

Abstract
Release of Ca2+ ions from sarcoplasmic reticulum (SR) into myocyte cytoplasm and their binding to troponin C is the final signal form myocardial contraction. Synchronous contraction of ventricular myocytes is necessary for efficient cardiac pumping function. This requires both shuttling of Ca2+ between SR and cytoplasm in individual myocytes, and organ-level synchronization of this process by means of electrical coupling among ventricular myocytes. Abnormal Ca2+ release from SR causes arrhythmias in the setting of CPVT (catecholaminergic polymorphic ventricular tachycardia) and digoxin toxicity.
Recent optical mapping data indicate that abnormal Ca2+ handling causes arrhythmias in models of both repolarization impairment and profound bradycardia. The mechanisms involve dynamic spatial heterogeneity of myocardial Ca2+ handling preceding arrhythmia onset, cell-synchronous systolic secondary Ca2+ elevation (SSCE), as well as more complex abnormalities of intracellular Ca2+ handling detected by subcellular optical mapping in Langendorff-perfused hearts. The regional heterogeneities in Ca2+ handling cause action potential (AP) heterogeneities through sodium-calcium exchange (NCX) activation and eventually overwhelm electrical coupling of the tissue.
Divergent Ca2+ dynamics among different myocardial regions leads to temporal instability of AP duration and – on the patient level – in T wave lability. Although T-wave alternans has been linked to cardiac arrhythmias, non-alternans lability is observed in pre-clinical models of the long QT syndrome (LQTS) and CPVT, and in LQTS patients. Analysis of T wave lability may provide a real-time window on the abnormal Ca2+ dynamics causing specific arrhythmias such as Torsade de Pointes (TdP).
Keywords: Action Potential, AP; AP duration, APD; Triggered Activity, TA; Long QT syndrome, LQTS; Long QT type 2, LQT2; Systolic Secondary Ca2+ Rel ease, SSCR; Early Afterdepolarization, EAD; Delayed Afterdepolarization, DAD; Simultaneous mapping of Voltage and intracellular free Ca2+; Subcellular Voltage and Ca2+ imaging
BACKGROUND
From a molecular perspective, the force generated during cardiac contraction is developed by the interaction between actin and myosin proteins and cross-bridge cycling in thousands of sarcomeres in each cardiomyocyte. For the heart to maintain blood circulation, individual sarcomeres have to contract in a synchronized pattern. Loss of temporal organization of this process results in circulatory arrest, as exemplified during the early phase of ventricular fibrillation, when the contractility of individual cardiomyocytes is still preserved.
At the organ level, the contraction of each myocyte is timed by a wavefront of cell membrane depolarization propagating through the cardiac chambers. For each myocyte, this electrical depolarizing signal is converted to a chemical signal, namely an increase in free Ca2+ concentration in the cytoplasm. Ca2+ binding to troponin C allows for cross-bridge formation between actin and myosin molecules, causing force generation along the longitudinal axis of individual sarcomeres and myocytes. Ca2+ enters the cytoplasm through voltage-gated L-type Ca2+ channels within approximately 10 milliseconds of membrane depolarization, but most of the Ca2+ ions responsible for cardiac contraction (80–85%)1 are released from internal stores, called sarcoplasmic reticulum (SR) through a mechanism described as Calcium-Induced Calcium Release (CICR). In cardiac muscle, the initial Ca2+ influx occurring via L-type Ca2+ channels on the T-tubule membrane leads to allosteric opening of Ca2+-release channels or cardiac ryanodine receptors (RyR2), located on the SR membrane across a gap of 10–15 nm between the T-tubule membrane and the terminal cisternae membrane of the SR.2 The RyR2 opening substantially amplifies the initial Ca2+ rise. After a contraction associated with elevation of cytoplasmic Ca2+, Ca2+ is removed from the cytoplasm to allow for myocardial relaxation and filling of cardiac chambers. Ca2+ released via RyR2 is transported back in the lumen of the SR against a concentration gradient by the cardiac Sarco (Endo) plasmic Reticulum Ca ATPase (SERCA2), a Ca2+ pump powered by ATP hydrolysis. Ca2+ influx from the extracellular via Ca2+ channels is returned to the extracellular space by the NCX1 countertransporter, a Na+/Ca2+ exchanger that exchanges 3 Na+ ions for each Ca2+ and is driven by the Na+ electrochemical gradient. Under steady state conditions, Ca2+ influx equals Ca2+ efflux in each subcellular compartment.3
Disturbances of electrical synchronization of cardiac contractions occur in many cardiac diseases and often lead to sudden cardiac death, a dramatic event which is usually caused by ventricular tachycardia (VT) or ventricular fibrillation (VF).4 A detailed understanding of the mechanisms triggering ventricular arrhythmias remains elusive. One reason for this is that ventricular arrhythmias can be produced by processes operating at different levels, ranging from point mutations in genes coding for ion channels,5, 6 which can cause repetitive firing in individual cells,7, 8 to reentry (continuous impulse propagation along a circular pathway), which requires spatially extended tissue and can occur around an anatomical obstacle such as a scar.9, 10
CALCIUM HANDLING AND TRIGGERED ACTIVITY
Triggered activity refers to abnormal generation of an AP, which is initiated (“triggered”) by the preceding (normal or abnormal) AP. In contrast to reentry, it does not require spatially extended tissue and can be observed in single cardiomyocytes. Most clinically relevant arrhythmias may involve more than one mechanism – reentry around a scar initiated by a triggered ectopic beat being an obvious example. However, study of arrhythmia models involving mostly triggered activity offer important insights into arrhythmogenesis in general.
Classically, triggered APs are categorized as either delayed afterdepolarizations (DADs) or early afterdepolarizations (EADs). EADs can be triggered during long ventricular AP durations (APDs) or during short atrial APDs and large amplitude Ca2+ transients as may occur in atrial fibrillation triggered by sympathovagal activity.11–14 DADs occur after completion of the preceding AP (i.e. the takeoff of the ectopic AP occurs from fully repolarized membrane potential values). EADs by definition occur before the completion of the preceding AP, from the plateau or repolarization phase. DADs and EADs are believed to occur in somewhat different settings. DADs typically develop in the setting of tachycardia or high extracellular Ca2+ concentration. Clinically, they have been linked to toxic levels of cardiac glycosides, adrenergic stimulation or catecholaminergic polymorphic ventricular tachycardia (CPVT). CPVT is a congenital arrhythmic syndrome associated with sudden cardiac death caused by mutations that destabilize Ca2+ release from the SR.15–19 The dominant factor in genesis of EADs is prolonged AP duration, along with bradycardia and adrenergic stimulation. EADs occur in a wide variety of clinical settings, including hypokalemia, hypocalcemia, toxicity of multiple cardiac and non-cardiac medications, and occasionally in patients with congenital long QT syndrome, a group of hereditary arrhythmic conditions defined by delayed ventricular repolarization. The typical clinical manifestation of EAD activity is a characteristic polymorphic ventricular tachycardia labeled torsade des pointes (TdP).20–22
The cellular mechanism of DAD genesis appears to be well understood. The conditions listed above in association with DADs all result in spontaneous SR Ca2+ release during diastole, which is not triggered by membrane depolarization. This spontaneous SR Ca2+ release is caused by an unusually high Ca2+ concentration in the junctional SR (jSR), in the case of digoxin toxicity, short cycle length or elevated extracellular Ca2+ which all promote RyR2 opening in the absence of a voltage trigger. This mechanism has been labeled Store-Overload Induced Calcium Release (SOICR).23 In many cases of CPVT, the threshold for SOICR is decreased because of mutation in the gene encoding the RyR itself, or in a gene encoding an interacting protein such as calsequestrin, allowing DAD formation despite normal or even low jSR Ca2+ load.17
The initial SOICR in cardiac myocytes is often fairly localized and propagates through the myocyte cytoplasm as a Ca2+ wave: Ca2+ ions diffuse from the region of elevated concentration (wave “crest”) into surrounding regions, triggering opening of neighboring RyRs in the direction of wave propagation through CICR. The regions in the “wake” of the wave are refractory with respect to Ca2+ release because of depletion of junctional SR Ca2+ or intrinsic refractoriness of RyRs following the previous Ca2+ release.24, 25
The Ca2+ released into cytoplasm that produce a Ca2+ wave is transported in part into the extracellular space by NCX1. Since this molecule transports one Ca2+ out of the cytoplasm in exchange for 3 Na+ ions into the cytoplasm in each transport cycle, its activity depolarizes cell membrane. In an isolated cell, AP is triggered if the threshold for activation of voltage-gated cardiac Na+ channels is reached. The question remains of how can Ca2+ waves occurring randomly in individual myocytes overcome the electrical load of the surrounding tissue to trigger a propagated AP? The question has been recently addressed by the elegant experiments of Wasserstrom et al.,16, 26 who used confocal microscopy of perfused rat hearts to study the mechanism of synchronization of Ca2+ waves. In their model of hypercalcemia and rapid pacing followed by a pause, both the coupling interval and timing dispersion of the Ca2+ wave appearance decreases with increasing degree of Ca2+ overload, eventually resulting in sufficient temporal overlap of depolarizations in individual myocytes to trigger tissue-wide depolarization.
Abnormal Ca2+ handling has been implicated in arrhythmias in acute myocardial ischemia,27 which results in marked AP shortening and CaT prolongation (setting the stage for diastolic depolarization by NCX current). It also plays a role in arrhythmogenesis during reperfusion, heart failure28 and atrial fibrillation. For example, simultaneous adrenergic and cholinergic stimulation of pulmonary vein cuffs causes triggered activity corresponding to EADs, which can be abolished by ryanodine administration, indicating role of intracellular Ca2+ handling. Interestingly, AP shortening rather than prolongation correlates with arrhythmia in this model.29, 30 Detailed discussion of these important topics is outside the scope of this review.
