

Abstract
Objective
The IκB kinase (IKK) is an enzyme complex that initiates the NF-κB transcription factor cascade, which is important in regulating multiple cellular responses. IKK alpha (IKKα) is directly associated with two major pro-survival pathways, PI3K/Akt and NF-κB, but its role in cell survival is not clear. Macrophages play critical roles in the pathogenesis of atherosclerosis, yet the impact of IKKα signaling on macrophage survival and atherogenesis remains unclear.
Approach and Results
Here we demonstrate that genetic IKKα deficiency, as well as pharmacologic inhibition of IKK, in mouse macrophages significantly reduces Akt S473 phosphorylation, which is accompanied by suppression of mTORC2 signaling. Moreover, IKKα null macrophages treated with lipotoxic palmitic acid exhibited early exhaustion of Akt signaling compared to WT cells. This was accompanied by a dramatic decrease in the resistance of IKKα−/− monocytes and macrophages to different pro-apoptotic stimuli compared to WT cells. In vivo, IKKα deficiency increased macrophage apoptosis in atherosclerotic lesions and decreased early atherosclerosis in both female and male LDLR−/− mice reconstituted with IKKα−/− hematopoietic cells and fed with the Western diet for 8 weeks compared to control LDLR−/− mice transplanted with WT cells.
Conclusions
Hematopoietic IKKα deficiency in mouse suppresses Akt signaling, compromising monocyte/macrophage survival and this decreases early atherosclerosis.
Keywords: Atherosclerosis, macrophages, apoptosis, Akt signaling
Introduction
Atherosclerosis is a progressive inflammatory disease and the underlying cause of heart attack and stroke1. Macrophage-derived foam cells are the major cell type in early atherosclerotic lesions and a key feature of unstable plaques2. The ability of macrophages to survive in atherosclerotic lesions is an important modulator of atherosclerotic lesion development3. For example, increased sensitivity of macrophages to apoptosis suppresses early lesion progression, whereas in advanced lesions, macrophage apoptosis promotes necrotic core formation, increasing plaque vulnerability and thrombosis4. Two major signaling pathways, PI3K/Akt and nuclear factor kappa-B (NF-κB), regulate cell survival5, 6. However the impact of these pathways on macrophage survival and atherosclerosis remains unclear.
NF-κB activation relies on inducible degradation of IκBα, which is mediated through its phosphorylation by the IκB kinase (IKK), a complex consisting of two catalytic subunits, IKKα and IKKβ, and a regulatory subunit, IKKγ. IKKβ initiates the well-defined classical NF-κB pathway, whereas IKKα has been implicated in the alternative pathway7. IKKα plays a key role in the negative feedback of NF-κB signaling to limit inflammatory gene activation in macrophages8, 9. In addition, IKKα is also required for B-cell maturation and formation of secondary lymphoid organs10. IKKα−/− mice die soon after birth due to dramatic abnormalities in morphogenesis11, 12. IKKβ knockout mice die as embryos (day F13), primarily due massive hepatocyte apoptosis13, 14. Furthermore, IKKα/IKKβ double knockout mice die at E12 due to enhanced apoptosis of the fetal liver during neurulation15. These data indicate that IKKα and IKKβ are essential for NF–κB activation, which plays a crucial role in protecting against excessive apoptosis. The results also suggest a possible link between the IKK pathway and the PI3K/Akt pathway, which is well known for its anti-apoptotic action.
Human and mouse macrophages express constitutively active p-Akt, which is vital for their survival16 and inhibition of PI3K or Akt induces apoptosis17. IKKα is known as a target in anti-apoptotic signaling of the PI3K/Akt pathway18–20. Recent reports indicate that IKKα directly regulates Akt signaling acting at several different levels and mediates crosstalk between the Akt and NF-κB pathways21–24. For example, both IKKα and IKKβ phosphorylate p85α promoting feedback inhibition of PI3K/Akt signaling in response to cellular starvation21. IKKα is also required for the mammalian target of rapamycin (mTOR) activity22, which forms two functionally distinct complexes: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). The mTORC1 is a nutrient/energy/redox sensor consisting of mTOR, regulatory associated protein of mTOR (Raptor), mammalian lethal with SEC13 protein 8 (mLST8), and Deptor, a negative regulator of mTOR. In contrast, mTORC2 controls activity of an important phosphorylation site for Akt signaling, Akt S473, and cell survival25. mTORC2 consists of mTOR, rapamycin-insensitive companion of mTOR (Rictor), stress-activated protein kinase-interacting protein (Sin1), mLST8, and Protor. IKKα directly phosphorylates mTOR to drive mTORC1 activation23 and interacts with rictor to regulate the function of mTORC224. These data suggest a complex regulation of Akt signaling by IKKα.
Recently, Telstam et al. have shown that hematopoietic knock-in of a non-activatable IKKα kinase mutant, in which the activation loop serines were replaced with alanines (IkkαAA/AA), in ApoE−/− mice had no effect on atherosclerosis26 However, it is important to note that IkkαAA/AA mice had no changes in IKKα activity and did not exhibit morphologic defects, which are specific for IKKα loss27. Therefore, the impact of complete loss of IKKα protein and activity in hematopoietic cells on atherogenesis remains to be examined.
In the present study, we found that loss of IKKα in macrophages significantly suppresses Akt S473 phosphorylation and this compromises cell survival. In addition, we show that IKKα deficiency in hematopoietic cells increases macrophage apoptosis and reduces atherosclerosis in vivo. Our data reveal a novel mechanism by which IKKα controls Akt signaling and macrophage survival with important implications in atherogenesis.
Materials and Methods
Materials and Methods are available in the online-only Data Supplement.
Loss of IKKalpha in macrophages suppresses Akt signaling, reduces cell viability and early atherosclerosis.
Results
IKKα deficiency in mouse macrophages inhibits Akt S473 phosphorylation
Recent studies reported that IKKα regulates Akt signaling acting at several different levels of the pathway21, 22, 23. Therefore, we analyzed whether loss of IKKα affects Akt signaling in mouse macrophages. Since IKKα−/− mice die soon after birth due to dramatic abnormalities in morphogenesis11, 12, we used a fetal liver cell (FLC) transplantation approach28 to generate C57BL/6 mice with IKKα−/− or wild-type (WT or IKKα+/+) macrophages to study Akt signaling.
First, we examined whether loss of macrophage IKKα impacts Akt activation in vitro. We found that recombinant mouse platelet-derived growth factor (PDGF) induced phosphorylation of p-Akt S473 in WT type (~2-fold increase), but not in IKKα−/− cells, whereas the content of Akt and β–actin remained comparable (Figure 1A,B). Similarly, IKKα−/− macrophages exhibited decreased levels of Akt S473 phosphorylation in response to insulin or Akt activator, SC79 (Supplemental Figure I A,B and C,D, respectively). These results indicate that Akt S473 phosphorylation is impaired in IKKα−/− macrophages.
Figure 1. Akt S473 phosphorylation and mTORC2 signaling are suppressed in IKKα−/− macrophages compared to WT cells.
