

Abstract
In situ hybridization is the technique by which specific RNA or DNA molecules are detected in cytological preparations. Basically it involves formation of a hybrid molecule between an endogenous single-stranded RNA or DNA in the cell and a complementary single-stranded RNA or DNA probe. In its original form the probe was labeled with 3H and the hybrid was detected by autoradiography. The first successful experiments in 1968 involved detection of the highly amplified ribosomal DNA in oocytes of the frog Xenopus, followed soon after by the reiterated “satellite DNA” in mouse and Drosophila chromosomes. Fluorescent probes were developed about ten years later.
My lifelong interest in giant chromosomes and what they might tell us about gene structure and function began about 1950, when I started graduate work at Yale in the laboratory of Donald Poulson. Poulson was a Drosophila geneticist who had trained at CalTech when T. H. Morgan, A. H. Sturtevant, and C. B. Bridges were still establishing Drosophila as the key organism for genetic analysis. Bridges had just completed his maps of the giant polytene chromosomes found in the larval salivary glands. These chromosomes were called polytene (many stranded) because they consisted of hundreds to thousands of copies of individual chromosomes lined up parallel to one another. With Poulson’s help I made “squash” preparations of Drosophila polytene chromosomes and familiarized myself with the exquisite banding pattern that allowed one to map the positions of individual genes. In the end, however, I could not think of anything new to do with these chromosomes.
As luck would have it, I stumbled onto the fact that giant chromosomes of another sort are found in the oocytes of frogs and salamanders. These are the so-called lampbrush chromosomes (LBCs), discovered in the late 19th century, but largely overlooked by cell biologists in the decades following their first description [1]. They were given the name lampbrush because of their fuzzy appearance, which was reminiscent of the brushes used at that time to clean the chimneys of oil lamps. I studied these chromosomes for my thesis, making use of the important discovery by William Duryee [2] that the amphibian oocyte nucleus and its giant chromosomes could both be isolated by hand from living oocytes. I was lucky that commercial phase contrast microscopes had just been introduced and so I was able to examine “living” LBCs for the first time at high magnification. About this time an investigator in Scotland, H.G. “Mick” Callan, also realized the advantages of LBCs. We quickly became friends and eventually collaborators. As we slowly learned, LBCs are the very opposite of polytene chromosomes, despite their enormous size. They are in prophase of the first meiotic division and each consists of exactly four chromatids, as in any other meiotic chromosome. What gives them their unique appearance are hundreds of looped out regions, where sister chromatids are not closely associated with each other. It gradually became clear from experiments that Mick Callan and I carried out that the paired loops are regions of active RNA synthesis.
After completing my Ph. D. at Yale, I took a position as Instructor at the University of Minnesota, where I remained for the next 11 years, rising up the academic ladder. Convinced that the bands of polytene chromosomes and the paired loops of LBCs corresponded to individual genes or small clusters of genes, I spent many hours wondering how we could use these chromosomes to identify specific gene loci. Sometime around this time I learned of an important paper recently published by two immunologists, Albert Coons and Melvin Kaplan [3]. They showed that individual proteins could be localized at the cellular level using an antibody coupled to fluorescein isocyanate. Of course, there were no commercial fluorescent antibodies available, so anyone wishing to make use of this new technique was faced with the rather daunting task of synthesizing their own probes. Moreover, epifluorescence microscopes were a thing of the future, so one also had to put together one’s own microscope using a wide-angle condenser designed for dark-field observations. Such a condenser was oiled to the bottom of the microscope slide on which the preparation was mounted. A mercury arc fitted with a deep blue filter supplied the necessary light for excitation of the fluorophore and a yellow filter in the eyepiece allowed one to observe the yellow-green fluorescence.
I decided to test the hypothesis that individual loops of the LBCs might carry gene-specific proteins that could be detected by the marvelous new technique of Coons and Kaplan. So I put together a fluorescence microscope and convinced a friend in the Chemistry Department to synthesize some fluorescein isocyanate for me (it was a dangerous synthesis involving phosgene, among other things). Antibody production was an equally daunting task for someone with no training in immunology. Fortunately, a graduate student in our department was making antibodies in chickens, so I was able to make use of his expertise (and chickens). I had no good idea what proteins to test, so I isolated and solubilized a large number of giant nuclei from salamander oocytes, injected them into chickens, and collected the serum. Equally naively, I conjugated the whole serum with fluorescein and applied the mixture to LBC preparations. As I remember - I cannot find any notes from these experiments - I never saw anything of interest. The fact that no publications resulted from this rather heroic foray into immunofluorescence is further evidence that there were no significant results. Nevertheless, these studies familiarized me with some basics of immunology and gave me a good working knowledge of fluorescence microscopy, both of which would be valuable in the future.
