM, confirming advantages to this moiety for anti-HSV activity. The relatively low activity of manicol derivatives 95 to 104 at 5 μM was unexpected given the strong anti-HSV activity of manicol itself (33). These compounds are known to undergo hydrolysis, and it is possible that some degree of degradation may have reduced their activity. Many of the synthetic α-hydroxytropolone derivatives of β-thujaplicinol remained highly active at a lower concentration of 5 μM; however, several of the less lipophilic molecules lost their activity, and trends began to emerge. For example, among the ketone-containing molecules, methyl and isopropyl ketone-containing α-hydroxytropolones 110 and 143 are inactive, whereas the activities of all the larger ketones are preserved (compounds 111, 118, 120, and 173). After these molecules were assessed at even lower concentrations (Fig. 1), the library was further narrowed down to two molecules that maintained activity at 0.5 μM (compounds 114 and 119) and three molecules that maintained activity at 0.17 μM (compounds 118, 115, and 146). EC50s were obtained for the latter three compounds plus compound 114. The two most potent compounds of this group were biphenyl compounds 115 and 118, which had EC50s for both HSV-1 and HSV-2 of ∼0.2 μM and 0.1 μM, respectively (Fig. 5). These two molecules, which are now the top anti-HSV troponoid leads, suggest advantages to the biaryl side chains and provide excellent starting points for future optimization-directed studies.
FIG 5.

Preliminary SAR for α-hydroxytropolones against HSV-1 and HSV-2. (A) Anti-HSV data from lead natural-product α-hydroxytropolones from previous studies (33). Also shown are tropolone natural-product analogs that were inactive against HSV-1 and HSV-2 at 5 μM. n.d., not determined. (B) Qualitative comparison of HSV-1 suppression (>3 log10 units) by closely related synthetic α-hydroxytropolones at 50 μM (+), 5 μM (++), and 0.5 μM (+++), highlighting a trend of increasing potency with larger substituents. (C) EC50 and CC50 values of synthetic α-hydroxytropolones, reported as averages of data from 2 runs.
Our studies indicate that α-hydroxytropolones are promising candidates for development as novel anti-HSV drugs. They provide initial guidance for chemical optimization, and they indicate promising avenues to maximize differences between the HSV target(s) and human RNase H1 and to address mitochondrial toxicity to avoid unacceptable side effects during therapy. The α-hydroxytropolone inhibitors may work synergistically with the nucleos(t)ide analogs because ACV and compound 118 synergistically suppress HSV replication (Fig. 4), and the two classes of inhibitors have different apparent mechanisms of action (Fig. 2) (33). Polymerase incorporation of the nucleoside analog ACV prevents chain elongation (69); however, the α-hydroxytropolone mechanism of action remains to be determined. Possible viral targets with NTS enzyme activity or homology include the polymerase (3′-to-5′ and 5′-to-3′ exonuclease activities), pUL12 nuclease, pUL15 terminase, and ICP8 single-stranded DNA binding protein (70–72, 76). Our observation that the α-hydroxytropolones suppress β-galactosidase expression under the control of an HSV immediate early gene promoter suggests that the compounds target a relatively early event in HSV replication, consistent with early effects previously seen in time-of-addition experiments (33). The synergy observed between compound 118 and ACV (Fig. 4) implies that NTS enzyme inhibitors would be good candidates for use in multidrug regimens to suppress HSV replication sufficiently to block viral shedding and, thus, transmission (15, 16). In addition, the two α-hydroxytropolones tested here plus β-thujaplicinol (33) suppressed acyclovir-resistant HSV-1 and HSV-2 mutants nearly as well as their cognate wild-type viruses. This capacity of NTS enzyme inhibitors to suppress the replication of acyclovir-resistant viruses also suggests their utility as salvage therapies in chronically infected patients whose HSV infection has developed resistance to one or more drugs (25, 61, 73–75).
Supplementary Material
ACKNOWLEDGMENTS
We are grateful for seed grants from the Saint Louis University Department of Molecular Microbiology and Immunology, the Saint Louis University School of Medicine, and the Saint Louis University Cancer Center.