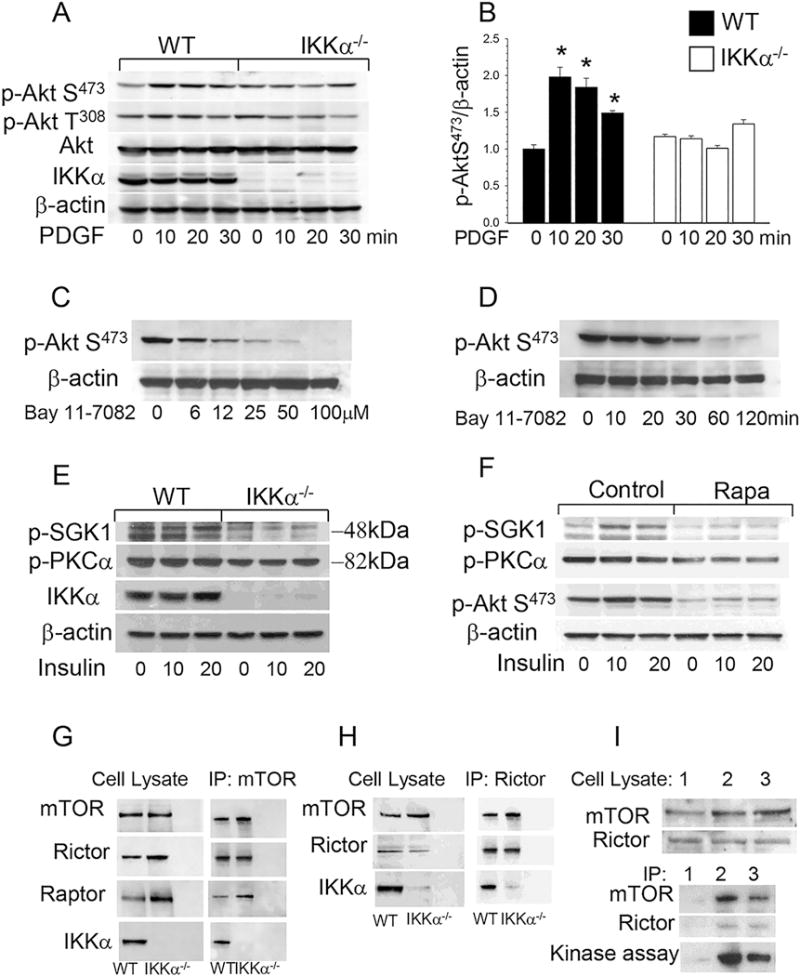
(A, B) Akt S473 phosphorylation in WT(■) and IKKα−/− (□) peritoneal macrophages treated with PDGF. Cells were incubated in serum free media for 16 hours and then treated with PDGF (20ng/ml) for 0 (control), 10, 15 and 20min. Proteins were extracted, resolved (60μg/well) and analyzed by western blot using antibodies against the proteins indicated. Graphs represent data (mean ± SEM) of three experiments (*p < 0.05 compared to control untreated group);
(C, D) An IKK inhibitor, Bay 11-7082, suppresses Akt S473 signaling in a dose- and time-dependent manner. WT peritoneal macrophages were treated with 0–100μM Bay 11-7082 for 30 min (C) or with 40μM Bay 11-7082 for 0-120min (D). Then proteins were extracted and subjected to Western blot analysis using antibodies to p-Akt (S473) and β-actin;
(E, F) mTORC2 targets, SGK1 and PKCα are suppressed in IKKα−/−macrophages and similar suppression is exhibited by WT macrophages treated with rapamycin overnight. WT and IKKα−/− macrophages were cultured in serum-free media alone (E) or with rapamycin (100nM; F) for 16 hours and then treated with insulin (100 nM) for 10 or 20 min. Extracted proteins were analyzed using antibodies to p-SGK (Ser422) and p-PKCα (Ser657);
(G–I) IKKα in cell precipitates formed by the antibody to mTOR (G), Rictor (H) or IKKα (I) from WT and IKKα−/− macrophage lysates. Note the presence of IKKα in mTOR complexes (G) and mTORC2 (H). I (Top Panel) Comparison of different antibodies including isotype control (1), antibody to Rictor (2), or antibody to IKKα (3); and (Lower Panel) kinase assays performed with precipitates from WT macrophages, antibodies to p-Akt (S473) and in the presence of full-length Akt1 protein as the substrate.
Next we tested whether pharmacologic IKK inhibition affects Akt signaling in macrophages. Treatment of WT macrophages with a specific IKK inhibitor, Bay 11-7082, suppressed the levels of p-Akt S473 in a dose- and time-dependent manner (Figure 1C,D). Thus, both genetic deficiency of IKKα and pharmacologic inhibition of IKK suppress Akt phosphorylation in macrophages.
Since Akt S473 activity is regulated by mTORC229, we examined whether IKKα plays an important role in mTORC2-mediated Akt signaling of mouse macrophages. First, we studied its down-stream targets including serum- and glucocorticoid-induced protein kinase-1 (SGK1) and protein kinase C alpha (PKCα). The protein levels of both of these downstream targets were significantly lower in IKKα−/− macrophages treated with insulin (36% p-SGK1 and 72% p-PKCα) than in WT cells (Figure 1E). It is known that prolonged rapamycin treatment inhibits the assembly of mTORC2 in different types of cells30. Here we demonstrate that WT macrophages treated with rapamycin overnight exhibited significant suppression of the mTORC2 targets, p-SGK1 (38%) and p-PKCα (67%), compared to untreated cells (Figure 1F). These results indicate that mTORC2 signaling is suppressed in IKKα−/− macrophages, and a similar effect can be induced in WT cells by inhibiting the assembly of mTORC2.
Finally, to analyze mTOR complexes in WT and IKKα−/− macrophages, we used an antibody to mTOR to precipitate the complexes. The precipitates contained the major components of the mTORC, including mTOR, Raptor and Rictor (Figure 1G). Surprisingly, IKKα was also present in the precipitates from WT macrophages (Figure 1G). Remarkably, mTORC2 precipitated by an antibody to Rictor also contained IKKα (Figure 1H). Therefore, we examined whether an antibody to IKKα is capable of precipitating mTORC2 compared with an isotype control and Rictor. The precipitates formed by the antibody to IKKα or Rictor, but not the isotype control, contained mTOR and Rictor (Figure 1I). Importantly, the precipitates also exhibited Akt kinase activity specific for mTORC2 (Figure 1I, lower panel). In contrast, loss of IKKα in macrophages prevents the precipitation of mTORC2 by the antibody to IKKα (Supplemental Figure II). These results indicate that IKKα plays an important role in mTORC2-mediated Akt signaling of mouse macrophages.
