

Abstract
Series of 4-amino-6-(arylamino)-1,3,5-triazine-2-carbohydrazides (3a–e) and N′-phenyl-4,6-bis(arylamino)-1,3,5-triazine-2-carbohydrazides (6a–e), for ease of readership, we will abbreviate our compound names as “ new triazines”, have been synthesized, based on the previously reported Rad6B-inhibitory diaminotriazinylmethyl benzoate anticancer agents TZ9 and 4-amino-N′-phenyl-6-(arylamino)-1,3,5-triazine-2-carbohydrazides. Synthesis of the target compounds was readily accomplished in two steps from either bis-aryl/aryl biguanides via reaction of phenylhydrazine or hydrazinehydrate with key 4-amino-6-bis(arylamino)/(arylamino)-1,3,5-triazine-2-carboxylate intermediates. These new triazine derivatives were evaluated for their abilities to inhibit Rad6B ubiquitin conjugation and in vitro anticancer activity against several human cancer cell lines: ovarian (OV90 and A2780), lung (H1299and A549), breast (MCF-7 and MDA-MB231) and colon (HT29) cancer cells by MTS assays. All the 10 new triazines exhibited superior Rad6B inhibitory activities in comparison to selective Rad6 inhibitor TZ9 that was reported previously. Similarly, new triazines also showed better IC50 values in survival assays of various tumor cell lines. Particularly, new triazines 6a–c, exhibited lower IC50 (3.3 to 22 μM) values compared to TZ9.
Keywords: Ubiquitination, E2 ubiquitin conjugating enzyme, Rad6B, anticancer, triazines
Graphical abstract
Series of 4-amino-6-(arylamino)-1,3,5-triazine-2-carbohydrazides (3a–e) and N′-phenyl-4,6-bis(arylamino)-1,3,5-triazine-2-carbohydrazides (6a–e) have been synthesized. New triazines showed better IC50 values in survival assays of various tumor cell lines. Particularly, new triazines 6a–c, exhibited much lower IC50 (3.3 to 22 μM) values compared to TZ9 (the previously reported Rad6B-inhibitory diaminotriazinylmethyl benzoate anticancer agent). Moreover, when compared to TZ9 in in vitro ubiquitin conjugation assays, compounds 6a, b, c, e exhibited superior Rad6B inhibitory properties, both in its conjugation to ubiquitin and to the substrate H2A.
The ubiquitin-proteasome system controls the turnover of regulatory proteins involved in critical cellular processes including cell cycle progression, cell development and differentiation, apoptosis, angiogenesis and cell signaling pathways 1–2. This system requires the action of three enzymes: E1 ubiquitin activating enzyme, E2 ubiquitin conjugating enzyme and E3 ubiquitin ligase 3. Firstly, the ubiquitin is activated by the E1 activating enzyme and once it is activated, it is then transferred to E2 conjugating enzyme. The final step is the formation of an isopeptide bond between a lysine of the target protein and the C-terminal glycine of ubiquitin (carried by E2). This step usually requires the action of an E3 ubiquitin ligase 4.
Interference with the proteasome activity was proven to be effective in cancer therapeutics since the clinical approval of bortezomib (Velcade®) as a proteasome inhibitor for treatment of relapsed multiple myeloma and mantle cell lymphoma 5. However, the requirement for more specific inhibiting targets like the design of potential E2 or E3 inhibitors, has appeared in order to reduce the side effects resulting from bortezomib 6. Recently, many E1 and E3 ligase inhibitors such as PYR-41, Nutlin-3a, P013222 and SCF-I2 have been successful and progressed to preclinical/clinical development. Also, the approved myeloma drug thalidomide has been recently identified as an E3 ligase inhibitor 7.