We thank Alina Taniuchi for technical assistance, Jian-Kang Jiang and Craig Thomas for the gift of compounds 95 to 104, and David Leib and Jim Smiley for their gifts of viruses.
L.A.M., R.P.M., and J.E.T. are inventors on U.S. provisional patent application USTL.P0062US.P1 that covers the inhibitors reported here.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02675-15.
REFERENCES
- 1.Roizman B, Knipe DM, Whitley RJ. 2013. Herpes simplex viruses, p 1823–1897. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Mark KE, Wald A, Magaret AS, Selke S, Olin L, Huang ML, Corey L. 2008. Rapidly cleared episodes of herpes simplex virus reactivation in immunocompetent adults. J Infect Dis 198:1141–1149. doi: 10.1086/591913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liesegang TJ. 2001. Herpes simplex virus epidemiology and ocular importance. Cornea 20:1–13. doi: 10.1097/00003226-200101000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Belshe RB, Leone PA, Bernstein DI, Wald A, Levin MJ, Stapleton JT, Gorfinkel I, Morrow RL, Ewell MG, Stokes-Riner A, Dubin G, Heineman TC, Schulte JM, Deal CD, Herpevac Trial for Women. 2012. Efficacy results of a trial of a herpes simplex vaccine. N Engl J Med 366:34–43. doi: 10.1056/NEJMoa1103151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernstein DI, Bellamy AR, Hook EW III, Levin MJ, Wald A, Ewell MG, Wolff PA, Deal CD, Heineman TC, Dubin G, Belshe RB. 2013. Epidemiology, clinical presentation, and antibody response to primary infection with herpes simplex virus type 1 and type 2 in young women. Clin Infect Dis 56:344–351. doi: 10.1093/cid/cis891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Horowitz R, Aierstuck S, Williams EA, Melby B. 2010. Herpes simplex virus infection in a university health population: clinical manifestations, epidemiology, and implications. J Am Coll Health 59:69–74. doi: 10.1080/07448481.2010.483711. [DOI] [PubMed] [Google Scholar]
- 7.Pena KC, Adelson ME, Mordechai E, Blaho JA. 2010. Genital herpes simplex virus type 1 in women: detection in cervicovaginal specimens from gynecological practices in the United States. J Clin Microbiol 48:150–153. doi: 10.1128/JCM.01336-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith PD, Roberts CM. 2009. American College Health Association annual Pap test and sexually transmitted infection survey: 2006. J Am Coll Health 57:389–394. doi: 10.3200/JACH.57.4.389-394. [DOI] [PubMed] [Google Scholar]
- 9.Wald A, Selke S, Warren T, Aoki FY, Sacks S, Diaz-Mitoma F, Corey L. 2006. Comparative efficacy of famciclovir and valacyclovir for suppression of recurrent genital herpes and viral shedding. Sex Transm Dis 33:529–533. doi: 10.1097/01.olq.0000204723.15765.91. [DOI] [PubMed] [Google Scholar]
- 10.Bradley H, Markowitz LE, Gibson T, McQuillan GM. 2014. Seroprevalence of herpes simplex virus types 1 and 2—United States, 1999-2010. J Infect Dis 209:325–333. doi: 10.1093/infdis/jit458. [DOI] [PubMed] [Google Scholar]
- 11.Looker KJ, Garnett GP, Schmid GP. 2008. An estimate of the global prevalence and incidence of herpes simplex virus type 2 infection. Bull World Health Organ 86:805–812. doi: 10.2471/BLT.07.046128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wald A, Link K. 2002. Risk of human immunodeficiency virus infection in herpes simplex virus type 2-seropositive persons: a meta-analysis. J Infect Dis 185:45–52. doi: 10.1086/338231. [DOI] [PubMed] [Google Scholar]