IKKα−/− macrophages have increased sensitivity to pro-apoptotic stimuli and express high levels of inflammatory genes in response to LPS
Since Akt signaling is the major pro-survival pathway that suppresses apoptosis5, we suspected that defective phosphorylation of Akt S473 in IKKα−/− macrophages may compromise cell survival. In addition, a recent report31 has demonstrated that ER stress inhibits mTORC2 activity and Akt signaling. Therefore, we hypothesized that IKKα−/− macrophages may be less resistant to ER stress conditions than WT cells. To test this hypothesis, WT and IKKα−/− peritoneal macrophages were treated with palmitic acid (PA), a lipotoxic factor that induces ER stress and triggers apoptosis32. Compared to WT cells, IKKα−/− macrophages had early exhaustion of Akt signaling, with suppression of p-AktS473 (Figure 2A,B) and p-Bad136 (Supplemental Figure III). The treatment with PA significantly increased the number of TUNEL-positive nuclei in both cell types (Figure 2C–F), but IKKα−/− macrophages had significantly higher levels of apoptosis than WT macrophages (Figure 2G). In addition, substantially more IKKα−/− macrophages underwent apoptotic changes when treated with oxidized LDL (80%) or acetylated LDL (63%) together with an ACAT inhibitor, Sandoz 58035, than did WT cells (Fig 2H). Compared to WT cells, IKKα−/− cells were predisposed to apoptosis (117%) in response to a specific inhibitor of IkBα phosphorylation, BAY 11-7082 (Figure 2I). Interestingly, treatment with wedelolactone (Wede), an inhibitor of IKK, alone had only a slight effect on apoptosis in both cell types, whereas the combination of Wede and PA significantly increased apoptosis in WT and IKKα−/− macrophages (Figure 2J), indicating the importance of Akt activity in the stress response mechanism. In contrast, the constitutively activated p-Akt in PTEN−/− macrophages significantly increased resistance to PA-induced stress (Supplemental Figure IVA,B) and protected the cells from apoptosis compared to WT macrophages (Supplemental Figure IVC,D). Taken together, these data support the concept that suppression of Akt signaling in IKKα−/− macrophages generates a cell phenotype with enhanced sensitivity to apoptosis.
Figure 2. IKKα−/− macrophages are less resistant to different pro-apoptotic stimuli than WT cells.
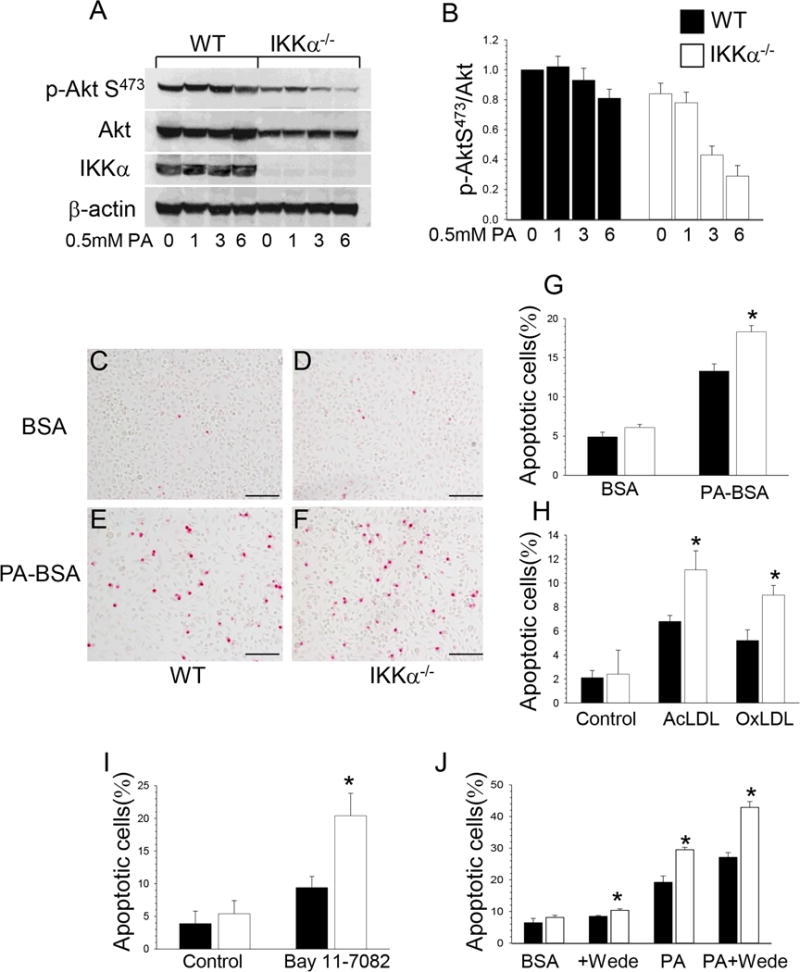
(A–B). ER stress inhibits Akt signaling faster in IKKα−/− compared to WT macrophages. WT (■) and IKKα−/− (□) peritoneal macrophages were treated with 0.5mM PA-BSA for 0, 1, 3 and 6 hours. Extracted proteins were used for analysis of Akt signaling. Graphs represent data (mean ± SEM) of three experiments;
(C–F). Detection of apoptosis by TUNEL assay in WT (C,E) and IKKα−/− (D,F) macrophages treated with BSA (C,D) or PA-BSA (E,F); Scale bars, 100μn;
(G–J). TUNEL+ cell numbers (mean ± SEM) in WT(■) and IKKα−/− (□) macrophages treated with BSA or 0.5mM PA-BSA (G) for 24 hours; with Ox-LDL (100mg/ml) or Ac-LDL (100mg/ml) plus an ACAT inhibitor, Sandoz 58035 (10mg/ml) for 48 hours (H) with Bay 11-7082 (20mM) (I), with Wede (50mM) alone or together with 0.5mM PA-BSA (J) for 24 hours. Each experiment was repeated three times (*p < 0.05).
In addition, WT and IKKα−/− peritoneal macrophages were treated with LPS, and the levels of inflammatory genes were measured by real-time PCR. Compared to WT cells, IKKα−/− macrophages showed increased expression of proinflammatory genes including Il1b, Il6 and Il12a (Figure 3A–C). In contrast, the levels of Il10 gene expression was significantly lower in IKKα−/− macrophages than in WT cells (Figure 3D). These results indicate that IKKα−/− macrophages express higher levels of inflammatory genes but low levels of the Il10 gene, representing the classical proinflammatory M1 phenotype.
Figure 3. IKKα−/− macrophages have increased inflammatory and decreased of Il-10 gene expression, changes in blood B-cells, neutrophils and monocytes of IKKα−/−→LDLR−/− mice compared to WT →LDLR−/− mice.
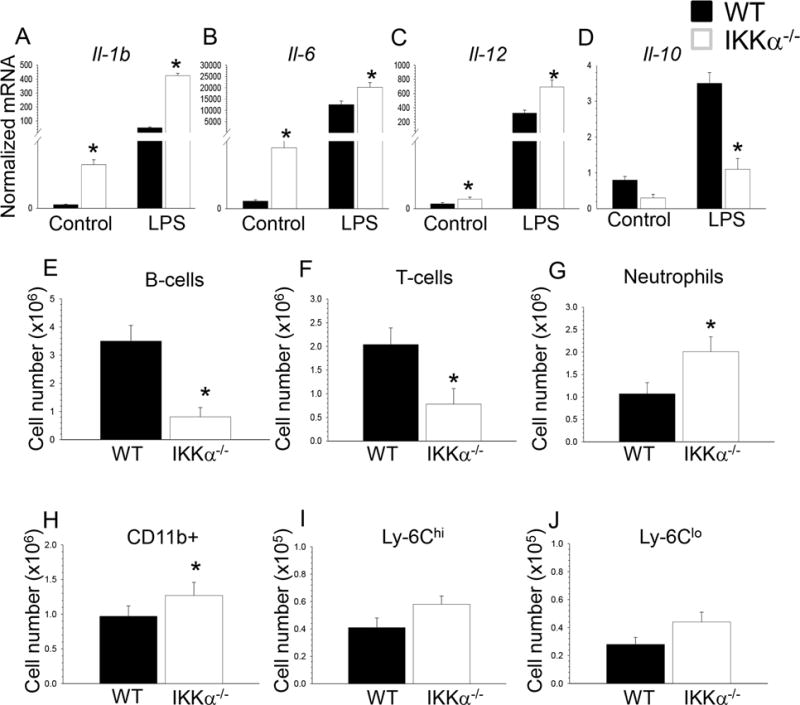
(A–D). WT and IKKα−/− peritoneal macrophages were incubated with media alone (control) or together with LPS (20ng/ml) for 6 hours and the gene-expression levels were measured by real-time PCR. Graphs represent data (mean ± SEM) obtained from the same numbers (n=3/group) of mice (*p < 0.05 compared to WT cells treated with LPS by Mann-Whitney rank sum test);
(E–J) Blood samples were collected from retro-orbital sinus of mice transplanted with WT and IKKα−/− FLC (n=4/group); Cells were incubated with antibodies to CD19, CD3, CD11b, CCR2, Ly-6G and Ly-6C, and analyzed by multicolor flow cytometry; Graphs represent data (mean ± SEM) of the experiments; *p < 0.05 by t-test.
