

Abstract
Objective
Understanding the mechanisms regulating normal and pathologic angiogenesis is of great scientific and clinical interest. In this report, we show that mutations in two different aminoacyl tRNA synthetases, threonyl tRNA synthetase (tarsy58) or isoleucyl tRNA synthetase (iarsy68), lead to similar increased branching angiogenesis in developing zebrafish.
Approach and Results
The Unfolded Protein Response (UPR) pathway is activated by aminoacyl tRNA synthetase deficiencies, and we show that UPR genes atf4, atf6, and xbp1, as well as the key pro-angiogenic ligand vascular endothelial growth factor (vegfaa), are all up-regulated in tarsy58 and iarsy68 mutants. Finally, we show that the PERK-ATF4 arm of the UPR pathway is necessary for both the elevated vegfaa levels and increased angiogenesis observed in tarsy58 mutants.
Conclusions
Our results suggest that endoplasmic reticulum (ER) stress acts as a pro-angiogenic signal via UPR pathway-dependent up-regulation of vegfaa.
Keywords: Zebrafish, Unfolded Protein Response, tRNA Synthetase, VEGF, Angiogenesis
INTRODUCTION
The complex and intricately ramified network of blood vessels making up the circulatory system of vertebrates plays a vital functional role, supplying oxygen and nutrients, removing wastes, and serving as the conduit for transport of immune and hormonal cells and factors. As might be expected, the formation, remodeling, and regression of blood vessels is a complex process exquisitely modulated by signals from the adjacent tissues that vessels serve, permitting rapid response to hypoxia or nutritional stress by promoting increased vessel growth. Vessel growth is regulated by an incompletely understood network of molecular signals and pathways. These include the key vascular endothelial growth factor (VEGF) signaling pathway. The pro-angiogenic ligand VEGF-A, secreted by a wide variety of non-endothelial cell types and tissues, regulates endothelial cell (EC) specification, proliferation, differentiation, migration, survival, as well as endothelial cell-cell adhesion and endothelial barrier function (reviewed in (1)).
The role of hypoxia in promoting VEGF-A ligand and VEGFR2 receptor production via induction of the HIF pathway in oxygen-deprived tissues has been well-documented (reviewed in (2)). Nutritional stress and metabolic stress are also potent pro-angiogenic stimuli promoting activation of VEGF signaling. Hypoxia or glucose deprivation each induce VEGF-A expression in glioma cells in spheroid culture, with VEGF-A levels reduced after the spheroids are implanted into nude mice and become vascularized (3). Activation of ER stress response pathways by nutritional stress or by treatment with chemical inhibitors of translation such as tunicamycin and thapsigargin also induces the expression of VEGF (4).
ER stress has been reported to up-regulate the Unfolded Protein Response (UPR) pathway, an adaptive pathway that either helps cells cope with an increased load of unfolded proteins or, in cases of irreversible ER stress, triggers apoptosis of damaged cells (reviewed in (5)). The UPR pathway involves ER-resident stress sensors IRE-1a, PERK, and ATF6, each of which transduces information about protein folding status to distinct intracellular signal transducers. In the IRE-1 arm of the UPR pathway, excess unfolded protein results in IRE-1a phosphorylation and dimerization, enabling a specific RNase activity of IRE-1a that processes un-spliced inactive X box-binding protein 1 (XBP1u) transcript to permit formation of spliced, active transcription factor XBP1s, which regulates transcription of a variety of genes involved in protein folding, ER-associated degradation, and protein and phospholipid synthesis. In the PERK arm of the UPR pathway, unfolded protein promotes PERK dimerization and activation, leading to increased production of ATF4 protein and modulation of protein synthesis (5). Several publications have reported that induction of either IRE1a-XBP1 and PERK-ATF4 can promote VEGF expression, with XBP1s and ATF4 binding to upstream sequences in the VEGF promoter (6–9). Recent data suggests that while ATF4 and XBP1s both bind the VEGF promoter, ATF4 may play a predominant role in regulating VEGF expression (8).
Amino acid deprivation is one potential source of ER stress (4). Reduced levels of specific amino acids can result from deficiencies in aminoacyl-transfer RNA (tRNA) synthetases (AARSs). AARS proteins are indispensible for protein synthesis, with specific AARSs catalyzing the ligation of particular amino acids to their cognate tRNAs. In addition to their canonical roles in protein synthesis, tRNA synthetase genes have also been reported to have a diversity of novel noncanonical functions in homeostatic regulation (reviewed in (10)), including the regulation of angiogenesis. Tyrosyl-tRNA synthetase (YARS) is cleaved by endogenous enzymes to generate a secreted N-terminal fragment that stimulates endothelial cell proliferation and migration (11, 12) by activating VEGF signaling through the VEGFR2 receptor (13). Other AARS protein derivatives can have anti-angiogenic or angiostatic activities. A C-terminal fragment of tryptophanyl-tRNA synthetase (WARS) induced by interferon-γ (IFN-γ) inhibits VEGF-induced EC migration via binding to the EC adhesion protein VE-cadherin (14–16), while Glutamyl-prolyl-tRNA synthetase (EPRS) inhibits angiogenesis via translational silencing of VEGF-A (17).