In contrast to DADs, the cellular mechanism of EAD generation remains controversial, although the involvement of Ca2+ handling is not disputed. The prevailing theory suggests that repolarization delay prolongs the time period which the L-type Ca2+ channels spend in the “window current” voltage range, when neither the activation nor the inactivation voltage gate of the channel is fully closed. The resulting increase in the inward current carried by L-type Ca2+ channels depolarize the membrane, further opening the activation gate and triggering the EAD. This scenario is supported by some mathematical models,31 and is consistent with experiments demonstrating that L-type channel opener promotes EAD formation, and L-type channel blockers suppress it.32 De Ferrari et al.33 directly observed Ca2+ release in isolated myocytes using a Ca2+-sensitive dye and found that Ca2+ waves are associated with DADs, but that that the Ca2+ rise related to EAD occurs relatively synchronously across the cell, a finding more consistent with the L-type channel window current theory than with ‘spontaneous’ SR Ca2+ release. Marban et al.34 reported that ryanodine (an alkaloid blocking RyR, the SR Ca2+ channel) failed to suppress EADs in ferret papillary muscle superfused with cesium, but that EADs could be suppressed by an L-type channel blocker.
On the other hand, Volders et al.,35 argued that spontaneous (non-voltage-triggered) SR Ca2+ release may be the cause of EADs, based on the observation that the beginning of cell contraction precedes EAD takeoff in isolated myocytes. According to their hypothesis, repolarization delay results in SR Ca2+ overload, and secondary SR Ca2+ release triggers an EAD by activating NCX, i.e. through a mechanism similar to DAD generation.11 The report by Milberg et al36 that NCX block suppresses EADs in a rabbit model of long QT syndrome supports this mechanism.
DUAL OPTICAL MAPPING DATA
The dual wavelength optical mapping technique has been used by our team and others to study the mechanisms of arrhythmogenesis in the setting of repolarization delay. This technique allows simultaneous recording of Vm and Cai signals with excellent spatial and temporal resolution. Moreover, it can be naturally applied to study perfused heart at physiological temperature, and can thus provide insights that cannot be readily obtained from isolated cell experiments.1
Simultaneous application of bradycardia, hypokalemia, hypomagnesemia, and block of IKr (the rapid component of the delayed rectifying K+ current) results in quick and reproducible TdP induction in a well-established rabbit heart model.37, 38 All these factors are known to promote TdP in humans.
The analysis of Vm and Cai signals simultaneously recorded during TdP using phase-plots suggested that at the site of EAD focus, that the rise of Cai rise precedes that of Vm, supporting the role of SR Ca2+ release as the driver of the arrhythmia.39 More detailed results were obtained by evaluation of Vm and Cai dynamics during transition from slow paced rhythm (after acute AV node destruction) to onset of TdP (caused by pharmacological IKr blockade).40 Invariably, gradual prolongation of AP duration was accompanied by change in Ca2+ dynamics, which preceded onset of EADs by minutes (Figure 1). Before the IKr block, the course of the Ca2+ transient (CaT) closely follows the course of AP. After IKr block, the APD prolongs markedly, but initially without the appearance of EADs. At this stage, CaT does not follow the AP by developing a smooth long plateau, but instead falls nearly as fast as it did before IKr block and then rises a second time during phase 2 or 3 of the AP, forming a second CaT peak which returns to baseline approximately at the same time as AP. The amplitude of this systolic secondary calcium elevation (SSCE) increases gradually in the span of a few minutes and eventually, electrical instabilities during the AP plateau develop to EADs. EADs are approximately synchronous with SSCEs, but are delayed by tens of milliseconds. The onset of SSCEs always precedes the onset of EAD at the earliest site of a propagated EAD.41 As the ectopic beat propagates away from the focus, AP upstroke overtakes the CaT upstroke, and the AP upstroke precedes the CaT upstroke as expected during the propagation of a normal AP (Figure 2). With further APD prolongation, the single SSCE is followed by multiple Ca2+ oscillations during the same AP plateau and each Cai peak tends to be accompanied by its own EAD.41 Finally, long AP plateaus with oscillations of both Vm and CaT are observed, which correspond to TdP runs on EKG tracing. Treatment with ryanodine and thapsigargin (SERCA inhibitor) prevented appearance of Ca oscillations and TdP following IKr block, although AP prolongation still developed. Treatment with K201 (1 μM to avoid off-target effects), a RyR stabilizer, had a similar effect.42 Taken together, these results provide compelling evidence that in this animal model, spontaneous systolic SR Ca2+ release, manifested as a SSCE, cause EADs. The observation that L-type Ca2+ channel blocker and low extracellular Ca2+ also prevent EAD appearance is not surprising, since both interventions are expected to deplete SR Ca2+ load.
Figure 1.
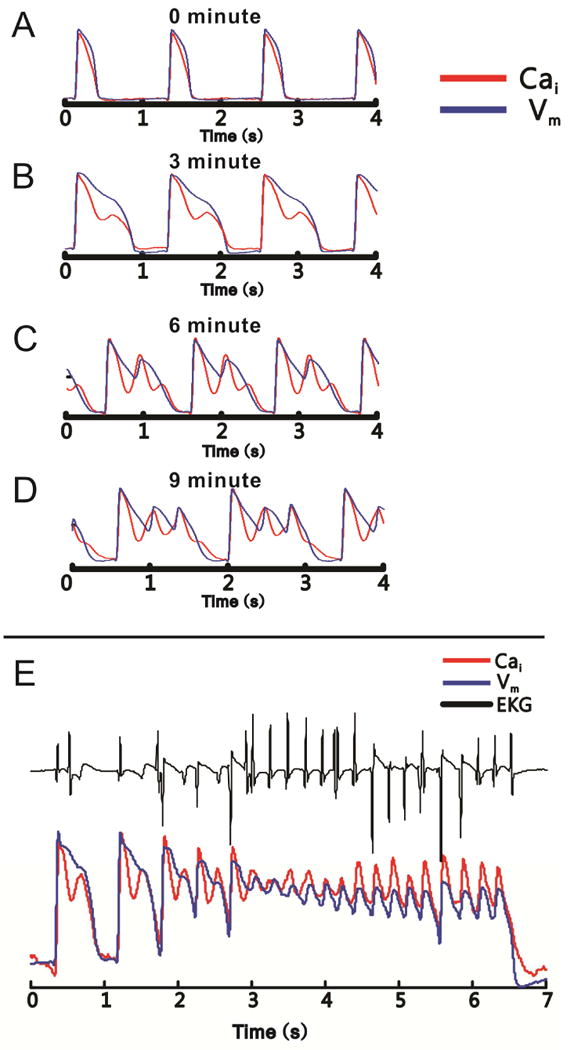
Development of SSCEs precedes the onset of TdP. Simultaneous recordings of membrane voltage (blue) and Ca2+ signals (red) are shown in all panels. A: During slow pacing before IKr block, the AP and CaT waveforms are monophasic and similar. B: After IKr block, but before the development of EADs, AP prolongation is accompanied by complex, biphasic CaT, with an SSCE following the initial rise of the CaT. C: With increasingly more robust IKr block, the amplitude of the SSCE increases and a corresponding EAD develops. Note that the rise of SSCEs precedes the EAD upstroke, and that a second SSCE of smaller amplitude develops during AP downstroke. D: Eventually, the SSCE increases in amplitude and develops its own EAD. E: Finally, long plateaus with superimposed oscillations of both membrane voltage and Ca2+ signal occur. They correspond to runs of polymorphic ventricular tachycardia on EKG (black). Adapted from41
Figure 2.
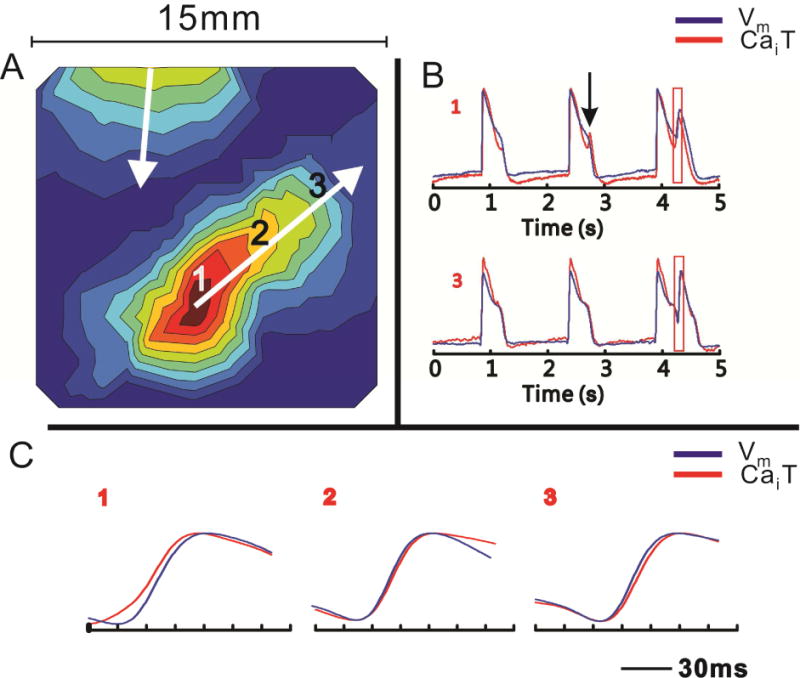
SSCE precedes EAD at the site of origin of the corresponding ectopic beat. A: An activation map of a propagated ectopic beat with an origin at site 1; the depolarization wavefront proceeds from red to blue regions. B: Simultaneous CaT and AP signals from the beat focus (site 1) and a distant site (site 3). Secondary CaT upstroke precedes secondary AP upstroke at site 1, but trails it at site 3. Note the tiny SSCE is not followed by an EAD on the seconds beat at site 1 (vertical arrow). C: Similar to panel B, but at displayed with better time resolution. Note the shift in relative positions of the CaT and AP upstrokes between sites 1 and 3. Adapted from41
SPATIAL HETEOGENEITY OF CALCIUM TRANSIENT
When the EAD model described above is studied at better spatial resolution (100×100 pixels, 150 μm per pixel), a striking spatial heterogeneity of the normalized CaT signal is easily appreciated.42 This is largely absent at baseline, when spatial the amplitude of both CaT and AP is relatively even across the epicardial surface at all times, but increases dramatically with the appearance of Ca oscillations. Intriguingly, the areas of high calcium oscillation amplitude are highly irregular, do not appear to follow obvious anatomic regions and differ among different experiments, although they generally remain relatively stable between subsequent beats. In fact, regions of elevated Cai as small as 1 mm can be resolved (Figure 3). Although the amplitudes of the Cai and Vm are well correlated and the spatial heterogeneity of Vm increases along with Cai, Vm heterogeneity increase is less pronounced and Vm distribution over the epicardial surface is smoother than that of Cai (Figure 4). This would be expected if Cai heterogeneity drives the process, since Cai diffusion is much slower than charge diffusion. Still, the spatial heterogeneity of Vm leads to appearance of steep spatial voltage gradients, which would be expected to generate electronic currents through the tissue. Such currents would depolarize a portion of cell membrane and could trigger propagated action potential. This seems to be an important arrhythmogenic mechanism, since all the propagated ectopic beats where the focus could be localized originated from regions of steep voltage gradients.