Hematopoietic IKKα deficiency changes blood cells decreasing monocyte viability and decreasing early atherosclerosis
To examine if macrophage IKKα deficiency has an impact on atherosclerosis in vivo, we conducted two separate experiments in female and male recipients due to the established impact of gender on atherosclerosis in mice. In the first experiment, eight-week-old female LDLR−/− mice were lethally irradiated and transplanted with female IKKα+/+ (control group; n=11) or IKKα−/− (n=10) FLC. After four weeks of the chow diet, the ratio of blood leukocytes was determined in recipient mice by flow cytometry. Compared to control WT→WT mice, IKKα−/−→WT mice had a dramatic decrease in blood B-cells and lower numbers of T-cells (Figure 3E, F). In contrast, numbers of neutrophils and monocytes were increased (Figure 3G,H) including both Ly-6Chi and Ly-6Clo subsets (Figure 3I,J). These blood cell changes were accompanied by enlargement of the spleen (0.11+0.01 vs. 0.08+0.01g; p=0.01) and altered spleen histology (Supplemental Figure V). In addition, IKKα−/−→WT mice had high levels of apoptosis in blood cells with no differences in neutrophils but with increased monocyte apoptosis (Figure 4A–C). To further investigate this phenomenon, we isolated blood monocytes and treated them with PA-BSA overnight. Analysis of apoptosis by Annexin V stain demonstrated that IKKα−/− monocytes had significantly increased apoptosis compared to WT cells (Figure 4D,E). Thus, IKKα−/− monocytes exhibit compromised resistance to apoptotic stimuli compared to WT cells.
Figure 4. Blood monocytes isolated from IKKα−/−→LDLR−/− mice are less resistant to pro-apoptotic stimuli than WT cells.
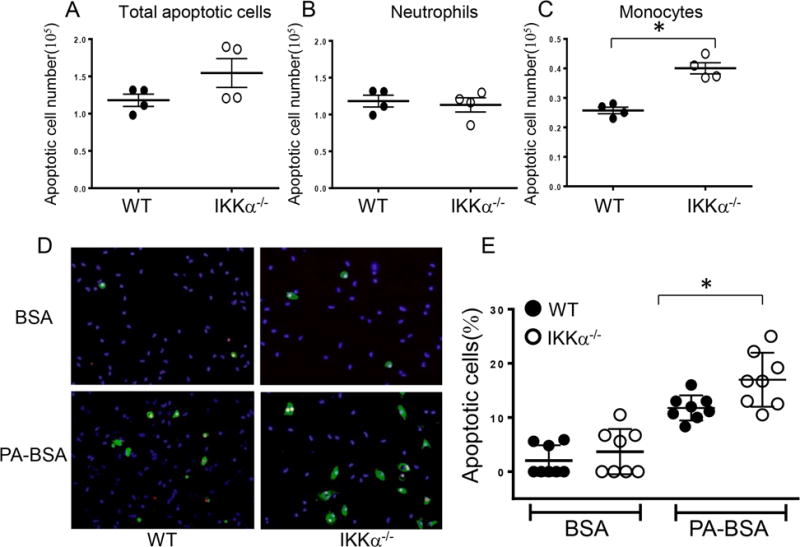
(A–C). Total apoptotic cell numbers (A), apoptotic neutrophils (B) and monocytes (C) in blood of LDLR−/− mice reconstituted with WT and IKKα−/− FLC; blood cells were treated with Alexa Flour 647 Annexin V together with antibodies to CD11b, Ly-6G, and analyzed by flow cytometry; Graphs represent data (mean ± SEM) of the experiments; *p < 0.05 by t-test;
(D,E) Detection of apoptotic (green) and dead cells (red) by the Alexa Flour 488 Annexin V/Dead cell apoptosis kit with nuclear counterstaining by DAPI (blue) (D) and percent of apoptotic cells in WT and IKKα−/− monocytes treated with BSA (A,B) or PA-BSA (C,D); monocytes were isolated from blood, and, two days later, treated with 0.3M PA-BSA in the presence of 3%FBS and 10ng/ml of mouse M-CSF overnight; apoptotic cells were detected by the Alexa Flour 488 Annexin V/Dead cell apoptosis kit; Scale bars, 50μm; Graphs represent data (mean ± SEM) of three experiments; *p < 0.05 by Mann-Whitney rank sum test.
After 8 weeks on the Western diet, no significant differences in body weight, serum total cholesterol and triglyceride levels were found between groups (Supplemental Table I). Remarkably, IKKα−/−→LDLR−/− mice had a 63.3% smaller atherosclerotic lesion area in the proximal aorta than mice transplanted with WT cells (Figure 5A–F,I; 98±15 vs. 269±25 ×103μm2; p=0.040). Similarly, IKKα−/−→LDLR−/− mice had smaller atherosclerotic lesions in pinned out aortas compared to mice transplanted with WT FLC (Figure 5G,H,J; 2.1±0.4 vs. 1.1±0.2%; p=0.023). The IKKα−/−→LDLR−/− mice also had significantly reduced MOMA2-positive area in their atherosclerotic lesions compared to WT→LDLR−/− mice (Figure 5K). Importantly, IKKα−/−→LDLR−/− mice had significantly more TUNEL-positive cells in their atherosclerotic lesions than control WT→LDLR−/− mice (Figure 5L). Next, we examined impact of loss IKKα on macrophage apoptosis and proliferation in the atherosclerotic lesions. Double staining of sections from the aortic sinus with the antibody to macrophage, MOMA-2, and anti-cleaved caspase 3 verified that these apoptotic cells are macrophage in origin (Supplemental Figure VI). At the same time, only a few macrophages in the atherosclerotic lesions expressed Ki-67, a cellular marker of all active phases (G1, S, G2 and mitosis) of cell proliferation, with no differences between WT→LDLR−/− and IKKα−/−→LDLR−/− mice (Supplemental Figure VII).
Figure 5. Female IKKα−/−→LDLR−/− mice had less atherosclerosis, more macrophage apoptosis and fewer macrophages in the lesion area than control WT → LDLR−/− mice.