- 13.Celum C, Levine R, Weaver M, Wald A. 2004. Genital herpes and human immunodeficiency virus: double trouble. Bull World Health Organ 82:447–453. [PMC free article] [PubMed] [Google Scholar]
- 14.Pinninti SG, Kimberlin DW. 2013. Maternal and neonatal herpes simplex virus infections. Am J Perinatol 30:113–119. doi: 10.1055/s-0032-1332802. [DOI] [PubMed] [Google Scholar]
- 15.Johnston C, Saracino M, Kuntz S, Magaret A, Selke S, Huang ML, Schiffer JT, Koelle DM, Corey L, Wald A. 2012. Standard-dose and high-dose daily antiviral therapy for short episodes of genital HSV-2 reactivation: three randomised, open-label, cross-over trials. Lancet 379:641–647. doi: 10.1016/S0140-6736(11)61750-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vere Hodge RA, Field HJ. 2013. Antiviral agents for herpes simplex virus. Adv Pharmacol 67:1–38. doi: 10.1016/B978-0-12-405880-4.00001-9. [DOI] [PubMed] [Google Scholar]
- 17.Bacon TH, Levin MJ, Leary JJ, Sarisky RT, Sutton D. 2003. Herpes simplex virus resistance to acyclovir and penciclovir after two decades of antiviral therapy. Clin Microbiol Rev 16:114–128. doi: 10.1128/CMR.16.1.114-128.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Field HJ, Vere Hodge RA. 2013. Recent developments in anti-herpesvirus drugs. Br Med Bull 106:213–249. doi: 10.1093/bmb/ldt011. [DOI] [PubMed] [Google Scholar]
- 19.Frisch S, Siegfried EC. 2011. The clinical spectrum and therapeutic challenge of eczema herpeticum. Pediatr Dermatol 28:46–52. doi: 10.1111/j.1525-1470.2010.01356.x. [DOI] [PubMed] [Google Scholar]
- 20.Frobert E, Burrel S, Ducastelle-Lepretre S, Billaud G, Ader F, Casalegno JS, Nave V, Boutolleau D, Michallet M, Lina B, Morfin F. 2014. Resistance of herpes simplex viruses to acyclovir: an update from a ten-year survey in France. Antiviral Res 111:36–41. doi: 10.1016/j.antiviral.2014.08.013. [DOI] [PubMed] [Google Scholar]
- 21.Reyes M, Shaik NS, Graber JM, Nisenbaum R, Wetherall NT, Fukuda K, Reeves WC, Task Force on Herpes Simplex Virus Resistance. 2003. Acyclovir-resistant genital herpes among persons attending sexually transmitted disease and human immunodeficiency virus clinics. Arch Intern Med 163:76–80. doi: 10.1001/archinte.163.1.76. [DOI] [PubMed] [Google Scholar]
- 22.Stranska R, Schuurman R, Nienhuis E, Goedegebuure IW, Polman M, Weel JF, Wertheim-Van Dillen PM, Berkhout RJ, van Loon AM. 2005. Survey of acyclovir-resistant herpes simplex virus in the Netherlands: prevalence and characterization. J Clin Virol 32:7–18. doi: 10.1016/j.jcv.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Wang Q, Zhu Q, Zhou R, Liu J, Peng T. 2011. Identification and characterization of acyclovir-resistant clinical HSV-1 isolates from children. J Clin Virol 52:107–112. doi: 10.1016/j.jcv.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 24.van Velzen M, Missotten T, van Loenen FB, Meesters RJW, Luider TM, Baarsma GS, Osterhaus ADME, Verjans GMGM. 2013. Acyclovir-resistant herpes simplex virus type 1 in intra-ocular fluid samples of herpetic uveitis patients. J Clin Virol 57:215–221. doi: 10.1016/j.jcv.2013.03.014. [DOI] [PubMed] [Google Scholar]
- 25.Duan R, de Vries RD, Osterhaus AD, Remeijer L, Verjans GM. 2008. Acyclovir-resistant corneal HSV-1 isolates from patients with herpetic keratitis. J Infect Dis 198:659–663. doi: 10.1086/590668. [DOI] [PubMed] [Google Scholar]
- 26.van Velzen M, van de Vijver DA, van Loenen FB, Osterhaus AD, Remeijer L, Verjans GM. 2013. Acyclovir prophylaxis predisposes to antiviral-resistant recurrent herpetic keratitis. J Infect Dis 208:1359–1365. doi: 10.1093/infdis/jit350. [DOI] [PubMed] [Google Scholar]