Figure 3.
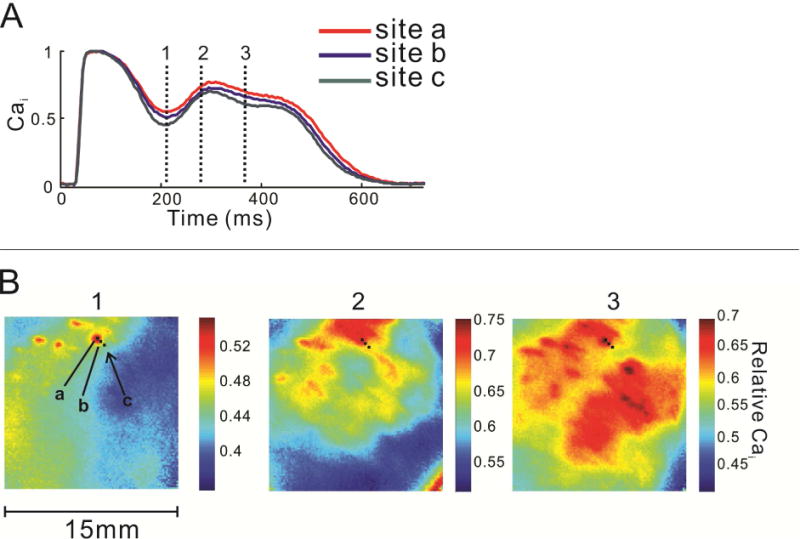
Spatial heterogeneity of CaT in LQTS prior to development of ectopic beats is demonstrated by 3 consecutive images of the anterior epicardial surface during a single paced heartbeat. A: Superimposed normalized CaTs from 3 pixels (a–c) indicate differences in Ca2+ concentration among closely spaced ventricular sites. B: The timing of the 3 images is indicated by the vertical lines in panel A. Note the highly irregular, non-contiguous “islands” of Ca2+ elevation which shift during the CaT. The color scale changes to facilitate visualization of the spatial heterogeneity. Adapted from42
Figure 4.
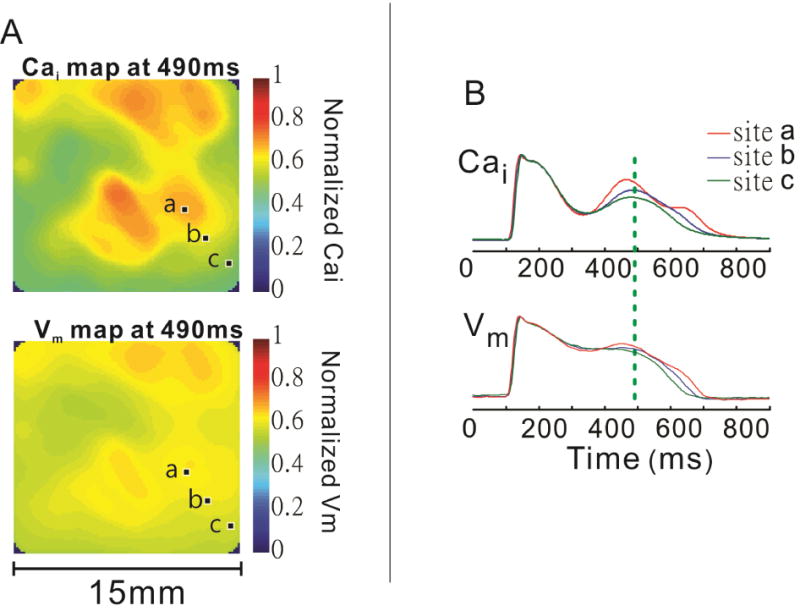
Comparison of spatial heterogeneity of Ca2+ and membrane voltage signals during paced rhythm and IKr block. A: Simultaneous images of Ca2+ (top) and membrane voltage (bottom) signals from the anterior epicardial surface at the time of SSCE. Areas of elevated Ca2+ correspond to more positive membrane potentials, but the voltage map is much smoother or homogeneous than the Ca2+ map. B: Superimposed Ca2+ and membrane voltage signals from 3 closely spaced pixels (a–c) indicated in the previous panel. The vertical line marks the timing of the images in A. Adapted from42
SEX-DIFFERENCES EXPLAIN SPATIAL HETEOGENEITIES OF CaTs and LOCATION OF EADs
An examination of the distribution of the sites that fired the first EADs produced a striking pattern. EADs could not be associated with specific anatomical features but were initiated around the base of the adult female rabbit heart.43 In contrast, the same LQT model failed to elicit EADs and TdP in adult male rabbit hearts. This sex difference in arrhythmia phenotype was reversed in pre-pubertal rabbits where IKr blockade elicited EADs and TdP in pre-pubertal males but not females.43, 44 Sex-differences in the vulnerability to TdP have long been acknowledged in humans for both congenital and drug-induced LQT type 2 (LQT2).45 Young adult women (pre-menopausal) are at greater risk of TdP compared to their male counter-part, but males under the age of 14 years old have a considerably greater risk of lethal arrhythmias in LQT2).45 These sex differences and the switch between pre-puberty and adulthood has been attributed to the surge of testosterone and estrogen.46
A comparison of the arrhythmia risk in LQTS was recently reviewed for various species (mouse, guinea pig, rat, rabbit and dog) and the New Zealand White rabbits most closely parallel human sex-differences in arrhythmia risk in adult and pre-puberty.46 Based on extensive studies in man and rabbit,46 the general consensus is that in LQT2, testosterone is protective of TdP and estrogen greatly enhances the risk of TdP. Ventricular myocytes were isolated from various regions (endo-epi, base and apex) of adult rabbit hearts and incubated with or without estrogen (1 nM) for 0 to 3 days. Estrogen treated myocytes from the base of the epicardium expressed increasing current densities of ICa,L and INCX as well as increasing levels of their mRNA and channel proteins (Cav1.2α and NCX1).47, 48 This genomic upregulation occurred during the first 2-days of estrogen treatment and only in female base myocytes isolated from the epicardium and not in male myocytes. Myocytes isolated from other regions of the ventricles did not respond to estrogen treatment and failed to exhibit EADs after IKr block.47, 48 In Langendorff rabbit hearts under LQT2 conditions, EADs were initiated near the base of the heart and from the epicardium because cryoablation of the ventricular chambers with liquid N2 did not alter the incidence of EADs.39 In these cryoablated hearts, only a 1 mm thick layer of epicardium survived which was the source of all EADs and provided compelling evidence that EADs did not originate from the conduction system, the endocardium or mid-myocardium.39 These measurements are consistent with the localization of the estrogen genomic upregulation of ICa,L and INCX. The modulation of cardiac channel by estrogen is localized to the base of the epicardium in female hearts which could be due to regional expression of the two estrogen receptors, ERα and ERβ. However, Western blot analysis from different regions of male and female rabbit hearts revealed a uniform estrogen receptor expression (ERα and ERβ) in hearts from both sexes and ERα and ERβ could not explain regional these heterogeneities.47 Table 1 summarizes sex and regional heterogeneities of ICa,L and INCX densities in rabbit hearts based on voltage clamp experiments. Differences in the transient outward current, It.o,. the rapid and slow delayed K+ rectifying currents, IKr and IKs and the inward rectifying K+ current, IK1 were compared between male and female rabbits but the regional heterogeneities have not been reported.
Table 1.
Sex differences in Ion Channel Expression in New Zealand White Rabbits
| Ionic current | ICa,L | INCX | It.o. | IKr | IKs | IK1 |
|---|---|---|---|---|---|---|
| Male Epicardium Base | ↔ | ↔ | M=F | M>F | M>F | M>F |
| Male Epicardium Apex | ↔ | ↔ | ||||
| Male Endocardium | ↔ | ↔ | ||||
| Female Epicardium Base | ↑ | ↑↑ | ||||
| Female Epicardium Apex | ↔ | ↔ | ||||
| Female Endocardium | ↔ | ↔ | ||||
| Pre-pubertal Male Epicardium Base | ↑ | ↑↑ | ND | ND | ND | ND |
| Pre-pubertal Male Epicardium Apex | ↔ | ↔ | ND | ND | ND | ND |
| Pre-pubertal Male Endocardium | ↔ | ↔ | ND | ND | ND | ND |
| Pre-pubertal Female Epicardium Base | ↔ | ↔ | ND | ND | ND | ND |
| Pre-pubertal Female Epicardium Apex | ↔ | ↔ | ND | ND | ND | ND |
| Pre-pubertal Female Endocardium | ↔ | ↔ | ND | ND | ND | ND |
Summary of sex and regional heterogeneities of ionic currents
Voltage-clamp data on sex and regional differences in L-type Ca2+ current density and Na-Ca exchange current density (ICa.L and INCX) are compiled from43, 47, 48 and preliminary data. Sex differences in IKr, It.o and IK1 currents are compiled from Liu et al., J. Pharmacology and Experimental Therapeutics, 199878 and IKs from Zhu et al., Pflugers Archives, 2013.79 Each arrow, ↑ represent ~30% increase in current density compared to other regions of the same heart and of the opposite sex.
At the molecular level, estrogen treatment (17β-estradiol at 0.3–1.0 nM) promotes the induction of EADs by upregulating the L-type Ca2+ current and the NCX current (ICa,L and INCX) by a genomic mechanism but only at the base of the female rabbit epicardium. In adult female rabbit hearts, mRNA, protein (Cav1.2α) and current density of ICa,L were ~ 30% greater at the base of the epicardium than the apex or endocardium or adult male myocytes.43 Treatment of freshly isolated female base, epicardial myocytes with an IKr blocker leads to cellular EADs when paced at long cycle lengths (1–2 s) but rarely elicited EADs in myocytes derived from other regions of the heart.43 Taken together, the female sex is more vulnerable to LQT2-related TdP because of estrogen upregulation of Ca2+ influx (ICa,L) and Ca2+ efflux (INCX) mechanisms which more readily results in Ca2+ overload, EADs and TdP under conditions of delayed repolarization.