In 1963 I returned to Yale for a sabbatical year. As things worked out, I was offered a professorship there, and I remained at Yale for the next 20 years. It soon became clear that I could not continue to make meaningful contributions to cell biology without learning the new techniques for analyzing RNA and DNA – what came to be called molecular biology. Over the next few years I continued to study the amphibian oocyte, but now with an emphasis on the molecular aspects of transcription. In those days – a decade before molecular cloning – the easiest molecules to study were the two RNA subunits of the ribosome, 18S and 28S ribosomal RNA (rRNA). Despite technical limitations, several groups made rapid progress in understanding these molecules in a variety of organisms. From the standpoint of cell biology, the most important finding was that newly synthesized rRNA appeared first in the nucleolus and that the genes coding for that RNA (rDNA) were reiterated and associated with the nucleolus in amphibians [4, 5] and in Drosophila [6]. These studies tied the new molecular information to cytological observations made three decades earlier by Barbara McClintock [7]. Somewhat oversimplified, McClintock’s “nucleolar organizer” corresponded to the rDNA in the chromosome, whereas the nucleolus itself represented the material being organized – rRNA and associated proteins.
This elegant understanding of the nucleolus and its important role in cell biology posed significant problems for those of us studying the amphibian oocyte. Since the 19th century it had been known that the gigantic nucleus inside the amphibian oocyte contained not one or two nucleoli but hundreds. Even more perplexing, these nucleoli were not attached to the chromosomes, but were localized primarily around the periphery of the nucleus near the nuclear envelope. How could they be involved in rRNA synthesis if they were not at the rDNA loci on the chromosomes? The easiest resolution of this dilemma would have been that these extrachromosomal “nucleoli” were not true nucleoli at all. That is, that they were not involved in rRNA synthesis, but represented some other kind of nuclear body, of which several were already known.
As things turned out, however, the multiple nucleoli are involved in rRNA synthesis. Each extrachromosomal nucleolus contains a small amount of rDNA, identical to the rDNA at the nucleolus organizer on the chromosomes. The history of “gene amplification,” as this phenomenon was named by Oscar Miller in 1965 [8], is quite complicated. In fact, gene amplification had been clearly described and correctly interpreted many years before, but had been largely ignored or forgotten [9, 10]. What was new was not the phenomenon, but rather the identification of rDNA as the specific molecule involved in amplification. Briefly, Oscar Miller [8] and Jim Kezer (cited in [11]) showed by DNase digestion that the free nucleoli in amphibian oocytes contained DNA, thereby confirming earlier claims based on staining with the Feulgen reagent. That this DNA was rDNA was demonstrated independently by me [12] and by Don Brown and Igor Dawid [13]. The most important fact for the subsequent development of in situ hybridization was the realization that the extra rDNA is synthesized early in meiosis and accumulates as a prominent cap on one side of the nucleus (Fig. 1). In other words, rDNA amplification is complete before the oocyte begins its growth phase and before the multiple nucleoli are formed.
Fig. 1.
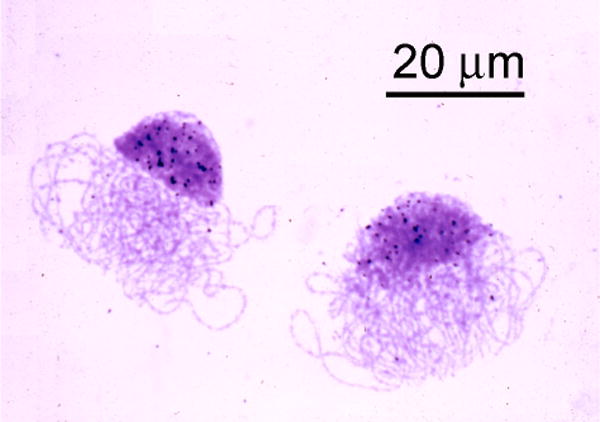
Autoradiograph of two oocyte nuclei in the pachytene stage of meiosis from a Xenopus ovary. In addition to the chromosomes in the characteristic “bouquet” arrangement, each nucleus contains a massive cap of amplified rDNA. The black dots over the cap are silver grains in the autoradiographic emulsion that covers the preparation. However, this is not an in situ hybridization. The silver grains were produced by radioactivity from 3H-thymidine incorporated into the DNA of these nuclei. The grains indicate that the rDNA in the cap is still replicating at the pachytene stage, long after the chromosomes have ceased replication.
