

Abstract
Hypersecretion of acid hydrolases is a hallmark feature of mucolipidosis II (MLII), a lysosomal storage disease caused by loss of carbohydrate-dependent lysosomal targeting. Inappropriate extracellular action of these hydrolases is proposed to contribute to skeletal pathogenesis, but the mechanisms that connect hydrolase activity to the onset of disease phenotypes remain poorly understood. Here we link extracellular cathepsin K activity to abnormal bone and cartilage development in MLII animals by demonstrating that it disrupts the balance of TGFß-related signaling during chondrogenesis. TGFß-like Smad2,3 signals are elevated and BMP-like Smad1,5,8 signals reduced in both feline and zebrafish MLII chondrocytes and osteoblasts, maintaining these cells in an immature state. Reducing either cathepsin K activity or expression of the transcriptional regulator Sox9a in MLII zebrafish significantly improved phenotypes. We further identify components of the large latent TGFß complex as novel targets of cathepsin K at neutral pH, providing a possible mechanism for enhanced Smad2,3 activation in vivo. These findings highlight the complexity of the skeletal disease associated with MLII and bring new insight to the role of secreted cathepsin proteases in cartilage development and growth factor regulation.
Keywords: CATHEPSINS, TGFß, MLII, ZEBRAFISH, CARTILAGE, LYSOSOMAL
Introduction
Carbohydrate-dependent sorting of acid hydrolases to lysosomes represents one of the best-studied mechanisms whereby oligosaccharides regulate essential cellular functions. Selective addition of mannose 6-phosphate (M6P) residues to the sugar chains of newly synthesized hydrolases allows them to bind M6P receptors within the trans-Golgi network and target to lysosomes. The Golgi-localized enzyme UDP-GlcNAc: lysosomal enzyme GlcNAc-1-phosphotransferase initiates the synthesis of M6P tags on the hydrolases.(1,2) This “phosphotransferase” enzyme is encoded by two genes: GNPTAB and GNPTG,(3,4) with the GNPTAB gene products responsible for both hydrolase recognition and catalytic function.(5,6)
The importance of carbohydrate-dependent lysosomal sorting to human health and development is underscored by the lysosomal storage disease mucolipidosis II (MLII; I-cell disease).(7,8) MLII arises from GNPTAB nonsense, frameshift, or splice site mutations that abolish mannose phosphorylation.(9,10) The clinical manifestations are severe and multisystemic, including impaired skeletal growth, progressive osteodystrophy, destructive bone lesions (ie, dysostosis multiplex), facial dysmorphism, cardiomyopathies, and early death.(9,11) Many MLII abnormalities are noted at birth, highlighting the impact of the disease on early developmental processes.(12,13) Loss of mannose phosphorylation causes secretion of lysosomal hydrolases, whose intracellular deficiency results in lysosomal storage and the proliferation of dense inclusions within cells of mesenchymal origin. The extent to which these two mechanisms contribute to phenotypes is not well understood. Moreover, the acute mechanisms that drive pathogenesis during development may differ from those causing chronic disease aspects such as neurodegeneration and bone loss.
Previous studies using a zebrafish model defined key aspects of MLII developmental pathogenesis.(14) Affected embryos showed impaired mannose phosphorylation of lysosomal hydrolases but lacked detectable lysosomal storage, pointing to other pathogenic mechanisms. Abnormal craniofacial cartilage development of MLII embryos was associated with sustained expression of the chondrogenic regulator Sox9 and its gene target type II collagen.(14) Parallel studies demonstrated increased and sustained activity of several cathepsin proteases, including the collagenase cathepsin K (Ctsk). Pharmacological or genetic inhibition of Ctsk activity reduced abnormal type II collagen expression and alleviated MLII craniofacial phenotypes.(15) The finding that ctsK inhibition reduced type II collagen was unexpected because decreased collagenolytic activity was predicted to cause collagen accumulation. This paradoxical relationship suggests a pathogenic function for cathepsins beyond collagen turnover.
Here we link excessive Ctsk activity to sustained type II collagen expression by showing that Ctsk alters the balance of the TGFß-related growth factor signals that regulate chondrocyte and osteoblast differentiation. In MLII zebrafish embryos, extracellular Ctsk perpetuates canonical TGFß Smad2,3 signaling while decreasing BMP Smad1,5,8 activation. This imbalance maintains cells in an immature state, impairing normal cartilage and bone development. Similar disruptions in growth factor signals and cellular maturity were observed in the bones of MLII kittens, extending the findings beyond the zebrafish system. This work uncovers a novel lysosomal disease mechanism by demonstrating the ability of secreted cathepsin proteases to impact growth factor latency and signaling.