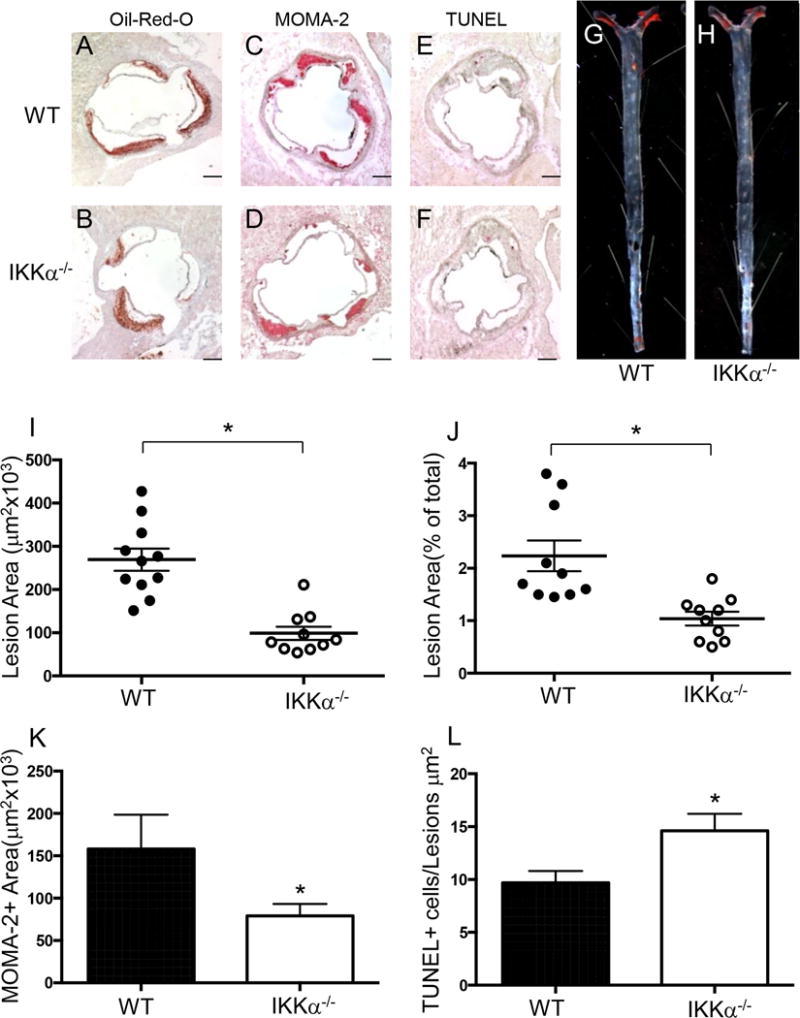
(A–H) Representative images of aortic sinus sections stained with Oil-Red-O/hematoxylin (A,B), MOMA-2 (C,D), TUNEL AP (E,F), and Sudan IV-stained en face preparation of aortas (G,H) from WT→LDLR−/− (A,C,E,G) and IKKα−/−→LDLR−/− (B,D,F,H) mice. Scale bars, 200μm; pin size, 10μm;
(I,J) The extent of atherosclerotic lesions in the proximal and distal aortas of LDLR−/− mice reconstituted with WT(●) or IKKα−/− (o) FLC; *p < 0.05 by Mann-Whitney rank sum test;
(K,L) Macrophage area stained with MOMA-2 and number of TUNEL+ cells in atherosclerotic lesions of mice with WT(■) or IKKα−/− (□) FLC; *p < 0.05 by t-test.
In a second experiment designed to exclude a gender–related difference in atherosclerosis33, 8-week-old male LDLR−/− mice were lethally irradiated and transplanted with IKKα+/+ (control group; n=7) or IKKα−/− (n=9) FLC. Again, there were no statistically significant differences in body weight and serum lipid levels (Supplemental Table I). These male recipients had smaller (45.6%) atherosclerotic lesions than control male WT→LDLR−/− mice (Figure 6A–F,I; 91±44 vs. 167±75 ×103μm2; p=0.001). Compared to control WT→LDLR−/− mice, IKKα−/−→LDLR−/− mice also had significantly reduced atherosclerotic lesions in pinned out aortas (Figure 6G,H,J; 0.29±0.02 vs. 0.37±0.03%; p=0.014), less MOMA2-positive area (Figure 6K) and more TUNEL-positive cells in the atherosclerotic lesion area (Figure 6L). These results demonstrate that elimination of IKKα in hematopoietic cells significantly increases macrophage apoptosis in atherosclerotic lesions and diminishes atherosclerosis in mice of both genders.
Figure 6. Male IKKα−/−→LDLR−/− mice had less atherosclerosis, more macrophage apoptosis and fewer macrophages in the lesion area than control WT →LDLR−/− mice.
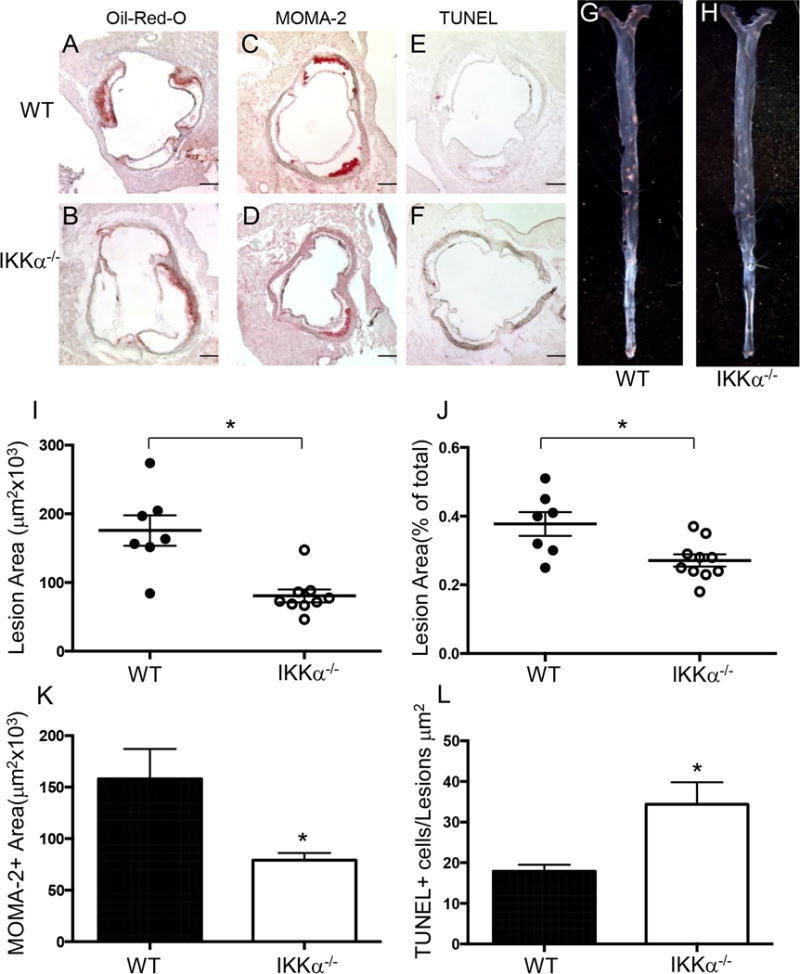
(A–H) Representative images of aortic sinus sections stained with Oil-Red-O/hematoxylin (A,B), MOMA-2 (C,D), TUNEL AP (E,F), and Sudan IV-stained en face preparation of aortas (G,H) from WT→LDLR−/− (A,C,E,G) and IKKα−/−→LDLR−/− (B,D,F,H) mice. Scale bars, 200μm; pin size, 10μm;
(I,J) Atherosclerotic lesions in the proximal and distal aortas of LDLR−/− mice reconstituted with WT(●) or IKKα−/− (o) FLC; *p < 0.05 by Mann-Whitney rank sum test;
(K,L) Macrophage area stained with MOMA-2 and number of TUNEL+ cells in atherosclerotic lesions of mice with WT(■) or IKKα−/− (□) FLC; *p < 0.05 by t-test.