Zebrafish mutants displaying increased angiogenesis were identified in several genetic screens, and cloning of these mutants revealed that they have defects in the Seryl tRNA synthetase (SARS or SerRS) gene (18–20). Further study indicated that SARS acts as an antiangiogenic or angiostatic factor via a noncanonical mechanism not related to its function in charging tRNAs (19, 20). The UNE-S domain appended to vertebrate SARS proteins contains a localization signal that allows these proteins to be imported into the nucleus, where they attenuate VEGF-A expression (21).
Here, we describe the isolation of two new zebrafish mutants displaying excess angiogenesis. We show that the vascular phenotypes in these mutants result from mutations in two different zebrafish tRNA synthetase genes, threonyl tRNA synthetase (tars) and isoleucyl tRNA synthetase (iars). Deficiencies in TARS or IARS function result in up-regulation of the UPR pathway with concomitant increased expression of VEGF, driving increased angiogenesis. Our results suggest that defects in either of these two tRNA synthetases lead to increased angiogenesis in vivo via UPR pathway activation.
MATERIALS AND METHODS
Materials and methods are available in the on-line only data supplement.
RESULTS
Identification of mutants exhibiting increased angiogenesis with defects in the threonyl and isoleucyl aminoacyl tRNA synthetase genes
We identified two ENU-induced recessive mutants, y58 and y68, with similar angiogenic phenotypes in a genetic screen for vascular mutants in the zebrafish. Initial endothelial specification and assembly of the primary trunk axial vessels (dorsal aorta and cardinal vein) and cranial primary vessels (lateral dorsal aorta and primordial hindbrain channel) by vasculogenesis occurs normally and circulatory flow is present in y58 and y68 homozygotes. However, by 3 days post-fertilization (dpf) y58 and y68 mutants show increased branching of angiogenic vessels in both the trunk (Figure 1A–D, H) and the head (Figure 1A, E–G, I, Supplemental Figure IC–D, Supplemental Movie I). Other tissues and organs appear to develop largely normally, although both mutants show decreased head, eye, and overall body size by 3 dpf indicative of some developmental delay in mutant animals (Figure 1J–L).
Fig 1. Zebrafish y58 and y68 mutants display excess angiogenesis.
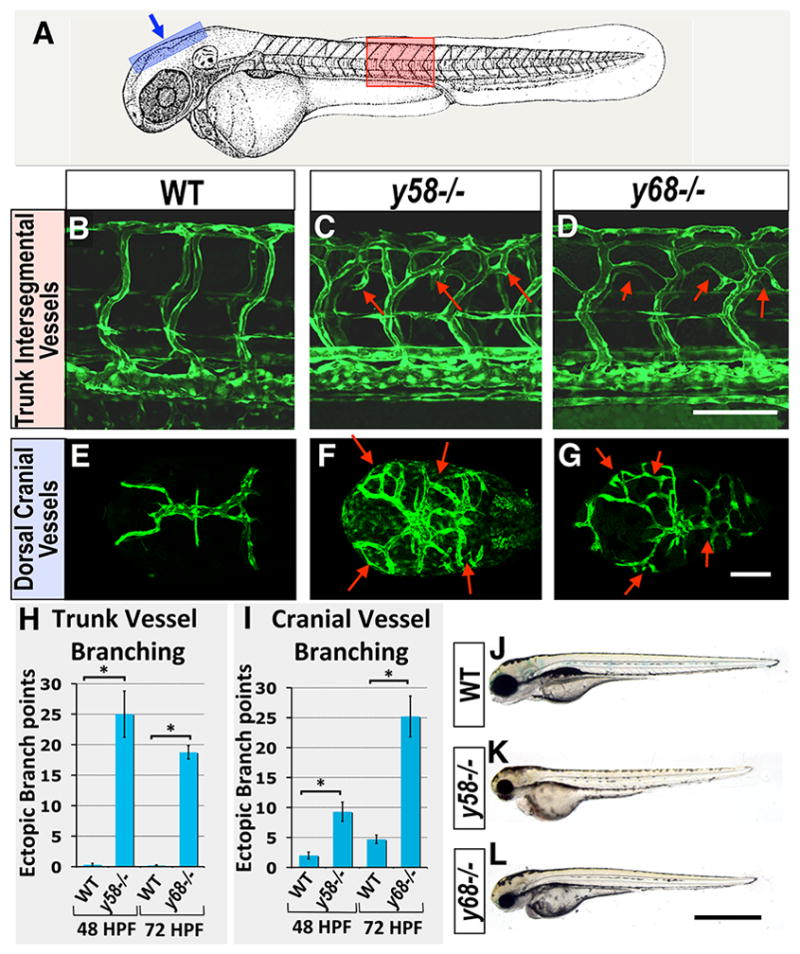
(A) Schematic diagram of a 2 dpf zebrafish (modified from (22)), with a red box showing the approximate position of images in B–D and a blue box showing the approximate position of images in E–G. (B–D) Confocal fluorescence micrographs of the mid-trunk vasculature of 3 dpf Tg(fli1a-EGFP)y1 transgenic wild type sibling (B), y58 mutant (C), and y68 mutant (D) animals. Lateral views, rostral to the left. (E–G) Confocal fluorescence micrographs of the dorsal cranial vasculature of 3 dpf Tg(kdrl:GFP)la116 transgenic wild type sibling (E), Tg(fli1a-EGFP)y1 y58 mutant (F), and Tg(kdrl:GFP)la116 y68 mutant (G) animals. Dorsal views, rostral to the left. Arrows in C, D, F, and G note ectopic vessel branches observed in y58 and y68 mutants. (H) Quantitation of the number of ectopic trunk intersegmental vessel branch points observed in y58 (N = 9)and y68 (N=23) mutants and their phenotypically wild type siblings (N = 19 for y58, N = 20 for y68). (I) Quantitation of the number of ectopic dorsal cranial vessel branch points observed in y58 (N = 6) and y68 (N = 6) mutants and their phenotypically wild type siblings (N = 6 for y58, N = 3 for y68). Ectopic branch points are quantitated at 48 hpf in y58 mutants and their phenotypically wild type siblings, and at 72 hpf in y68 mutants and their phenotypically wild type siblings in both panels H and I. (J–L) Transmitted light images of 3 dpf wild type sibling (J), y58 mutant (K), and y68 mutant (L) animals. Scale bars = 100 μm (B–G), 500 μm (J–L). Significance at P < 0.05 is noted with an asterisk in panels H and I.