Materials and Methods
Zebrafish strains and husbandry
Animals were maintained according to standard protocols. Zebrafish strains were obtained from Fish 2U (F2U; Gibsonton, FL, USA) and the Zebrafish International Resource Center (ZIRC, Eugene, OR, USA) (TL, AB, sox9ahi1134Tg/+, Tg(fli1a:EGFP)y1,(16) and osx:EGFP (sp7).(17) BRE:EGFP was provided by Dr Brian Link (MCW, Milwaukee, WI, USA).(18) col2a1:mCherry BAC line was provided by Dr Chrissy Hammond (University of Bristol, Bristol, UK).(19) Staging was according to established criteria.(20) In some cases, 0.003% 1-phenyl 2-thiourea (PTU) was added to embryo medium to block pigmentation. Handling and euthanasia of fish for all experiments complied with the University of Georgia policies, as approved by the UGA Institutional Animal Care and Use Committee (permit #A2013-8-144).
Morpholinos and inhibition of gene expression
Morpholino knockdown of Glc-NAc-1-phosphotransferase and ctsk were performed and assessed as previously described.(14,15) MO-based reduction of col2a1a was performed and assessed as previously described.(21)
In situ hybridization
Analyses were performed according to Thisse and Thisse protocol.(22) Col1a2, col2a1a, col10a1, osx, runx2a, sox9a, and sox9b probes were generated as described.(23) Probe constructs provided by Dr Henry Roehl (University of Sheffield, Sheffield, UK) and Dr John Postlethwait (University of Oregon, Eugene, OR, USA). For acana, a portion of the coding region was isolated from 3dpf embryos by RT-PCR. The 3′ primer contained the T3 polymerase binding site. The staining pattern for this probe matched another published probe.(24) The decorin (dcn) probe was also PCR-generated using the following primers: forward TTCCGATGCCAGTGCCATCT GC; Reverse TGTAATACGACTCACTATAGGGCTCCA CCACGCTGATCTC. Embryos were imaged on an Olympus SZ16 microscope with a Retiga 2000R camera and Q-capture software.
Reverse transcriptase and qRT-PCR analyses
Quantitative PCR analyses were performed as described.(15) Data were analyzed as described.(15) Specific primers are listed in Supplemental Fig. S1.
Histochemistry and immunohistochemistry
Alcian blue and Alizarin red staining were used as previously described.(17,25) Immunohistochemistry was performed on 30 μM sections from fli1a:EGFP+ cartilages. See Supplemental Methods for a list of primary antibodies. Samples were imaged on an FV-100 laser scanning confocal microscope with a 60× (N.A.1.4) oil immersion lens. Image projections and 3D reconstruction/rotations were generated with either Image J or Slidebook 5.0 (3i, Denver, CO, USA) software. Final images were processed in Adobe Photoshop Extended.
Analyses of feline bones
The animals were housed at the School of Veterinary Medicine, University of Pennsylvania, under the National Institutes of Health and the US Department of Agriculture guidelines for the care and use of animals in research and approved by the University Animal Care and Use Committee. MLII-affected animals were determined by PCR genotyping. See Supplemental Methods for details of histology.
FACS analyses of GFP+ cells
Osx:EGFP embryos were dissociated into single-cell suspensions as previously described.(15) GFP+ cells were sorted and counted as previously described.(14) Enzyme assays and western analyses were performed as previously described.(15)
In vitro analyses and reporter assays
In vitro experiments used recombinant human LAP dimer, TGFß ligand, and BMP2 ligand. Twenty to 40 μg of protein was digested for 30 minutes at RT with human recombinant CTSK at different pHs. CTSK activities were normalized using fluorescent substrates. For reporter experiments, HEL cells were differentiated for 72 hours with PMA and LLC-enriched media collected for 24 hours. Spin concentrated media samples were protease digested for 30 minutes and/or heat activated for 10 minutes at 75 °C or 80 °C. Heat-activated or protease-treated media concentrates were added to the reporter cells for 16 hours and luciferase induction measured. Detailed descriptions of the in vitro and reporter cell methods are in Supplemental Materials.
Statistical analyses
In cases where numerical or quantitative data were generated, standard deviations were calculated using Microsoft Excel (version 14.1.0.). Unless otherwise indicated, a two-tailed Student t test, also calculated in Excel, was used to assess statistical significance. In Fig. 7D, where multiple experimental conditions were compared, a one-way ANOVA with Dunnett’s post test was performed using GraphPad InStat version 3.10 for Windows (GraphPad Software, San Diego, CA, USA). In cases where expression levels or staining patterns were assessed visually, representative embryos are shown and the number of animals from multiple experimental samples that resembled those pictured was calculated.