Among the E2 ubiquitin conjugating enzyme family, Rad6B is of special interest since it is found to be over-expressed in many human cancer cell lines and tumors 8–9. Constitutive over-expression of Rad6B in the non-transformed human breast epithelial cell line MCF 10A induces a number of adverse effects associated with cancer progression such as formation of multinucleated cells, centrosome amplification, abnormal mitosis, aneuploidy, and transformation 10. Most importantly, Rad6B has been shown to positively regulate β-catenin stabilization and activity that drives the malignant progression of breast cancer cells 11–13. Since β-catenin-mediated signaling has been implicated in many human malignancies, including lung, colon, breast, and ovarian, it has been an important therapeutic target. Furthermore, Rad6B plays a central role in regulation of multiple DNA repair pathways through its interactions with different E3 ubiquitin ligases. For example, Rad6 partners with the E3 ubiquitin ligase Rad18 and monoubiquitinates PCNA in response to replication for k-stalling lesions to promote translesion synthesis (TLS) or the DNA damage tolerance pathway 14–17. Rad18/Rad6 ubiquitin ligase complex is also important in the activation of the Fanconi anemia tumor suppressor pathway, which plays critical roles in genome integrity and tumor resistance to a variety of chemotherapeutic agents, including those that induce DNA crosslinks and DNA double strand breaks 17–18. Rad6 has also been shown to associate with RNF168 to monoubiquitinate histone H1.2 thereby enabling chromatin relaxation and allowing DNA damage response factors access to damage sites 19. Moreover, increased expression or activation of these DNA damage response (DDR) signaling and repair genes accounts for tumor resistance to chemotherapy 9, 20–22. Therefore, development of DNA damage response and repair signal inhibitors are important to effectively treat these tumors.
We have recently reported [4-amino-6-(arylamino)-1,3,5-triazin-2-yl]methyl 4-nitrobenzoates TZ8–TZ9 (Fig. 1) as novel and selective Rad6B-inhibitory lead compounds 23. These inhibitors were identified by virtual screening of a pharmacophore model generated from the conserved key residues stabilizing the E2-ubiquitin thioester intermediate against a pre-prepared database using drug-like filters which determined the substituted diaminotriazine core structure as a starting point for analogue synthesis. Triazine analogue synthesis coupled to in vitro anticancer evaluation led to the identification of lead compounds TZ8–TZ9 23.
Fig. 1.

Chemical structures of Rad6B-inhibitory lead compounds TZ8 and TZ9
Using a molecular modeling approach to guide the design of new derivatives of TZ8 and TZ9, we reported 4-amino-N′-phenyl-6-(arylamino)-1,3,5-triazine-2-carbohydrazides (Fig. 2) with IC50 values (2.48–4.79 μM) superior to those of TZ8 and TZ9 when tested on the Rad6B-expressing MDA-MB-231 cell line 24. In docking studies, such triazinecarbohydrazide derivatives were found to be incorporated deep inside the Rad6B binding pocket, making key interactions between the hydrazine nitrogen atoms and the Rad6B active site residues Cys88 and Asp90. Additional interactions between the aniline nitrogens and Asn119/Gln93 and between the phenyl (hydrazide) ring and Leu89 were also apparent from our docking analysis. The importance of these active site residues to the allosteric effect on Rad6B induced by E3 ligases, and the observation that no other E2 family members (with the exception of Rad6A) have residues corresponding to Gln93 or Asn119, suggest that these triazinecarbohydrazides could be selective Rad6B inhibitors 24.
Fig. 2.

4-amino-N′-phenyl-6-(arylamino)-1,3,5-triazine-2-carbohydrazides
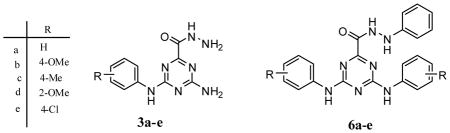
In the current work, we studied the effect of removal of the phenyl (hydrazide) ring (3a–e) or the addition of aryl substituent to the free amino group (6a–e) of our previously reported 4-amino-N′-phenyl-6-(arylamino)-1,3,5-triazine-2-carbohydrazides (Fig. 2) on the Rad6B inhibitory activity and cellular anticancer activity. (Fig. 3)
Fig. 3.

