

Abstract
Chlorhexidine is a bisbiguanide antiseptic used for infection control. Vancomycin-resistant E. faecium (VREfm) is among the leading causes of hospital-acquired infections. VREfm may be exposed to chlorhexidine at supra- and subinhibitory concentrations as a result of chlorhexidine bathing and chlorhexidine-impregnated central venous catheter use. We used RNA sequencing to investigate how VREfm responds to chlorhexidine gluconate exposure. Among the 35 genes upregulated ≥10-fold after 15 min of exposure to the MIC of chlorhexidine gluconate were those encoding VanA-type vancomycin resistance (vanHAX) and those associated with reduced daptomycin susceptibility (liaXYZ). We confirmed that vanA upregulation was not strain or species specific by querying other VanA-type VRE. VanB-type genes were not induced. The vanH promoter was found to be responsive to subinhibitory chlorhexidine gluconate in VREfm, as was production of the VanX protein. Using vanH reporter experiments with Bacillus subtilis and deletion analysis in VREfm, we found that this phenomenon is VanR dependent. Deletion of vanR did not result in increased chlorhexidine susceptibility, demonstrating that vanHAX induction is not protective against chlorhexidine. As expected, VanA-type VRE is more susceptible to ceftriaxone in the presence of sub-MIC chlorhexidine. Unexpectedly, VREfm is also more susceptible to vancomycin in the presence of subinhibitory chlorhexidine, suggesting that chlorhexidine-induced gene expression changes lead to additional alterations in cell wall synthesis. We conclude that chlorhexidine induces expression of VanA-type vancomycin resistance genes and genes associated with daptomycin nonsusceptibility. Overall, our results indicate that the impacts of subinhibitory chlorhexidine exposure on hospital-associated pathogens should be further investigated in laboratory studies.
INTRODUCTION
Enterococcus faecium and Enterococcus faecalis are Gram-positive bacteria and gastrointestinal tract colonizers that opportunistically colonize wounds and the bloodstream, causing life-threatening infections, including bacteremia and endocarditis (1, 2). They are particularly associated with central-line associated bloodstream infection (CLABSI), a type of hospital-acquired infection (HAI) that arises from central venous catheter use. Enterococci are associated with 18% of CLABSIs in the United States (3).
Of particular concern for CLABSI treatment are vancomycin-resistant enterococci (VRE), which are resistant to the glycopeptide antibiotic vancomycin. Vancomycin forms complexes with the terminal d-alanyl–d-alanine (d-Ala–d-Ala) residues of peptidoglycan precursors, thereby halting peptidoglycan synthesis (4, 5). VanA- and VanB-type VRE have an alternate pathway of cell wall synthesis due to their acquisition of transposons containing vancomycin resistance genes. The genes enable enterococci to form modified peptidoglycan precursors that terminate in d-alanyl–d-lactate (d-Ala–d-Lac) instead of d-Ala–d-Ala (6–8). Vancomycin has a lower affinity for d-Ala–d-Lac termini (9), and cross-links in the cell wall can be formed using these precursors. By this mechanism, the enterococcal cell wall becomes highly resistant to the action of vancomycin.
To attempt to reduce the number of hospital-acquired infections, including those caused by VRE, infection control practices are implemented by health care facilities. Chlorhexidine is a bisbiguanide antiseptic (10) that is incorporated into a number of infection control products, including chlorhexidine- and silver-impregnated central venous catheters (11, 12). The practice of chlorhexidine bathing is recommended for all acute-care hospitals to reduce CLABSI occurrence (13). For chlorhexidine bathing, patients are bathed daily with a no-rinse chlorhexidine preparation or chlorhexidine-impregnated washcloths (14). The chlorhexidine remains on the skin, providing an antimicrobial coating that is replenished with each bathing. Chlorhexidine is amphipathic, and it likely interacts with both phospholipids and proteins on the bacterial cell surface (15, 16). Its interaction with the membrane is reported to be similar to that of antimicrobial peptides (15). These interactions disrupt membrane integrity and potential, leading to leakage of cytoplasmic constituents; at high chlorhexidine concentrations, cytoplasm congealing and complete breakdown of the cell membrane occur, conferring a bactericidal effect (17–19). For Bacillus subtilis, a rod-shaped Gram-positive bacterium, chlorhexidine at the MIC induces the formation of dented spots on the cell surface near the cell poles, leading to the hypothesis that chlorhexidine preferentially interacts with anionic lipids located at the B. subtilis cell poles (20).
A recent clinical trial that reported no impact of chlorhexidine bathing on hospital-acquired infection occurrence (21) raised concerns about the effects of chlorhexidine bathing on hospital-associated pathogens, including selection for reduced chlorhexidine susceptibility and for cross-resistance to antibiotics in clinical use (22, 23). A recent study semiquantitatively evaluated chlorhexidine levels on the skin of 20 patients pre- and post-chlorhexidine bathing, finding that the levels varied depending on body site and time postbath (24). Levels within the reported range of chlorhexidine MIC for enterococci (25–29) were detected on patient skin (24). In another study, chlorhexidine susceptibilities were monitored for CLABSI enterococcal isolates obtained from hospital wards using chlorhexidine bathing (30). It was observed that the chlorhexidine MIC increased significantly in those isolates compared to CLABSI isolates from nonbathing wards (30). The results of both studies indicate that enterococci are exposed to subinhibitory chlorhexidine concentrations in clinical settings as a result of chlorhexidine bathing.
Motivated by studies indicating that VRE are exposed to subinhibitory levels of chlorhexidine, in this study, we used RNA sequencing to assess the global transcriptional responses of E. faecium 1,231,410, a VanA-type vancomycin-resistant E. faecium (VREfm) strain, to exposure to a chlorhexidine gluconate (CHG)-containing consumer product. To our knowledge, this is the first study to evaluate E. faecium global transcriptional response to an antiseptic. We observed a potent induction of VanA-type vancomycin resistance genes and genes associated with daptomycin resistance upon exposure to the MIC of CHG. Induction of vancomycin resistance genes by CHG was found to be dependent upon VanR, and resulted in increased susceptibility to ceftriaxone in the presence of subinhibitory CHG. Our results suggest that the long-term impact of chlorhexidine bathing on HAI pathogens such as VRE should be further investigated in laboratory studies.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Bacterial strains used in this study are shown in Table 1. E. faecium and E. faecalis were cultured at 37°C on brain heart infusion (BHI) agar or in BHI broth without agitation unless otherwise stated. Bacillus subtilis was cultured at 37°C on lysogeny broth (LB) with agar or in LB broth with shaking at 225 rpm unless otherwise stated. Escherichia coli strains were cultured on LB. Antibiotics were used at the following concentrations: chloramphenicol, 15 μg/ml for E. coli and E. faecium and 34 μg/ml for B. subtilis, and erythromycin, 10 μg/ml for B. subtilis. Vancomycin concentrations used are stated for specific experiments below.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Description | Reference |
|---|---|---|
| Bacterial strains | ||
| E. faecium 1,231,410 | Skin and soft tissue infection isolate; VanA-type VRE | 35 |
| E. faecium 1,231,502 | Bloodstream isolate; VanA-type VRE | 35 |
| E. faecium 1,230,933 | Wound isolate; VanA-type VRE | 35 |
| E. faecium TUH4-64 | Human clinical isolate; VanB-type VRE | 77 |
| E. faecalis HIP11704 | VanA-type VRE; coisolated with vancomycin-resistant Staphylococcus aureus | 78 |
| E. faecalis V583 | Bloodstream isolate; VanB-type VRE | 79 |
| PB103 | E. faecium 1,231,410 transformed with pPB101 | This study |
| PB104 | E. faecium 1,231,410 transformed with pPB102 | This study |
| PB221 | E. faecium 1,231,410 with hexahistidine coding sequence integrated upstream of vanX (EFTG_02040) stop codon | This study |
| PB222 | E. faecium 1,231,410 ΔvanR (EFTG_02044) | This study |
| PB223 | E. faecium 1,231,410 ΔvanRS (EFTG_02043-44) | This study |
| B. subtilis BAU-101 | B. subtilis harboring vanH::lacZ cassette integrated into the amyE locus of chromosome | 34 |
| B. subtilis BAU-102 | BAU-101 harboring vanRS cassette inserted 81 bp downstream of the cat-86 promoter on plasmid pHB201 | 34 |
| B. subtilis BAU-103 | BAU-101 harboring vanR cassette inserted 81 bp downstream of the cat-86 promoter on plasmid pHB201 | 34 |
| B. subtilis BAU-104 | BAU-101 harboring vanS cassette inserted 81 bp downstream of the cat-86 promoter on plasmid pHB201 | 34 |
| E. coli EC1000 | E. coli cloning host; provides repA in trans; F− araD139 (ara ABC-leu)7679 galU galK lacX74 rspL thi; repA of pWV01 in glgB; Km | 80 |
| E. coli DH5α | E. coli cloning host; F− endA1 glnV44 thi-1 recA1 relA1 gyrA96 deoR nupG Φ80dlacZΔM15 Δ(lacZYA-argF)U169 hsdR17(rK−mK+) λ− | |
| E. coli BW23474 | Cloning host for pTCV-lac and pPB101; Δlac-169 robA1 cre C510 hsdR514 endA recA1 ΔuidA::pir-116 | 81 |
| Plasmids | ||
| pLT06 | Markerless, counterselectable exchange plasmid; confers chloramphenicol resistance | 82 |
| pHOU2 | Derivative of pCJK47 in which the erm(C) gene was replaced by aph-2′-ID and cat was incorporated in the cloning site for allelic replacements; confers gentamicin resistance | 83 |
| pHA101 | pLT06 plasmid with oriT from pHOU2 inserted at PstI; confers chloramphenicol resistance | This study |
| pTCV-lac | Expression vector for Gram-positive bacteria; confers kanamycin and erythromycin resistance | 33 |
| pPB101 | pTCV-lac-cat; expression vector for Gram-positive bacteria; confers kanamycin, erythromycin, and chloramphenicol resistance | This study |
| pPB102 | pPB101 containing 248-bp EcoRI/BamHI-digested vanH (EFTG_02042) promoter region | This study |
| pPB201 | pHA101 containing a 2.043-kb EcoRI/EcoRI-digested fragment flanking upstream and downstream of E. faecium 410 vanX gene EFTG_02040 | This study |
| pPB202 | pHA101 containing a 2.028-kb BamHI/BamHI-digested fragment flanking upstream and downstream of E. faecium 410 vanR EFTG_02044 | This study |
| pPB203 | pHA101 containing a 2.028-kb BamHI/BamHI-digested fragment flanking upstream and downstream of vanRS EFTG_02043-44 | This study |
Routine molecular biology techniques.
E. faecium genomic DNA (gDNA) was isolated using a previously published protocol (31). Electroporation of E. faecium is described in the supplemental material. Plasmids were purified using the Qiagen Miniprep kit. DNA fragments were purified using the Qiagen QIAquick PCR purification kit. Taq polymerase (New England BioLabs [NEB]) was used for routine PCRs. Phusion polymerase (Fisher) was used for cloning applications. Restriction endonuclease and T4 DNA ligase reactions were performed per the manufacturer's instructions (NEB). Routine DNA sequencing was performed by the Massachusetts General Hospital DNA core facility (Boston, MA). Primers used in this study are shown in Table S1 in the supplemental material.
MIC determinations.