Discussion
Macrophages are uniquely resistant to pro-apoptotic stimuli and the ability of macrophages to survive is a key determinant of the development of atherosclerotic lesions. IKKα is associated with two pro-survival pathways, the PI3K/Akt and NF-κB, but its role in macrophage survival and atherosclerosis remains unclear. Here we demonstrate that IKKα deficiency in mouse macrophages significantly suppresses Akt phosphorylation and this radically compromises cell resistance to pro-apoptotic stimuli. Furthermore, our results show that IKKα plays a crucial role in mTORC2-mediated Akt signaling in macrophages. We also provide in vivo evidence that male and female LDLR−/− mice reconstituted with IKKα−/− hematopoietic cells have smaller atherosclerotic lesions that are enriched in apoptotic macrophages compared to control mice transplanted with WT cells. Thus, IKKα plays an important role in Akt phosphorylation and IKKα deficiency reduces macrophage survival and suppresses early atherosclerosis.
NF-κB inducing kinase acting through IKKα triggers the alternative NF-κB pathway, which is crucial for the generation of B and T lymphocytes and the development of lymphoid organs34. In addition, IKKα limits the NF-κB-mediated expression of inflammatory genes in macrophages, and macrophage IKKα deficiency enhances inflammation8, 9. Our results are consistent with this role for IKKα in that IKKα−/−→LDLR−/− mice exhibited monocytosis and more Ly-6Chi monocytes and M1 macrophages. However, loss of IKKα also powerfully suppressed anti-apoptotic Akt signaling in both blood monocytes and macrophages, dramatically decreasing their viability compared to control WT cells. Since macrophages play a key role in atherosclerotic lesion formation2, the compromised ability of IKKα−/− monocytes/macrophages to survive markedly modulates atherogenesis diminishing early lesion development. Thus, we believe that the suppression of Akt due to loss of IKKα increases macrophage apoptosis, playing a dominant role in reducing atherosclerosis.
In addition, we found decreased numbers of T-cells in blood of IKKα−/−→LDLR−/− mice and these data support the view that IKKα is an essential component of T-cell regulatory function35. Finally, our IKKα−/−→LDLR−/− mice had significantly diminished numbers of blood B-cells and this is in harmony with the notion that IKKα is essential for B-cell maturation10. A similar B-cell reduction was described in kinase-dead IKKα knock-in mice36. Importantly, loss of B-cells has been reported to both increase37, 38 and decrease39 the extent of atherosclerosis. More recent studies suggest that traditional T cell-driven B2 cell responses appear to be atherogenic, while innate B1 cells appear to exert a protective action via the secretion of naturally occurring antibodies40. Therefore, it is uncertain whether the decreased numbers of T- or B-cells due to IKKα deficiency impacted the development of atherosclerosis, and future studies will be required to address this issue.
Our data implicating mTORC2 assembly in IKKα–mediated Akt signaling are consistent with a recent study24 reporting that Rictor directly interacts with both IKKα and IKKβ. Chemical inhibition of IKK, knockdown of IKK by siRNA, or expression of kinase-dead IKK significantly inhibited the kinase activity of mTORC2 suppressing Akt S473 phosphorylation24. At present, the precise sites of IKKα–mediated phosphorylation are unknown. IKKα likely interacts with mTORC2 essential components, Rictor and/or Sin1, which form a heterodimer that determines the mutual stability of both proteins41. Rictor contains 21 unique phosphorylation sites clustered in the C-terminal end, and this sequence is highly conserved among vertebrates42. Phosphorylation of Sin1, triggered by several different growth factors, suppresses mTORC2 kinase activity by dissociating Sin1 from the complex43. Taken together our results strongly indicate that IKKα regulates Akt signaling in macrophages, and this suggests an exciting opportunity to target this pathway in order to impact function of the innate immune system cells, which are critically involved in a variety of pathological processes.
It is important to note that assembly of mTORC2 is a complex process, and its impairment results in suppression of Akt S473 activity. For example, both Rictor and mLST8 are required for Akt signaling, and genetic deletion of either of them is associated with complete loss of Akt S473 activity44. Similarly, genetic ablation of another key component of mTORC2, Sin1, eliminated Akt S473 phosphorylation, whereas T308 activity was preserved41, 45, 46. Even a single Rictor point mutation (G934E) markedly reduced Akt S473 phosphorylation47. Importantly, loss of Sin1 and consequently p-Akt S473 significantly increased cell susceptibility to stress-induced apoptosis41 supporting the concept that Akt signaling is anti-apoptotic. Thus, our current results provide experimental support for this concept by demonstrating that loss of IKKα reduces Akt activity in macrophages, increasing their sensitivity to apoptosis.
The current results also demonstrate that deficiency of IKKα in hematopoietic cells reduces early atherosclerosis in vivo. The atherosclerotic lesions of IKKα−/−→LDLR−/− mice had dramatically increased macrophage apoptosis and reduced macrophage area compared to lesions of IKKα+/+→LDLR−/− mice. These data are consistent with the report48 demonstrating that Akt S473 phosphorylation was markedly reduced in macrophages lacking the PI3-kinase110γ and this significantly diminished atherosclerosis in apoE−/− mice. In contrast, Tilstam et al26 have shown recently that apoE−/− mice reconstituted with IKKα knockin (IkkαAA/AA) bone marrow cells had less B- and T-cells but no differences in atherosclerosis compared to control mice transplanted with apoE−/− marrow. Importantly, IkkαAA/AA bone marrow-derived macrophages did not exhibit changes in NF-κB activity or cytokine expression26. These results are consistent with the initial report27 indicating that IkkαAA/AA mice do not exhibit morphologic defects specific for loss of IKKα11, 12 with the only exception being that females displayed a severe lactation defect due impaired proliferation of mammary epithelial cells. Similar to macrophages, IkkαAA/AA embryonic fibroblast did not show any changes in IKK or NF-κB activation induced by TNFα, IL-1 or endotoxin27. Thus, IkkαAA/AA cells presumably have preserved IKKα kinase activity, which likely underlies the difference in our results regarding the role of IKKα in atherosclerosis. In contrast, we report that genetic ablation of IKKα in the bone marrow, which results in complete loss of IKKα protein and activity in hematopoietic cells, dramatically reduces atherosclerosis in LDLR−/− mice, demonstrating a crucial role for expression of IKKα in hematopoietic cells in atherogenesis.