- 27.Chilukuri S, Rosen T. 2003. Management of acyclovir-resistant herpes simplex virus. Dermatol Clin 21:311–320. doi: 10.1016/S0733-8635(02)00093-1. [DOI] [PubMed] [Google Scholar]
- 28.Cury K, Valin N, Gozlan J, Jacquier I, Boutolleau D, Guegan S, Flejou JF, Girard PM. 2010. Bipolar hypertrophic herpes: an unusual presentation of acyclovir-resistant herpes simplex type 2 in a HIV-infected patient. Sex Transm Dis 37:126–128. doi: 10.1097/OLQ.0b013e3181bcaf91. [DOI] [PubMed] [Google Scholar]
- 29.Hardy WD. 1992. Foscarnet treatment of acyclovir-resistant herpes simplex virus infection in patients with acquired immunodeficiency syndrome: preliminary results of a controlled, randomized, regimen-comparative trial. Am J Med 92(2A):30S–35S. doi: 10.1016/0002-9343(92)90335-9. [DOI] [PubMed] [Google Scholar]
- 30.Sbidian E, Battistella M, Legoff J, Lafaurie M, Bezier M, Agbalika F, Simon F, Bouscarat F, Cayuela JM, Carcelain G, Houhou N, Bagot M, Molina JM, Janier M, Bachelez H. 2013. Recalcitrant pseudotumoral anogenital herpes simplex virus type 2 in HIV-infected patients: evidence for predominant B-lymphoplasmocytic infiltration and immunomodulators as effective therapeutic strategy. Clin Infect Dis 57:1648–1655. doi: 10.1093/cid/cit592. [DOI] [PubMed] [Google Scholar]
- 31.Nowotny M. 2009. Retroviral integrase superfamily: the structural perspective. EMBO Rep 10:144–151. doi: 10.1038/embor.2008.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang W, Steitz TA. 1995. Recombining the structures of HIV integrase, RuvC and RNase H. Structure 3:131–134. doi: 10.1016/S0969-2126(01)00142-3. [DOI] [PubMed] [Google Scholar]
- 33.Tavis JE, Wang H, Tollefson AE, Ying B, Korom M, Cheng X, Cao F, Davis KL, Wold WS, Morrison LA. 2014. Inhibitors of nucleotidyltransferase superfamily enzymes suppress herpes simplex virus replication. Antimicrob Agents Chemother 58:7451–7461. doi: 10.1128/AAC.03875-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu Y, Cheng X, Cao F, Huang A, Tavis JE. 2013. Beta-thujaplicinol inhibits hepatitis B virus replication by blocking the viral ribonuclease H activity. Antiviral Res 99:221–229. doi: 10.1016/j.antiviral.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 35.Meck C, D'Erasmo MP, Hirsch DR, Murelli RP. 2014. The biology and synthesis of alpha-hydroxytropolones. Medchemcomm 5:842–852. doi: 10.1039/c4md00055b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sugawara K, Ohbayashi M, Shimizu K, Hatori M, Kamei H, Konishi M, Oki T, Kawaguchi H. 1988. BMY-28438 (3,7-dihydroxytropolone), a new antitumor antibiotic active against B16 melanoma. I. Production, isolation, structure and biological activity. J Antibiot (Tokyo) 41:862–868. [DOI] [PubMed] [Google Scholar]
- 37.Beilhartz GL, Wendeler M, Baichoo N, Rausch J, Le Grice S, Gotte M. 2009. HIV-1 reverse transcriptase can simultaneously engage its DNA/RNA substrate at both DNA polymerase and RNase H active sites: implications for RNase H inhibition. J Mol Biol 388:462–474. doi: 10.1016/j.jmb.2009.