MICs were determined by broth microdilution. Twofold serial dilutions of drug were made with BHI broth in a 96-well microtiter plate. An overnight culture of bacteria was diluted to an optical density at 600 nm (OD600) of 0.01, and 5 μl of the diluted culture was used to inoculate wells of the plate. The OD600 of the cultures was monitored every 30 min for 24 h using a microtiter plate reader (Synergy MX; Biotek). The MIC was defined as the lowest drug concentration at which the OD600 of the well matched the OD600 of the negative-control well (uninoculated BHI medium).
Growth kinetic assays.
An over-the-counter chlorhexidine gluconate (CHG) product, Hibiclens (4% [wt/vol] CHG with 4% isopropyl alcohol solution, referred to as H-CHG here), was used for growth kinetic assays and RNA sequencing experiments. Overnight cultures of E. faecium 1,231,410 were diluted to an OD600 of 0.01 in BHI broth and incubated at 37°C with agitation at 100 rpm until the OD600 reached 0.4 to 0.5. Twenty-five milliliters of the culture was added to equal volumes of prewarmed BHI broth containing different concentrations of H-CHG such that final concentrations of 0×, 0.5×, 1× or 2× MIC of H-CHG were attained. OD600 values were monitored for 24 h. For viability counts, 100 μl of culture obtained at each time point was serially diluted in 1× phosphate-buffered saline, and appropriate dilutions were spread on BHI agar plates.
RNA sequencing.
RNA was harvested from E. faecium 1,231,410 cultures, then treated with DNase, and verified for integrity (see the supplemental material). RNA samples were submitted for RNA sequencing to the Tufts University Core Facility (Boston, MA). Library preparation for Illumina HiSeq 2000 sequencing was performed using the Illumina TruSeq Stranded RNA Sample Preparation kit with RiboZero treatment for rRNA removal. The kit allowed for strand-specific transcript detection. Fifty base reads were obtained from single-end sequencing. The RNA sequencing experiment was independently performed twice.
RNA sequencing results were analyzed using the CLC Genomics Workbench version 7.5. Raw sequencing reads were mapped to E. faecium DO (GenBank accession number NC_017960) rRNA and tRNA genes using default parameters. Next, the remaining unassembled reads were mapped to the E. faecium 1,231,410 draft reference genome (whole-genome sequencing [WGS]; GenBank accession number NZ_ACBA00000000.1) using default parameters. Read mappings for control and test cultures were compared using the RNA-Seq analysis algorithm using default parameters. The gene expression values were quantified on the basis of RPKM (reads per kilobase of transcript per millions of reads mapped), and these values were compared between control and H-CHG treatment conditions to calculate fold change. Kal's Z-test was used to calculate P value. Genes upregulated in H-CHG-treated cultures with a fold change of ≥10 and P value of <0.05 in each of the two trials were considered for further analysis in this study.
RT-qPCR.
One hundred nanograms of RNA was used to synthesize cDNA with Superscript II (Life Technologies) and random hexamers according to the manufacturer's instructions. RNase H (NEB) was added to remove RNA, and cDNA was purified using the QIAquick PCR purification kit (Qiagen). Five nanograms of cDNA was used as the template in quantitative reverse transcription-PCR (RT-qPCR) with primers to amplify internal regions of vanA, vanB, or clpX (see Table S1 in the supplemental material). Primers for RT-qPCR were designed using NCBI Primer-BLAST (32). RT-qPCR was performed with a Cepheid Smart Cycler and SYBR green I (Sigma-Aldrich). vanA and -B gene expression was internally normalized to clpX. Threshold cycle (CT) values were used to calculate the fold change of vanA and -B gene expression between H-CHG-treated cultures and control cultures according to the formula FC = 2−(ΔΔCT), where ΔΔCT = (CT of vanA or -B in H-CHG-treated cultures − CT of clpX in H-CHG-treated cultures) − (CT of vanA or -B in control cultures − CT of clpX in control cultures). The expression of vanA and -B in the control culture was set to 1. The relative fold changes in vanA and -B expression from two independent experiments (trials 1 and 2) were quantified.
Assessment of vanHA promoter activity in E. faecium 1,231,410.
E. faecium 1,231,410 vanH promoter activity was evaluated using the expression plasmid pTCV-lac (33) modified to express chloramphenicol resistance (pPB101) (see the supplemental material). A 248-bp region containing the vanH promoter region was amplified from E. faecium 1,231,410 gDNA, digested, and ligated with EcoRI- and BamHI-digested pPB101, resulting in plasmid pPB102. pPB101 and pPB102 were introduced into E. faecium 1,231,410 via electroporation, resulting in strains PB103 and PB104, respectively. β-Galactosidase assays were performed to assess vanH promoter activity upon exposure to vancomycin and different concentrations of H-CHG (see the supplemental material). The activity was measured in duplicate for each time point, and the experiment was performed independently four times.
Assessment of VanX levels in E. faecium 1,231,410 cultures.
A hexahistidine tag was added in-frame to the C-terminal end of VanX (EFTG_02040) by knock-in of DNA sequence into the E. faecium 1,231,410 genome, generating strain E. faecium PB221 (see the supplemental material). An overnight culture of E. faecium PB221 was diluted into BHI broth and incubated with shaking at 100 rpm until an OD600 of 0.4 to 0.5 was reached. The culture was split into BHI broth with different concentrations of H-CHG or vancomycin such that final concentrations of 0×, 1/4×, 1/2×, or 1× MIC of H-CHG or 20 μg/ml of vancomycin were attained. Cultures were sampled for analysis after 1.5 and 2 h of incubation. Total soluble protein was isolated from each sample as described in the supplemental material. Equal amounts (250 μg) of total soluble protein from each culture sample were loaded onto 100 μl of washed nickel-nitrilotriacetic acid (Ni-NTA) agarose beads (Qiagen). The beads were washed twice with 1 ml of wash buffer supplemented with 45 mM imidazole. The proteins and beads were incubated together for 2 h at 4°C. After incubation, the beads were centrifuged (13,300 × g for 2 min at room temperature) and washed twice with 1 ml of wash buffer supplemented with 75 mM imidazole to remove nonspecific proteins. Next, 6× SDS loading dye was added directly to the beads and boiled for 10 min vigorously. The samples were analyzed by 12% SDS-PAGE, and VanX protein levels were evaluated by Western blotting on a polyvinylidene difluoride (PVDF) membrane with an alkaline phosphatase-conjugated monoclonal anti-polyhistidine clone His-1 antibody (Sigma-Aldrich) and Western Blue stabilized substrate for alkaline phosphatase (Promega) per the manufacturer's instructions to confirm the presence of His-tagged VanX proteins. VanX protein levels were quantified by calculating the integrated density value (IDV) of the protein bands using the Alphaimager spot density tool.
B. subtilis reporter assays.
A previously developed vanH promoter reporter system in the heterologous host Bacillus subtilis 168 (34) was used to test vanH promoter responsiveness to specific components of H-CHG as well as the roles of vanR and vanS in induction. For qualitative β-galactosidase assays, 0.3 ml of an overnight culture of each reporter strain was spread on LB agar containing 5-bromo-4-chloro-3-indoyl-β-d-galactopyranoside (X-Gal; 40 μg/ml) with chloramphenicol for BAU-101, chloramphenicol and erythromycin for BAU-102, and erythromycin for BAU-103 and BAU-104. Paper discs containing different amounts of H-CHG (1× or 2× MIC) or 5 μl of a 40-mg/ml vancomycin stock (positive control) or water or a 40-mg/ml kanamycin stock (negative controls) were placed on the plates. The plates were incubated overnight at 37°C and on the next day were transferred to 4°C for the complete development of blue color around the discs. Chlorhexidine diacetate solution (Sigma-Aldrich), chlorhexidine powder (Sigma-Aldrich), sodium d-gluconate salt (Sigma-Aldrich), and isopropyl alcohol were also assessed for their abilities to induce the vanH promoter.
Assessment of vanA expression in E. faecium 1,231,410 ΔvanR and ΔvanRS.
The vanR (EFTG_02044) and vanRS genes (EFTG_02043-44) were deleted in frame utilizing plasmid pHA101 (see the supplemental material). Broth microdilution in BHI broth was used to determine the vancomycin and H-CHG MICs for the E. faecium 1,231,410 ΔvanR and ΔvanRS strains. RNA was isolated from cultures treated with 0× and 1× MIC of H-CHG for 15 min as described above. RNA was also isolated from cultures treated with 50 μg/ml of vancomycin for 2 h. RT-qPCR was performed as described above to assess vanA and clpX expression.
Synergy assays.
Broth microdilution was utilized to test for synergism between CHG and ceftriaxone or vancomycin. For synergy tests with ceftriaxone, 2-fold serial dilutions of a fresh 1-mg/ml stock of ceftriaxone disodium salt in water (TCI) for E. faecalis and 50 mg/ml for E. faecium were made in BHI broth (control), BHI broth supplemented with 2, 5, or 20 μg/ml of vancomycin (positive control), and BHI broth supplemented with different concentrations of H-CHG or chlorhexidine digluconate solution (Sigma-Aldrich; diluted to 5% prior to use) in 96-well microtiter plates. For synergy tests with vancomycin, 2-fold dilutions of a fresh 40-mg/ml vancomycin stock were made in BHI broth or BHI broth supplemented with different concentrations of H-CHG. Overnight cultures of E. faecalis and E. faecium were diluted to an OD600 of 0.01, and 5 μl of the diluted culture was used to inoculate wells of the plate. The OD600 of the cultures was measured after 24 h of incubation at 37°C.
Sequence accession number.
Raw Illumina RNA sequencing data generated in this study are available in the Sequence Read Archive under accession number SRP065084.
RESULTS
Chlorhexidine MICs for enterococci used in this study.
E. faecium 1,231,410 was isolated in 2005 and is a clade A1 skin and soft tissue infection isolate harboring VanA-type vancomycin resistance genes (35, 36). E. faecium 1,231,410 was the model strain used for our chlorhexidine experiments.
Previous studies have reported that E. faecium and E. faecalis chlorhexidine MICs range from 0.5 to 16 μg/ml and that chlorhexidine MICs for VRE and vancomycin-sensitive enterococci are similar (25–29). The over-the-counter chlorhexidine gluconate product Hibiclens (referred to as H-CHG here) was selected for our studies because we sought to evaluate a widely available chlorhexidine-containing consumer product. The H-CHG MIC for E. faecium 1,231,410 was determined to be a 1/8,192 dilution of H-CHG, corresponding to 4.9 μg/ml of chlorhexidine. H-CHG MICs for all enterococci queried (Table 1) ranged from 2.5 to 9.8 μg/ml. These values are in the range expected based on previous literature (25–29). For further confirmation, the MIC of a chlorhexidine digluconate solution from Sigma-Aldrich was determined for E. faecium 1,231,410 and found to be identical to the H-CHG MIC.
Growth kinetics of E. faecium 1,231,410 with H-CHG.
Next, the growth kinetics of E. faecium 1,231,410 cultures exposed to different concentrations of H-CHG were studied. For these experiments, an exponentially growing culture of E. faecium 1,231,410 in BHI broth was split into flasks containing BHI with H-CHG such that final concentrations of 1/2× MIC, 1× MIC, and 2× MIC of H-CHG were attained. The OD600 of cultures exposed to 1× MIC and 2× MIC decreased compared to that of cultures not exposed to H-CHG (Fig. 1A). The OD600 of the cultures growing in 1/2× MIC decreased for 30 min after H-CHG exposure but began to increase afterwards. After 24 h of incubation, the OD600 of E. faecium 1,231,410 cultures exposed to 1× MIC of H-CHG were equivalent to those of control cultures; E. faecium 1,231,410 cultures exposed to 2× MIC did not recover (data not shown).