In conclusion, our data demonstrate that loss of IKKα in macrophages suppresses mTORC2-mediated Akt S473 signaling and this compromises cell survival and decreases early atherosclerosis (Supplemental Figure VIII). These results support an inverse relationship between macrophage apoptosis and lesion size in early atherosclerotic lesions4. Our findings also stress a crucial role of IKKα in mTORC2-mediated Akt signaling and provide new therapeutic targets for the regulation of macrophage survival and the prevention of atherosclerosis.
Supplementary Material
Significance.
Macrophages are the major type of cells of atherosclerotic lesions and they play critical roles in the pathogenesis of atherosclerosis. Macrophages are uniquely resistant to pro-apoptotic stimuli and their ability to survive is a key determinant of atherosclerotic lesion formation. IKKα is associated with two major pro-survival pathways, the PI3K/Akt and NF-κB. however the role of IKKα in macrophage survival and atherosclerosis remains unclear. Here we demonstrated for the first time that genetic loss of IKKα as well as IKK inhibition significantly suppresses Akt signaling in macrophages and this markedly compromises monocyte and macrophage viability compared to WT cells. In addition, our results show that IKKα plays a crucial role in mTORC2-mediated Akt signaling in macrophages. In our atherosclerosis experiments, female and male LDLR−/− mice reconstituted with IKKα null bone marrow exhibited significantly smaller atherosclerotic lesions than control mice with WT marrow. Our findings provide new IKKα related therapeutic targets for the regulation of macrophage survival and the development of atherosclerosis.
Acknowledgments
The authors are grateful to Michael Karin from University of California at San Diego School of Medicine for providing IKKα+/− mice on the C57BL/6J background.
Sources of Funding: This work was supported by National Institutes of Health grants HL105375, HL116263, DK50435, DK59637 (Lipid, Lipoprotein and Atherosclerosis Core of the Vanderbilt Mouse Metabolic Phenotype Centers).
Abbreviations
- IKK
IκB kinase
- Akt
protein kinase B
- LDLR
LDL-receptor
- FLC
fetal liver cells
Footnotes
Conflict of Interests: The authors declare no conflicts of interest.
SUPPLEMENTAL DATA: Supplemental data contain 8 Figures and a Table.
References
- 1.Hansson GK, Libby P. The immune response in atherosclerosis: A double-edged sword. Nat Rev Immunol. 2006;6:508–519. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- 2.Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: A dynamic balance. Nat Rev Immunol. 2013;13:709–721. doi: 10.1038/nri3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kockx MM, Herman AG. Apoptosis in atherosclerosis: Beneficial or detrimental? Cardiovasc Res. 2000;45:736–746. doi: 10.1016/s0008-6363(99)00235-7. [DOI] [PubMed] [Google Scholar]
- 4.Seimon T, Tabas I. Mechanisms and consequences of macrophage apoptosis in atherosclerosis. J Lipid Res. 2009;50:S382–S387. doi: 10.1194/jlr.R800032-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duronio V. The life of a cell: Apoptosis regulation by the pi3k/pkb pathway. Biochem J. 2008;415:333–344. doi: 10.1042/BJ20081056. [DOI] [PubMed] [Google Scholar]
- 6.Manning BD, Cantley LC. Akt/pkb signaling: Navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karin M. The ikappab kinase – a bridge between inflammation and cancer. Cell Res. 2008;18:334–342. doi: 10.1038/cr.2008.30. [DOI] [PubMed] [Google Scholar]
- 8.Lawrence T, Bebien M, Liu GY, Nizet V, Karin M. Ikk[alpha] limits macrophage nf-[kappa]b activation and contributes to the resolution of inflammation. Nature. 2005;434:1138–1143. doi: 10.1038/nature03491. [DOI] [PubMed] [Google Scholar]
- 9.Li Q, Lu Q, Bottero V, Estepa G, Morrison L, Mercurio F, Verma IM. Enhanced nf-kb activation and cellular function in macrophages lacking ikkb kinase 1 (ikk1) Proc Natl Acad Sci USA. 2005;102:12425–12430. doi: 10.1073/pnas.0505997102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Senftleben U, Cao Y, Xiao G, Greten FR, Krähn G, Bonizzi G, Chen Y, Hu Y, Fong A, Sun S-C, Karin M. Activation by ikkα of a second, evolutionary conserved, nf-κb signaling pathway. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- 11.Hu Y, Baud V, Delhase M, Zhang P, Deerinck T, Ellisman M, Johnson R, Karin M. Abnormal morphogenesis but intact ikk activation in mice lacking the ikk subunit of ib kinase. Science. 1999;284:316–320. doi: 10.1126/science.284.5412.316. [DOI] [PubMed] [Google Scholar]
- 12.Takeda K, Takeuchi O, Tsujimura T, Itami S, Adachi O, Kawai T, Sanjo H, Yoshikawa K, Terada N, Akira S. Limb and skin abnormalities in mice lacking ikk. Science. 1999;284:313–316. doi: 10.1126/science.284.5412.313. [DOI] [PubMed] [Google Scholar]
- 13.Li Q, Antwerp DV, Mercurio F, Lee K-F, Verma IM. Severe liver degeneration in mice lacking the iκb kinase 2 gene. Science. 1999;284:321–325. doi: 10.1126/science.284.5412.321. [DOI] [PubMed] [Google Scholar]
- 14.Li Z-W, Chu W, Hu Y, Delhase M, Deerinck T, Ellisman M, Johnson R, Karin M. The ikkβ subunit of iκb kinase (ikk) is essential for nuclear factor κb activation and prevention of apoptosis. J Exp Med. 1999;189:1839–1845. doi: 10.1084/jem.189.11.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Q, Estepa G, Memet S, Israel A, Verma IM. Complete lack of nf-κb activity in ikk1 and ikk2 double-deficient mice: Additional defect in neurulation. Genes & Development. 2000;14:1729–1733. [PMC free article] [PubMed] [Google Scholar]
- 16.Liu H, Perlman H, Pagliari LJ, Pope RM. Constitutively activated akt-1 is vital for the survival of human monocyte-differentiated macrophages: Role of mcl-1, independent of nuclear factor (nf)-{kappa}b, bad, or caspase activation. J Exp Med. 2001;194:113–126. doi: 10.1084/jem.194.2.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Babaev VR, Chew JD, Ding L, Davis S, Breyer MD, Breyer RM, Oates JA, Fazio S, Linton MF. Macrophage ep4 deficiency increases apoptosis and suppresses early atherosclerosis. Cell Metabolism. 2008;8:492–501. doi: 10.1016/j.cmet.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Madrid LV, Wang C-Y, Guttridge DC, Schottelius AJG, Baldwin AS, Jr, Mayo MW. Akt suppresses apoptosis by stimulating the transactivation potential of the rela/p65 subunit of nf-kappa b. Mol Cell Biol. 2000;20:1626–1638. doi: 10.1128/mcb.20.5.1626-1638.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. Nf-kappab activation by tumour necrosis factor requires the akt serine-threonine kinase. Nature. 1999;401:82–85. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- 20.Romashkova JA, Makarov SS. Nf-[kappa]b is a target of akt in anti-apoptotic pdgf signalling. Nature. 1999;401:86. doi: 10.1038/43474. [DOI] [PubMed] [Google Scholar]