Although I spent most of my time on the molecular biology of rRNA, I did not forget my hope to develop some way to visualize the genes on polytene and lampbrush chromosomes. Fluorescent antibodies had been a flop, but now the thought of nucleic acid hybridization seemed a possibility. The melting and reassociation of DNA was demonstrated in the early 1960s as well as the hybridization of RNA to complementary DNA sequences. Many of these experiments were done with 32P-labeled molecules, the reassociated molecules or hybrids being detected by scintillation counting. One particularly useful technique was filter hybridization. Here one attached denatured, single-stranded DNA to a nitrocellulose filter and then incubated the filter in a solution of radioactive RNA. This technique, developed especially by Gillespie and Spiegelman [14], proved extremely useful for molecular analysis of RNA-DNA hybrids. I used this technique in many of my experiments on rRNA and it occurred to me that it should be possible to adapt the method to cytological preparations. The idea was to make a flat tissue squash, denature the DNA in the nuclei on the slide, and then hybridize with 3H-labeled rRNA. Detection would be by autoradiography rather than scintillation counting. I had done a good deal of autoradiography, so that didn’t pose any problem, but the question was whether one could denature the DNA in a preparation without entirely losing the morphology. On several occasions during the late 1960s I made preparations of amphibian red blood cells and other tissues on microscope slides, dried them to attach the material tightly, denatured the preparation with alkali, incubated with 3H-labeled rRNA and covered with autoradiographic emulsion. I hoped to see silver grains above the nucleoli. But unfortunately I never saw anything, even after weeks of autoradiographic exposure. In retrospect, it became clear that the major problem was the very low specific activity of the 3H-labeled rRNA and, of course, the relatively small amount of rDNA in each diploid nucleus.
With the discovery of the amplified rDNA cap, however, I knew I had the perfect system for testing in situ hybridization. But I did not begin experiments immediately. Shortly after writing up my work on rDNA amplification and submitting it to the PNAS [12], I left for a sabbatical term in St. Andrews, Scotland. There I continued to work on rDNA amplification with my close friends and colleagues, Mick Callan and Herbert Macgregor. It was clear in retrospect that gene amplification had been seen by early investigators, not only in amphibians but also in many other organisms as far back as the turn of the century. I wanted to confirm one of the more spectacular examples, which occurred in oocytes of the water beetle Dytiscus. Fortunately, these beetles were abundant in a swampy area near St. Andrews. Herbert and I, along with graduate student Elizabeth Kidston, quickly confirmed the classical morphological account. We then showed by molecular experiments that a massive rDNA amplification occurs during early oogenesis of water beetles, more or less as in Xenopus [15].
Returning to Yale in the fall of 1968, I decided to give in situ hybridization one more go, this time using the amplified rDNA in oocytes as the test system. I made standard cytological squash preparations of ovaries from young Xenopus froglets. Because I was worried that the denaturation step would lead to extensive loss of DNA, I coated the preparations with a thin layer of agar before denaturing the DNA with a dilute solution of NaOH. Finally, the preparations were incubated with 3H-labeled rRNA and covered with autoradiographic emulsion. After a few weeks of exposure the slides were developed. Unfortunately, most of the very weak β-particles from the 3H decay could not reach the emulsion through the agar layer. But there was just enough signal above some of the oocyte nuclei to convince me that the method had worked.
About this time my student Mary Lou Pardue joined me on this project. We made rapid progress, in particular finding that the morphology of “good” squashes, i.e., very flat ones, was largely unchanged by alkali denaturation, even without protection by an agar coat. Now all cells on the slide were in close contact with the autoradiographic emulsion, and it was easy to distinguish labeled from unlabeled nuclei. Another important improvement was production of 3H-rRNA of very high specific activity, which we did by labeling cultured cells with large amounts of high specific activity 3H-uridine.