Genetic mapping and positional cloning were carried out to identify the defective loci in the y58 and y68 mutants (Supplemental Figure II). The y58 and y68 mutants map to the threonyl tRNA synthetase and isoleucyl tRNA synthetase genes, respectively. These genes encode enzymes responsible for charging tRNAs for protein synthesis by catalyzing the addition of their cognate amino acids. The y58 mutant has a C- to- T mutation changing a Glutamine to a nonsense codon at position 615 of the TARS peptide sequence, resulting in an early termination prior to the anticodon binding domain of TARS (Supplemental Figure IIA). The y68 mutant has a C- to- T mutation changing a conserved Threonine to a Methionine at position 570 of the IARS peptide sequence, within part of the core catalytic domain of IARS (Supplemental Figure IIB). Throughout the rest of the text we refer to the y58 and y68 mutants as tarsy58 and iarsy68, respectively.
We performed additional experiments to further verify that the mutations we discovered in the tars and iars genes are indeed responsible for the mutant phenotypes of tarsy58 and iarsy68 mutants (Figure 2). tars and iars are both expressed in cranial tissues and in the trunk somites adjacent to sites of vessel growth during development of early angiogenic vascular networks (Figure 2A,B). The vascular defects in tarsy58 and iarsy68 mutants can be rescued by injection of Tol2(beta-actin:tarsWT-2A-mCherry) or Tol2(beta-actin:iarsWT-2A-mCherry) transgene constructs expressing the full-length length wild type tars or iars (respecively) under the control of the beta actin promoter, but not by injection of a Tol2(beta-actin:mCherry) construct driving expression of mCherry alone (Figure 2C–I).
Fig 2. The y58 and y68 mutations are defective in threonyl tRNA synthetase (tarsy58) and isoleucyl tRNA synthetase (iarsy68), respectively.

(A,B) Whole mount in situ hybridization of 24 hpf embryos probed for threonyl tRNA synthetase (tars, panel A) and isoleucyl tRNA synthetase (iars, panel B), showing expression in the somites. (C–E) Confocal fluorescence micrographs of the mid-trunk vasculature of 2 dpf Tg(fli1a-EGFP)y1 transgenic tarsy58−/− mutant larvae that were either un-injected (C), injected with Tol2(beta-actin:mCherry) transgene DNA (D), or injected with Tol2(beta-actin:tarsWT-2A-mCherry) transgene DNA (E). (F–H) Confocal fluorescence micrographs of the mid-trunk vasculature of 3 dpf Tg(fli1a-EGFP)y1 transgenic iarsy68−/− mutant larvae that were either un-injected (F), injected with Tol2(beta-actin:mCherry) transgene DNA (G), or injected with Tol2(beta-actin:iarsWT-2A-mCherry) transgene DNA (H). Panels C–H show lateral views of the trunk, rostral to the left. (I) Quantitation of the number of ectopic trunk intersegmental vessel branch points observed in phenotypically wild type (WT) siblings (Column 1, N = 10), Tol2(beta-actin:mCherry)-injected tarsy58−/− mutants (Column 2, N = 7), Tol2(beta-actin:tarsWT-2A-mCherry)-injected tarsy58−/− mutants (Column 3, N = 4), Tol2(beta-actin:mCherry)-injected iarsy68−/− mutants (Column 4, N = 9), or Tol2(beta-actin:iarsWT-2A-mCherry)-injected iarsy68−/− mutants (Column 5, N = 10). (J–M) Confocal fluorescence micrographs of the dorsalmost cranial vasculature of 3 dpf Tg(kdrl:GFP)la116 uninjected (J,L), 2.5 ng of tars ATG morpholino-injected (K), or 2.5 ng of iars ATG morpholino-injected (M) animals. Panels J–M show dorsal views, rostral to the left. (N) Quantitation of the number of ectopic dorsal cranial vessel branch points observed in phenotypically wild type siblings (Column 1, N = 4), tarsy58−/− mutants (Column 2, N = 6), or wild type animals injected with 2.5 ng of tars ATG morpholino (Column 3, N = 6). (O) Quantitation of the number of ectopic dorsal cranial vessel branch points observed in phenotypically wild type siblings (Column 1, N = 3), iarsy68−/− mutants (Column 2, N = 5), or wild type animals injected with 2.5 pg of iars ATG morpholino (Column 3, N = 5). Ectopic cranial branch points are quantitated at 48 hpf in tars mutants and morphants, and at 72 hpf in iars mutants and morphants. Scale bars = 100 μm. Significance at P < 0.05 is noted with an asterisk in panels I, N, O (NS = not significant).