FIG 1.
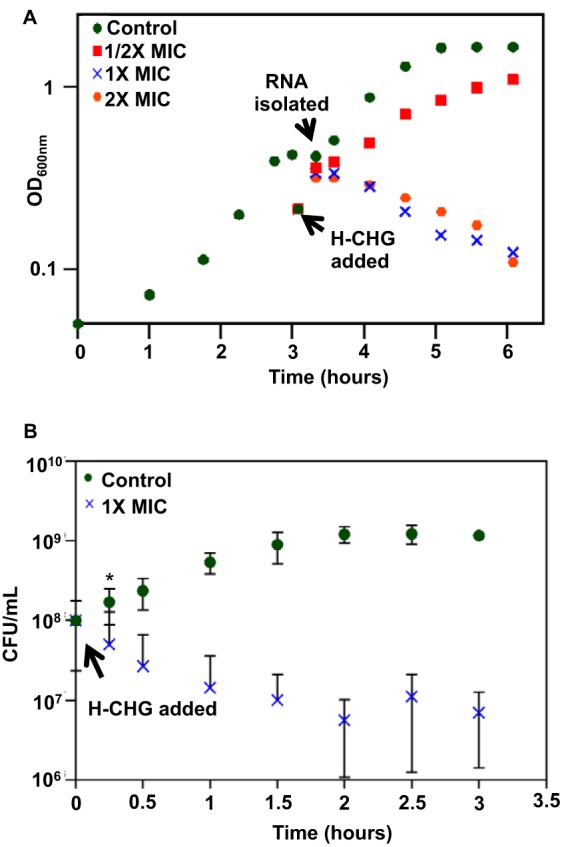
Growth kinetics of E. faecium 1,231,410 after H-CHG treatment. (A) Representative optical density curve. E. faecium was grown at 37°C in BHI with shaking at 100 rpm until the optical density at 600 nm (OD600) reached 0.3 to 0.4. Twenty-five milliliters of culture was added to an equal volume of prewarmed BHI containing different concentrations of H-CHG such that final concentrations of 0× (control; green circles), 1/2× (red squares), 1× (blue crosses), or 2× MIC (orange hexagons) were attained. The cultures were incubated at 37°C with shaking, and OD600 was monitored. (B) Viability curve. Viable cell count (CFU per milliliter) for 1× MIC-treated cultures (blue crosses) and control cultures (green circles) was assessed. Error bars indicate standard deviations from 3 independent experiments. *, P < 0.05 by the Student one-tailed t test; significance was assessed only for the 15-min time point. For transcriptomic analyses, RNA was harvested from cultures exposed to 0× (control) and 1× MIC of H-CHG for 15 min.
The viability of cultures treated with 1× MIC of H-CHG and control cultures was assessed, and a significant reduction in viable cells was observed after 15 min of exposure to H-CHG (Fig. 1B). The average CFU per milliliter for control cultures at this time point was 1.7 × 108, while for H-CHG-treated cultures it was 5.0 × 107.
RNA sequencing analysis of E. faecium 1,231,410 response to H-CHG.
Illumina RNA sequencing was performed for cultures exposed to 0× (control) and 1× (test) MIC of H-CHG for 15 min, indicated by an arrow in Fig. 1A. Genes differentially expressed shortly after H-CHG exposure may be representative of the E. faecium response to contact with patient skin contaminated with subbactericidal CHG, which is a point of interest for our research.
The RNA sequencing experiment was performed twice independently. We identified 35 genes upregulated ≥10-fold in H-CHG-exposed cells in both of the two RNA sequencing trials (Table 2). We focused on highly upregulated genes in this study because we reasoned that these genes could be protective for H-CHG exposure. We segregated the 35 genes into 4 groups: vancomycin resistance genes, extracytoplasmic stress-associated genes, predicted transport systems, and miscellaneous genes. Data Set S1 in the supplemental material is an expanded version of Table 2 showing read mapping data, cotranscription predictions, and conserved domain analysis of protein sequences.
TABLE 2.
Differentially upregulated genes in cultures treated with 1× MIC of H-CHG versus control cultures identified by two independent RNA sequencing trials
| E. faecium ORF | V583 orthologa | Trial 1 FCb | Trial 2 FCb | Description | Overlapc |
|---|---|---|---|---|---|
| Predicted transport systems | |||||
| EFTG_01192 | EF1057 | 11.4 | 14.6 | mntH2; natural resistance-associated macrophage protein | □α |
| EFTG_01315 | 13.2 | 26.7 | Zinc/iron permease | ||
| EFTG_02616 | EF1262 | 27.5 | 43.3 | Conserved hypothetical protein | ϕα |
| EFTG_02617 | 45.5 | 29.7 | Conserved hypothetical protein | ||
| EFTG_02287 | EF2226 | 22.0 | 21.8 | ABC transporter | □ |
| EFTG_02288 | EF2227 | 16.0 | 18.6 | ABC transporter | □ |
| EFTG_02514 | EF0575 | 20.5 | 63.2 | Cationic ABC transporter | α |
| EFTG_02515 | 25.6 | 48.0 | Transposase | ||
| EFTG_02516 | EF0576 | 10.4 | 39.3 | Cation ABC transporter | α |
| EFTG_02517 | EF0577 | 14.7 | 61.2 | Adhesion lipoprotein | α |
| EFTG_02518 | EF0578 | 13.5 | 55.9 | Iron-dependent repressor | α |
| EFTG_02519 | EF0579 | 11.7 | 17.8 | Conserved hypothetical protein | α |
| EFTG_02682 | 43.7 | 28.2 | Extracellular solute-binding protein family 1 | ||
| EFTG_02683 | 29.7 | 29.1 | Conserved hypothetical protein | ||
| EFTG_02684 | 26.7 | 11.3 | ABC transporter permease | ||
| EFTG_02685 | 26.7 | 21.6 | Predicted protein | ||
| EFTG_02686 | 25.1 | 17.9 | ABC transporter system ATP-binding protein | ||
| Vancomycin resistance genes | |||||
| EFTG_02038 | 19.0 | 8.4d | vanZ; VanZ protein | ||
| EFTG_02039 | 15.4 | 8.4d | vanY; d-alanyl–d-alanine carboxypeptidase | ||
| EFTG_02040 | EF2293 | 104.3 | 61.1 | vanX; d-Ala–d-Ala dipeptidase | |
| EFTG_02041 | EF2294 | 78.7 | 37.1 | vanA; d-Ala–d-lactate ligase | |
| EFTG_02042 | EF2295 | 82.6 | 58.9 | vanH; d-lactate dehydrogenase | |
| EFTG_02043 | 6.4d | 7.6d | vanS; sensor histidine kinase | ||
| EFTG_02044 | 7.1d | 9.8d | vanR; vancomycin response regulator | ||
| Extracytoplasmic stress-associated genes | |||||
| EFTG_00421 | EF1533 | 10.5 | 19.3 | Conserved hypothetical protein | □ϕ |
| EFTG_00736 | EF2698 | 9.8d | 28.0 | telA; toxic anion resistance protein | △ϕ |
| EFTG_00737 | EF2697 | 35.8 | 55.9 | xpaC; conserved hypothetical protein | △ϕ |
| EFTG_00904 | EF2477 | 11.9 | 10.5 | Conserved hypothetical protein | ϕ |
| EFTG_00974 | EF1006 | 15.9 | 15.2 | Conserved hypothetical protein | ϕ |
| EFTG_01178 | EF1753 | 69.0 | 117.9 | liaX; conserved hypothetical protein | △ϕ |
| EFTG_01179 | EF1752 | 32.0 | 28.5 | liaY; conserved hypothetical protein | △□ϕ |
| EFTG_01180 | EF1751 | 31.1 | 33.1 | liaZ; integral membrane protein | ϕ |
| EFTG_01316 | EF0026 | 52.6 | 34.6 | Predicted protein | ϕ |
| EFTG_01545 | EF3027 | 19.9 | 15.2 | htrA; serine protease | ϕ |
| EFTG_01950 | EF0932 | 23.1 | 10.7 | Conserved hypothetical protein | □ϕ |
| Miscellaneous genes | |||||
| EFTG_00189 | 11.0 | 30.8 | spx; arsenate reductase | ||
| EFTG_01407 | 58.2 | 45.1 | Conserved hypothetical protein | ||
| EFTG_01778 | EF0466 | 14.7 | 15.1 | nagB; glucosamine-6-phosphate isomerase | |
| EFTG_01890 | 10.6 | 13.4 | Predicted protein | ||
| EFTG_02731 | 18.9 | 12.5 | Predicted protein |
Identified by previous comparative genome analysis (35).
Fold change (FC) in gene expression in H-CHG cells relative to control cells as assessed by RNA sequencing.
See Data Set S1 in the supplemental material and the text for references. Triangles indicate that the gene is associated with daptomycin nonsusceptibility. Squares indicate that the gene is associated with stress response to overexpression of the Fst toxin. Alpha symbols indicate that the gene is associated with metal stress response. Phi symbols indicate that the gene is upregulated in response to cell wall-active antibiotics.
Fold changes of <10.
Among the most highly upregulated genes in H-CHG-treated cultures were the VanA-type vancomycin resistance gene cluster (Table 2; see also Data Set S1). vanHAX, whose expression is regulated by the two-component system VanRS (37), includes the structural genes required for vancomycin resistance (reviewed in reference 5). The VanH dehydrogenase converts pyruvate into d-lactate, which is utilized by the VanA ligase to form d-Ala–d-Lac cell wall precursors. d-Ala–d-Ala generated by the chromosomally encoded d-Ala–d-Ala ligase, Ddl, is hydrolyzed by the d-Ala–d-Ala dipeptidase VanX. The accessory genes vanY and vanZ encode a carboxypeptidase that cleaves d-Ala from late cell wall precursors terminating in d-Ala–d-Ala and a protein of unknown function, respectively (5). The induction of these genes in the presence of H-CHG suggests that chlorhexidine and/or chlorhexidine-induced stress induces VanA-type vancomycin resistance gene expression in E. faecium.
Other highly upregulated genes have previously been implicated in the enterococcal extracytoplasmic stress response (Table 2; see also Data Set S1). We identified overlap between the H-CHG transcriptomic response in E. faecium 1,231,410 with the transcriptomic response of E. faecalis OG1RF to the cell wall-active antibiotics ampicillin, bacitracin, cephalothin, and vancomycin (38) and with the E. faecalis OG1X response to the plasmid-encoded Fst toxin, which likely interacts with the OG1X cell membrane, causing stress (39, 40). Further, mutations in four genes in our data set are associated with daptomycin nonsusceptibility in E. faecalis and E. faecium (41–44). Of specific interest, the liaXYZ genes, which are directly regulated by the cell envelope stress regulator LiaR in E. faecalis (45), are highly upregulated in H-CHG-treated E. faecium cells. Finally, htrA, encoding a predicted membrane-anchored cell surface serine protease, was also upregulated. HtrA family proteins are important for the perception and turnover of misfolded and mislocalized proteins in phylogenetically diverse organisms (46). The E. coli HtrA family protein DegS participates in the activation of RpoE, an extracytoplasmic function sigma factor, in response to mislocalized proteins (46). Upregulation of htrA suggests that the perception and/or turnover of misfolded or mislocalized proteins on the cell surface is important for the H-CHG stress response. Overall, these results indicate that within 15 min of H-CHG exposure, E. faecium 1,231,410 mounts a transcriptional response to extracytoplasmic stress. This is consistent with chlorhexidine causing cell wall and/or cell membrane damage in E. faecium 1,231,410.