SAR modifications of previous lead compounds
The synthesis of the 4-amino-6-(arylamino)-1,3,5-triazine-2 carbohydrazide (3a–e) was accomplished in two steps from arylbiguanide hydrochloride salts (1a–e), which were prepared from commercially available substituted aniline and dicyandiamide according to previously reported procedures 25–26. Neutralization of the arylbiguanide hydrochloride salt using sodium methoxide/methanol was followed by reaction with dimethyloxalate in refluxing methanol. This gave the intermediate methyl 4-amino-6-(arylamino)-1,3,5-triazine-2-carboxylates (2a–e) in 83–92% isolated yield following recrystallization from methanol. Reaction of intermediates (2a–e) with hydrazinehydrate in refluxing ethanol produced the target new triazines (3a–e) in high yield (81–92%) following recrystallization from ethanol (Scheme 1).
Scheme 1.

Synthetic pathway for compounds 3a–e
Reagents and conditions: (i) NaOCH3, CH3OH, room temp. 3 h; (ii) dimethyloxalate, CH3OH, reflux, 4 h; (iii) hydrazine hydrate, EtOH, reflux 3 h.
A similar strategy was used to synthesize the N′-phenyl-4,6-bis(arylamino)-1,3,5-triazine-2-carbohydrazides (6a–e) from bis-arylbiguanide hydrochloride salts (4a–e), which were prepared from commercially available substituted aniline and sodium dicyanamide according to previously reported procedures 25–26. Neutralization of the bis-arylbiguanide hydrochloride salt using sodium methoxide/methanol was followed by reaction with dimethyloxalate in refluxing methanol. This produced the intermediate methyl 4,6-bis(arylamino)-1,3,5-triazine-2-carboxylates (5a–e) in 72–87% isolated yield following recrystallization from methanol. Reaction of intermediates (5a–e) with phenylhydrazine in refluxing ethanol, catalyzed by glacial acetic acid produced the target new triazines (6a–e) in good yield (76–86%) following recrystallization from ethanol: water (3:1) (Scheme 2).
Scheme 2.

Synthetic pathways for compounds 6a–e
Reagents and conditions: (i) NaOCH3, CH3OH, room temp. 3 h; (ii) dimethyloxalate, CH3OH, NaOCH3 reflux, 12 h; (iii) phenylhydrazine, EtOH, AcOH, reflux 18 h.
Evaluation of newly synthesized 4-amino-6-(arylamino)-1,3,5-triazine-2-carbohydrazide (3a–e) and N′-phenyl-4,6-bis(arylamino)-1,3,5-triazine-2-carbohydrazide (6a–e) was carried out in the human cancer cell lines OV90 and A2780 (ovarian), H1299 and A549 (lung), MCF-7 and MDA-MB231 (breast), and HT29 (colon) using MTS assay reagent (Promega) to assess the cell viability (Table 1). TZ9 was used as a positive control and aqueous DMSO as a negative control.
Table 1.
IC50 Values for New Triazine Analogues.
| Name of Compound | Formula Weight | OV 90 | A2780 | MCF-7 | MDA-MB231 | A549 | H1299 | HT-29 |
|---|---|---|---|---|---|---|---|---|
| TZ9 | 366.3 | 60 | 7.8 | 5 | 4.6 | 7.2 | 45 | 8.3 |
| 3a | 245.24 | 75 | 7.3 | 5.9 | 2.6 | 7.8 | 65 | 3.3 |
| 3b | 275.27 | 80 | 14.7 | 6.2 | 7.6 | 5.2 | 65 | 5.4 |
| 3c | 259.27 | 60 | 5.5 | 16 | 12.7 | 9.3 | 40 | 4.5 |
| 3d | 275.27 | 90 | 5.6 | 11.8 | 15.7 | 7.5 | 50 | 8.6 |
| 3e | 279.69 | 85 | 36.6 | 8.8 | 24.6 | 7.5 | 30 | 4.6 |
| 6a | 397.43 | 8 | 7.1 | 6 | 2.5 | 14.6 | 11 | 9.5 |
| 6b | 457.48 | 12 | 6.3 | 7.2 | 4.2 | 10.8 | 5 | 5.8 |
| 6c | 425.49 | 5 | 3.6 | 4.2 | 3.5 | 11.6 | 22 | 5.2 |
| 6d | 457.48 | 10 | 5 | 7.2 | 3.9 | 6.9 | 26 | 8.9 |
| 6e | 466.32 | 60 | 8.2 | 4.2 | 3.5 | 7.8 | 15 | 4.1 |
As described in the methods, the MTS assay was used to evaluate cell survival in the presence of each new triazine analogue in the range of 0 to 125 μM concentrations. For each cell line, the MTS assays were performed at least three independent experiments and each time in triplicates. The results presented in the table are average values from multiple experiments. The IC50 values were calculated from the data of % survival vs. drug concentration using Microsoft Excel 2010.