SUBCELLULAR Ca2+ DYNAMICS
Clearly, study of subcellular Ca2+ dynamics underlying EAD generation in the beating heart may fundamentally improve the understanding of the arrhythmogenic mechanisms. If SR Ca2+ release is indeed responsible for EADs, does it occur in the form of propagated Ca2+ waves as observed in the DAD models?
The subcellular Ca2+ dynamics in the beating heart has been studied with confocal microscopy by several research teams.16, 49–51 The advantage of confocal microscopy is its high z-axis resolution which comes at the price of low light levels due to confocal apertures, slow scan rates which limit these studies to line scans and the inability to correlate CaT events in different regions of a myocyte let alone adjacent myocytes. With conventional fluorescence microscopy and a high aperture, water-immersion 40X objective, allows excellent spatial and temporal resolution, with a z-axis resolution considerably smaller than the diameter of ventricular myocytes. The movie files from our subcellular experiments have spatial resolution of 1.5 μm per pixel (100×100 pixels) and a frame rate of 200 to 500 frames per second. Continuous perfusion with blebbistatin and custom-designed perfusion chamber were used to diminish contraction artifacts.
The subcellular optical recordings show that the secondary systolic Ca2+ oscillations corresponding to EADs occur synchronously throughout myocyte cytoplasm, in agreement with the report by De Ferrari et al.,33 in isolated cells. This contrasted with propagated diastolic Ca2+ waves, which were clearly observed in many of the same experiments (Figure 5).
Figure 5.

Contrasting subcellular patterns of systolic and diastolic Ca2+ elevations in LQTS. A: Superimposed CaT tracings from 2 pixels of in the same myocyte. The SSCE occurs nearly simultaneously, and there is a low level of spatial heterogeneity of Ca2+ signal. B: In contrast, a wave of diastolic Ca2+ elevation propagates from top right to bottom left of the images, and can be easily seen in the left and middle image. Adapted from42
Other, previously unreported forms of subcellular Ca2+ dynamics were also observed. These included brief diastolic Ca2+ elevations that did not propagate through the cell, low-amplitude Ca2+ oscillations (“ripples”) that occurred in on top of long CaT plateaus and in contrast to the typical Ca2+ oscillation, were not synchronous across the cell. On rare occasions, chaotic intracellular Ca2+ dynamics was observed, with multiple simultaneous wavefronts propagating through the cytoplasm of individual cell in various directions, with continuous collision, annihilation and generation of new wavefronts (Figure 6), with an overall pattern superficially resembling depolarization wavefront propagation through the ventricular myocardium during VF. These spatially dyssynchronous modes of subcellular of Ca2+ dynamics cannot be explained by activation of sarcolemmal Ca2+ channels.
Figure 6.
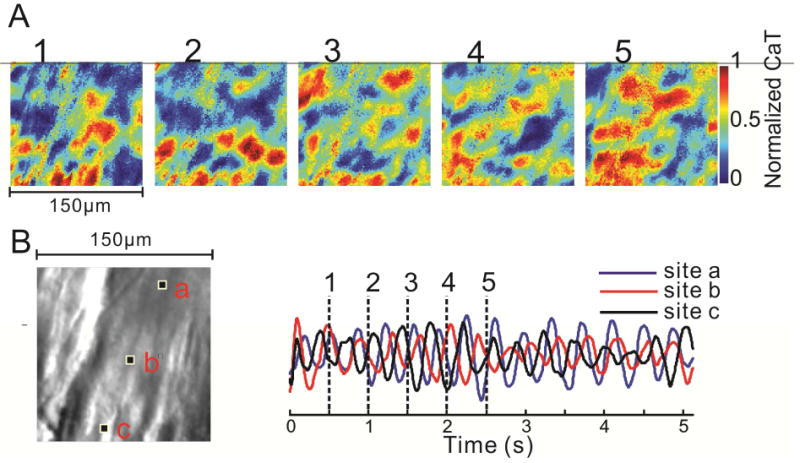
An example of disorganized subcellular Ca2+ dynamics. A: Five consecutive subcellular Cai images approximately 500 ms apart illustrate the presence of multiple intracellular waves. These waves may terminate with collisions, but new waves continue to arise and a stable state of low Ca2+ concentration is never achieved. B: Superposition of 3 Cai signal tracings (right) from the 3 pixels marked in the microscopic image (left). Vertical lines indicate the timing of the images in A. Adapted from42
Overall, these results indicate that SSCE is a necessary precondition for EADs in our rabbit model, and the reactivation of L-type Ca2+current contributes to EADs after the initial depolarization caused by INCX.
DISCUSSION
The results of the optical mapping experiments described above lead us to suspect that the mechanisms of EAD generation share many similarities with DAD arrhythmogenesis, namely cellular Ca2+ overload, SOICR and depolarization mediated by NCX activity. In contrast to situations associated with DADs, repolarization delay has not been firmly linked to SR overload. However, there are good reasons to believe that repolarization delay increases SR Ca2+ load, since the influx of Ca2+ into the cell during each AP increases compared to baseline because the incomplete inactivation of the L-type Ca2+ channels due to the component of voltage-dependent inactivation during prolonged depolarization.52 At the same time, the efflux of Ca2+ out of the cell by NCX is attenuated because of decreased driving force during the long AP plateau. During long APs, SERCA continues to pump excess of Ca2+ ions in the lumen of the SR. Ca2+ transported in the SR increases with prolonged APD. After the initial Ca2+ release from the junctional SR (jSR) caused by the normal voltage-triggered CICR, the jSR is gradually replenished because of finite speed of SERCA transport and the diffusion limitation between non-junctional SR (where most SERCA pumps are located) and jSR – the site of RyR and Ca2+ release. We believe that in LQTS, jSR is replenished to a point which triggers SOICR during the prolonged AP plateau, causing the calcium oscillation (Figure 7). The rise of Ca2+ concentration in the cytoplasm then activates NCX. The resulting membrane depolarization triggers EAD by increasing L-type open channel probability. It is likely that in electrically connected tissue, the electrotonic currents related to Vm gradients contribute to this final step. The often invoked argument that NCX cannot play a major role in EAD triggering because EAD takeoff voltage is relatively close to NCX equilibrium potential may not be valid, since the this potential depends on cytoplasmic Ca2+ concentration and moves to more positive values when cytoplasmic Ca2+ increases, as is likely to be the case in our EAD model.
Figure 7.
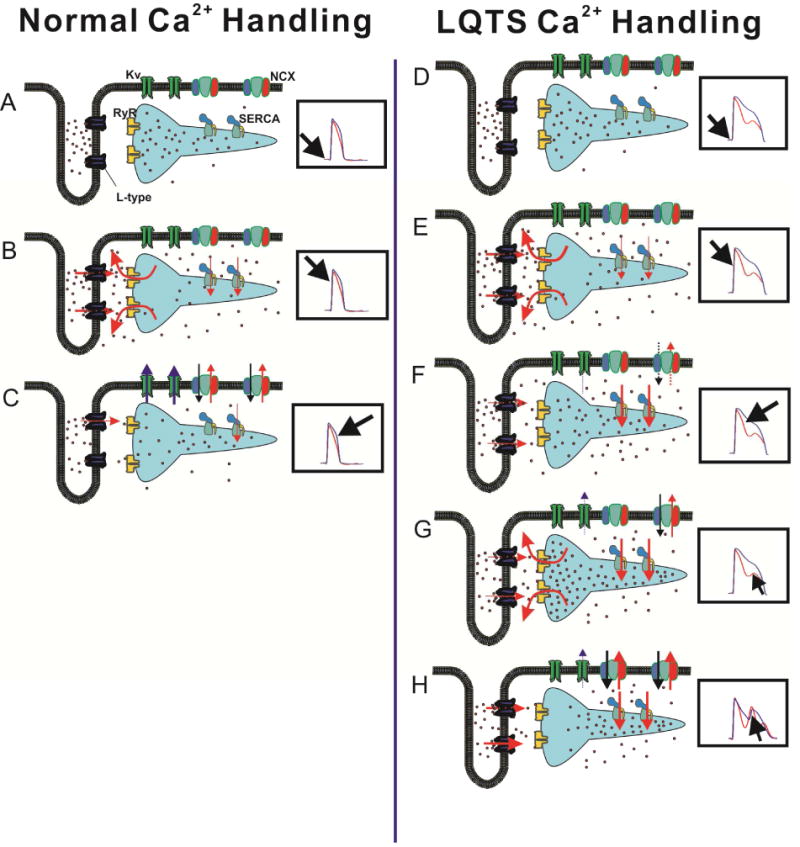
Proposed mechanism of Ca2+ handling underlying EAD generation in repolarization delay (panels D–H); normal Ca2+ handling is shown in panels (A–C). Each panel shows a cartoon of T-tubule and SR cisterna, with selected Ca2+ handling molecules (labeled in A; Ca2+ ions are schematized as red dots). The corresponding phase of cardiac cycle is indicated in the insets, which show simultaneous membrane voltage signals in blue and Ca2+ signals in red. A In diastole, Ca2+ concentration in cytoplasm is low, RyRs are closed and Ca2+ fills both junctional and nonjunctional SR. B Depolarization of sarcolemma opens L-type Ca2+, which in turn open RyRs through CICR mechanism, leading to emptying of junctional SR and increase in cytoplasmic Ca2+. C During AP phase 3, opening of K+ channels repolarizes cell membrane, resulting in gradual closure of L-type channel. RyRs have closed, and SERCA and NCX transport Ca2+ back to SR and extracellular fluid. D and E In repolarization delay, CICR follows AP upstroke and opening of L-type Ca2+ channels, similar to A and B. F During the prolonged plateau phase, deactivation of L-type Ca2+ channels is delayed, and inactivation is incomplete, leading to persistent Ca2+ influx into cytoplasm. NCX activity is attenuated because of decreased driving force at depolarized membrane potentials. This indirectly accentuates the SERCA-mediated transport of Ca2+ onto SR compartment. G Diffusion of Ca2+ ions from the nonjunctional SR eventually increases junctional SR load to a level sufficient to cause SOICR; this occurs before CaT and membrane potential return to diastolic levels. H The cell-wide Ca2+ release results in increase of inward current carried by NCX, depolarizing sarcolemma and reactivating L-type Ca2+ channels, i.e. triggering EAD.