Several predicted membrane transport systems were highly upregulated in response to H-CHG exposure (Table 2; see also Data Set S1 in the supplemental material). Seven of these genes have previously been implicated in metal stress response in E. faecalis; specifically, they are upregulated in response to zinc (47, 48). In addition, the ortholog of EFTG_01192 in E. faecalis V583, referred to as EF1057, is downregulated in response to iron chloride excess (49). Orthologs of another predicted transport system, encoded by EFTG_02287-02288 in E. faecium 1,231,410 and EF2226-EF2227 in E. faecalis V583, were upregulated in Fst toxin-treated E. faecalis OG1X (39), indicating overlap with the extracytoplasmic stress response.
A putative transport system encoded by a predicted 5-gene operon (EFTG_02682-EFTG_02686) was highly upregulated in E. faecium 1,231,410 exposed to H-CHG (Table 2; see also Data Set S1). Interestingly, this operon is not present in 18 E. faecalis or 3 clade B (commensal clade) E. faecium genomes previously compared by whole-genome analysis (35), nor is it present in E. faecium DO, a common reference strain for E. faecium studies. However, the operon is present on the 131-kbp p1 plasmid present in the VanB-type VRE E. faecium bloodstream isolate Aus0085 (open reading frames [ORFs] EFAU085_p1045 to EFAU085_p1041) (50) and is present in 8 clade A1 and 6 clade A2 E. faecium isolates from a recent comparative genome study (36). This result is significant because it demonstrates that additional genes in the auxiliary (i.e., noncore) E. faecium genome, other than the VanA-type vancomycin resistance genes, are responsive to H-CHG exposure. As for function, conserved domain analysis indicates that the operon codes for an ATP-binding cassette (ABC) transport system, potentially transporting polyamines or iron (see Data Set S1).
Other upregulated genes include EFTG_00189, encoding a putative redox-responsive transcriptional regulator, and EFTG_01890, encoding a protein with a rhodanese-like domain that may also be involved in redox stress response. EFTG_01778, encoding a putative glucosamine-6-phosphate deaminase, is also upregulated. This gene is likely involved in N-acetylglucosamine metabolism in E. faecium (51).
vanA upregulation in response to H-CHG occurs in other VanA-type VRE and is not strain or species specific.
By RNA sequencing, we observed up to 104-fold upregulation of vancomycin resistance genes (vanHAX) in E. faecium 1,231,410 exposed to 1× MIC of H-CHG for 15 min. Because of the clinical significance of vancomycin resistance in enterococci, we further studied vanHAX induction by H-CHG. RT-qPCR for vanA expression was performed to confirm the RNA sequencing results (Fig. 2). vanA expression was internally normalized to the housekeeping gene clpX, which encodes the ATPase subunit of the housekeeping ClpXP protease (52), and was not found to be differentially regulated in RNA sequencing trials (data not shown). Using RT-qPCR, E. faecium 1,231,410 vanA was 64-fold upregulated in cultures exposed to 1× MIC of H-CHG for 15 min, compared to unexposed cultures (Fig. 2), confirming the RNA sequencing results. vanA was up to 47-fold and 18-fold upregulated in cultures exposed to 1/2× MIC and 1/4× MIC of H-CHG, respectively, for 15 min (Fig. 2).
FIG 2.
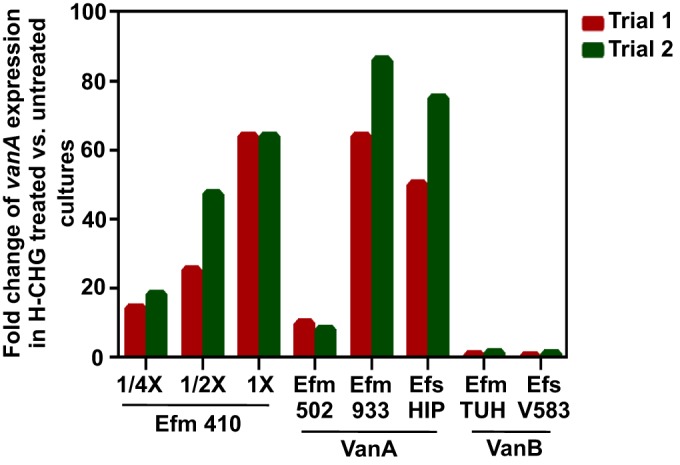
H-CHG induces VanA-type vancomycin resistance genes in E. faecium and E. faecalis. RT-qPCR was used to quantify the expression of vanA or vanB upon exposure to H-CHG for 15 min compared to control conditions. Expression of vanA and -B was internally normalized to clpX as described in the text. Expression of vanA or -B in control cultures was set to 1 (not shown). The fold change in vanA or -B expression in cultures treated with 1× MIC of H-CHG relative to the control was quantified for two independent experiments for all strains queried. E. faecium 1,231,410 vanA expression upon exposure to 1/4× or 1/2× MIC of H-CHG for 15 min was additionally queried. Efm, E. faecium; Efs, E. faecalis; HIP, HIP11704; TUH, TUH4-64.
We next evaluated whether H-CHG induction of vanA was strain or species specific by RT-qPCR analysis of VanA-type VRE E. faecium 1,231,502, the VanA-type VRE E. faecium 1,230,933, and the VanA-type VRE E. faecalis HIP11704. Induction of vanA in response to H-CHG was observed for all three strains, although the fold upregulation of vanA in E. faecium 1,231,502 was modest (at least 8.6-fold) compared to that in the other strains (at least 50-fold) (Fig. 2). From these results, we conclude that vanA induction by H-CHG is not strain or species specific.
Interestingly, no upregulation of vanB was observed after exposure to 1× MIC of H-CHG for the VanB-type VRE isolate E. faecium TUH4-64 or in the VanB-type VRE isolate E. faecalis V583 (Fig. 2). The maximum fold change in vanB expression observed was 1.3 for one of the TUH4-64 trials. A key difference between VanA- and VanB-type systems is specificity of induction. VanB-type systems are induced only by vancomycin, while VanA-type systems are induced by vancomycin, teicoplanin, and other compounds (discussed further below). This difference in specificity is linked to their respective VanRS regulatory two-component systems, which share little amino acid sequence identity (53). The induction of vanA but not vanB by H-CHG suggests that the VanA-type VanRS system is responsive to chlorhexidine and/or chlorhexidine-induced stress.
The E. faecium 1,231,410 vanH promoter is responsive to H-CHG.
The vanHAX genes have a common promoter upstream of vanH (37, 54). We sought to determine whether increased vanH promoter activity contributed to vanHAX induction in response to H-CHG exposure. The previously identified vanH transcription start site, predicted sigma factor binding sites, and inverted repeat sequences and predicted VanR binding sites upstream of the vanH coding region (37, 54) are conserved in E. faecium 1,231,410 (data not shown). However, a partial IS1251 sequence is inserted at position −102 relative to the vanH transcription start site. This insertion disrupts the 5′ 15 bp of the ∼80-bp phosphorylated VanR DNA footprint previously identified by Holman et al. (54), although sigma factor and predicted regulator binding sites are intact. The sequence occurring between the IS1251 insertion and vanH was amplified and used to generate a reporter construct. Plasmid pPB102 contains a transcriptional fusion of the vanH promoter to the lacZ gene in pPB101 (pTCV-lac-cat).
β-Galactosidase activities of E. faecium 1,231,410 strains harboring pPB101 or pPB102 were assessed in the presence of vancomycin and in the presence of different concentrations of H-CHG at 0, 30, 60, 90, and 120 min postexposure (Fig. 3). For E. faecium 1,231,410 harboring pPB102, vanH promoter activity increased over the growth curve under control conditions, as expected based on previous studies of vanH promoter activity (55). Addition of vancomycin stimulated vanH promoter activity, as expected. Addition of 1/4× MIC of H-CHG to the cultures resulted in a significant increase in vanH promoter activity with time compared to that of the control (Fig. 3). β-Galactosidase activity was equivalent to that of the control for cultures treated with 1/2× MIC of H-CHG and was negligible for cultures treated with 1× MIC of H-CHG (data not shown). These results are likely due to the inhibitory action of H-CHG on growth of the reporter strain at 1/2× and 1× MIC (Fig. 3A). No β-galactosidase activity was detected for E. faecium 1,231,410 transformed with pPB101 (data not shown). We conclude from these results that vanH promoter activity is directly impacted by the addition of H-CHG, leading to increased vanHAX transcription. These results indicate that vanHAX induction by H-CHG is dependent on the VanRS two-component system.
FIG 3.
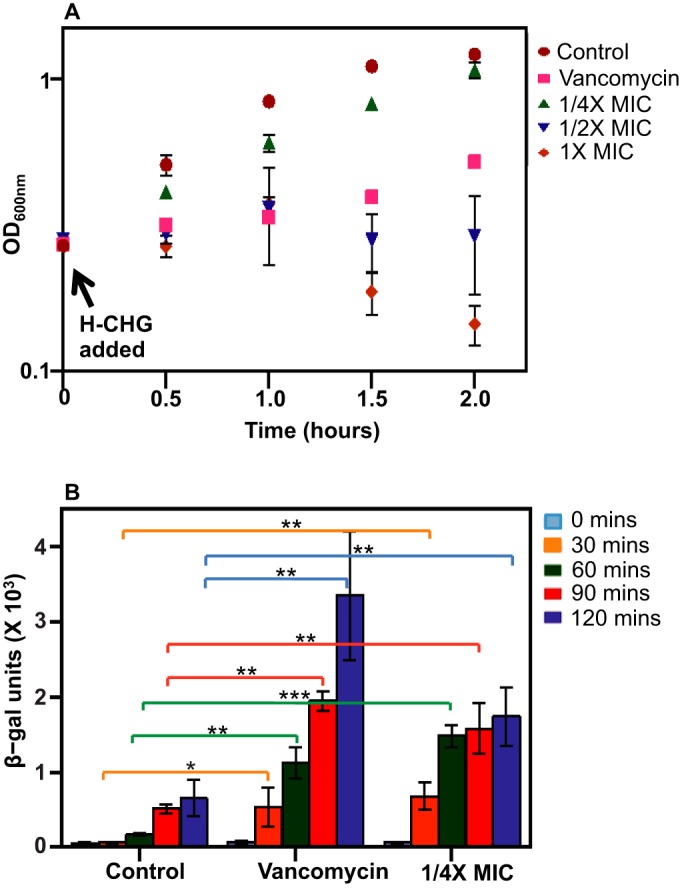
vanH promoter activity in E. faecium 1,231,410. (A) Average growth curve of E. faecium PB104. Aliquots of an exponentially growing E. faecium PB104 culture at an OD600 of 0.4 to 0.5 were added to 25 ml of BHI-chloramphenicol (control; red circles) or BHI-chloramphenicol containing H-CHG or vancomycin such that 1/4× MIC of H-CHG (green triangles), 1/2× MIC of H-CHG (blue triangles), 1× MIC of H-CHG (orange diamonds), or 2 μg/ml of vancomycin (pink squares) were attained. Cultures were incubated with shaking at 100 rpm at 37°C, and the OD600 was monitored. Error bars indicate standard deviations from 4 independent experiments. (B) β-Galactosidase activity assays. Enzyme activity assays were performed as described in the text. Samples were harvested from E. faecium PB104 cultures at multiple time points after exposure to H-CHG, vancomycin, or control conditions. Error bars indicate standard deviations from 4 independent experiments. The one-tailed Student t test was used to assess significance. *, P < 0.05; **, P < 0.005; ***, P < 0.0005.