To further confirm that defects in tars or iars function lead to angiogenic defects, we performed knockdown of tars and iars by injecting morpholino antisense oligonucleotides (morpholinos, (22)) targeting the translation start sites of each of the two genes. Injection of tars or iars ATG morpholinos causes excess vascular branching phenotypes similar to those observed in tarsy58 or iarsy68 mutants, respectively (Figure 2J–O). Together, these results indicate that loss of either threonyl tRNA synthetase or isoleucyl tRNA synthetase leads to increased angiogenesis.
As noted above, recent work has shown that many aminoacyl-tRNA synthetases possess non-canonical activities that do not depend on the tRNA charging function of these enzymes (reviewed in (23)), including both pro- and anti-angiogenic functions for “extra” protein domains of tRNA synthetases (11–13, 15, 16, 19–21). We examined whether, in similar fashion, the aminoacylation activity of tars is dispensable for its anti-angiogenic activity. We used site-directed mutagenesis to alter a residue in the active site of tars (Supplemental Figure IIIA) previously shown to be essential in the aminoacylation reaction (24). As expected, the mutated tars(H365A) product was inactive in an in vitro aminoacylation assay (Supplemental Figure IIIB). When wild type tars or mutated tars(H365A) DNA was injected into tarsy58 mutant zebrafish, the mutant protein was was unable to significantly rescue the defect in tarsy58 mutants (Supplemental Figure IIIC), unlike the wild type DNA, suggesting that unlike other tRNA synthetases with clear non-canonical roles in angiogenesis (reviewed in (10)), the anti-angiogenic activity of tars requires its aminoacylation activity.
The Unfolded Protein Response pathway and vegfaa are up-regulated in tarsy58 and iarsy68 mutants
We wished to further examine the molecular basis for the increased angiogenesis observed in tarsy58 and iarsy68 mutants and the relationship between increased angiogenesis and reduced levels of these two aminoacyl tRNA synthetases. Previous work has shown that deficits in protein synthesis can lead to up-regulation of the Unfolded Protein Response (UPR) pathway, a cellular stress response pathway that senses improperly folded proteins and responds by activating pro-survival response genes, apoptotic genes, or both sets of genes (5). As noted above, the atf4, atf6, and xbp1 genes are all key components of different arms of the UPR pathway, and all three genes are up-regulated upon activation of this pathway (5). To determine whether the UPR pathway is activated in tarsy58 and iarsy68 mutants, we used whole mount in situ hybridization (WISH) and quantitative RT-PCR to examine whether expression levels of atf4, atf6, and xbp1 are increased in mutant animals (Figure 3). Indeed, WISH staining showed that cranial and trunk expression of atf4 (Figure 3A, D, G), atf6 (Figure 3B, E, H), and xbp1 (Figure 3C, F, I) are all increased in tarsy58 and iarsy68 mutants. Quantitative RT-PCR measurement revealed that the three UPR genes are each elevated between 2.5- and 3.6-fold in tarsy58 mutants (Figure 3J) and slightly less, between 1.7- and 1.9–fold, in iarsy68 mutants (Figure 3K).
Fig 3. Unfolded Protein Response (UPR) pathway genes are up-regulated in tarsy58 and iarsy68 mutant animals.

(A–I) Whole mount in situ hybridization of 48 hpf wild-type (A, B, C), tarsy58 −/− (D, E, F), and iarsy68 −/− (G, H, I) mutant embryos probed for atf4 (A, D, G), atf6 (B, E, H), and xbp1 (C, F, I). All images are lateral views, rostral to the left. (J–L) TaqMan analysis of analysis of atf4, atf6, xbp1, and vegfaa transcript levels in 48 hpf tarsy58 and iarsy68 embryos. Target transcript levels are normalized to the reference gene eef1a1|1 and to levels in wild type sibling controls, which are set to 1. All graphs show mean ± SEM, and include statistical significance calculated by paired t-test. N = 3 pooled biological replicates of >/= 25 embryos, with 3 technical replicates per N. Significance at P < 0.05 is noted with an asterisk in panels J, K, L. Scale bar = 500 μm.
Previous studies have also reported that expression of the key pro-angiogenic ligand VEGF-A is up-regulated by UPR pathway activation (6, 7). We used quantitative RT-PCR to examine expression of vegfaa, the zebrafish ortholog of VEGF-A responsible for production of the two lower molecular weight VEGF-A isoforms. We found that vegfaa is increased approximately 3.9-fold in tarsy58 and 2.6-fold in iarsy68 mutants (Figure 3L), suggesting that increased vegfaa stimulation may be the cause of the increased angiogenesis observed in mutant animals.
We also tested for potential activation of several other stress pathways using Western blotting of wild type and tarsy58 mutant protein extracts and probing for (i) phospho-p38 vs. total p38, (ii) nuclear vs. cytoplasmic relA, and (iii) phospho-AKT vs. total AKT (Supplemental Figure IV). We did not see significant change in p38 phosphorylation, relA localization, or AKT phosphoryation in mutants compared to their phenotypically wild type siblings. Together, our results suggest that increased VEGF expression and increased intersegmental vessel angiogenesis in the zebrafish trunk is a likely consequence of UPR pathway induction due to reduced levels of TARS or IARS.