In December 1968 I attended an international symposium on “Nuclear Physiology and Differentiation” held in Belo Horizonte, Brazil. The manuscript I prepared for this symposium dealt largely with rDNA amplification. Nevertheless, the in situ hybridization experiments were quite conclusive by this time and I presented our results in my talk at the meeting and included images in the manuscript [16]. Mary Lou and I then wrote a more detailed paper on the technique, which was submitted to the PNAS in early 1969 [17]. The symposium volume took some time to appear in print, so it actually came out after the PNAS paper. The microscope slide from which the black and white images in the PNAS paper were taken is still in good condition. I have reimaged the same nuclei in color for this paper (Fig. 2).
Fig. 2.
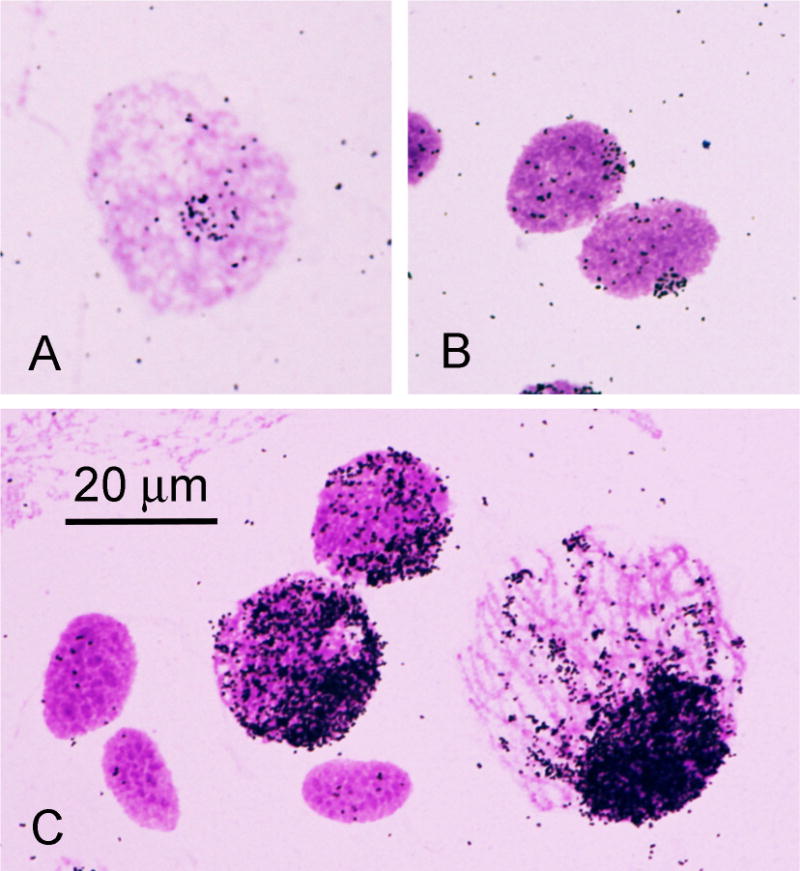
Autoradiographs of nuclei from a Xenopus ovary after in situ hybridization with 3H-labeled ribosomal RNA. A) Oogonial nucleus. B) Two leptotene nuclei. C) Three unlabeled follicle nuclei and three pachytene nuclei showing the progressive increase in amplified rDNA as the nucleus enlarges. Recent color images taken from one of the first in situ hybridization preparations made in 1968 by the author and Mary Lou Pardue, originally published as Fig. 1 in Pardue and Gall [17].
Ken Jones and colleagues in Edinburgh carried out in situ hybridization experiments on the amplified rDNA of Xenopus oocytes more or less identical to ours; they submitted their work [18] about the same time that we submitted our PNAS paper. Successful in situ hybridization was developed independently by Buongiorno-Nardelli and Amaldi in Rome using 3H-labeled rRNA on sections of paraffin-embedded Chinese hamster tissues [19].
Our experiments with the amplified rDNA of Xenopus were a clear “proof of principle,” but the extension of in situ hybridization to other systems posed two major problems: sensitivity and the availability of probes. Because rRNA genes occurred in large clusters in the genomes of several organisms and their sequence was highly conserved, it was reasonable to examine these genes in another system, using the same probe that worked in Xenopus. We chose the salivary gland chromosomes of Drosophila and two other flies, Sciara and Rhynchosciara, where we hoped that polytenization of the chromosomes would lead to even higher levels of rDNA. We were able to detect the rDNA in the salivary gland chromosomes and fortunately the morphology of bands and interbands survived the somewhat harsh conditions of denaturation and hybridization [20].