Fig. 7.
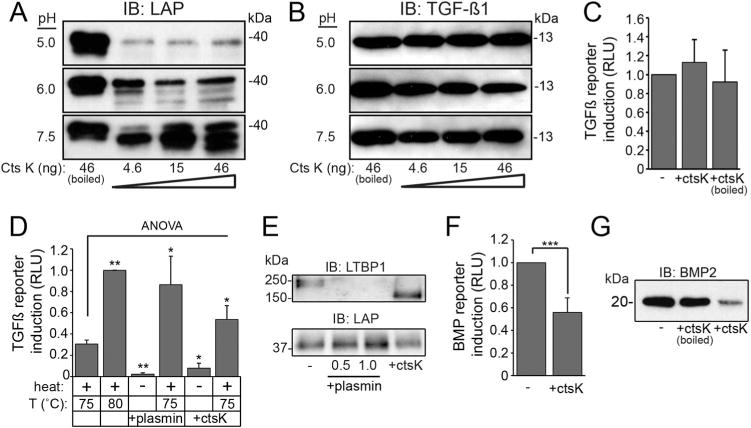
Differential sensitivity of latent TGFß complex components toward CtsK lowers the barrier for ligand activation. (A, B) Western blot of human LAP (A) and TGFß ligand (B) digested with CTSK. n = 4 experiments. (C) Average luciferase induction of TGFß reporter cells using CTSK-digested recombinant TGFß. n = 5 experiments. (D) Average luciferase induction in TGFß reporter cells by LLC after plasmin or CTSK digestion and heat treatment. n = 4 repeated experiments. Error bars = SD. Significance was calculated by a one-way ANOVA with Dunnett’s post-test; *p < 0.05; **p < 0.01 (see Methods). (E) Western blot of enzyme-digested LLC using antibodies against LTBP and LAP; n = 4 repeated experiments. (F) Luciferase induction in C2C12 BMP reporter cells using CTSK-digested human proBMP2; n = 3 repeated experiments. Significance calculated by Student t test; ***p < 0.001. (G) Western blot of CTSK-digested proBMP2; n = 3 repeated experiments.
Results
Loss of gnptab expression disrupts chondrocyte maturation
Chondrocyte maturation is characterized by phases of alternate extracellular matrix (ECM) protein expression.(26) Previous work on gnptab-deficient zebrafish demonstrated that altered cartilage morphology was associated with increased expression of type II collagen, one of the earliest markers of chondrocyte differentiation.(14) To ask if this reflected disrupted chondrocyte maturation, in situ hybridization (ISH) was used to locally assess expression of several additional markers. Consistent with previous findings, col2a1a transcript abundance was higher in MLII cartilage (Fig. 1A). In contrast, aggrecan (acana), a chondroitin sulfate proteoglycan (CSPG) expressed by more mature chondrocytes, was reduced in MLII structures (Fig. 1B). This was matched by a decrease of another CSPG, decorin (dcn) (Fig. 1C). Differences in col2a1a, acana, and dcn expression were confirmed by quantitative PCR (qRT-PCR) (Fig. 1D). Immunohistochemically stained fli1a:EGFP cartilage sections confirmed altered CSPG expression (Fig. 1E), with co-injection of WT gnptab mRNA restoring normal staining >60% of the time.
Fig. 1.