VanX protein levels are elevated after H-CHG exposure.
We sought to determine whether increased transcription of vanHAX resulted in increased levels of van-encoded proteins. Previous studies indicated that increased vanHAX promoter activity did not necessarily result in vancomycin resistance, since high-level expression of the genes is required (56, 57). Since vancomycin is also an inducer of vanHAX, it was not possible to use vancomycin resistance as a phenotypic output. To assess translation resulting from increased vanHAX transcription with H-CHG, an 18-bp hexahistidine coding sequence was knocked into the E. faecium 1,231,410 genome, upstream of the vanX stop codon. Previous studies have reported using a VanX dipeptidase enzyme assay (56–58) or a VanX-specific antibody (59) for detecting increased VanX activity or protein levels in cultures treated with vancomycin or other test compounds. Therefore, there is a precedent for VanX detection as a proxy for vancomycin resistance.
We first compared the growth kinetics and vancomycin MIC of the hexahistidine knock-in strain, E. faecium PB221, compared to the wild-type strain. These assays were performed to verify that the sequence knock-in did not affect the vancomycin resistance of the strain or confer a growth defect. No difference in growth rate or yield was observed during growth in BHI media and BHI media supplemented with vancomycin (see Fig. S1 in the supplemental material), and the vancomycin MICs for the two strains were the same (312.5 μg/ml). The H-CHG MIC was also unaffected. Next, Western blotting was used to detect VanX levels in E. faecium PB221 cultures exposed to vancomycin, H-CHG, or water for 1.5 or 2 h (Fig. 4). These time points were chosen to allow for translation of the Van proteins after exposure to a promoter inducer. A representative Western blot is shown in Fig. 4A; the 23-kDa VanX-His6 protein is indicated by an arrow. The average VanX band intensity values (integrated density values) for three independent experiments are shown in Fig. 4B. The results demonstrate that significantly more VanX protein was produced in vancomycin- and H-CHG-treated cells than in control cells.
FIG 4.

Detection and quantification of VanX protein levels in E. faecium cultures. (A) Representative Western blot. A total of 250 μg of protein extracted from E. faecium PB221 cultures was analyzed by Western blotting with anti-polyhistidine antibody as described in the text. A 23-kDa protein (indicated by arrow) was detected in cultures treated with H-CHG and vancomycin. Lanes: 1, control cells after 1.5 h of incubation; 2, control cells after 2 h of incubation; 3, cells after 1.5 h of incubation with 1/4× MIC of H-CHG; 4, cells after 2 h of incubation with 1/4× MIC of H-CHG; 5, cells after 1.5 h of incubation with 1/2× MIC of H-CHG; 6, cells after 2 h of incubation with 1/2× MIC of H-CHG; 7, cells after 1.5 h of incubation with 1× MIC of H-CHG; 8, cells after 2 h of incubation with 1× MIC of H-CHG; 9, cells after 2 h of incubation with 20 μg/ml of vancomycin. (B) Quantification of VanX protein levels. The amount of VanX protein was quantified by calculating the IDV (integrated density value) of each of the protein band by using Alphaimager spot density tool. Average values are shown. Error bar represent the standard deviations from 3 experiments. The one-tailed Student t test was used to assess significance. *, P < 0.05.
vanHAX promoter induction by H-CHG requires VanR in a B. subtilis heterologous expression system.
To further extend the study of the induction of vancomycin resistance genes by H-CHG, we used a previously developed vanH promoter reporter system in B. subtilis 168 (34). In strain BAU-101, the vanH promoter from the VanA-type VRE E. faecium A624 is fused to lacZ and chromosomally integrated. Derivatives of this strain express plasmid-borne vanR and/or vanS genes (Table 1). If a compound is an inducer of the vanH promoter, in the presence of a chromogenic indicator for β-galactosidase activity, a blue halo will be observed around the zone of inhibition.
We used the B. subtilis BAU reporter strain series to assess vanH promoter activity in response to vancomycin (positive control), kanamycin or water (negative controls), and H-CHG (Table 3; see also Fig. S2 in the supplemental material). For the reporter strains BAU-102 and BAU-103, which express vanRS or vanR, respectively, blue haloes were observed around H-CHG and vancomycin zones of inhibition. The faint blue haloes observed around H-CHG and vancomycin zones of inhibition for strain BAU-103 could result from cross talk between VanR and heterologous two-component membrane sensors and/or gene dosage effects of vanR expression from a multicopy plasmid.
TABLE 3.
Bacillus subtilis reporter strain results
| B. subtilis strain | Promoter activity in response toa,b: |
|||||||
|---|---|---|---|---|---|---|---|---|
| Van | Kan | Water | H-CHG | CDA | CHX | NaG | Isopropanol | |
| BAU-101 | − | − | − | − | ND | ND | ND | ND |
| BAU-102 | + | − | − | + | + | + | − | − |
| BAU-103 | + | − | − | + | + | + | − | − |
| BAU-104 | − | − | − | − | ND | ND | ND | ND |
Abbreviations: Van, vancomycin (positive control); Kan, kanamycin (negative control); H-CHG, Hibiclens; CDA, chlorhexidine diacetate salt; NaG, sodium gluconate; CHX, chlorhexidine powder.
+, blue halo observed around the compound; −, no blue halo observed; ND, not determined.
The B. subtilis BAU reporter strain series was also used to determine the specific component of H-CHG responsible for vanH promoter induction (Table 3; see also Fig. S2). A chlorhexidine-containing solution (chlorhexidine diacetate) and individual components of the H-CHG antiseptic (powdered chlorhexidine, isopropyl alcohol, and sodium d-gluconate salt) were tested for the ability to induce blue halo formation in reporter strains BAU-102 and BAU-103. H-CHG, powdered chlorhexidine, and chlorhexidine diacetate were the only substances tested that induced the vanH promoter.
Induction of vanA by H-CHG is VanR dependent.
To confirm that induction of vancomycin resistance genes by H-CHG is dependent upon VanR, we constructed E. faecium 1,231,410 vanR and vanRS deletion mutants. Because deletion of vanS alone leads to constitutive expression of vancomycin resistance (60), the contribution of VanR in a VanS deletion mutant was not assessed. The vancomycin MICs for the ΔvanR and ΔvanRS mutants were reduced compared to that for the wild-type E. faecium 1,231,410 (2.4 μg/ml versus 312.5 μg/ml), but the H-CHG MIC was unaltered.
RT-qPCR was used to assess vanA expression in cultures treated with vancomycin for 2 h (Fig. 5A) or 1× MIC of H-CHG for 15 min (Fig. 5B). As expected, vanA expression was reduced in the ΔvanR and ΔvanRS mutants relative to the wild-type strain for both vancomycin- and H-CHG-treated cultures. For Fig. 5A, the difference in vanA expression observed between the ΔvanR and ΔvanRS strains suggests that VanS plays some role in activation of vanA expression in the absence of VanR. However, this gene expression pattern does not result in vancomycin resistance, since, as noted above, the ΔvanR and ΔvanRS mutants have identical vancomycin MICs. From results shown in Fig. 5B, we conclude that H-CHG induction of vanA is VanR dependent. Additionally, since the H-CHG MIC is unaffected by vanR or vanRS deletion, induction of VanA-type vancomycin resistance genes is not protective against H-CHG.
FIG 5.

RT-qPCR analysis of vanA gene expression in response to vancomycin and H-CHG. RT-qPCR was used to quantify the expression of vanA ligase upon exposure to vancomycin (50 μg/ml) for 2 h (A) and 1× MIC H-CHG for 15 min (B) versus unexposed cultures for the E. faecium 410 wild type and ΔvanR and ΔvanRS deletion mutants. The expression of the vanA gene was internally normalized to clpX. The expression of vanA in control cultures was set to 1 (not shown), and relative fold change expression in vancomycin and H-CHG treated cultures from two independent experiments was quantified (trial 1, red bars; trial 2, green bars).
E. faecalis HIP11704 is resensitized to ceftriaxone in the presence of sub-MIC chlorhexidine.
Enterococci are intrinsically resistant to most cephalosporins, which are β-lactam antibiotics. β-Lactam antibiotics inhibit cell wall biosynthesis by binding to penicillin-binding proteins (PBPs), which cross-link peptidoglycan precursors (61). The mechanism for cephalosporin resistance in enterococci is multifactorial and incompletely understood, but production of PBP5, which has a low affinity for β-lactams, is involved (61). Previous studies on vancomycin-resistant enterococci have reported synergism between β-lactams and vancomycin (62–65). Induction of the vancomycin resistance genes, resulting in carboxypeptidase expression and hydrolysis of d-Ala–d-Ala termini from peptidoglycan precursors, is required for this synergy (64). In this study, we have shown that H-CHG induces vancomycin resistance gene expression in VanA-type E. faecium and E. faecalis (Fig. 2). Therefore, an expected phenotype of H-CHG-treated VRE is increased cephalosporin susceptibility.
We tested VanA-type E. faecium 1,231,410 and E. faecalis HIP11704 for synergism with ceftriaxone, a broad-spectrum cephalosporin, using a broth microdilution assay (Table 4). Interestingly, a reduction in ceftriaxone MIC (from 500 μg/ml to 2 μg/ml) was observed for E. faecalis HIP11704 in the presence of subinhibitory H-CHG or CHG, demonstrating synergism between chlorhexidine and ceftriaxone. As expected (64), vancomycin also induced ceftriaxone susceptibility in E. faecalis HIP11704.
TABLE 4.
Ceftriaxone MIC for VanA-type VRE
| Strain | Ceftriaxone MIC (mg/ml)a |
|||||
|---|---|---|---|---|---|---|
| No Van | +Van,2 μg/ml | +Van,20 μg/ml | +1/4×MIC | +1/2×MIC | +0.75×MIC | |
| E. faecium 410 | >50 | ND | 12.5 | 25 | 6.25 | No growth |
| E. faecalis HIP11704 | 0.50 | 0.002 | ND | 0.25 | 0.002 | 0.002 |
Average MICs were determined in BHI media after 24 h of incubation from 3 independent biological replicates. ND, not determined.
The ceftriaxone MIC was significantly higher for E. faecium 1,231,410 than for E. faecalis HIP11704. As noted previously (66), E. faecium 1,231,410 has an elevated ampicillin MIC (>4 μg/ml), which may be due to sequence variations in pbp5 and other, as-yet-unidentified loci. While the ceftriaxone MIC did decrease for E. faecium 1,231,410 in the presence of either vancomycin or H-CHG (Table 4), the extent of the reduction is likely of little clinical significance.
E. faecium 1,231,410 is more susceptible to vancomycin in the presence of sub-MIC chlorhexidine.
We performed a second set of synergy assays to assess whether vancomycin and H-CHG could act additively to increase the vancomycin resistance of E. faecium 1,231,410. Unexpectedly, the strain became more sensitive to vancomycin in the presence of sub-MIC H-CHG (Table 5). A drop in vancomycin MIC of similar magnitude was not observed for the E. faecium 1,231,410 ΔvanR mutant. This result indicates that upregulation of the vancomycin resistance genes is required for vancomycin sensitization in the presence of chlorhexidine.