However, my ultimate hope of mapping specific genes to the bands of polytene chromosomes and to the loops of LBCs was not immediately possible, primarily because we did not have the necessary probes - gene cloning was still a few years in the future. Instead, I decided to use our new technique to shed light on another issue, the so-called “satellite” DNA of the mouse.
At that time a standard way of analyzing DNA was by buoyant density centrifugation in a CsCl gradient, the technique introduced by Meselson and Stahl in their famous demonstration of semi-conservative replication of DNA [21]. It had been shown that mouse DNA separated into two major fractions when centrifuged in CsCl. About 90% of the DNA had a mean buoyant density of 1.701, whereas about 10% was lighter with a density of 1.690 [22]. Molecular analysis indicated that this lighter “satellite” DNA consisted of about a million repeats of a relatively short sequence, some 300–400 nucleotides in length [23]. There were very few clues as to its significance. Mary Lou and I prepared radioactive mouse satellite DNA and hybridized it to cultured mouse cells [24]. The culture had been treated with colchicine to arrest cells in metaphase of mitosis. We were delighted when we saw that hybridization was limited to one end of each chromosome (Fig. 3), except for the Y chromosome, which was unlabeled. It was known that mouse chromosomes were “telocentric,” that is, their centromeres were at or near one end of each chromosome. Next to the centromere was a block of differentially staining material knows to cytologists as “heterochromatin” or more specifically as “centromeric heterochromatin.” Clearly, the satellite sequences were located in the heterochromatin. Because the heterochromatin made up about 10% of the chromosome length and the satellite about 10% of the total DNA, it seemed reasonable to conclude that the heterochromatin was made primarily of this simple-sequence DNA. Our preparations of mouse cells had been stained with Giemsa stain, a standard histological technique used to distinguish different types of blood cells. We noticed that the combination of alkali denaturation followed by Giemsa stain gave a sharp differentiation between the euchromatin and heterochromatin. By accident we had discovered a useful staining technique that is still used for chromosome analysis.
Fig. 3.
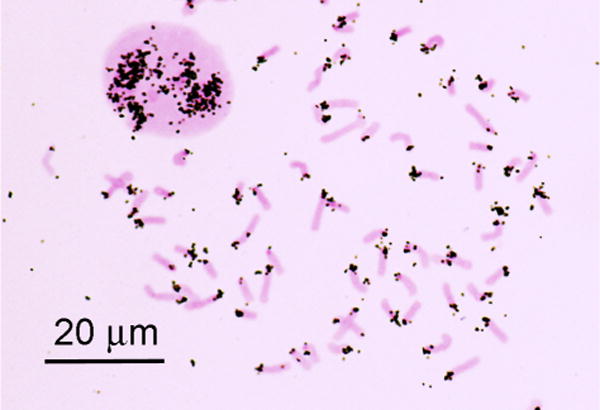
Autoradiograph of an interphase nucleus and mitotic chromosomes of the mouse after in situ hybridization with 3H-labeled RNA transcribed in vitro from mouse satellite DNA. Recent color image taken from a slide made in 1969 by the author and Mary Lou Pardue.
The experiments with mouse chromosomes immediately suggested that we should look at Drosophila, where the heterochromatin had been studied exhaustively by both cytologists and geneticists. From the earliest days of genetics, remarkably even before the rediscovery of the giant polytene chromosomes in the 1930s, it had been shown that almost none of the known genes were located in the heterochromatic regions next to the centromeres. But it was not known why these regions were gene poor. The experiments with the mouse satellite suggested a plausible explanation: these regions must consist of simple sequence DNA.
CsCl fractionation quickly showed that DNA from D. melanogaster, like that from the mouse, contained a satellite fraction of low buoyant density. Early cytological studies had shown that another species of Drosophila, D. virilis, had even more prominent segments of heterochromatin in its mitotic chromosomes. By CsCl centrifugation we found that roughly 40% of all D. virilis DNA consisted of satellite sequences. Satellite sequences from each species hybridized in situ to the heterochromatin of its own mitotic chromosomes. As with the mouse satellite DNA, these experiments demonstrated that gene-poor heterochromatic regions of the genome consisted largely of simple repetitive sequences [25]. An additional bit of information came from these experiments: satellite sequences were barely detectable in larval salivary glands by CsCl centrifugation, and they hybridized only weakly to the centromere regions in squashes of the polytene chromosomes. These molecular data confirmed what had been known for some time from cytology, that the heterochromatin was not replicated during formation of the giant polytene chromosomes in the salivary glands.