Unfolded Protein Response pathway signaling is necessary for increased angiogenesis and vegfaa up-regulation in tarsy58 mutants
To further explore the role of UPR signaling in promoting angiogenesis we carried out additional experiments to determine whether specifically activating UPR pathway components alone can induce pro-angiogenic phenotypes. As noted above, recent evidence suggests that protein kinase RNA-like endoplasmic reticulum (ER) kinase (PERK) activation of transcription factor ATF4 may play a major role in regulating VEGF expression downstream from the UPR pathway (8). To examine whether atf4 function is necessary for the increased angiogenesis and elevated vegfaa levels observed in tarsy58 mutant zebrafish embryos, we injected morpholinos targeting atf4 into tarsy58 mutant embryos or their wild type siblings (Figure 4A–F). Injection of atf4 morpholinos into wild type embryos did not cause developmental delay or changes in gross morphology (Supplemental Figure V), and did not result in angiogenic phenotypes or alteration in vegfaa levels (Figure 4A,C,E,F). However, injection of atf4 MO into tarsy58 mutants resulted in nearly complete reversal of vessel branching phenotypes (Figure 4B,D,E) and strong reduction in excess vegfaa expression (Figure 4F), suggesting that UPR pathway induction is necessary for both vegfaa expression and increased angiogenesis in tars-deficient animals.
Fig 4. The atf4 gene is required for increased angiogenesis and vegfaa up-regulation in tarsy58 mutants.

(A–D) Confocal fluorescence micrographs of the mid-trunk vasculature of 2 dpf wild type sibling (A,C) or tarsy58−/− mutant (B,D) Tg(fli1a-EGFP)y1 larvae that were either un-injected (A,B) or injected with 2.5 pg of atf4 morpholino (C,D). (E) Quantitation of the number of ectopic trunk intersegmental vessel branch points observed in 2 dpf Tg(fli1a-EGFP)y1 transgenic animals comparable to those shown in panels A through D (N = 10). (F) TaqMan analysis of vegfaa transcript levels in 48 hpf animals, with conditions as in panels A–D. Target transcript levels are normalized to the reference gene eef1a1|1 and to levels in un-injected wild type controls, which are set to 1. N = 2 pooled pooled biological replicates of >/= 25 embryos, with 3 technical replicates per N. All columns show mean ± SEM, and include statistical significance calculated by paired t-test. Significance at P < 0.05 is noted with an asterisk in panels E and F. Scale bar = 100 μm.
DISCUSSION
The zebrafish has emerged as an important new model for uncovering novel genes and pathways regulating vessel formation during development, in large part through forward-genetic screens for mutants with vascular phenotypes (25, 26). Many of the genes identified are also important players in vascular pathologies (27–32). We carried out a genetic screen using zebrafish with fluorescently labeled blood vessels (Tg(fli1a-EGFP)y1) to identify mutants displaying increased growth and branching of angiogenic vessels in the trunk (intersegmental vessels) and head (cranial central arteries). Molecular cloning of two of these mutants identified mutational alterations in two different aminoacyl tRNA synthetases, threonyl tRNA synthetase (tarsy58) and isoleucyl tRNA synthetase (iarsy68). Defects in either gene result in excess trunk and cranial angiogenesis in developing zebrafish, although these defects become apparent somewhat later in iarsy68 mutants. We verified that the mutant defects in tars and iars were indeed causing the excess angiogenesis phenotypes by phenocopying the mutant defects using morpholinos targeting each gene, and by rescuing the defects in each mutant with the corresponding wild-type gene product, by injection of tars and iars containing transgenes (the relatively later appearance of the phenotype in mutants necessitated using a transgenic approach rather then short-lived mRNA). The expression of Unfolded Protein Response (UPR) pathway genes is up-regulated in both tarsy58 and iarsy68 mutants, as is the key pro-angiogenic ligand vascular endothelial growth factor (VEGF), and we show that UPR pathway activation induces vegfaa expression and angiogenesis in wild type animals. ATF4, a transcription factor functioning downstream from PERK in the UPR pathway, is required for both the increased angiogenesis and elevated levels of vegfaa observed in tarsy58 mutant animals. Together, these results suggest that defects in either tars or iars result in increased angiogenesis via up-regulation of the UPR pathway.