Several questions about this process remain to be answered. One of them is the reason for the difference between the regime of SR Ca2+ release underlying DADs (propagated Ca2+ waves) and EADs (cell-synchronous release). This difference could be caused by the relatively high cytoplasmic Ca2+ concentration at the time of SSCE takeoff. The RyR2 channels can be opened either by Ca2+ binding in a cytoplasmic site (possibly formed by the EF hand motif at the central domain of the cytoplasmic portion of the channel),53, 54 or by the Ca2+ binding to the luminal site at the S6 transmembrane domain of the channel.55, 56 Although Ca2+ binding to either site can open RyR and trigger Ca2+ release, they are likely to interact, i.e. increased Ca2+ concentration in the cytoplasm decreases the threshold for SOICR. One could then expect that during diastole, when cytoplasmic Ca2+ is low, the SOICR threshold might be only reached at one or a few sites, resulting in a Ca2+ propagating relatively slowly from that focus. Prior to systolic Ca2+ oscillation, the cytoplasmic Ca2+ is much higher and SOICR threshold might be substantially lower and may thus can be simultaneously reached at many junctional sites at approximately the same time.
Another puzzling observation is the unexpected degree of spatial heterogeneity in the amplitude of SSCE, with discontinuous, irregular and initially found in small “islands” of SSCE. Pre-existing differences in density of ion channels or expression of Ca2+-handling proteins most likely contribute these local effects. The findings of Sims et al., provide compelling evidence that only cells from specific locations on the heart exhibit EADs after IKr block.43 Moreover, these cells prone to EADs are characterized by their higher levels of ICa,L and INCX.47, 48 Besides differences in ion channel expression, local differences in signaling could amplify small initial perturbations by a positive feedback mechanism. CaMKII phosphorylation could contribute to an increase the incidence of SSCEs through the phosphorylation of L-type Ca2+ channel, RyR2 and phospholamban and the increase in Cai could in turn augment CaMKII activity. Such a mechanism could explain why CaMKII inhibition suppresses EADs in rabbits with chronic heart block (prone to TdP).57
Isolated cell experiments were used to study the mechanism of EAD generation. In a related paper by Horvath et al.,58 experiments on myocytes with drug-induced LQT3 (gain of function of the late Na+ current) demonstrated that EADs are preceded by a rise of Cai. Moreover, EADs were eliminated when Cai was buffered with BAPTA even though AP duration remained prolonged. This study supports the primary role of non-voltage gated SR Ca2+ release as the cause of EADs (Figure 8).
Figure 8.
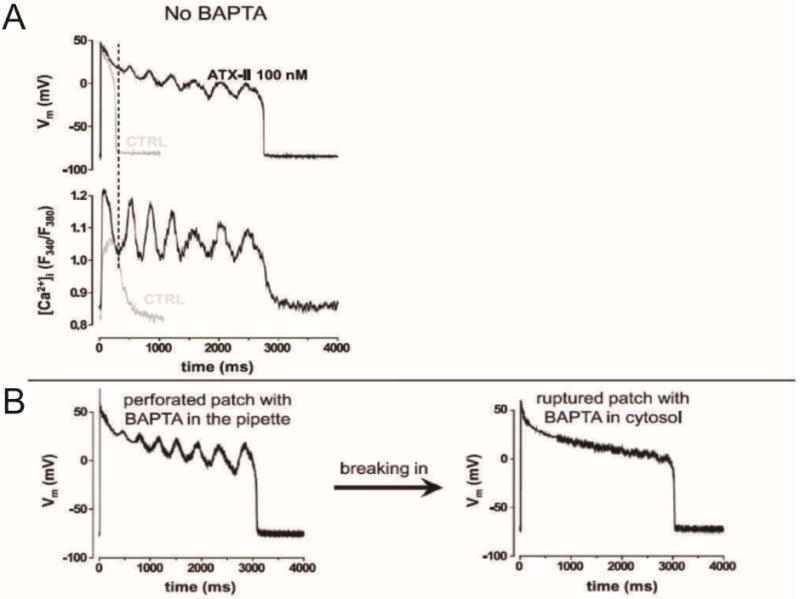
Patch-clamping experiments from a rabbit cell superfused with anemonetoxin II (ATX-II; an inhibitor of cardiac sodium channel inactivation) indicated that CaT oscillations are required to generate EADs. A: Simultaneous membrane voltage (top) and calcium signal (bottom) tracings in the whole cell, current clamp configuration, without calcium buffering. In the absence of ATX-II (gray tracings), both AP and CaT are short and monophasic. With ATX-II (black tracings), marked AP prolongation with multiple EADs occurs. The simultaneous CaT tracing demonstrates Ca2+ oscillations. Note that the takeoff of Ca2+ oscillations precedes the EAD rise. B: When the Ca2+ chelator BAPTA is present in the pipette solution, EADs are present in the perforated patch configuration (when BAPTA does not have access to the cytoplasm). With patch rupture and buffering of cytoplasmic Ca2+, EADs disappear, although AP duration remains long. Adapted from58
The acquired long QT syndrome model we studied is likely to be clinically relevant – bradycardia and hypokalemia occur frequently in patients with a wide range of conditions, and scores of FDA-approved drugs block IKr to some degree. However, it is conceivable that EADs may form through a different mechanism in different situations. For example, in the study of isolated rabbit ventricular myocytes by Zhao et al.,59 the EADs induced by a combination of isoproterenol and L-type Ca2+ channel opener and exhibited similar properties to those we observed in perfused hearts.59 Significantly, systolic Ca2+ oscillations persisted even after forcing for a normal APD and morphology using the AP clamp technique, such that the highest CaT peak occurred after the termination of the clamped AP. These Ca2+ dynamics provide robust evidence against the Ca2+ “window current” mechanism as an EAD trigger.
Interestingly, EADs induced by oxidative stress with a high concentration of hydrogen peroxide exhibited a different behavior: the CaT rise followed Vm rise, and Ca2+ oscillations could be eliminated by an AP clamp to force a short AP, and not by blocking SR Ca2+ release. These properties implicate a Ca2+ “window current” mechanism. It has even been suggested that in mice (which have short AP duration and utilize different K+ currents from rabbits and humans to achieve ventricular repolarization), non-equilibrium gating of the cardiac Na+ current could play an important role in EAD generation.60 Nevertheless, our results are mostly concordant with the data reported by Maruyama et al.61 in a similar rabbit model. Although these authors did not report subcellular data, they demonstrated highly irregular regions of CaT elevation during plateau and repolarization, and CaT rise preceding EAD takeoff.
It has been proposed62, 63 that AP prolongation gives rise to deterministic chaos in a certain range of cycle lengths and that electrotonic coupling of the cells operating in the “chaotic” regime results in spatial dispersion of APD and leads to ventricular arrhythmia. We have not tested this ingenious hypothesis in our experimental model, but we would argue that a mathematical framework based solely on dynamics of sarcolemmal currents, not including the intracellular Ca2+ handling dynamics, is likely incomplete. The inclusion of SOICR and SSCE in the whole-heart level mathematical models would certainly be interesting and probably quite challenging, since the relevant phenomena occur on both μm and cm scales.
TRANSLATIONAL ASPECTS
How do these findings fit into the clinical picture of TdP treatment, and what are the implications for arrhythmia management and SCD risk stratification in general? First, the model we propose predicts the protective effect of rapid pacing (~90 beats per minute), recommended in patients with runs of TdP. Regular and frequent “unloading” of jSR through the normal CICR mechanism expected during relatively rapid pacing may minimize the chance that jSR will fill over the SOICR threshold. On the other hand, a long pause may increase the chance of such an event, consistent with the “long-short” sequence which often initiates TdP. SSCEs of small amplitude typically cause a delay in CaT downstroke, can be observed in profound bradycardia in the absence of IKr block, increase APD dispersion and occasionally initiate arrhythmia.64 High adrenergic tone could further increase SR Ca2+ load and promote arrhythmias during profound bradycardia in the clinical setting.
If SOICR and NCX activation are required to generate EADs, could NCX blockade and RyR stabilization have a role in TdP management? It is certainly possible, but the effect at the level of the whole organism may be difficult to predict. For example, partial NCX blockade might initially protect against EADs, but eventually aggravate SR overload unless AP duration shortens, with unpredictable effects. Similarly, stabilizers of RyR are potentially promising antiarrhythmic agents, and K201 suppressed TdP in our model, but it should be kept in mind that RyR2 mutation which profoundly decreases SOICR is actually associated with ventricular fibrillation in affected patients,65, 66 probably because it increases jSR load during β-adrenergic stimulation.
The abnormal Ca2+ handling in LQTS is reflected by changes on a surface EKG. The local Ca2+ handling dynamics – i.e. the amplitude, precise timing and number of SSCEs – differ among different ventricular regions before arrhythmia onset. The SSCE pattern changes over the course of several seconds, presumably reflecting variable capacity of different ventricular regions to respond to increasing SR load. Although different myocardial regions are electrically coupled, resulting in partial smoothing of CaT through the AP effects on Ca2+ handling, the local heterogeneities of Ca2+ handling eventually overcome the tissue coupling. This dynamic interplay between CaT and AP before TdP onset is reflected in beat-to-beat changes of local CaT and AP, and globally as beat-to-beat changes of the T wave morphology. Beat-to-beat repolarization variability increases prior to TdP onset in LQTS40 and produces non-alternans patterns of repolarization or T-wave lability. T-wave lability has been reported prior to TdP onset in patients with congenital LQTS (Figure 9),67 in a canine model of acquired LQTS involving chronic complete heart block,68 and was found to be predictive of arrhythmia in a rabbit heart exposed to a wide array of repolarization-prolonging agents.69 In congenital LQTS patients, aperiodic T wave lability during catecholamine infusion correlates better with clinical status than corrected QT interval duration.67 Interestingly, non-alternans repolarization lability has been also seen in a murine CPVT model,70 indicating that it may not be unique to long QT syndrome. The question whether aperiodic T wave lability precedes arrhythmia onset in more common cardiac conditions and might provide a general risk-stratification tool for SCD remains unanswered at this time, but non-alternans repolarization variability prior to onset of ventricular arrhythmias has been reported in patients with structural heart disease and left ventricular dysfunction in ambulatory EKG tracings71 and in intra-cardiac electrograms stored by implantable defibrillators.72 In principle, repolarization lability might allow noninvasive, real-time monitoring of the arrhythmogenic process minutes before arrhythmia onset at least in some clinical settings.