I am occasionally asked why we used the cumbersome technique of autoradiography for these first in situ hybridization experiments and indeed for all our experiments during the next few years. Fluorescent in situ hybridization (FISH) seems such a natural choice now, with its superior localization and ease of probe synthesis and detection. The answer is actually quite simple: there were no fluorescent nucleic acid probes available at that time. Even if there had been, fluorescence microscopy was still in its infancy and microscopes with epifluorescent illumination were not in general use. When David Ward and his student Pennina Langer showed me their first biotin-labeled FISH slides in the late 1970’s, I was still using the same awkward fluorescence microscope I had put together in the 1950s. They had purchased a commercial epifluorescence microscope that was considerably easier to use and much more efficient.
Fluorescent probes for in situ hybridization were introduced by three groups. The first method [26] involved an antibody against poly(rA)-poly(dT). An unlabeled 5S rRNA was hybridized to Drosophila polytene chromosomes, which were then reacted with the antibody. This antibody, in turn, was detected with a rhodamine-labeled secondary antibody, as in the Coons and Kaplan technique. The major drawback of this early FISH technique was the requirement for a high-titer antibody against poly(rA)-poly(dT).
A more generally applicable technique involved direct chemical labeling of the RNA (or DNA) molecule used in the hybridization reaction. Either the probe itself or a secondary probe was labeled with a fluorescent molecule. Success was obtained in the late 1970s and early 1980s with fluorochrome labels [27] and with a combination of biotin and avidin [28]. In these early experiments mouse satellite sequences and polytene chromosomes remained favorite test subjects because of the multiplicity of the target sequence (Figs. 4 and 5). Although I had no direct role in developing FISH probes, I closely followed the thesis work of Pennina Langer, a student with David Ward in the Human Genetics Department at Yale. Fig. 5 of polytene chromosomes from a Drosophila salivary gland is taken from her thesis, of which I still have a copy. The same figure was published in Langer-Safer et al. [28]. She also demonstrated hybridization of a repetitive satellite sequence to the RNA of a newt lampbrush chromosome preparation that I supplied. The result of that experiment is mentioned in her thesis and in her PNAS paper, but no image was published. Fig. 6 shows a FISH preparation made some time later using the same repetitive satellite sequence as probe.
Fig. 4.
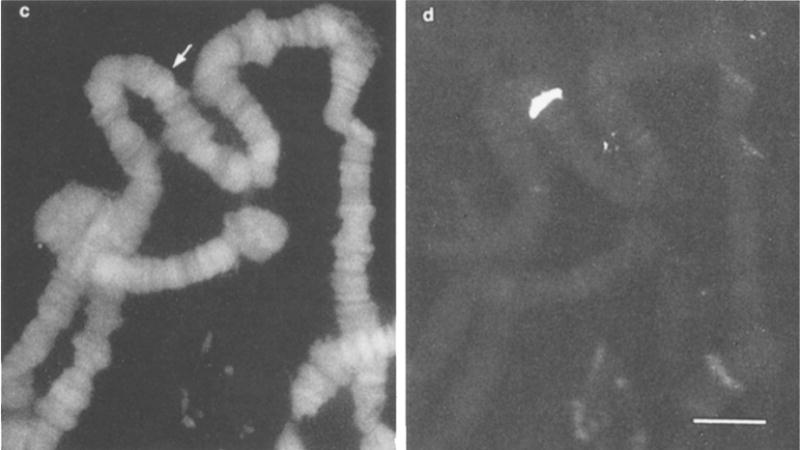
Polytene chromosomes of Drosophila hydei after in situ hybridization with a rhodamine-labeled 5S RNA probe. Panel c shows DAPI staining of all the chromosome bands (blue channel). Panel d shows specific staining of the single band that contains 5S DNA (red channel). Copy of figure 1 (c, d) from Bauman et al. [29].
Fig. 5.