TABLE 5.
Vancomycin MIC for E. faecium 1,231,410
| Strain | Vancomycin MIC (μg/ml)a |
||
|---|---|---|---|
| No H-CHG | +1/2× MIC H-CHG | +1× MIC H-CHG | |
| E. faecium 1,231,410 | 312.5 | 0.6–4.8 | No growth |
| ΔvanR strain | 1.2–2.4 | 0.3–0.6 | No growth |
MICs were determined in BHI media after 24 h of incubation from 3 (wild-type strain) and 4 (ΔvanR strain) independent biological replicates. The range of MICs observed is shown where applicable.
DISCUSSION
The goal of this study was to investigate the transcriptional responses of E. faecium 1,231,410, a vancomycin-resistant clinical isolate, to MIC levels of a CHG-containing consumer product (H-CHG). Among the highly upregulated genes after 15 min of exposure to the product was the VanA-type vancomycin resistance gene cluster. Because of the clinical relevance of vancomycin resistance, the rest of the current study focused on this aspect of the E. faecium 1,231,410 transcriptional response. However, other genes of note were induced by H-CHG exposure. These include liaXYZ, which are associated with daptomycin nonsusceptibility in enterococci (41–44), as well as other genes associated with extracytoplasmic stress. Presumably, these genes are upregulated in response to chlorhexidine-induced cell surface stress. Other highly upregulated genes include predicted metal transport systems and a predicted ABC transport system of unknown function that appears to be encoded by a mobile genetic element. The specific roles of these transport systems in the H-CHG stress response remain to be determined; possibilities include chlorhexidine efflux (note that a new family of chlorhexidine efflux proteins was recently identified for Gram-negative bacteria [67, 68]), transport of metals to maintain redox balance in the cell, or transport of cell wall-related metabolites. Future studies will compare the transcriptomic response of E. faecium to H-CHG to that for CHG and sodium gluconate, which will help to determine which specific components of H-CHG are responsible for the transcriptional changes observed.
We observed that MIC and sub-MIC levels of H-CHG induced expression of the VanA-type vancomycin resistance gene cluster in E. faecium 1,231,410. Induction of vanA by H-CHG occurred in VanA-type E. faecalis and E. faecium, but vanB was not induced by H-CHG in VanB-type E. faecalis and E. faecium. Using a vanH promoter reporter, we determined that exposure to H-CHG resulted in increased vanH promoter activity. An E. faecium 1,231,410 derivative expressing a hexahistidine-tagged VanX protein was used in Western blotting experiments to show that increased vanH promoter activity with H-CHG resulted in significantly increased VanX protein levels. Using a combination of approaches in E. faecium 1,231,410 and the heterologous host B. subtilis, we determined that VanR is required for induction of vanHAX by H-CHG. Experiments with B. subtilis demonstrated that chlorhexidine is the specific component of H-CHG responsible for vanH promoter induction. Importantly, expression of the vancomycin resistance genes is not protective against chlorhexidine, since the H-CHG MIC is unaffected by vanR or vanRS deletion.
Collectively, our results indicate that the VanRS two-component system senses either chlorhexidine or chlorhexidine-induced cell surface stress and activates vanHAX expression. Induction of VanA-type vancomycin resistance genes by compounds other than vancomycin has been well studied, although with conflicting results (34, 55, 56, 69, 70). These conflicting results could be attributed to the different methods used to assess induction (multicopy expression plasmids, expression of van genes outside or inside their native context, resistance phenotype, and VanX enzymatic activity). For this reason, we endeavored to use multiple approaches to demonstrate that chlorhexidine is an inducer of VanA-type vancomycin resistance. Despite the conflicts noted above, there is consensus that vancomycin, teicoplanin, and moenomycin are inducers of VanA-type vancomycin resistance, while only vancomycin is an inducer of VanB-type resistance. For VanB-type vancomycin resistance (responsive only to vancomycin), direct binding of vancomycin to the VanS sensor has been experimentally demonstrated for a Streptomyces host (71). For VanA-type vancomycin resistance, an alternative model has been proposed to explain its relaxed specificity of induction relative to the VanB-type resistance. This model posits that the VanA-type VanS protein is responsive to cell surface destabilization and, specifically, inhibition of transglycosylation (56, 69). This model is supported by the observation that structurally unrelated drug classes activate VanA-type resistance expression, including glycopeptides, moenomycin, and now chlorhexidine. Further study is required to elucidate the specific aspect of chlorhexidine or chlorhexidine-induced stress that leads to induction of VanA-type resistance.
As expected for VRE actively expressing vancomycin resistance (64), VanA-type VRE were more sensitive to ceftriaxone in the presence of subinhibitory CHG. The magnitude of this effect was small for E. faecium 1,231,410, which has elevated ceftriaxone (Table 4) and ampicillin (66) MICs. Unexpectedly, sub-MIC H-CHG increased E. faecium 1,231,410 susceptibility to vancomycin, but a similar increase in vancomycin susceptibility was not observed for a ΔvanR derivative (Table 5). This suggests that this effect is dependent upon expression of the vancomycin resistance genes. We identified two explanations for vancomycin sensitivity of E. faecium 1,231,410 in the presence of both vancomycin and chlorhexidine. The first explanation is that in the presence of both vancomycin and chlorhexidine, E. faecium 1,231,410 synthesizes peptidoglycan precursors that terminate in neither d-Ala–d-Ala (synthesized by Ddl) nor d-Ala–d-Lac (synthesized by VanA). Instead, precursors that terminate in a structure that is sensitive to vancomycin binding are synthesized by VanA. The mechanism underlying this would be the relaxed substrate specificity of the VanA ligase (72, 73) combined with chlorhexidine-dependent gene expression changes that alter substrate pools and lead to the incorporation of amino or short acids that weaken the cell wall, as has been previously reported (74). Based on an analysis of predicted amino acid racemases and d-isomer-specific dehydrogenases (see Data Set S2 in the supplemental material), this hypothesis is not supported by our RNA sequencing data. An alternative explanation is that in the presence of both vancomycin and chlorhexidine, E. faecium 1,231,410 synthesizes peptidoglycan precursors that terminate in d-Ala–d-Lac, as expected. However, these termini fail to be cross-linked due to chlorhexidine-induced changes in transpeptidase expression. Although not achieving our ≥10-fold change threshold used in this study, several transpeptidases (encoded by pbpF, ddcP, and pbpA) are upregulated after 15 min of exposure to chlorhexidine (see Data Set S2). These data suggest that transpeptidase ratios are altered in the presence of chlorhexidine, which supports the second hypothesis. Analyses of E. faecium 1,231,410 peptidoglycan structure are of interest for future work, as are deletion of pbpF, ddcP, and pbpA to assess their roles in the vancomycin sensitization we observe for E. faecium 1,231,410 in the presence of chlorhexidine. Understanding this mechanism could be informative for novel strategies to treat VREfm infections.
What are possible clinical impacts of VRE exposures to sub-MIC chlorhexidine? Based on our results, E. faecium and E. faecalis isolates harboring VanA-type resistance genes will synthesize modified cell walls in response to subbactericidal levels of chlorhexidine. Glycopeptide-dependent VRE have been isolated from patients undergoing vancomycin therapy (5). These isolates have mutations in ddl and depend upon the exogenous presence of vancomycin to induce vanA or vanB such that a cell wall can be formed (75). It would be of interest to investigate whether sub-MIC chlorhexidine exposure can rescue VanA-type glycopeptide-dependent VRE. It is also significant that genes protective against daptomycin (liaXYZ) are highly upregulated in response to chlorhexidine. It was recently demonstrated that deletion of liaR, encoding an activator of liaXYZ expression (45), restores daptomycin susceptibility to daptomycin-nonsusceptible enterococci (76). Further studies will be required to determine whether gene expression “priming” by chlorhexidine impacts treatment outcomes with daptomycin, or if frequent exposure to subinhibitory chlorhexidine is selective for strains that constitutively activate liaXYZ.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by startup funds from the University of Texas at Dallas to K.L.P.
We gratefully acknowledge Bernie Weisblum for providing the B. subtilis BAU strain series. We thank Juan González and Corey Slape for assistance with RT-qPCR experiments. We thank Marinelle Rodrigues, Karthik Hullahalli, Ajit Shah, and all Palmer lab members for helpful discussions. We thank Hannah Adams for contributing pHA101.
P.B. and K.L.P. designed experiments, P.B. and E.Z. performed experiments, P.B. and K.L.P. analyzed data, and P.B. and K.L.P. wrote the manuscript.
Funding Statement
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02595-15.