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Budihas SR, Gorshkova I, Gaidamakov S, Wamiru A, Bona MK, Parniak MA, Crouch RJ, McMahon JB, Beutler JA, Le Grice SF. 2005. Selective inhibition of HIV-1 reverse transcriptase-associated ribonuclease H activity by hydroxylated tropolones. Nucleic Acids Res 33:1249–1256. doi: 10.1093/nar/gki268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chung S, Himmel DM, Jiang JK, Wojtak K, Bauman JD, Rausch JW, Wilson JA, Beutler JA, Thomas CJ, Arnold E, Le Grice SF. 2011. Synthesis, activity, and structural analysis of novel alpha-hydroxytropolone inhibitors of human immunodeficiency virus reverse transcriptase-associated ribonuclease H. J Med Chem 54:4462–4473. doi: 10.1021/jm2000757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Didierjean J, Isel C, Querre F, Mouscadet JF, Aubertin AM, Valnot JY, Piettre SR, Marquet R. 2005. Inhibition of human immunodeficiency virus type 1 reverse transcriptase, RNase H, and integrase activities by hydroxytropolones. Antimicrob Agents Chemother 49:4884–4894. doi: 10.1128/AAC.49.12.4884-4894.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Himmel DM, Maegley KA, Pauly TA, Bauman JD, Das K, Dharia C, Clark AD Jr, Ryan K, Hickey MJ, Love RA, Hughes SH, Bergqvist S, Arnold E. 2009. Structure of HIV-1 reverse transcriptase with the inhibitor beta-thujaplicinol bound at the RNase H active site. Structure 17:1625–1635. doi: 10.1016/j.str.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu G, Lomonosova E, Cheng X, Moran EA, Meyers MJ, Le Grice SF, Thomas CJ, Jiang JK, Meck C, Hirsch DR, D'Erasmo MP, Suyabatmaz DM, Murelli RP, Tavis JE. 2015. Hydroxylated tropolones inhibit hepatitis B virus replication by blocking viral ribonuclease H activity. Antimicrob Agents Chemother 59:1070–1079. doi: 10.1128/AAC.04617-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iwatsuki M, Takada S, Mori M, Ishiyama A, Namatame M, Nishihara-Tsukashima A, Nonaka K, Masuma R, Otoguro K, Shiomi K, Omura S. 2011. In vitro and in vivo antimalarial activity of puberulic acid and its new analogs, viticolins A-C, produced by Penicillium sp. FKI-4410. J Antibiot (Tokyo) 64:183–188. doi: 10.1038/ja.2010.124. [DOI] [PubMed] [Google Scholar]
- 44.Allen NE, Alborn WE Jr, Hobbs JN Jr, Kirst HA. 1982. 7-Hydroxytropolone: an inhibitor of aminoglycoside-2″-O-adenylyltransferase. Antimicrob Agents Chemother 22:824–831. doi: 10.1128/AAC.22.5.824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martin SF, Follows BC, Hergenrother PJ, Franklin CL. 2000. A novel class of zinc-binding inhibitors for the phosphatidylcholine-preferring phospholipase C from Bacillus cereus. J Org Chem 65:4509–4514. doi: 10.1021/jo9915731. [DOI] [PubMed] [Google Scholar]
- 46.Piettre SR, Ganzhorn A, Hoflack J, Islam K, Hornsperger JM. 1997. Alpha-hydroxytropolones: a new class of potent inhibitors of inositol monophosphatase and other bimetallic enzymes. J Am Chem Soc 119:3201–3204. doi: 10.1021/ja9634278. [DOI] [Google Scholar]
- 47.Semenova EA, Johnson AA, Marchand C, Davis DA, Yarchoan R, Pommier Y. 2006. Preferential inhibition of the magnesium-dependent strand transfer reaction of HIV-1 integrase by alpha-hydroxytropolones. Mol Pharmacol 69:1454–1460. doi: 10.1124/mol.105.020321. [DOI] [PubMed] [Google Scholar]
- 48.Takeshita H, Mori A, Kusaba T. 1986. An improved synthesis of 2,7-dihydroxytropolone (2-hydroxytrolone). Synthesis 7:578–579. [Google Scholar]