To assess the efficacy of these compounds in cell cultures, MTS assays were performed using multiple tumor cell lines from different tissue origins. We have randomly chosen these cell lines as many of these tumors were previously shown to either over express Rad6 or β-catenin mediated signaling. The IC50 values for each of the compounds against the individual cell line were calculated from their respective dose response curves. As shown in Table 1, almost all the compounds inhibited survival of cancer cells, although their IC50 concentrations and specificities to the cell lines varied. For example, compounds 3a–e, 6a–e and TZ9 showed IC50 values in the low micromolar range (3.3–16 μM) when tested on HT-29 and MCF-7 cell lines, whereas only compounds 6a–d show low micromolar IC50 values (3.6–12 μM) when tested on OV 90 and A2780 cell lines. These values compare favorably with the studies on TZ9. The compound 6b shows the lowest IC50 value (5 μM) when tested on the H1299 cell line. However, the reasons for these biological selectivities to particular cell lines and differences in IC50 values of the compounds need further evaluation. Moreover, these variations could be attributed to differences in functional groups of these compounds and inherent molecular signatures of the tumor cells, which may include their Rad6B status and their inherent dependence on its mediated signaling networks.
To evaluate the efficacy of newly synthesized 4-amino-6-(arylamino)-1,3,5-triazine-2-carbohydrazide (3a–e) and N′-phenyl-4,6-bis(arylamino)-1,3,5-triazine-2-carbohydrazide (6a–e) analogues for their Rad6 inhibitory activities, in vitro ubiquitin conjugation assays were performed in comparison with our previously reported Rad6 inhibitor TZ9. In these assays (Fig. 4A and 4B), ubiquitin conjugation to Rad6 enzyme readily occurs in the positive control (lane 3 in Fig. 4A and lane 2 in Fig. 5A), but not in the negative controls, which lacks either Rad6 (lane 1 in Fig. 4A.) or ubiquitin (lane 2 in Fig. 4A and lane 1 in Fig. 5A) in these assays. Consistent with the previous data, pretreatment of Rad6 with TZ9 inhibited its ability to conjugate with ubiquitin 23. Interestingly, in the initial screening experiments, all the new triazines exhibited Rad6 inhibitory activity (Fig 4A and 4B). As shown in the figures (4B and 5B) most of the new triazines are better Rad6 inhibitors than TZ9 at the equimolar concentration (25 nM), for both inhibition of Rad6 conjugation to ubiquitin and its substrate H2A ubiquitination in these assays.
Figure 4.

New triazine analogues inhibit conjugation of ubiquitin to Rad6B more effectively than TZ9. A). Representative western blot of in vitro ubiquitin conjugation experiment showing all new triazine analogues in comparison with TZ9, and probed with Rad6 antibody. Rad6-; negative control with all experimental components except Rad6B. Ub-; negative control with all experimental components except ubiquitin. Control: positive control with all experimental components that shows Rad6B conjugation to ubiquitin. Experiment was repeated for reproducibility at a minimum of two times. B). Densitometry calculations of Rad6B-Ub bands were made using ImageJ software on the western blot shown in (A).