Figure 9.
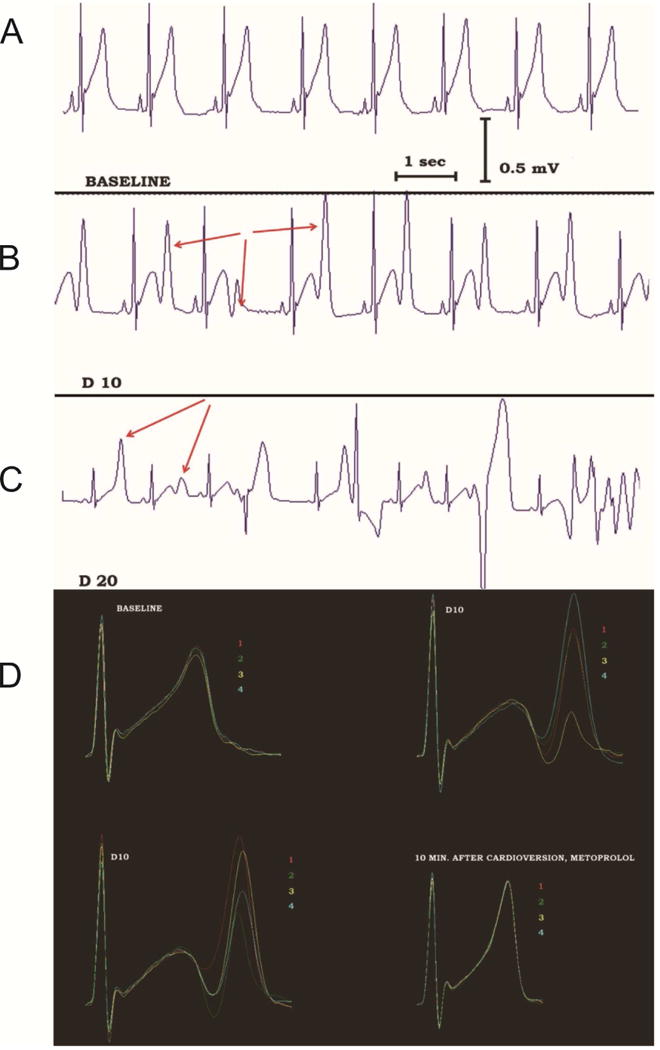
Non-alternans repolarization lability precedes TdP onset in a patient with congenital LQTS during dobutamine infusion. A: Before dobutamine infusion, sinus rhythm without obvious repolarization lability is recorded. B: During dobutamine infusion (dose of 10 μg/kg/minute), sinus rhythm is still present without ectopic beats, but the terminal component of the T-wave fluctuates dramatically in amplitude (red arrows) without an alternans pattern. C: With increased dobutamine dose (20 μg/kg/minute), TdP develops, preceded by polymorphic ventricular ectopy. D: The repolarization lability can be easily appreciated when 4 consecutive sinus beats are superimposed and color-coded. At baseline (top left), the repolarization lability is minimal. Striking T wave lability is evident during dobutamine infusion (top right and bottom left). This disappears after electrical cardioversion of TdP, cessation of dobutamine infusion and β-blocker administration (bottom right). Similar repolarization lability is observed in the rabbit LQTS model, when rises along with lability of Cai and Vm signals (not shown). Adapted from67
A most interesting question is whether arrhythmia mechanisms involving SOICR play a role in ventricular arrhythmias associated with coronary ischemia or heart failure, which are much more complex than pure repolarization delay. However, various forms of repolarization impairment and abnormal Ca2+ handling are invariably present in heart failure models. One of the better defined pathways involves activation of CaMKII and phosphorylation of β2a subunit of the L-type Ca2+ channels (augmenting Ca2+ influx),73 RyR2 (promoting Ca2+ release)74, 75 and the Nav1.5 Na+ channel (augmenting the late sodium current and prolonging repolarization).76, 77 At this moment, the extent of similarities in arrhythmogenic mechanisms between LQTS and heart failure remains uncertain.
CONCLUSION
Normal cardiac function involves propagation of electrical signals through the cardiac chambers; local action potential leads to Ca2+ entry into the cell and release from SR in individual cardiomyocytes, resulting in synchronized contraction. The main mechanism of Ca2+ removal from myocytes is the electrogenic NCX exchanger. Therefore, disturbance of myocyte Ca2+ handling can in turn affect membrane potential via changes in NCX current and cause arrhythmia. This appears to be the case in long QT syndrome, as prolonged action potential increases the amount of Ca2+ entering SR compartment during each cardiac cycle. Multiple forms of abnormal Ca2+ handling regimes occur on the subcellular level in this setting. It possible that arrhythmogenic mechanisms involved in long QT syndrome play some role in sudden cardiac death associated with more common cardiac conditions.
Acknowledgments
Funding Sources: Supported in part by NHLBI HL-70722 and HL-093074 to GS and AHA to JK
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Disclosures:
None
Editors’ Note:
Please see also related communications in this issue by AUTHOR-1 et al. (2015) and AUTHOR-2 et al. (2015)
References
- 1.Choi BR, Salama G. Simultaneous maps of optical action potentials and calcium transients in guinea-pig hearts: Mechanisms underlying concordant alternans. J Physiol. 2000;529(Pt 1):171–188. doi: 10.1111/j.1469-7793.2000.00171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sommer J, Johnson E. Ultrastructure of cardiac muscle. In: Berne R, editor. The cardiovascular system. 1979. pp. 113–186. [Google Scholar]
- 3.Suzuki T, Shioya T, Murayama T, Sugihara M, Odagiri F, Nakazato Y, Nishizawa H, Chugun A, Sakurai T, Daida H, Morimoto S, Kurebayashi N. Multistep ion channel remodeling and lethal arrhythmia precede heart failure in a mouse model of inherited dilated cardiomyopathy. PloS one. 2012;7:e35353. doi: 10.1371/journal.pone.0035353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aiba T, Hesketh GG, Barth AS, Liu T, Daya S, Chakir K, Dimaano VL, Abraham TP, O’Rourke B, Akar FG, Kass DA, Tomaselli GF. Electrophysiological consequences of dyssynchronous heart failure and its restoration by resynchronization therapy. Circulation. 2009;119:1220–1230. doi: 10.1161/CIRCULATIONAHA.108.794834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chopra N, Knollmann BC. Genetics of sudden cardiac death syndromes. Curr Opin Cardiol. 2011;26:196–203. doi: 10.1097/HCO.0b013e3283459893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morita H, Wu J, Zipes DP. The qt syndromes: Long and short. Lancet. 2008;372:750–763. doi: 10.1016/S0140-6736(08)61307-0. [DOI] [PubMed] [Google Scholar]
- 7.Li GR, Lau CP, Ducharme A, Tardif JC, Nattel S. Transmural action potential and ionic current remodeling in ventricles of failing canine hearts. Am J Physiol Heart Circ Physiol. 2002;283:H1031–1041. doi: 10.1152/ajpheart.00105.2002. [DOI] [PubMed] [Google Scholar]
- 8.Zicha S, Xiao L, Stafford S, Cha TJ, Han W, Varro A, Nattel S. Transmural expression of transient outward potassium current subunits in normal and failing canine and human hearts. J Physiol. 2004;561:735–748. doi: 10.1113/jphysiol.2004.075861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lacroix D, Gluais P, Marquie C, D’Hoinne C, Adamantidis M, Bastide M. Repolarization abnormalities and their arrhythmogenic consequences in porcine tachycardia-induced cardiomyopathy. Cardiovasc Res. 2002;54:42–50. doi: 10.1016/s0008-6363(02)00236-5. [DOI] [PubMed] [Google Scholar]
- 10.Akar FG, Wu RC, Juang GJ, Tian Y, Burysek M, Disilvestre D, Xiong W, Armoundas AA, Tomaselli GF. Molecular mechanisms underlying k+ current downregulation in canine tachycardia-induced heart failure. Am J Physiol Heart Circ Physiol. 2005;288:H2887–2896. doi: 10.1152/ajpheart.00320.2004. [DOI] [PubMed] [Google Scholar]
- 11.Szabo B, Kovacs T, Lazzara R. Role of calcium loading in early afterdepolarizations generated by cs+ in canine and guinea pig purkinje fibers. J Cardiovasc Electrophysiol. 1995;6:796–812. doi: 10.1111/j.1540-8167.1995.tb00356.x. [DOI] [PubMed] [Google Scholar]
- 12.Pogwizd SM, McKenzie JP, Cain ME. Mechanisms underlying spontaneous and induced ventricular arrhythmias in patients with idiopathic dilated cardiomyopathy. Circulation. 1998;98:2404–2414. doi: 10.1161/01.cir.98.22.2404. [DOI] [PubMed] [Google Scholar]
- 13.Patterson E, Jackman WM, Beckman KJ, Lazzara R, Lockwood D, Scherlag BJ, Wu R, Po S. Spontaneous pulmonary vein firing in man: Relationship to tachycardia-pause early afterdepolarizations and triggered arrhythmia in canine pulmonary veins in vitro. J Cardiovasc Electrophysiol. 2007;18:1067–1075. doi: 10.1111/j.1540-8167.2007.00909.x. [DOI] [PubMed] [Google Scholar]
- 14.Patterson E, Po SS, Scherlag BJ, Lazzara R. Triggered firing in pulmonary veins initiated by in vitro autonomic nerve stimulation. Heart Rhythm. 2005;2:624–631. doi: 10.1016/j.hrthm.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 15.Hwang HS, Nitu FR, Yang Y, Walweel K, Pereira L, Johnson CN, Faggioni M, Chazin WJ, Laver D, George AL, Jr, Cornea RL, Bers DM, Knollmann BC. Divergent regulation of ryanodine receptor 2 calcium release channels by arrhythmogenic human calmodulin missense mutants. Circ Res. 2014;114:1114–1124. doi: 10.1161/CIRCRESAHA.114.303391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aistrup GL, Kelly JE, Kapur S, Kowalczyk M, Sysman-Wolpin I, Kadish AH, Wasserstrom JA. Pacing-induced heterogeneities in intracellular ca2+ signaling, cardiac alternans, and ventricular arrhythmias in intact rat heart. Circ Res. 2006;99:e65–73. doi: 10.1161/01.RES.0000244087.36230.bf. [DOI] [PubMed] [Google Scholar]
- 17.Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, Knollmann BE, Horton KD, Weissman NJ, Holinstat I, Zhang W, Roden DM, Jones LR, Franzini-Armstrong C, Pfeifer K. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. 2006;116:2510–2520. doi: 10.1172/JCI29128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eldar M, Pras E, Lahat H. A missense mutation in the casq2 gene is associated with autosomal-recessive catecholamine-induced polymorphic ventricular tachycardia. Trends in cardiovascular medicine. 2003;13:148–151. doi: 10.1016/s1050-1738(03)00025-2. [DOI] [PubMed] [Google Scholar]
- 19.Postma AV, Denjoy I, Hoorntje TM, Lupoglazoff JM, Da Costa A, Sebillon P, Mannens MM, Wilde AA, Guicheney P. Absence of calsequestrin 2 causes severe forms of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2002;91:e21–26. doi: 10.1161/01.res.0000038886.18992.6b. [DOI] [PubMed] [Google Scholar]