- 21.Comb William C, Hutti Jessica E, Cogswell P, Cantley Lewis C, Baldwin Albert S. P85α sh2 domain phosphorylation by ikk promotes feedback inhibition of pi3k and akt in response to cellular starvation. Mol Cell. 2012;45:719–730. doi: 10.1016/j.molcel.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dan HC, Baldwin AS. Differential involvement of ikappab kinases alpha and beta in cytokine- and insulin-induced mammalian target of rapamycin activation determined by akt. J Immunol. 2008;180:7582–7589. doi: 10.4049/jimmunol.180.11.7582. [DOI] [PubMed] [Google Scholar]
- 23.Dan HC, Ebbs A, Pasparakis M, Van Dyke T, Basseres DS, Baldwin AS. Akt-dependent activation of mtorc1 involves phosphorylation of mtor by ikkalpha. J Biol Chem. 2014 doi: 10.1074/jbc.M114.554881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu Y, Lai E, Liu J, Lin J, Yang C, Jia C, Li Y, Bai X, Li M. Ikk interacts with rictor and regulates mtorc2. Cell Signalling. 2013;25:2239–2245. doi: 10.1016/j.cellsig.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 25.Laplante M, Sabatini DM. Mtor signaling at a glance. J Cell Sci. 2009;122:3589–3594. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tilstam PV, Gijbels MJ, Habbeddine M, et al. Bone marrow-specific knock-in of a non-activatable ikkα kinase mutant influences haematopoiesis but not atherosclerosis in apoe-deficient mice. PLoS ONE. 2014;9:e87452. doi: 10.1371/journal.pone.0087452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cao Y, Bonizzi G, Seagroves TN, Greten FR, Johnson R, Schmidt EV, Karin M. Ikkα provides an essential link between rank signaling and cyclin d1 expression during mammary gland development. Cell. 2001;107:763–775. doi: 10.1016/s0092-8674(01)00599-2. [DOI] [PubMed] [Google Scholar]
- 28.Babaev VR, Fazio S, Gleaves LA, Carter KJ, Semenkovich CF, Linton MF. Macrophage lipoprotein lipase promotes foam cell formation and atherosclerosis in vivo. J Clin Invest. 1999;103:1697–1705. doi: 10.1172/JCI6117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of akt/pkb by the rictor-mtor complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 30.Sarbassov DD, Ali SM, Sengupta S, Sheen J-H, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mtorc2 assembly and akt/pkb. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 31.Chen C-H, Shaikenov T, Peterson TR, Aimbetov R, Bissenbaev AK, Lee S-W, Wu J, Lin H-K, Sarbassov DD. Er stress inhibits mtorc2 and akt signaling through gsk-3{beta}-mediated phosphorylation of rictor. Sci Signal. 2011;4:ra10. doi: 10.1126/scisignal.2001731. [DOI] [PubMed] [Google Scholar]
- 32.Borradaile NM, Han X, Harp JD, Gale SE, Ory DS, Schaffer JE. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J Lipid Res. 2006;47:2726–2737. doi: 10.1194/jlr.M600299-JLR200. [DOI] [PubMed] [Google Scholar]
- 33.Paigen B, Holmes PA, Mitchell D, Albee D. Comparison of atherosclerotic lesions and hdl-lipid levels in male, female, and testosterone-treated female mice from strains c57bl/6, balb/c, and c3h. Atherosclerosis. 1987;64:215–221. doi: 10.1016/0021-9150(87)90249-8. [DOI] [PubMed] [Google Scholar]
- 34.Sun S-C. The noncanonical nf-κb pathway. Immunol Rev. 2012;246:125–140. doi: 10.1111/j.1600-065X.2011.01088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen X, Willette-Brown J, Wu X, Hu Y, Howard OMZ, Hu Y, Oppenheim JJ. Ikkα is required for the homeostasis of regulatory t cells and for the expansion of both regulatory and effector cd4 t cells. FASEB J. 2015;29:443–454. doi: 10.1096/fj.14-259564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Balkhi MY, Willette-Brown J, Zhu F, Chen Z, Liu S, Guttridge DC, Karin M, Hu Y. Ikkα-mediated signaling circuitry regulates early b lymphopoiesis during hematopoiesis. Blood. 2012;119:5467–5477. doi: 10.1182/blood-2012-01-401547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caligiuri G, Nicoletti A, Poirier B, Hansson GK. Protective immunity against atherosclerosis carried by b cells of hypercholesterolemic mice. J Clin Invest. 2002;109:745–753. doi: 10.1172/JCI07272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Major AS, Fazio S, Linton MF. B-lymphocyte deficiency increases atherosclerosis in ldl receptor–null mice. Arterioscler Thromb Vasc Biol. 2002;22:1892–1898. doi: 10.1161/01.atv.0000039169.47943.ee. [DOI] [PubMed] [Google Scholar]
- 39.Ait-Oufella H, Herbin O, Bouaziz J-D, Binder CJ, Uyttenhove C, Laurans L, Taleb S, Van Vré E, Esposito B, Vilar J, Sirvent J, Van Snick J, Tedgui A, Tedder TF, Mallat Z. B cell depletion reduces the development of atherosclerosis in mice. J Expl Med. 2010;207:1579–1587. doi: 10.1084/jem.20100155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ammirati E, Moroni F, Magnoni M, Camici PG. The role of t and b cells in human atherosclerosis and atherothrombosis. Clin Exp Immun. 2015;179:173–187. doi: 10.1111/cei.12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. Sin1/mip1 maintains rictor-mtor complex integrity and regulates akt phosphorylation and substrate specificity. Cell. 2006;127:125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 42.Dibble CC, Asara JM, Manning BD. Characterization of rictor phosphorylation sites reveals direct regulation of mtor complex 2 by s6k1. Mol Cell Biol. 2009;29:5657–5670. doi: 10.1128/MCB.00735-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu P, Gan W, Inuzuka H, Lazorchak AS, et al. Sin1 phosphorylation impairs mtorc2 complex integrity and inhibits downstream akt signalling to suppress tumorigenesis. Nat Cell Biol. 2013;15:1340–1350. doi: 10.1038/ncb2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, Sabatini DM. Ablation in mice of the mtorc components raptor, rictor, or mlst8 reveals that mtorc2 is required for signaling to akt-foxo and pkcα, but not s6k1. Develop Cell. 2006;11:859–871. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 45.Shiota C, Woo J-T, Lindner J, Shelton KD, Magnuson MA. Multiallelic disruption of the rictor gene in mice reveals that mtor complex 2 is essential for fetal growth and viability. Develop Cell. 2006;11:583–589. doi: 10.1016/j.devcel.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 46.Yang Q, Inoki K, Ikenoue T, Guan K-L. Identification of sin1 as an essential torc2 component required for complex formation and kinase activity. Genes Develop. 2006;20:2820–2832. doi: 10.1101/gad.1461206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aimbetov R, Chen CH, Bulgakova O, Abetov D, Bissenbaev AK, Bersimbaev RI, Sarbassov DD. Integrity of mtorc2 is dependent on the rictor gly-934 site. Oncogene. 2011 doi: 10.1038/onc.2011.404. Online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chang JD, Sukhova GK, Libby P, Schvartz E, Lichtenstein AH, Field SJ, Kennedy C, Madhavarapu S, Luo J, Wu D, Cantley LC. Deletion of the phosphoinositide 3-kinase p110γ gene attenuates murine atherosclerosis. Proc Nat Acad Sci. 2007;104:8077–8082. doi: 10.1073/pnas.0702663104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.