Figure 5.

Rad6-mediated ubiquitination of H2A is inhibited by new triazine analogues. A). Western blot showing Rad6-mediated ubiquitination of histone H2A in the presence of new triazine analogues probed with ubiquitin, H2A and Rad6 antibodies. B). Represents densitometric values of western blot shown in (A), were calculated using ImageJ software and presented as relative to positive control. All experiments were repeated at least twice to verify reproducibility.
To further assess their inhibitory effects on Rad6B ability to transfer ubiquitin to the substrate, in vitro ubiquitination assays were performed using H2A as substrate 23. We have selected four compounds 6a, b, c, e that exhibited more favorable IC50 values and better inhibitory effects on Rad6 conjugation to ubiquitin in the in vitro ubiquitin assays compared to TZ9. As shown in Figure 5A and 5B, compounds 6a, b, c, e exhibited superior Rad6B inhibitory properties, both in its conjugation to ubiquitin and substrate ubiquitination (H2A-Ub) when compared to TZ9. Together, these studies report the synthesis of new triazines with better Rad6B inhibitory activities and anticancer properties compared to previously reported Rad6B inhibitor TZ9 in in vitro evaluations.
In summary, Series of novel 4-amino-6-(arylamino)-1,3,5-triazine-2-carbohydrazides (3a–e) and N′-phenyl-4,6-bis(arylamino)-1,3,5-triazine-2-carbohydrazides (6a–e) have been synthesized. Compared to the previously reported Rad6B-inhibitor TZ9, new triazines showed better IC50 values in survival assays of various tumor cell lines. Particularly, new triazines 6a–c exhibited much lower IC50 (3.3 to 22 μM) values.
Moreover, when compared to TZ9 in in vitro ubiquitin conjugation assays, compounds 6a, b, c, e exhibited superior Rad6B inhibitory properties, both in its conjugation to ubiquitin and to the substrate H2A.
Supplementary Material
Highlights.
Novel 4-amino-6-(arylamino)-1,3,5-triazine-2-carbohydrazides (3a–e) and N′-phenyl-4,6-bis(arylamino)-1,3,5-triazine-2-carbohydrazides (6a–e) have been synthesized.
To evaluate the efficacy of newly synthesized triazines in Rad6 inhibitory activities, in vitro ubiquitin conjugation assays were performed in comparison with our previously reported Rad6 inhibitor TZ9 as well as their in vitro anticancer activity against several human cancer cell lines.
Our new triazines showed better Rad6B inhibitory activities and anticancer properties compared to previously reported Rad6B inhibitor TZ9 in in vitro evaluations.
Acknowledgments
This work is supported by Zagazig University to HK, in part by NIH grant R01GM098956, and Abraham Mitchell Cancer Research Scholar Endowment grant to K.P.
Footnotes
Supplementary Data available:
Experimental procedure ; full compound characterisation data (m.p., 1H, 13C NMR spectroscopy, mass spectrometry, % CHN analysis); In vitro ubiquitin conjugating assays and MTS assay and IC50 calculations.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hochstrasser M. Curr Opin Cell Biol. 1995;7:215. doi: 10.1016/0955-0674(95)80031-x. [DOI] [PubMed] [Google Scholar]
- 2.Ciechanover A. Cell. 1994;79:13. doi: 10.1016/0092-8674(94)90396-4. [DOI] [PubMed] [Google Scholar]
- 3.Hershko A, Ciechanover A. Annu Rev Biochem. 1998;67:425. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 4.Nandi D, Tahiliani P, Kumar A, Chandu D. J Biosci. 2006;31:137. doi: 10.1007/BF02705243. [DOI] [PubMed] [Google Scholar]