Aminoacyl-transfer RNA (tRNA) synthetases (AARSs) play an essential role in protein synthesis, catalyzing ligation of amino acids to their cognate tRNAs. In addition to this canonical role, recent studies have also documented a variety of important noncanonical functions for AARS proteins, including both pro- and anti-angiogenic activities (10–12, 14–16, 19, 20, 23, 33–36). In many cases specific pro- or anti-angiogenic activities have been attributed to accessory domains or cleavage fragments of tRNA synthetases, including the pro-angiogenic effects of an N-terminal fragment of tyrosyl-tRNA synthetase (YARS) (11–13) or the anti-angiogenic effects of a C-terminal fragment of tryptophanyl-tRNA synthetase (WARS) (14–16). These have been reported to act via different mechanisms to modulate VEGF signaling, VE-cadherin activation, or other endothelial cell functions. Some of these effects have been attributed to cell autonomous mechanisms, while in other cases non-cell autonomous effects have been reported for AARS protein fragments, although the mechanism of export or release of the fragments mediating these non-cell autonomous effects has not been elucidated in all cases. Previous genetic screens in the zebrafish identified mutants in Seryl tRNA synthetase (SARS or SerRS) (18–20). Like the IARS and TARS mutants described here, zebrafish SARS mutants also display ectopic branching of cranial and trunk vessels and up-regulation of vegfaa. Injection of morpholinos targeting VEGF pathway components or treatment with VEGF pathway inhibitors suggests that excess branching is VEGF signaling dependent (19, 20). An enzymatically dead form of SARS is able to suppress both excess branching and vegfaa induction, suggesting the angiostatic effects of SARS are independent of its aminoacylation activity (19). In contrast, we find that the aminoacylation activity of TARS is required for its role in moderating angiogenesis via UPR pathway-mediated up-regulation of VEGF expression. Since VEGF is expressed predominately in medial portions of the somites in the developing zebrafish trunk (37–39), the effects of UPR activation on the endothelium and angiogenesis are presumably non-autonomous, reflecting the “indirect” effects of UPR-induced, somite-produced VEGF on the adjacent intersomitic vessel endothelial cells. Additional experiments in zebrafish using tissue-specific mosaic gene expression or cell transplantation using combinations of TARS mutant and wild type donor and host animals would be needed to provide definitive validation of this, however. Injection of DNA for tars(H365A), a point mutant that has lost its aminoacylation activity, fails to significantly rescue tarsy58 mutants. This suggests that up-regulation of UPR and vegfaa expression in TARS-deficient animals is likely downstream from loss of aminoacylation activity. Since vegfaa is also elevated in SARS mutants (19, 20), as it is in both TARS- and IARS-deficient animals, it will be useful to determine whether the as-yet-unidentified noncanonical activity of SARS mediating vegfaa induction and/or angiogenesis also acts via the UPR pathway. Zebrafish intersegmental vessel overbranching phenotypes similar to those we show in TARS mutants were also described in a recently published report utilizing morpholino knockdown of TARS(40), although in vitro experiments using pharmacologic inhibitors of TARS in the same report suggested a non-canonical pro-angiogenic role for TARS and the authors proposed that TARS normally plays a proangiogenic “guidance” role for intersegmental vessels. Interestingly, another recent publication has also reported that VEGF signaling can itself “feed back” to activate UPR mediators ATF6 and PERK via PLCg-mediated crosstalk with the mTORC1 complex (41), suggesting that there are additional complexities to be uncovered in the interplay between VEGF and UPR signaling.
Taken together, our results and previous studies suggest that multiple complex mechanisms act together to mediate linkage between aminoacyl tRNA synthetase gene function and angiogenesis. Some of these mechanisms involve non-canonical functions of “accessory” domains or fragments of AARS proteins, while others engage molecular mechanisms downstream from the canonical aminoacylation activity of AARS proteins. This emphasizes the exquisite regulatory control of endothelial responses to the physiological state of the cells and tissues that blood vessels innervate and supply, and reinforces other recent work highlighting metabolic regulation of angiogenesis. For example, deficiency of lipoproteins was recently shown to lead to increased angiogenesis via down regulation of VEGFR1, a high-affinity non-signaling VEGF decoy receptor that is thought to act as a “sink” for VEGF ligand (42). Our in vivo data support the idea that up-regulation of cellular stress pathways by amino acid deprivation acts as a powerful angiogenic stimulus, but our data also have substantial implications beyond the specific genetic defects in aminoacyl tRNA synthetases described here. Cellular stress and UPR pathway induction are associated with a variety of pathological conditions including metabolic disease, inflammation, neurodegenerative disorders, and cancer (43). Angiogenic phenotypes are associated with many or all of these pathologies, hinting that UPR pathway-mediated up-regulation of VEGF might be a factor in their etiology.
Supplementary Material
SIGNIFICANCE.
The Unfolded Protein Response (UPR) has been shown to stimulate angiogenesis, the growth of new blood vessels from preexisting vessels, but the mechanism behind this phenomenon has been difficult to study in vivo. Here we show that mutations in either of two tRNA synthetase genes cause excess angiogenesis in developing zebrafish embryos through the activation of the UPR. Although tRNA synthetases have been shown to be involved in angiogenic regulation through non-canonical mechanisms, this is the first evidence showing that disrupting the canonical function of tRNA synthetase genes can cause excess angiogenesis through the activation of the UPR. This study also highlights the usefulness of the developing zebrafish as an in vivo model for studying metabolic regulation of angiogenesis.
Acknowledgments
We would like to thank the NICHD RAMB staff for their excellent zebrafish husbandry.