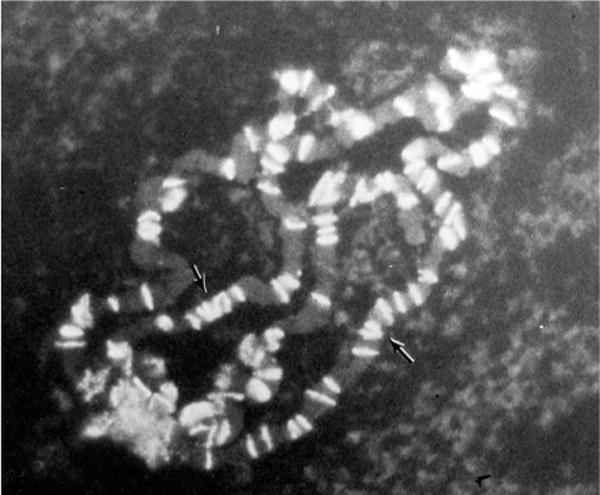
Polytene chromosomes of Drosophila melanogaster after in situ hybridization with a biotinylated probe against the roo transposable element (clone pAC104). Image copied from Pennina Langer’s thesis. The same image appears in Langer-Safer et al. [28].
Fig. 6.
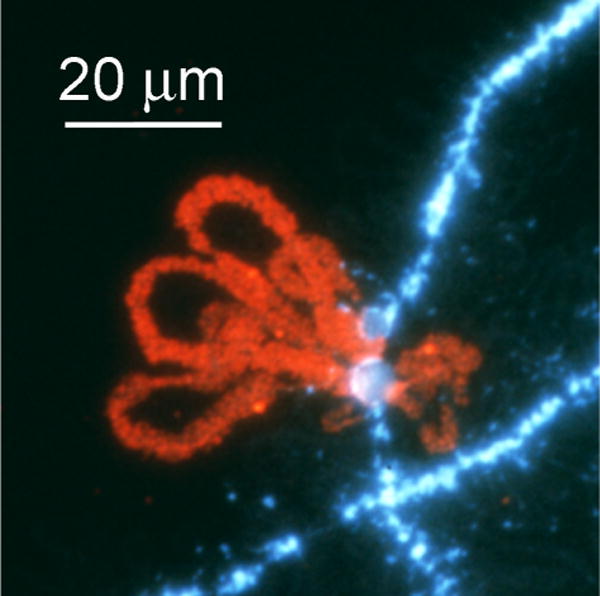
Lampbrush chromosome loops at the histone gene locus of the newt Notophthalmus viridescens, hybridized with a fluorescent probe against a 222 bp sequence. This sequence is repeated many times as a spacer between the multiple histone gene clusters. Image from Zheng’an Wu (Carnegie Institution for Science).
FISH rapidly became the technique of choice for demonstrating RNA and DNA molecules at the cellular and subcellular levels. Even so, for quite a while detection of specific RNA or DNA sequences required the presence of multiple copies in close proximity. For DNA this condition was met by polytene chromosomes and by multiple copies of a sequence in a localized region, as in heterochromatin or telomeres. For RNA, there were many cases where high concentrations of specific sequences occurred in the cytoplasm or even on the chromosomes. Eventually, improvements in probes increased the sensitivity of the technique many fold, so that today single molecule studies are routine. Other papers in this volume outline the steps in this history and the many uses that FISH now makes possible.
The first in situ hybridization in 1968 involved a 3H-labeled ribosomal RNA probe.
The first target of in situ hybridization was the amplified rDNA of frog oocytes.
Mouse and Drosophila satellite DNAs were localized to centromeric heterochromatin.
Fluorescent probes for in situ hybridization (FISH) were developed around 1980.