REFERENCES
- 1.Agudelo Higuita NI, Huycke MM. 2014. Enterococcal disease, epidemiology, and implications for treatment, p 45–70. In Gilmore MS, Clewell DB, Ike Y, Shankar N (ed), Enterococci: from commensals to leading causes of drug resistant infection. Massachusetts Eye and Ear Infirmary, Boston, MA. [Google Scholar]
- 2.Arias CA, Murray BE. 2012. The rise of the Enterococcus: beyond vancomycin resistance. Nat Rev Microbiol 10:266–278. doi: 10.1038/nrmicro2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sievert DM, Ricks P, Edwards JR, Schneider A, Patel J, Srinivasan A, Kallen A, Limbago B, Fridkin S, National Healthcare Safety Network (NHSN) Team and Participating NHSN Facilities. 2013. Antimicrobial-resistant pathogens associated with healthcare-associated infections: summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2009-2010. Infect Control Hosp Epidemiol 34:1–14. doi: 10.1086/668770. [DOI] [PubMed] [Google Scholar]
- 4.Reynolds PE. 1989. Structure, biochemistry and mechanism of action of glycopeptide antibiotics. Eur J Clin Microbiol Infect Dis 8:943–950. doi: 10.1007/BF01967563. [DOI] [PubMed] [Google Scholar]
- 5.Courvalin P. 2006. Vancomycin resistance in gram-positive cocci. Clin Infect Dis 42(Suppl 1):S25–S34. doi: 10.1086/491711. [DOI] [PubMed] [Google Scholar]
- 6.Handwerger S, Pucci MJ, Volk KJ, Liu J, Lee MS. 1992. The cytoplasmic peptidoglycan precursor of vancomycin-resistant Enterococcus faecalis terminates in lactate. J Bacteriol 174:5982–5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allen NE, Hobbs JN Jr, Richardson JM, Riggin RM. 1992. Biosynthesis of modified peptidoglycan precursors by vancomycin-resistant Enterococcus faecium. FEMS Microbiol Lett 77:109–115. [DOI] [PubMed] [Google Scholar]
- 8.Messer J, Reynolds PE. 1992. Modified peptidoglycan precursors produced by glycopeptide-resistant enterococci. FEMS Microbiol Lett 73:195–200. [DOI] [PubMed] [Google Scholar]
- 9.Liu J, Volk KJ, Lee MS, Pucci M, Handwerger S. 1994. Binding studies of vancomycin to the cytoplasmic peptidoglycan precursors by affinity capillary electrophoresis. Anal Chem 66:2412–2416. doi: 10.1021/ac00086a031. [DOI] [PubMed] [Google Scholar]
- 10.Davies GE, Francis J, Martin AR, Rose FL, Swain G. 1954. 1:6-Di-4′-chlorophenyldiguanidohexane (“Hibitane”). Laboratory investigation of a new antibacterial agent of high potency. Br J Pharmacol Chemother 9:192–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Darouiche RO, Raad II, Heard SO, Thornby JI, Wenker OC, Gabrielli A, Berg J, Khardori N, Hanna H, Hachem R, Harris RL, Mayhall G. 1999. A comparison of two antimicrobial-impregnated central venous catheters. N Engl J Med 340:1–8. doi: 10.1056/NEJM199901073400101. [DOI] [PubMed] [Google Scholar]
- 12.Rupp ME, Lisco SJ, Lipsett PA, Perl TM, Keating K, Civetta JM, Mermel LA, Lee D, Dellinger EP, Donahoe M, Giles D, Pfaller MA, Maki DG, Sherertz R. 2005. Effect of a second-generation venous catheter impregnated with chlorhexidine and silver sulfadiazine on central catheter-related infections: a randomized, controlled trial. Ann Intern Med 143:570–580. doi: 10.7326/0003-4819-143-8-200510180-00007. [DOI] [PubMed] [Google Scholar]
- 13.Marschall J, Mermel LA, Fakih M, Hadaway L, Kallen A, O'Grady NP, Pettis AM, Rupp ME, Sandora T, Maragakis LL, Yokoe DS, Society for Healthcare Epidemiology of America. 2014. Strategies to prevent central line-associated bloodstream infections in acute care hospitals: 2014 update. Infect Control Hosp Epidemiol 35:753–771. doi: 10.1086/676533. [DOI] [PubMed] [Google Scholar]
- 14.Supple L, Kumaraswami M, Kundrapu S, Sunkesula V, Cadnum JL, Nerandzic MM, Tomas M, Donskey CJ. 2015. Chlorhexidine only works if applied correctly: use of a simple colorimetric assay to provide monitoring and feedback on effectiveness of chlorhexidine application. Infect Control Hosp Epidemiol 36:1095–1097. doi: 10.1017/ice.2015.124. [DOI] [PubMed] [Google Scholar]
- 15.Komljenović I, Marquardt D, Harroun TA, Sternin E. 2010. Location of chlorhexidine in DMPC model membranes: a neutron diffraction study. Chem Phys Lipids 163:480–487. doi: 10.1016/j.chemphyslip.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 16.Koontongkaew S, Jitpukdeebodintra S. 1995. Interaction of chlorhexidine with cytoplasmic membranes of Streptococcus mutans GS-5. Caries Res 29:413–417. doi: 10.1159/000262101. [DOI] [PubMed] [Google Scholar]
- 17.Hugo WB, Longworth AR. 1964. Some aspects of the mode of action of chlorhexidine. J Pharm Pharmacol 16:655–662. doi: 10.1111/j.2042-7158.1964.tb07384.x. [DOI] [PubMed] [Google Scholar]
- 18.Hugo WB, Longworth AR. 1965. Cytological aspects of the mode of action of chlorhexidine diacetate. J Pharm Pharmacol 17:28–32. doi: 10.1111/j.2042-7158.1965.tb07562.x. [DOI] [PubMed] [Google Scholar]
- 19.Hugo WB, Longworth AR. 1966. The effect of chlorhexidine on the electrophoretic mobility, cytoplasmic constituents, dehydrogenase activity and cell walls of Escherichia coli and Staphylococcus aureus. J Pharm Pharmacol 18:569–578. doi: 10.1111/j.2042-7158.1966.tb07935.x. [DOI] [PubMed] [Google Scholar]
- 20.Cheung HY, Wong MM, Cheung SH, Liang LY, Lam YW, Chiu SK. 2012. Differential actions of chlorhexidine on the cell wall of Bacillus subtilis and Escherichia coli. PLoS One 7:e36659. doi: 10.1371/journal.pone.0036659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Noto MJ, Domenico HJ, Byrne DW, Talbot T, Rice TW, Bernard GR, Wheeler AP. 2015. Chlorhexidine bathing and health care-associated infections: a randomized clinical trial. JAMA 313:369–378. doi: 10.1001/jama.2014.18400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pittet D, Angus DC. 2015. Daily chlorhexidine bathing for critically ill patients: a note of caution. JAMA 313:365–366. doi: 10.1001/jama.2014.18482. [DOI] [PubMed] [Google Scholar]
- 23.Noto MJ, Wheeler AP. 2015. Understanding chlorhexidine decolonization strategies. Intensive Care Med 41:1351–1354. doi: 10.1007/s00134-015-3846-6. [DOI] [PubMed] [Google Scholar]
- 24.Popovich KJ, Lyles R, Hayes R, Hota B, Trick W, Weinstein RA, Hayden MK. 2012. Relationship between chlorhexidine gluconate skin concentration and microbial density on the skin of critically ill patients bathed daily with chlorhexidine gluconate. Infect Control Hosp Epidemiol 33:889–896. doi: 10.1086/667371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suller MT, Russell AD. 1999. Antibiotic and biocide resistance in methicillin-resistant Staphylococcus aureus and vancomycin-resistant Enterococcus. J Hosp Infect 43:281–291. doi: 10.1016/S0195-6701(99)90424-3. [DOI] [PubMed] [Google Scholar]
- 26.Beier RC, Duke SE, Ziprin RL, Harvey RB, Hume ME, Poole TL, Scott HM, Highfield LD, Alali WQ, Andrews K, Anderson RC, Nisbet DJ. 2008. Antibiotic and disinfectant susceptibility profiles of vancomycin-resistant Enterococcus faecium (VRE) isolated from community wastewater in Texas. Bull Environ Contam Toxicol 80:188–194. doi: 10.1007/s00128-007-9342-0. [DOI] [PubMed] [Google Scholar]
- 27.Kõljalg S, Naaber P, Mikelsaar M. 2002. Antibiotic resistance as an indicator of bacterial chlorhexidine susceptibility. J Hosp Infect 51:106–113. doi: 10.1053/jhin.2002.1204. [DOI] [PubMed] [Google Scholar]
- 28.Koburger T, Hubner NO, Braun M, Siebert J, Kramer A. 2010. Standardized comparison of antiseptic efficacy of triclosan, PVP-iodine, octenidine dihydrochloride, polyhexanide and chlorhexidine digluconate. J Antimicrob Chemother 65:1712–1719. doi: 10.1093/jac/dkq212. [DOI] [PubMed] [Google Scholar]
- 29.Barry AL, Fuchs PC, Brown SD. 1999. Lack of effect of antibiotic resistance on susceptibility of microorganisms to chlorhexidine gluconate or povidone iodine. Eur J Clin Microbiol Infect Dis 18:920–921. doi: 10.1007/s100960050434. [DOI] [PubMed] [Google Scholar]
- 30.Suwantarat N, Carroll KC, Tekle T, Ross T, Maragakis LL, Cosgrove SE, Milstone AM. 2014. High prevalence of reduced chlorhexidine susceptibility in organisms causing central line-associated bloodstream infections. Infect Control Hosp Epidemiol 35:1183–1186. doi: 10.1086/677628. [DOI] [PubMed] [Google Scholar]
- 31.Adams HM, Li X, Mascio C, Chesnel L, Palmer KL. 2015. Mutations associated with reduced surotomycin susceptibility in Clostridium difficile and Enterococcus species. Antimicrob Agents Chemother 59:4139–4147. doi: 10.1128/AAC.00526-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL. 2012. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 13:134. doi: 10.1186/1471-2105-13-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Poyart C, Trieu-Cuot P. 1997. A broad-host-range mobilizable shuttle vector for the construction of transcriptional fusions to beta-galactosidase in gram-positive bacteria. FEMS Microbiol Lett 156:193–198. doi: 10.1016/S0378-1097(97)00423-0. [DOI] [PubMed] [Google Scholar]
- 34.Ulijasz AT, Grenader A, Weisblum B. 1996. A vancomycin-inducible lacZ reporter system in Bacillus subtilis: induction by antibiotics that inhibit cell wall synthesis and by lysozyme. J Bacteriol 178:6305–6309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Palmer KL, Godfrey P, Griggs A, Kos VN, Zucker J, Desjardins C, Cerqueira G, Gevers D, Walker S, Wortman J, Feldgarden M, Haas B, Birren B, Gilmore MS. 2012. Comparative genomics of enterococci: variation in Enterococcus faecalis, clade structure in E. faecium, and defining characteristics of E. gallinarum and E. casseliflavus. mBio 3:e00318-11. doi: 10.1128/mBio.00318-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lebreton F, van Schaik W, McGuire AM, Godfrey P, Griggs A, Mazumdar V, Corander J, Cheng L, Saif S, Young S, Zeng Q, Wortman J, Birren B, Willems RJ, Earl AM, Gilmore MS. 2013. Emergence of epidemic multidrug-resistant Enterococcus faecium from animal and commensal strains. mBio 4:e00534-13. doi: 10.1128/mBio.00534-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arthur M, Molinas C, Courvalin P. 1992. The VanS-VanR two-component regulatory system controls synthesis of depsipeptide peptidoglycan precursors in Enterococcus faecium BM4147. J Bacteriol 174:2582–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abranches J, Tijerina P, Aviles-Reyes A, Gaca AO, Kajfasz JK, Lemos JA. 2014. The cell wall-targeting antibiotic stimulon of Enterococcus faecalis. PLoS One 8:e64875. doi: 10.1371/journal.pone.0064875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brinkman CL, Bumgarner R, Kittichotirat W, Dunman PM, Kuechenmeister LJ, Weaver KE. 2013. Characterization of the effects of an rpoC mutation that confers resistance to the Fst peptide toxin-antitoxin system toxin. J Bacteriol 195:156–166. doi: 10.1128/JB.01597-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Göbl C, Kosol S, Stockner T, Ruckert HM, Zangger K. 2010. Solution structure and membrane binding of the toxin Fst of the par addiction module. Biochemistry 49:6567–6575. doi: 10.1021/bi1005128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Humphries RM, Kelesidis T, Tewhey R, Rose WE, Schork N, Nizet V, Sakoulas G. 2012. Genotypic and phenotypic evaluation of the evolution of high-level daptomycin nonsusceptibility in vancomycin-resistant Enterococcus faecium. Antimicrob Agents Chemother 56:6051–6053. doi: 10.1128/AAC.01318-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Palmer KL, Daniel A, Hardy C, Silverman J, Gilmore MS. 2011. Genetic basis for daptomycin resistance in enterococci. Antimicrob Agents Chemother 55:3345–3356. doi: 10.1128/AAC.00207-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Diaz L, Tran TT, Munita JM, Miller WR, Rincon S, Carvajal LP, Wollam A, Reyes J, Panesso D, Rojas NL, Shamoo Y, Murray BE, Weinstock GM, Arias CA. 2014. Whole-genome analyses of Enterococcus faecium with diverse daptomycin MICs. Antimicrob Agents Chemother 58:4527–4534. doi: 10.1128/AAC.02686-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miller C, Kong J, Tran TT, Arias CA, Saxer G, Shamoo Y. 2013. Adaptation of Enterococcus faecalis to daptomycin reveals an ordered progression to resistance. Antimicrob Agents Chemother 57:5373–5383. doi: 10.1128/AAC.01473-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Davlieva M, Shi Y, Leonard PG, Johnson TA, Zianni MR, Arias CA, Ladbury JE, Shamoo Y. 2015. A variable DNA recognition site organization establishes the LiaR-mediated cell envelope stress response of enterococci to daptomycin. Nucleic Acids Res 43:4758–4773. doi: 10.1093/nar/gkv321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clausen T, Kaiser M, Huber R, Ehrmann M. 2011. HtrA proteases: regulated proteolysis in protein quality control. Nat Rev Mol Cell Biol 12:152–162. doi: 10.1038/nrm3065. [DOI] [PubMed] [Google Scholar]