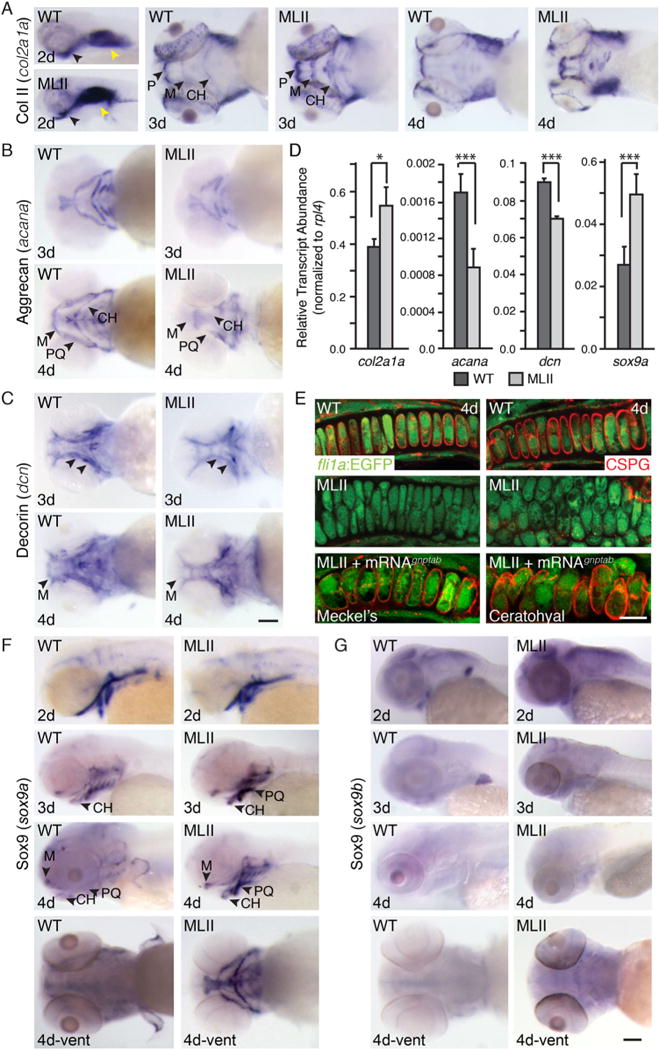
MLII chondrocyte maturation is reduced. (A–C) ISH of col2a1a, acana, and dcn in WT and MLII embryos. Arrowheads indicate regions of difference. (D) qPCR of maturation markers in 4 dpf embryonic heads. n = 30 embryos/sample, 4 experiments. All values were normalized to ribosomal protein L4 (rpl4). *p < 0.05; ***p < 0.001. (E) CSPG immunohistochemistry in 4 dpf fli1a:EGFP WT, MLII, and MLII+gnptab mRNA embryos. n = 25 embryos/condition. (F, G) ISH of sox9a and sox9b in WT and MLII. For all ISH, n > 100 embryos/condition. Scale bars = ISH 100 μm, CSPG 10 μm. M = Meckel’s; P = palate; CH = ceratohyal; PQ = palatoquadrate. Error = SD.
Sox9a is increased in MLII
Altered expression of ECM proteins was matched by increases in the chondrogenic regulator Sox9a (Fig. 1F). Sox9, originally designated sex determining region Y–box9, is an HMG-box transcription factor essential for chondrogenesis.(27,28) Consistent with previous analyses,(14) MLII sox9a transcript levels were higher (see arrows, Fig. 1F), whereas levels of the less prominent sox9b(29) were similar between WT and morphant embryos (Fig. 1G). Although required for early aspects of differentiation, sustained Sox9a activity inhibits chondrogenic and osteogenic maturation.(30) These data demonstrate a disruption in MLII chondrogenesis, which maintains cells in an early developmental state.
Loss of gnptab expression alters osteoblast maturation
We next assessed whether loss of gnptab also affected osteogenesis (Fig. 2). ISH analyses of the atypical cartilage and bone marker, type X collagen (col10a1), revealed significant changes in its expression. Although mammalian systems only express type X collagen in hypertrophic chondrocytes, in zebrafish it’s present in both osteoblasts and chondrocytes,(23) primarily marking osteoblasts 3 to 4 dpf. Although strong col10a1 expression was observed in WT embryos, it was severely reduced in MLII structures (Fig. 2A, see arrows), most notably the parasphenoid (Ps) and opercle (Op) bones, which develop by intramembranous rather than endochondral ossification. Two additional osteogenic regulators, the runt-related transcription factor runx2a and the essential factor osterix (osx), were also reduced in MLII structures (Fig. 2B, C, arrows). This was matched by decreased expression of type I collagen (col1a2) (Fig. 2D). Reduced expression of col10a1, runx2a, and col1a2 was quantitatively confirmed by qRT-PCR (Fig. 2E). Alizarin red staining for mineralized matrix further demonstrated diminished calcium deposition in the majority of dermal bones 5 dpf (Fig. 2F, arrows). Staining was also absent from the “bone collars” surrounding MLII ceraothyal structures, which at this early stage are the only endochondral-based osteoblasts undergoing mineralization (Fig. 2F, asterisks). In 63% of the animals, co-injected WT gnptab mRNA restored Alizarin-positive staining to multiple structures. To determine whether reduced osteogenesis reflected altered osteoblast development or cellular loss, gnptab morpholino (MO) was injected into an osx:EGFP (sp7) transgenic line(17) and embryos assessed by live confocal microscopy (Fig. 2G). The shape and distribution of morphant cells were noticeably altered, and although the number of GFP+ osteoblasts was similar at 2 dpf, it was slightly lower in 3 dpf MLII embryos. FACScans (fluorescent-activated cells scan) of dissociated WT and MLII cells showed that at 2 dpf 2.3% ± 0.3% and 2.4% ± 0.3%, respectively, of the cells were GFP+, whereas at 3 dpf 2.5% ± 0.4% and 1.9% ± 0.3% of the cells were GFP+ (p<0.05), suggesting that osteoblast number may also be affected in MLII.