Acknowledgments
The author’s research is currently supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R01 GM33397. The content is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health. Earlier research of the author mentioned in this review was supported by various grants from the National Institutes of Health (National Cancer Institute and National Institute of General Medical Sciences) and from the American Cancer Society. JGG is American Cancer Society Professor of Developmental Genetics.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Callan HG. Lampbrush Chromosomes. Springer-Verlag; Berlin: 1986. [Google Scholar]
- 2.Duryee WR. Arch Exp Zellforsch. 1937;19:171–176. [Google Scholar]
- 3.Coons AH, Kaplan MH. J Exp Med. 1950;91:1–13. doi: 10.1084/jem.91.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wallace H, Birnstiel ML. Biochim Biophys Acta. 1966;114:296–310. doi: 10.1016/0005-2787(66)90311-x. [DOI] [PubMed] [Google Scholar]
- 5.Brown DD, Gurdon JB. Proc Natl Acad Sci USA. 1964;51:139–146. doi: 10.1073/pnas.51.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ritossa FM, Spiegelman S. Proc Natl Acad Sci USA. 1965;53:737–745. doi: 10.1073/pnas.53.4.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McClintock B. Zeitschrift Zellforschung. 1934;21:294–328. [Google Scholar]
- 8.Miller OL., Jr National Cancer Institute Monograph. 1966;23:53–66. [PubMed] [Google Scholar]
- 9.Gall JG. In: Harvey Lectures. Zabriskie J, editor. Academic Press; New York: 1978. pp. 55–70. [PubMed] [Google Scholar]
- 10.Painter TS, Taylor AN. Proc Natl Acad Sci USA. 1942;28:311–317. doi: 10.1073/pnas.28.8.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peacock WJ. National Cancer Institute Monograph. 1965;18:101–131. [PubMed] [Google Scholar]
- 12.Gall JG. Proc Natl Acad Sci USA. 1968;60:553–560. doi: 10.1073/pnas.60.2.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown DD, Dawid IB. Science. 1968;160:272–280. doi: 10.1126/science.160.3825.272. [DOI] [PubMed] [Google Scholar]
- 14.Gillespie D, Spiegelman S. J Mol Biol. 1965;12:829–842. doi: 10.1016/s0022-2836(65)80331-x. [DOI] [PubMed] [Google Scholar]
- 15.Gall JG, Macgregor HC, Kidston ME. Chromosoma. 1969;26:169–187. doi: 10.1007/BF00326453. [DOI] [PubMed] [Google Scholar]
- 16.Gall JG. Genetics. 1969;61(Suppl):121–132. [PubMed] [Google Scholar]
- 17.Gall JG, Pardue ML. Proc Natl Acad Sci USA. 1969;63:378–383. doi: 10.1073/pnas.63.2.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.John HA, Birnstiel ML, Jones KW. Nature. 1969;223:582–587. doi: 10.1038/223582a0. [DOI] [PubMed] [Google Scholar]
- 19.Buongiorno-Nardelli M, Amaldi F. Nature. 1970;225:946–948. doi: 10.1038/225946a0. [DOI] [PubMed] [Google Scholar]
- 20.Pardue ML, Gerbi SA, Eckhardt RA, Gall JG. Chromosoma. 1970;29:268–290. doi: 10.1007/BF00325943. [DOI] [PubMed] [Google Scholar]
- 21.Meselson M, Stahl FW. Proc Natl Acad Sci USA. 1958;44:671–682. doi: 10.1073/pnas.44.7.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kit S. J Mol Biol. 1961;3:711–716. doi: 10.1016/s0022-2836(61)80075-2. [DOI] [PubMed] [Google Scholar]
- 23.Waring M, Britten RJ. Science. 1966;154:791–794. doi: 10.1126/science.154.3750.791. [DOI] [PubMed] [Google Scholar]
- 24.Pardue ML, Gall JG. Science. 1970;168:1356–1358. doi: 10.1126/science.168.3937.1356. [DOI] [PubMed] [Google Scholar]
- 25.Gall JG, Cohen EH, Polan ML. Chromosoma. 1971;33:319–344. doi: 10.1007/BF00284948. [DOI] [PubMed] [Google Scholar]
- 26.Rudkin GT, Stollar BD. Nature. 1977;265:472–473. doi: 10.1038/265472a0. [DOI] [PubMed] [Google Scholar]
- 27.Bauman JGJ, Wiegant J, Borst P, van Duijn P. Exp Cell Res. 1980;128:485–490. doi: 10.1016/0014-4827(80)90087-7. [DOI] [PubMed] [Google Scholar]
- 28.Langer-Safer PR, Levine M, Ward DC. Proc Natl Acad Sci USA. 1982;79:4381–4385. doi: 10.1073/pnas.79.14.4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bauman JGJ, Wiegant J, Van Duijn P, Lubsen NH, Sondermeijer PJA, Hennig W, Kubli E. Chromosoma. 1981;84:1–18. doi: 10.1007/BF00293359. [DOI] [PubMed] [Google Scholar]