- 20.Locati EH, Maison-Blanche P, Dejode P, Cauchemez B, Coumel P. Spontaneous sequences of onset of torsade de pointes in patients with acquired prolonged repolarization: Quantitative analysis of holter recordings. J Am Coll Cardiol. 1995;25:1564–1575. doi: 10.1016/0735-1097(95)00100-i. [DOI] [PubMed] [Google Scholar]
- 21.Roden DM. Torsade de pointes. Clin Cardiol. 1993;16:683–686. doi: 10.1002/clc.4960160910. [DOI] [PubMed] [Google Scholar]
- 22.Dessertenne F. ventricular tachycardia with 2 variable opposing foci. Arch Mal Coeur Vaiss. 1966;59:263–272. [PubMed] [Google Scholar]
- 23.Jiang D, Xiao B, Yang D, Wang R, Choi P, Zhang L, Cheng H, Chen SR. Ryr2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced ca2+ release (soicr) Proc Natl Acad Sci U S A. 2004;101:13062–13067. doi: 10.1073/pnas.0402388101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen W, Wasserstrom JA, Shiferaw Y. Role of coupled gating between cardiac ryanodine receptors in the genesis of triggered arrhythmias. American journal of physiology Heart and circulatory physiology. 2009;297:H171–180. doi: 10.1152/ajpheart.00098.2009. [DOI] [PubMed] [Google Scholar]
- 25.Shiferaw Y, Aistrup GL, Wasserstrom JA. Intracellular ca2+ waves, afterdepolarizations, and triggered arrhythmias. Cardiovasc Res. 2012;95:265–268. doi: 10.1093/cvr/cvs155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wasserstrom JA, Shiferaw Y, Chen W, Ramakrishna S, Patel H, Kelly JE, O’Toole MJ, Pappas A, Chirayil N, Bassi N, Akintilo L, Wu M, Arora R, Aistrup GL. Variability in timing of spontaneous calcium release in the intact rat heart is determined by the time course of sarcoplasmic reticulum calcium load. Circ Res. 2010;107:1117–1126. doi: 10.1161/CIRCRESAHA.110.229294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lakireddy V, Bub G, Baweja P, Syed A, Boutjdir M, El-Sherif N. The kinetics of spontaneous calcium oscillations and arrhythmogenesis in the in vivo heart during ischemia/reperfusion. Heart Rhythm. 2006;3:58–66. doi: 10.1016/j.hrthm.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 28.London B, Baker LC, Lee JS, Shusterman V, Choi BR, Kubota T, McTiernan CF, Feldman AM, Salama G. Calcium-dependent arrhythmias in transgenic mice with heart failure. American journal of physiology Heart and circulatory physiology. 2003;284:H431–441. doi: 10.1152/ajpheart.00431.2002. [DOI] [PubMed] [Google Scholar]
- 29.Schwartz P, Locati E, Napolitano C, Priori S. The long qt syndrome. Philadelphia, Pa: WB Saunders; 1995. [Google Scholar]
- 30.Keating MT, Sanguinetti MC. Molecular genetic insights into cardiovascular disease. Science. 1996;272:681–685. doi: 10.1126/science.272.5262.681. [DOI] [PubMed] [Google Scholar]
- 31.Luo CH, Rudy Y. A dynamic model of the cardiac ventricular action potential. Ii. Afterdepolarizations, triggered activity, and potentiation. Circ Res. 1994;74:1097–1113. doi: 10.1161/01.res.74.6.1097. [DOI] [PubMed] [Google Scholar]
- 32.January CT, Riddle JM. Early afterdepolarizations: Mechanism of induction and block. A role for l-type ca2+ current. Circ Res. 1989;64:977–990. doi: 10.1161/01.res.64.5.977. [DOI] [PubMed] [Google Scholar]
- 33.De Ferrari GM, Viola MC, D’Amato E, Antolini R, Forti S. Distinct patterns of calcium transients during early and delayed afterdepolarizations induced by isoproterenol in ventricular myocytes. Circulation. 1995;91:2510–2515. doi: 10.1161/01.cir.91.10.2510. [DOI] [PubMed] [Google Scholar]
- 34.Marban E, Robinson SW, Wier WG. Mechanisms of arrhythmogenic delayed and early afterdepolarizations in ferret ventricular muscle. J Clin Invest. 1986;78:1185–1192. doi: 10.1172/JCI112701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Volders PG, Vos MA, Szabo B, Sipido KR, de Groot SH, Gorgels AP, Wellens HJ, Lazzara R. Progress in the understanding of cardiac early afterdepolarizations and torsades de pointes: Time to revise current concepts. Cardiovascular research. 2000;46:376–392. doi: 10.1016/s0008-6363(00)00022-5. [DOI] [PubMed] [Google Scholar]
- 36.Milberg P, Pott C, Fink M, Frommeyer G, Matsuda T, Baba A, Osada N, Breithardt G, Noble D, Eckardt L. Inhibition of the na+/ca2+ exchanger suppresses torsades de pointes in an intact heart model of long qt syndrome-2 and long qt syndrome-3. Heart Rhythm. 2008;5:1444–1452. doi: 10.1016/j.hrthm.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 37.Zabel M, Hohnloser SH, Behrens S, Li YG, Woosley RL, Franz MR. Electrophysiologic features of torsades de pointes: Insights from a new isolated rabbit heart model. J Cardiovasc Electrophysiol. 1997;8:1148–1158. doi: 10.1111/j.1540-8167.1997.tb01001.x. [DOI] [PubMed] [Google Scholar]
- 38.Eckardt L, Haverkamp W, Borggrefe M, Breithardt G. Experimental models of torsade de pointes. Cardiovasc Res. 1998;39:178–193. doi: 10.1016/s0008-6363(98)00043-1. [DOI] [PubMed] [Google Scholar]
- 39.Choi BR, Burton F, Salama G. Cytosolic ca2+ triggers early afterdepolarizations and torsade de pointes in rabbit hearts with type 2 long qt syndrome. J Physiol. 2002;543:615–631. doi: 10.1113/jphysiol.2002.024570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pertsov AM, Davidenko JM, Salomonsz R, Baxter WT, Jalife J. Spiral waves of excitation underlie reentrant activity in isolated cardiac muscle. Circ Res. 1993;72:631–650. doi: 10.1161/01.res.72.3.631. [DOI] [PubMed] [Google Scholar]
- 41.Nemec J, Kim JJ, Gabris B, Salama G. Calcium oscillations and t-wave lability precede ventricular arrhythmias in acquired long qt type 2. Heart Rhythm. 2010;7:1686–1694. doi: 10.1016/j.hrthm.2010.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim JJ, Nemec J, Li Q, Salama G. Synchronous systolic subcellular ca2+-elevations underlie ventricular arrhythmia in drug-induced long qt type 2. Circulation Arrhythmia and electrophysiology. 2015;8:703–712. doi: 10.1161/CIRCEP.114.002214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sims C, Reisenweber S, Viswanathan PC, Choi BR, Walker WH, Salama G. Sex, age, and regional differences in l-type calcium current are important determinants of arrhythmia phenotype in rabbit hearts with drug-induced long qt type 2. Circ Res. 2008;102:e86–100. doi: 10.1161/CIRCRESAHA.108.173740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu T, Choi BR, Drici MD, Salama G. Sex modulates the arrhythmogenic substrate in prepubertal rabbit hearts with long qt 2. J Cardiovasc Electrophysiol. 2005;16:516–524. doi: 10.1046/j.1540-8167.2005.40622.x. [DOI] [PubMed] [Google Scholar]
- 45.Winfree AT. Electrical instability in cardiac muscle: Phase singularities and rotors. Journal of theoretical biology. 1989;138:353–405. doi: 10.1016/s0022-5193(89)80200-0. [DOI] [PubMed] [Google Scholar]
- 46.Salama G, Bett GC. Sex differences in the mechanisms underlying long qt syndrome. American journal of physiology Heart and circulatory physiology. 2014;307:H640–648. doi: 10.1152/ajpheart.00864.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang X, Chen G, Papp R, Defranco DB, Zeng F, Salama G. Oestrogen upregulates l-type ca(2)(+) channels via oestrogen-receptor- by a regional genomic mechanism in female rabbit hearts. J Physiol. 2012;590:493–508. doi: 10.1113/jphysiol.2011.219501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen G, Yang X, Alber S, Shusterman V, Salama G. Regional genomic regulation of cardiac sodium-calcium exchanger by oestrogen. J Physiol. 2011;589:1061–1080. doi: 10.1113/jphysiol.2010.203398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aistrup GL, Shiferaw Y, Kapur S, Kadish AH, Wasserstrom JA. Mechanisms underlying the formation and dynamics of subcellular calcium alternans in the intact rat heart. Circ Res. 2009;104:639–649. doi: 10.1161/CIRCRESAHA.108.181909. [DOI] [PubMed] [Google Scholar]
- 50.Atiga WL, Calkins H, Lawrence JH, Tomaselli GF, Smith JM, Berger RD. Beat-to-beat repolarization lability identifies patients at risk for sudden cardiac death. J Cardiovasc Electrophysiol. 1998;9:899–908. doi: 10.1111/j.1540-8167.1998.tb00130.x. [DOI] [PubMed] [Google Scholar]
- 51.Kaab S, Dixon J, Duc J, Ashen D, Nabauer M, Beuckelmann DJ, Steinbeck G, McKinnon D, Tomaselli GF. Molecular basis of transient outward potassium current downregulation in human heart failure: A decrease in kv4.3 mrna correlates with a reduction in current density. Circulation. 1998;98:1383–1393. doi: 10.1161/01.cir.98.14.1383. [DOI] [PubMed] [Google Scholar]
- 52.Mahajan A, Sato D, Shiferaw Y, Baher A, Xie LH, Peralta R, Olcese R, Garfinkel A, Qu Z, Weiss JN. Modifying l-type calcium current kinetics: Consequences for cardiac excitation and arrhythmia dynamics. Biophys J. 2008;94:411–423. doi: 10.1529/biophysj.106.98590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Efremov RG, Leitner A, Aebersold R, Raunser S. Architecture and conformational switch mechanism of the ryanodine receptor. Nature. 2015;517:39–43. doi: 10.1038/nature13916. [DOI] [PubMed] [Google Scholar]