- 47.Abrantes MC, Lopes Mde F, Kok J. 2011. Impact of manganese, copper and zinc ions on the transcriptome of the nosocomial pathogen Enterococcus faecalis V583. PLoS One 6:e26519. doi: 10.1371/journal.pone.0026519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abrantes MC, Kok J, Lopes Mde F. 2013. EfaR is a major regulator of Enterococcus faecalis manganese transporters and influences processes involved in host colonization and infection. Infect Immun 81:935–944. doi: 10.1128/IAI.06377-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.López G, Latorre M, Reyes-Jara A, Cambiazo V, Gonzalez M. 2012. Transcriptomic response of Enterococcus faecalis to iron excess. Biometals 25:737–747. doi: 10.1007/s10534-012-9539-5. [DOI] [PubMed] [Google Scholar]
- 50.Lam MM, Seemann T, Tobias NJ, Chen H, Haring V, Moore RJ, Ballard S, Grayson LM, Johnson PD, Howden BP, Stinear TP. 2013. Comparative analysis of the complete genome of an epidemic hospital sequence type 203 clone of vancomycin-resistant Enterococcus faecium. BMC Genomics 14:595. doi: 10.1186/1471-2164-14-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alvarez-Añorve LI, Calcagno ML, Plumbridge J. 2005. Why does Escherichia coli grow more slowly on glucosamine than on N-acetylglucosamine? Effects of enzyme levels and allosteric activation of GlcN6P deaminase (NagB) on growth rates. J Bacteriol 187:2974–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baker TA, Sauer RT. 2012. ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim Biophys Acta 1823:15–28. doi: 10.1016/j.bbamcr.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Evers S, Courvalin P. 1996. Regulation of VanB-type vancomycin resistance gene expression by the VanS(B)-VanR (B) two-component regulatory system in Enterococcus faecalis V583. J Bacteriol 178:1302–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Holman TR, Wu Z, Wanner BL, Walsh CT. 1994. Identification of the DNA-binding site for the phosphorylated VanR protein required for vancomycin resistance in Enterococcus faecium. Biochemistry 33:4625–4631. doi: 10.1021/bi00181a024. [DOI] [PubMed] [Google Scholar]
- 55.Lai MH, Kirsch DR. 1996. Induction signals for vancomycin resistance encoded by the vanA gene cluster in Enterococcus faecium. Antimicrob Agents Chemother 40:1645–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baptista M, Depardieu F, Courvalin P, Arthur M. 1996. Specificity of induction of glycopeptide resistance genes in Enterococcus faecalis. Antimicrob Agents Chemother 40:2291–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arthur M, Depardieu F, Reynolds P, Courvalin P. 1996. Quantitative analysis of the metabolism of soluble cytoplasmic peptidoglycan precursors of glycopeptide-resistant enterococci. Mol Microbiol 21:33–44. doi: 10.1046/j.1365-2958.1996.00617.x. [DOI] [PubMed] [Google Scholar]
- 58.Arthur M, Depardieu F, Courvalin P. 1999. Regulated interactions between partner and non-partner sensors and response regulators that control glycopeptide resistance gene expression in enterococci. Microbiology 145:1849–1858. doi: 10.1099/13500872-145-8-1849. [DOI] [PubMed] [Google Scholar]
- 59.Hill CM, Krause KM, Lewis SR, Blais J, Benton BM, Mammen M, Humphrey PP, Kinana A, Janc JW. 2010. Specificity of induction of the vanA and vanB operons in vancomycin-resistant enterococci by telavancin. Antimicrob Agents Chemother 54:2814–2818. doi: 10.1128/AAC.01737-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Arthur M, Depardieu F, Gerbaud G, Galimand M, Leclercq R, Courvalin P. 1997. The VanS sensor negatively controls VanR-mediated transcriptional activation of glycopeptide resistance genes of Tn1546 and related elements in the absence of induction. J Bacteriol 179:97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kristich CJ, Rice LB, Arias CA. 2014. Enterococcal infection—treatment and antibiotic resistance, p 87–134. In Gilmore MS, Clewell DB, Ike Y, Shankar N (ed), Enterococci: from commensals to leading causes of drug resistant infection. Massachusetts Eye and Ear Infirmary, Boston, MA. [PubMed] [Google Scholar]
- 62.Leclercq R, Bingen E, Su QH, Lambert-Zechovski N, Courvalin P, Duval J. 1991. Effects of combinations of beta-lactams, daptomycin, gentamicin, and glycopeptides against glycopeptide-resistant enterococci. Antimicrob Agents Chemother 35:92–98. doi: 10.1128/AAC.35.1.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gutmann L, al-Obeid S, Billot-Klein D, Guerrier ML, Collatz E. 1994. Synergy and resistance to synergy between beta-lactam antibiotics and glycopeptides against glycopeptide-resistant strains of Enterococcus faecium. Antimicrob Agents Chemother 38:824–829. doi: 10.1128/AAC.38.4.824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kristich CJ, Djoric D, Little JL. 2014. Genetic basis for vancomycin-enhanced cephalosporin susceptibility in vancomycin-resistant enterococci revealed using counterselection with dominant-negative thymidylate synthase. Antimicrob Agents Chemother 58:1556–1564. doi: 10.1128/AAC.02001-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shlaes DM, Etter L, Gutmann L. 1991. Synergistic killing of vancomycin-resistant enterococci of classes A, B, and C by combinations of vancomycin, penicillin, and gentamicin. Antimicrob Agents Chemother 35:776–779. doi: 10.1128/AAC.35.4.776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Galloway-Peña JR, Rice LB, Murray BE. 2011. Analysis of PBP5 of early U.S. isolates of Enterococcus faecium: sequence variation alone does not explain increasing ampicillin resistance over time. Antimicrob Agents Chemother 55:3272–3277. doi: 10.1128/AAC.00099-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hassan KA, Jackson SM, Penesyan A, Patching SG, Tetu SG, Eijkelkamp BA, Brown MH, Henderson PJ, Paulsen IT. 2013. Transcriptomic and biochemical analyses identify a family of chlorhexidine efflux proteins. Proc Natl Acad Sci U S A 110:20254–20259. doi: 10.1073/pnas.1317052110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hassan KA, Liu Q, Henderson PJ, Paulsen IT. 2015. Homologs of the Acinetobacter baumannii AceI transporter represent a new family of bacterial multidrug efflux systems. mBio 6:e01982-14. doi: 10.1128/mBio.01982-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Handwerger S, Kolokathis A. 1990. Induction of vancomycin resistance in Enterococcus faecium by inhibition of transglycosylation. FEMS Microbiol Lett 58:167–170. [DOI] [PubMed] [Google Scholar]
- 70.Grissom-Arnold J, Alborn WE Jr, Nicas TI, Jaskunas SR. 1997. Induction of VanA vancomycin resistance genes in Enterococcus faecalis: use of a promoter fusion to evaluate glycopeptide and nonglycopeptide induction signals. Microb Drug Resist 3:53–64. doi: 10.1089/mdr.1997.3.53. [DOI] [PubMed] [Google Scholar]
- 71.Koteva K, Hong HJ, Wang XD, Nazi I, Hughes D, Naldrett MJ, Buttner MJ, Wright GD. 2010. A vancomycin photoprobe identifies the histidine kinase VanSsc as a vancomycin receptor. Nat Chem Biol 6:327–329. doi: 10.1038/nchembio.350. [DOI] [PubMed] [Google Scholar]
- 72.Bugg TD, Dutka-Malen S, Arthur M, Courvalin P, Walsh CT. 1991. Identification of vancomycin resistance protein VanA as a D-alanine:D-alanine ligase of altered substrate specificity. Biochemistry 30:2017–2021. doi: 10.1021/bi00222a002. [DOI] [PubMed] [Google Scholar]
- 73.Bugg TD, Wright GD, Dutka-Malen S, Arthur M, Courvalin P, Walsh CT. 1991. Molecular basis for vancomycin resistance in Enterococcus faecium BM4147: biosynthesis of a depsipeptide peptidoglycan precursor by vancomycin resistance proteins VanH and VanA. Biochemistry 30:10408–10415. doi: 10.1021/bi00107a007. [DOI] [PubMed] [Google Scholar]
- 74.Zarlenga LJ, Gilmore MS, Sahm DF. 1992. Effects of amino acids on expression of enterococcal vancomycin resistance. Antimicrob Agents Chemother 36:902–905. doi: 10.1128/AAC.36.4.902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Van Bambeke F, Chauvel M, Reynolds PE, Fraimow HS, Courvalin P. 1999. Vancomycin-dependent Enterococcus faecalis clinical isolates and revertant mutants. Antimicrob Agents Chemother 43:41–47. doi: 10.1093/jac/43.suppl_1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Panesso D, Reyes J, Gaston E, Deal M, Londono A, Nigo M, Munita JM, Miller W, Shamoo Y, Tran TT, Arias CA. 2015. Deletion of liaR reverses daptomycin resistance in Enterococcus faecium independent of the genetic background. Antimicrob Agents Chemother 59:7327–7334. doi: 10.1128/AAC.01073-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dahl KH, Simonsen GS, Olsvik O, Sundsfjord A. 1999. Heterogeneity in the vanB gene cluster of genomically diverse clinical strains of vancomycin-resistant enterococci. Antimicrob Agents Chemother 43:1105–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Weigel LM, Clewell DB, Gill SR, Clark NC, McDougal LK, Flannagan SE, Kolonay JF, Shetty J, Killgore GE, Tenover FC. 2003. Genetic analysis of a high-level vancomycin-resistant isolate of Staphylococcus aureus. Science 302:1569–1571. doi: 10.1126/science.1090956. [DOI] [PubMed] [Google Scholar]
- 79.Sahm DF, Kissinger J, Gilmore MS, Murray PR, Mulder R, Solliday J, Clarke B. 1989. In vitro susceptibility studies of vancomycin-resistant Enterococcus faecalis. Antimicrob Agents Chemother 33:1588–1591. doi: 10.1128/AAC.33.9.1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Leenhouts K, Buist G, Bolhuis A, ten Berge A, Kiel J, Mierau I, Dabrowska M, Venema G, Kok J. 1996. A general system for generating unlabelled gene replacements in bacterial chromosomes. Mol Gen Genet 253:217–224. [DOI] [PubMed] [Google Scholar]
- 81.Haldimann A, Prahalad MK, Fisher SL, Kim SK, Walsh CT, Wanner BL. 1996. Altered recognition mutants of the response regulator PhoB: a new genetic strategy for studying protein-protein interactions. Proc Natl Acad Sci U S A 93:14361–14366. doi: 10.1073/pnas.93.25.14361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Thurlow LR, Thomas VC, Hancock LE. 2009. Capsular polysaccharide production in Enterococcus faecalis and contribution of CpsF to capsule serospecificity. J Bacteriol 191:6203–6210. doi: 10.1128/JB.00592-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Panesso D, Montealegre MC, Rincon S, Mojica MF, Rice LB, Singh KV, Murray BE, Arias CA. 2011. The hylEfm gene in pHylEfm of Enterococcus faecium is not required in pathogenesis of murine peritonitis. BMC Microbiol 11:20. doi: 10.1186/1471-2180-11-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.