- 5.Chen D, Frezza M, Schmitt S, Kanwar J, Dou QP. Curr Cancer Drug Targets. 2011;11:239. doi: 10.2174/156800911794519752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen D, Dou QP. Curr Protein Pept Sci. 2010;11:459. doi: 10.2174/138920310791824057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edelmann MJ, Nicholson B, Kessler BM. Expert Rev Mol Med. 2011;13:e35. doi: 10.1017/S1462399411002031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosner K, Mehregan DR, Kirou E, Abrams J, Kim S, Campbell M, Frieder J, Lawrence K, Haynes B, Shekhar MP. J Skin Cancer. 2014;2014:439205. doi: 10.1155/2014/439205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lyakhovich A, Shekhar MP. Oncogene. 2004;23:3097. doi: 10.1038/sj.onc.1207449. [DOI] [PubMed] [Google Scholar]
- 10.Shekhar MP, Lyakhovich A, Visscher DW, Heng H, Kondrat N. Cancer Res. 2002;62:2115. [PubMed] [Google Scholar]
- 11.Shekhar MP, Gerard B, Pauley RJ, Williams BO, Tait L. Cancer Res. 2008;68:1741. doi: 10.1158/0008-5472.CAN-07-2111. [DOI] [PubMed] [Google Scholar]
- 12.Gerard B, Tait L, Nangia-Makker P, Shekhar MP. J Mol Signal. 2011;6:6. doi: 10.1186/1750-2187-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gerard B, Sanders MA, Visscher DW, Tait L, Shekhar MP. Biochim Biophys Acta. 2012;1823:1686. doi: 10.1016/j.bbamcr.2012.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hibbert RG, Huang A, Boelens R, Sixma TK. Proc Natl Acad Sci U S A. 2011;108:5590. doi: 10.1073/pnas.1017516108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsuji Y, Watanabe K, Araki K, Shinohara M, Yamagata Y, Tsurimoto T, Hanaoka F, Yamamura K, Yamaizumi M, Tateishi S. Genes Cells. 2008;13:343. doi: 10.1111/j.1365-2443.2008.01176.x. [DOI] [PubMed] [Google Scholar]
- 16.Watanabe K, Tateishi S, Kawasuji M, Tsurimoto T, Inoue H, Yamaizumi M. EMBO J. 2004;23:3886. doi: 10.1038/sj.emboj.7600383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song IY, Palle K, Gurkar A, Tateishi S, Kupfer GM, Vaziri C. J Biol Chem. 2010;285:31525. doi: 10.1074/jbc.M110.138206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palle K, Vaziri C. Cell Cycle. 2011;10:1625. doi: 10.4161/cc.10.10.15617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu C, Wang D, Wu J, Keller J, Ma T, Yu X. J Cell Sci. 2013;126:2042. doi: 10.1242/jcs.122945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo Y, Fu P, Zhu H, Reed E, Remick SC, Petros W, Mueller MD, Yu JJ. Oncol Rep. 2012;27:286. doi: 10.3892/or.2011.1483. [DOI] [PubMed] [Google Scholar]
- 21.Meng E, Mitra A, Tripathi K, Finan MA, Scalici J, McClellan S, Madeira da Silva L, Reed E, Shevde LA, Palle K, Rocconi RP. PLoS One. 2014;9:e107142. doi: 10.1371/journal.pone.0107142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. Nat Rev Cancer. 2008;8:193. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 23.Sanders MA, Brahemi G, Nangia-Makker P, Balan V, Morelli M, Kothayer H, Westwell AD, Shekhar MP. Mol Cancer Ther. 2013;12:373. doi: 10.1158/1535-7163.MCT-12-0793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kothayer H, Elshanawani AA, Abu Kull ME, El-Sabbagh OI, Shekhar MP, Brancale A, Jones AT, Westwell AD. Bioorg Med Chem Lett. 2013;23:6886. doi: 10.1016/j.bmcl.2013.09.087. [DOI] [PubMed] [Google Scholar]
- 25.LeBel O, Maris T, Duval H, Wuest JD. Can J Chem. 2005;83:615. [Google Scholar]
- 26.Kothayer H, Morelli M, Brahemi G, Elshanawani AA, Abu Kull ME, El-Sabbagh OI, Shekhar MPV, Westwell AD. Tetrahedron Lett. 2014;55:7015. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.