- 54.Zalk R, Clarke OB, des Georges A, Grassucci RA, Reiken S, Mancia F, Hendrickson WA, Frank J, Marks AR. Structure of a mammalian ryanodine receptor. Nature. 2015;517:44–49. doi: 10.1038/nature13950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen W, Wang R, Chen B, Zhong X, Kong H, Bai Y, Zhou Q, Xie C, Zhang J, Guo A, Tian X, Jones PP, O’Mara ML, Liu Y, Mi T, Zhang L, Bolstad J, Semeniuk L, Cheng H, Zhang J, Chen J, Tieleman DP, Gillis AM, Duff HJ, Fill M, Song LS, Chen SR. The ryanodine receptor store-sensing gate controls ca2+ waves and ca2+-triggered arrhythmias. Nature medicine. 2014;20:184–192. doi: 10.1038/nm.3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yan Z, Bai XC, Yan C, Wu J, Li Z, Xie T, Peng W, Yin CC, Li X, Scheres SH, Shi Y, Yan N. Structure of the rabbit ryanodine receptor ryr1 at near-atomic resolution. Nature. 2015;517:50–55. doi: 10.1038/nature14063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Qi X, Yeh YH, Chartier D, Xiao L, Tsuji Y, Brundel BJ, Kodama I, Nattel S. The calcium/calmodulin/kinase system and arrhythmogenic afterdepolarizations in bradycardia-related acquired long-qt syndrome. Circulation Arrhythmia and electrophysiology. 2009;2:295–304. doi: 10.1161/CIRCEP.108.815654. [DOI] [PubMed] [Google Scholar]
- 58.Horvath B, Banyasz T, Jian Z, Hegyi B, Kistamas K, Nanasi PP, Izu LT, Chen-Izu Y. Dynamics of the late na(+) current during cardiac action potential and its contribution to afterdepolarizations. Journal of molecular and cellular cardiology. 2013;64:59–68. doi: 10.1016/j.yjmcc.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhao Z, Wen H, Fefelova N, Allen C, Baba A, Matsuda T, Xie LH. Revisiting the ionic mechanisms of early afterdepolarizations in cardiomyocytes: Predominant by ca waves or ca currents? American journal of physiology Heart and circulatory physiology. 2012;302:H1636–1644. doi: 10.1152/ajpheart.00742.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Edwards AG, Grandi E, Hake JE, Patel S, Li P, Miyamoto S, Omens JH, Heller Brown J, Bers DM, McCulloch AD. Nonequilibrium reactivation of na+ current drives early afterdepolarizations in mouse ventricle. Circulation Arrhythmia and electrophysiology. 2014;7:1205–1213. doi: 10.1161/CIRCEP.113.001666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maruyama M, Lin SF, Xie Y, Chua SK, Joung B, Han S, Shinohara T, Shen MJ, Qu Z, Weiss JN, Chen PS. Genesis of phase 3 early afterdepolarizations and triggered activity in acquired long-qt syndrome. Circulation Arrhythmia and electrophysiology. 2011;4:103–111. doi: 10.1161/CIRCEP.110.959064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sato D, Xie LH, Sovari AA, Tran DX, Morita N, Xie F, Karagueuzian H, Garfinkel A, Weiss JN, Qu Z. Synchronization of chaotic early afterdepolarizations in the genesis of cardiac arrhythmias. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:2983–2988. doi: 10.1073/pnas.0809148106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shimizu W, Antzelevitch C. Cellular basis for the ecg features of the lqt1 form of the long-qt syndrome: Effects of beta-adrenergic agonists and antagonists and sodium channel blockers on transmural dispersion of repolarization and torsade de pointes. Circulation. 1998;98:2314–2322. doi: 10.1161/01.cir.98.21.2314. [DOI] [PubMed] [Google Scholar]
- 64.Kim JJ, Nemec J, Papp R, Strongin R, Abramson JJ, Salama G. Bradycardia alters ca(2+) dynamics enhancing dispersion of repolarization and arrhythmia risk. American journal of physiology Heart and circulatory physiology. 2013;304:H848–860. doi: 10.1152/ajpheart.00787.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jiang D, Chen W, Wang R, Zhang L, Chen SR. Loss of luminal ca2+ activation in the cardiac ryanodine receptor is associated with ventricular fibrillation and sudden death. Proc Natl Acad Sci U S A. 2007;104:18309–18314. doi: 10.1073/pnas.0706573104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhao YT, Valdivia CR, Gurrola GB, Powers PP, Willis BC, Moss RL, Jalife J, Valdivia HH. Arrhythmogenesis in a catecholaminergic polymorphic ventricular tachycardia mutation that depresses ryanodine receptor function. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:E1669–1677. doi: 10.1073/pnas.1419795112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nemec J, Hejlik JB, Shen WK, Ackerman MJ. Catecholamine-induced t-wave lability in congenital long qt syndrome: A novel phenomenon associated with syncope and cardiac arrest. Mayo Clinic Proc. 2003;78:40–50. doi: 10.4065/78.1.40. [DOI] [PubMed] [Google Scholar]
- 68.Thomsen MB, Volders PG, Beekman JD, Matz J, Vos MA. Beat-to-beat variability of repolarization determines proarrhythmic outcome in dogs susceptible to drug-induced torsades de pointes. Circulation. 2006;48:1268–1276. doi: 10.1016/j.jacc.2006.05.048. [DOI] [PubMed] [Google Scholar]
- 69.Hondeghem LM, Carlsson L, Duker G. Instability and triangulation of the action potential predict serious proarrhythmia, but action potential duration prolongation is antiarrhythmic. Circulation. 2001;103:2004–2013. doi: 10.1161/01.cir.103.15.2004. [DOI] [PubMed] [Google Scholar]
- 70.Mezu UL, Singh P, Shusterman V, Hwang HS, Knollmann BC, Nemec J. Accelerated junctional rhythm and nonalternans repolarization lability precede ventricular tachycardia in casq2−/− mice. J Cardiovasc Electrophysiol. 2012 doi: 10.1111/j.1540-8167.2012.02406.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shusterman V, Goldberg A, London B. Upsurge in t-wave alternans and nonalternating repolarization instability precedes spontaneous initiation of ventricular tachyarrhythmias in humans. Circulation. 2006;113:2880–2887. doi: 10.1161/CIRCULATIONAHA.105.607895. [DOI] [PubMed] [Google Scholar]
- 72.Krokhaleva Y, Patel D, Shah H, Shusterman V, Saba S, Nemec J. Increased non-alternans repolarization variability precedes onset of spontaneous ventricular tachycardia in patients with implantable defibrillators. Journal of the American College of Cardiology. 2015;65:A440. [Google Scholar]
- 73.Koval OM, Guan X, Wu Y, Joiner ML, Gao Z, Chen B, Grumbach IM, Luczak ED, Colbran RJ, Song LS, Hund TJ, Mohler PJ, Anderson ME. Cav1.2 beta-subunit coordinates camkii-triggered cardiomyocyte death and afterdepolarizations. Proc Natl Acad Sci U S A. 2010;107:4996–5000. doi: 10.1073/pnas.0913760107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG, Li N, Santonastasi M, Muller FU, Schmitz W, Schotten U, Anderson ME, Valderrabano M, Dobrev D, Wehrens XH. Calmodulin kinase ii-mediated sarcoplasmic reticulum ca2+ leak promotes atrial fibrillation in mice. The Journal of clinical investigation. 2009;119:1940–1951. doi: 10.1172/JCI37059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tsuji Y, Hojo M, Voigt N, El-Armouche A, Inden Y, Murohara T, Dobrev D, Nattel S, Kodama I, Kamiya K. Ca(2+)-related signaling and protein phosphorylation abnormalities play central roles in a new experimental model of electrical storm. Circulation. 2011;123:2192–2203. doi: 10.1161/CIRCULATIONAHA.110.016683. [DOI] [PubMed] [Google Scholar]
- 76.Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P, Maier SK, Zhang T, Hasenfuss G, Brown JH, Bers DM, Maier LS. Ca2+/calmodulin-dependent protein kinase ii regulates cardiac na+ channels. The Journal of clinical investigation. 2006;116:3127–3138. doi: 10.1172/JCI26620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Glynn P, Musa H, Wu X, Unudurthi SD, Little S, Qian L, Wright PJ, Radwanski PB, Gyorke S, Mohler PJ, Hund TJ. Voltage-gated sodium channel phosphorylation at ser571 regulates late current, arrhythmia, and cardiac function in vivo. Circulation. 2015;132:567–577. doi: 10.1161/CIRCULATIONAHA.114.015218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu XK, Katchman A, Drici MD, Ebert SN, Ducic I, Morad M, Woosley RL. Gender difference in the cycle length-dependent qt and potassium currents in rabbits. The Journal of pharmacology and experimental therapeutics. 1998;285:672–679. [PubMed] [Google Scholar]
- 79.Zhu Y, Ai X, Oster RA, Bers DM, Pogwizd SM. Sex differences in repolarization and slow delayed rectifier potassium current and their regulation by sympathetic stimulation in rabbits. Pflugers Archiv: European journal of physiology. 2013;465:805–818. doi: 10.1007/s00424-012-1193-9. [DOI] [PMC free article] [PubMed] [Google Scholar]