Sources of Funding - This research was supported by the intramural program of the NICHD, NIH (to BMW), the Leducq Foundation (to BMW), and by University of Maryland start up funds (to PJP). The Funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
ABREVIATIONS
- VEGF
vascular endothelial growth factor
- UPR
unfolded protein response
- ER
endoplasmic reticulum
- EC
endothelial cell
- TARS
threonyl tRNA synthetase
- IARS
isoleucyl tRNA synthetase
- PERK
protein kinase RNA-like endoplasmic reticulum kinase
- ATF4
activating transcription factor 6
- ATF1
activating transcription factor 1
- IRE1
Inositol-requiring enzyme-1
- XBP1
Xbox binding protein
- AARS
aminoacyl-transfer RNA (tRNA) synthetase
Footnotes
Disclosures - none
References
- 1.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 2.Fong GH. Regulation of angiogenesis by oxygen sensing mechanisms. J Mol Med (Berl) 2009;87:549–560. doi: 10.1007/s00109-009-0458-z. [DOI] [PubMed] [Google Scholar]
- 3.Shweiki D, Neeman M, Itin A, Keshet E. Induction of vascular endothelial growth factor expression by hypoxia and by glucose deficiency in multicell spheroids: implications for tumor angiogenesis. Proc Natl Acad Sci U S A. 1995;92:768–772. doi: 10.1073/pnas.92.3.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abcouwer SF, Marjon PL, Loper RK, Vander Jagt DL. Response of VEGF expression to amino acid deprivation and inducers of endoplasmic reticulum stress. Invest Ophthalmol Vis Sci. 2002;43:2791–8. [PubMed] [Google Scholar]
- 5.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 6.Drogat B, Auguste P, Nguyen DT, et al. IRE1 signaling is essential for ischemia-induced vascular endothelial growth factor-A expression and contributes to angiogenesis and tumor growth in vivo. Cancer Res. 2007;67:6700–6707. doi: 10.1158/0008-5472.CAN-06-3235. [DOI] [PubMed] [Google Scholar]
- 7.Ghosh R, Lipson KL, Sargent KE, et al. Transcriptional regulation of VEGF-A by the unfolded protein response pathway. PLoS One. 2010;5:e9575. doi: 10.1371/journal.pone.0009575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pereira ER, Frudd K, Awad W, Hendershot LM. Endoplasmic reticulum (ER) stress and hypoxia response pathways interact to potentiate hypoxia-inducible factor 1 (HIF-1) transcriptional activity on targets like vascular endothelial growth factor (VEGF) J Biol Chem. 2014;289:3352–3364. doi: 10.1074/jbc.M113.507194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pereira ER, Liao N, Neale GA, Hendershot LM. Transcriptional and post-transcriptional regulation of proangiogenic factors by the unfolded protein response. PLoS One. 2010;5:e12521. doi: 10.1371/journal.pone.0012521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo M, Schimmel P. Homeostatic mechanisms by alternative forms of tRNA synthetases. Trends Biochem Sci. 2012;37:401–403. doi: 10.1016/j.tibs.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wakasugi K, Schimmel P. Two distinct cytokines released from a human aminoacyl-tRNA synthetase. Science. 1999;284:147–151. doi: 10.1126/science.284.5411.147. [DOI] [PubMed] [Google Scholar]
- 12.Wakasugi K, Slike BM, Hood J, Ewalt KL, Cheresh DA, Schimmel P. Induction of angiogenesis by a fragment of human tyrosyl-tRNA synthetase. J Biol Chem. 2002;277:20124–20126. doi: 10.1074/jbc.C200126200. [DOI] [PubMed] [Google Scholar]
- 13.Greenberg Y, King M, Kiosses WB, et al. The novel fragment of tyrosyl tRNA synthetase, mini-TyrRS, is secreted to induce an angiogenic response in endothelial cells. FASEB J. 2008;22:1597–1605. doi: 10.1096/fj.07-9973com. [DOI] [PubMed] [Google Scholar]
- 14.Kise Y, Lee SW, Park SG, et al. A short peptide insertion crucial for angiostatic activity of human tryptophanyl-tRNA synthetase. Nat Struct Mol Biol. 2004;11:149–156. doi: 10.1038/nsmb722. [DOI] [PubMed] [Google Scholar]
- 15.Tzima E, Reader JS, Irani-Tehrani M, Ewalt KL, Schwartz MA, Schimmel P. VE-cadherin links tRNA synthetase cytokine to anti-angiogenic function. J Biol Chem. 2005;280:2405–2408. doi: 10.1074/jbc.C400431200. [DOI] [PubMed] [Google Scholar]
- 16.Wakasugi K, Slike BM, Hood J, et al. A human aminoacyl-tRNA synthetase as a regulator of angiogenesis. Proc Natl Acad Sci U S A. 2002;99:173–177. doi: 10.1073/pnas.012602099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ray PS, Fox PL. A post-transcriptional pathway represses monocyte VEGF-A expression and angiogenic activity. EMBO J. 2007;26:3360–3372. doi: 10.1038/sj.emboj.7601774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amsterdam A, Nissen RM, Sun Z, Swindell EC, Farrington S, Hopkins N. Identification of 315 genes essential for early zebrafish development. Proc Natl Acad Sci U S A. 2004;101:12792–12797. doi: 10.1073/pnas.0403929101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fukui H, Hanaoka R, Kawahara A. Noncanonical activity of seryl-tRNA synthetase is involved in vascular development. Circ Res. 2009;104:1253–1259. doi: 10.1161/CIRCRESAHA.108.191189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herzog W, Muller K, Huisken J, Stainier DY. Genetic evidence for a noncanonical function of seryl-tRNA synthetase in vascular development. Circ Res. 2009;104:1260–1266. doi: 10.1161/CIRCRESAHA.108.191718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu X, Shi Y, Zhang HM, et al. Unique domain appended to vertebrate tRNA synthetase is essential for vascular development. Nat Commun. 