- 49.Hirsch DR, Cox G, D'Erasmo MP, Shakya T, Meck C, Mohd N, Wright GD, Murelli RP. 2014. Inhibition of the ANT(2″)-Ia resistance enzyme and rescue of aminoglycoside antibiotic activity by synthetic alpha-hydroxytropolones. Bioorg Med Chem Lett 24:4943–4947. doi: 10.1016/j.bmcl.2014.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meck C, Mohd N, Murelli RP. 2012. An oxidopyrylium cyclization/ring-opening route to polysubstituted alpha-hydroxytropolones. Org Lett 14:5988–5991. doi: 10.1021/ol302892g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Williams YD, Meck C, Mohd N, Murelli RP. 2013. Triflic acid-mediated rearrangements of 3-methoxy-8-oxabicyclo[3.2.1]octa-3,6-dien-2-ones: synthesis of methoxytropolones and furans. J Org Chem 78:11707–11713. doi: 10.1021/jo401617r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Korom M, Wylie KM, Wang H, Davis KL, Sangabathula MS, Delassus GS, Morrison LA. 2013. A proautophagic antiviral role for the cellular prion protein identified by infection with a herpes simplex virus 1 ICP34.5 mutant. J Virol 87:5882–5894. doi: 10.1128/JVI.02559-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McDermott MR, Smiley JR, Leslie P, Brais J, Rudzroga HE, Bienenstock J. 1984. Immunity in the female genital tract after intravaginal vaccination of mice with an attenuated strain of herpes simplex virus type 2. J Virol 51:747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Davido DJ, Leib DA, Schaffer PA. 2002. The cyclin-dependent kinase inhibitor roscovitine inhibits the transactivating activity and alters the posttranslational modification of herpes simplex virus type 1 ICP0. J Virol 76:1077–1088. doi: 10.1128/JVI.76.3.1077-1088.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smibert CA, Smiley JR. 1990. Differential regulation of endogenous and transduced beta-globin genes during infection of erythroid cells with a herpes simplex virus type 1 recombinant. J Virol 64:3882–3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Knipe DM, Spang AE. 1982. Definition of a series of stages in the association of two herpesviral proteins with the cell nucleus. J Virol 43:314–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Morrison LA, Knipe DM. 1996. Mechanisms of immunization with a replication-defective mutant of herpes simplex virus 1. Virology 220:402–413. doi: 10.1006/viro.1996.0328. [DOI] [PubMed] [Google Scholar]
- 58.Chou TC. 2010. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 59.Elion GB, Furman PA, Fyfe JA, de Miranda P, Beauchamp L, Schaeffer HJ. 1977. Selectivity of action of an antiherpetic agent, 9-(2-hydroxyethoxymethyl) guanine. Proc Natl Acad Sci U S A 74:5716–5720. doi: 10.1073/pnas.74.12.5716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Duan R, de Vries RD, van Dun JM, van Loenen FB, Osterhaus AD, Remeijer L, Verjans GM. 2009. Acyclovir susceptibility and genetic characteristics of sequential herpes simplex virus type 1 corneal isolates from patients with recurrent herpetic keratitis. J Infect Dis 200:1402–1414. doi: 10.1086/606028. [DOI] [PubMed] [Google Scholar]
- 61.Morfin F, Thouvenot D. 2003. Herpes simplex virus resistance to antiviral drugs. J Clin Virol 26:29–37. doi: 10.1016/S1386-6532(02)00263-9. [DOI] [PubMed] [Google Scholar]
- 62.Corona A, Schneider A, Schweimer K, Rosch P, Wohrl BM, Tramontano E. 2014. Inhibition of foamy virus reverse transcriptase by human immunodeficiency virus type 1 RNase H inhibitors. Antimicrob Agents Chemother 58:4086–4093. doi: 10.1128/AAC.00056-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bohn-Wippert K, Schmidt S, Runtze A, Zell R, Sauerbrei A. 2015. Resistance testing of clinical herpes simplex virus type 2 isolates collected over 4 decades. Int J Med Microbiol 305:644–651. doi: 10.1016/j.ijmm.2015.08.014. [DOI] [PubMed] [Google Scholar]
- 64.Gupta R, Hill EL, McClemon D, Davis G, Selke S, Corey L, Wald A. 2005. Acyclovir sensitivity of sequential herpes simplex virus type 2 isolates from the genital mucosa of immunocompetent women. J Infect Dis 192:1102–1107. doi: 10.1086/432766. [DOI] [PubMed] [Google Scholar]