2012;3:681. doi: 10.1038/ncomms1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nasevicius A, Larson J, Ekker SC. Distinct requirements for zebrafish angiogenesis revealed by a VEGF-A morphant. Yeast. 2000;17:294–301. doi: 10.1002/1097-0061(200012)17:4<294::AID-YEA54>3.0.CO;2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo M, Yang XL, Schimmel P. New functions of aminoacyl-tRNA synthetases beyond translation. Nat Rev Mol Cell Biol. 2010;11:668–674. doi: 10.1038/nrm2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sankaranarayanan R, Dock-Bregeon AC, Romby P, et al. The structure of threonyl-tRNA synthetase-tRNA(Thr) complex enlightens its repressor activity and reveals an essential zinc ion in the active site. Cell. 1999;97:371–381. doi: 10.1016/s0092-8674(00)80746-1. [DOI] [PubMed] [Google Scholar]
- 25.Kamei M, Isogai S, Pan W, Weinstein BM. Imaging blood vessels in the zebrafish. Methods Cell Biol. 2010;100:27–54. doi: 10.1016/B978-0-12-384892-5.00002-5. [DOI] [PubMed] [Google Scholar]
- 26.McKinney MC, Weinstein BM. Chapter 4. Using the zebrafish to study vessel formation. Methods Enzymol. 2008;444:65–97. doi: 10.1016/S0076-6879(08)02804-8. [DOI] [PubMed] [Google Scholar]
- 27.Buchner DA, Su F, Yamaoka JS, et al. pak2a mutations cause cerebral hemorrhage in redhead zebrafish. Proc Natl Acad Sci U S A. 2007;104:13996–14001. doi: 10.1073/pnas.0700947104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Childs S, Weinstein BM, Mohideen MA, Donohue S, Bonkovsky H, Fishman MC. Zebrafish dracula encodes ferrochelatase and its mutation provides a model for erythropoietic protoporphyria. Curr Biol. 2000;10:1001–1004. doi: 10.1016/s0960-9822(00)00653-9. [DOI] [PubMed] [Google Scholar]
- 29.Gore AV, Lampugnani MG, Dye L, Dejana E, Weinstein BM. Combinatorial interaction between CCM pathway genes precipitates hemorrhagic stroke. Dis Model Mech. 2008;1:275–281. doi: 10.1242/dmm.000513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lawson ND, Mugford JW, Diamond BA, Weinstein BM. phospholipase C gamma-1 is required downstream of vascular endothelial growth factor during arterial development. Genes Dev. 2003;17:1346–1351. doi: 10.1101/gad.1072203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu J, Fraser SD, Faloon PW, et al. A betaPix Pak2a signaling pathway regulates cerebral vascular stability in zebrafish. Proc Natl Acad Sci U S A. 2007;104:13990–13995. doi: 10.1073/pnas.0700825104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roman BL, Pham VN, Lawson ND, et al. Disruption of acvrl1 increases endothelial cell number in zebrafish cranial vessels. Development. 2002;129:3009–3019. doi: 10.1242/dev.129.12.3009. [DOI] [PubMed] [Google Scholar]
- 33.Otani A, Slike BM, Dorrell MI, et al. A fragment of human TrpRS as a potent antagonist of ocular angiogenesis. Proc Natl Acad Sci U S A. 2002;99:178–183. doi: 10.1073/pnas.012601899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ray PS, Jia J, Yao P, Majumder M, Hatzoglou M, Fox PL. A stress-responsive RNA switch regulates VEGFA expression. Nature. 2009;457:915–919. doi: 10.1038/nature07598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tzima E, Reader JS, Irani-Tehrani M, Ewalt KL, Schwartz MA, Schimmel P. Biologically active fragment of a human tRNA synthetase inhibits fluid shear stress-activated responses of endothelial cells. Proc Natl Acad Sci U S A. 2003;100:14903–14907. doi: 10.1073/pnas.2436330100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tzima E, Schimmel P. Inhibition of tumor angiogenesis by a natural fragment of a tRNA synthetase. Trends Biochem Sci. 2006;31:7–10. doi: 10.1016/j.tibs.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 37.Lawson ND, Vogel AM, Weinstein BM. sonic hedgehog and vascular endothelial growth factor act upstream of the Notch pathway during arterial endothelial differentiation. Dev Cell. 2002;3:127–136. doi: 10.1016/s1534-5807(02)00198-3. [DOI] [PubMed] [Google Scholar]
- 38.Liang D, Chang JR, Chin AJ, et al. The role of vascular endothelial growth factor (VEGF) in vasculogenesis, angiogenesis, and hematopoiesis in zebrafish development. Mech Dev. 2001;108:29–43. doi: 10.1016/s0925-4773(01)00468-3. [DOI] [PubMed] [Google Scholar]
- 39.Liang D, Xu X, Chin AJ, et al. Cloning and characterization of vascular endothelial growth factor (VEGF) from zebrafish, Danio rerio. Biochim Biophys Acta. 1998;1397:14–20. doi: 10.1016/s0167-4781(97)00233-9. [DOI] [PubMed] [Google Scholar]
- 40.Mirando AC, Fang P, Williams TF, et al. Aminoacyl-tRNA synthetase dependent angiogenesis revealed by a bioengineered macrolide inhibitor. Sci Rep. 2015;5:13160. doi: 10.1038/srep13160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karali E, Bellou S, Stellas D, Klinakis A, Murphy C, Fotsis T. VEGF Signals through ATF6 and PERK to promote endothelial cell survival and angiogenesis in the absence of ER stress. Mol Cell. 2014;54:559–572. doi: 10.1016/j.molcel.2014.03.022. [DOI] [PubMed] [Google Scholar]
- 42.Avraham-Davidi I, Grunspan M, Yaniv K. Lipid signaling in the endothelium. Exp Cell Res. 2013;319:1298–1305. doi: 10.1016/j.yexcr.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 43.Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol. 2012;197:857–867. doi: 10.1083/jcb.201110131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.