- 65.Cerritelli SM, Frolova EG, Feng C, Grinberg A, Love PE, Crouch RJ. 2003. Failure to produce mitochondrial DNA results in embryonic lethality in Rnaseh1 null mice. Mol Cell 11:807–815. doi: 10.1016/S1097-2765(03)00088-1. [DOI] [PubMed] [Google Scholar]
- 66.Ruhanen H, Ushakov K, Yasukawa T. 2011. Involvement of DNA ligase III and ribonuclease H1 in mitochondrial DNA replication in cultured human cells. Biochim Biophys Acta 1813:2000–2007. doi: 10.1016/j.bbamcr.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu H, Sun H, Liang X, Lima WF, Crooke ST. 2013. Human RNase H1 is associated with protein P32 and is involved in mitochondrial pre-rRNA processing. PLoS One 8:e71006. doi: 10.1371/journal.pone.0071006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nakagawa Y, Tayama K. 1998. Mechanism of mitochondrial dysfunction and cytotoxicity induced by tropolones in isolated rat hepatocytes. Chem Biol Interact 116:45–60. doi: 10.1016/S0009-2797(98)00078-7. [DOI] [PubMed] [Google Scholar]
- 69.McGuirt PV, Furman PA. 1982. Acyclovir inhibition of viral DNA chain elongation in herpes simplex virus-infected cells. Am J Med 73:67–71. doi: 10.1016/0002-9343(82)90066-3. [DOI] [PubMed] [Google Scholar]
- 70.Bryant KF, Yan Z, Dreyfus DH, Knipe DM. 2012. Identification of a divalent metal cation binding site in herpes simplex virus 1 (HSV-1) ICP8 required for HSV replication. J Virol 86:6825–6834. doi: 10.1128/JVI.00374-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Boehmer PE, Lehman IR. 1997. Herpes simplex virus DNA replication. Annu Rev Biochem 66:347–384. doi: 10.1146/annurev.biochem.66.1.347. [DOI] [PubMed] [Google Scholar]
- 72.Schumacher AJ, Mohni KN, Kan Y, Hendrickson EA, Stark JM, Weller SK. 2012. The HSV-1 exonuclease, UL12, stimulates recombination by a single strand annealing mechanism. PLoS Pathog 8:e1002862. doi: 10.1371/journal.ppat.1002862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schubert A, Gentner E, Bohn K, Schwarz M, Mertens T, Sauerbrei A. 2014. Single nucleotide polymorphisms of thymidine kinase and DNA polymerase genes in clinical herpes simplex virus type 1 isolates associated with different resistance phenotypes. Antiviral Res 107:16–22. doi: 10.1016/j.antiviral.2014.03.015. [DOI] [PubMed] [Google Scholar]
- 74.Suzutani T, Ishioka K, De Clercq E, Ishibashi K, Kaneko H, Kira T, Hashimoto K, Ogasawara M, Ohtani K, Wakamiya N, Saijo M. 2003. Differential mutation patterns in thymidine kinase and DNA polymerase genes of herpes simplex virus type 1 clones passaged in the presence of acyclovir or penciclovir. Antimicrob Agents Chemother 47:1707–1713. doi: 10.1128/AAC.47.5.1707-1713.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang LX, Takayama-Ito M, Kinoshita-Yamaguchi H, Kakiuchi S, Suzutani T, Nakamichi K, Lim CK, Kurane I, Saijo M. 2013. Characterization of DNA polymerase-associated acyclovir-resistant herpes simplex virus type 1: mutations, sensitivity to antiviral compounds, neurovirulence, and in-vivo sensitivity to treatment. Jpn J Infect Dis 66:404–410. doi: 10.7883/yoken.66.404. [DOI] [PubMed] [Google Scholar]
- 76.Selvarajan Sigamani S, Zhao H, Kamau YN, Baines JD, Tang L. 2013. The structure of the herpes simplex virus DNA-packaging terminase pUL15 nuclease domain suggests an evolutionary lineage among eukaryotic and prokaryotic viruses. J Virol 87:7140–7148. doi: 10.1128/JVI.00311-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.