Fig. 2.
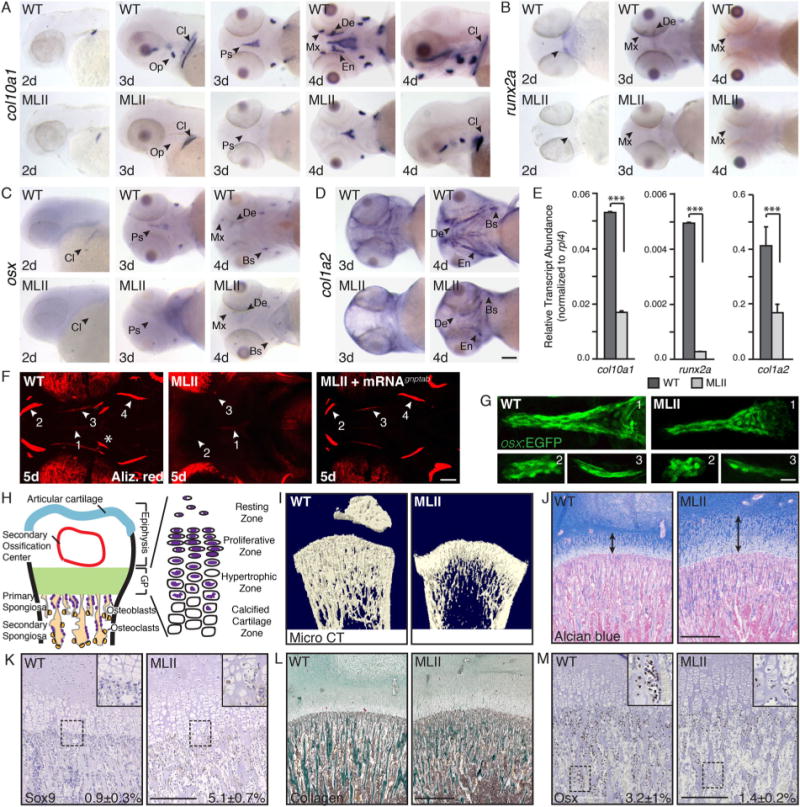
MLII animals exhibit reduced osteogenesis of dermal bones. ISH of (A) col10a1, (B) runx2a, (C) osx, and (D) col1a2 show reduced osteogenesis in MLII zebrafish. n = 100 embryos/condition. Scale bar = 100 μm. Bs = brachiostegal ray; Cl = cleithrum; De = dentary; En = entopterygoid; Mx = maxilla; Op = opercle; Ps = parasphenoid; (E) qPCR of embryonic heads 4 dpf. n = 30 embryos/sample, 4 experiments. ***p < 0.001. (F) Live Alizarin red–stained 5 dpf WT, MLII, and MLII+gnptab mRNA embryos. Arrows denote regions of interest (ROI 1–4, where 1 = Ps, 2 = Mx/De, 3 = En, 4 = Bs). Asterisks show endochondral bone collars. n = 30 embryos. Scale bar = 50 μm. (G) Confocal analyses of osx(sp7):EGFP embryos. Regions (ROI 1–3, where 1 = Ps, 2 = Mx, 3 = En) shown. n = 30 embryos/condition, 3 experiments. Scale bar = 20 μm. (H) Schematic of growing mammalian femur. (I) Distal femurs of MLII kittens (n = 3, aged 0 to 2 months) and WT littermates (n = 6) analyzed by X-ray micro–CT scan show delayed ossification and osteopenia. Femur sections stained with Alcian blue (J) or for Sox9 (K) show widened growth plates (arrows) and increased Sox9 expression and cartilaginous matrix in primary spongiosa. Femur sections stained with Masson’s trichrome (L) or for Osx (M) show reduced Osx and collagen in primary spongiosa. Representative sections from 15-day-old animals. For J–M, WT n = 4–7, MLII n = 2–3 different kittens. The percent values listed equal the number of cells stained positively for a given marker. (J, L) Scale bar = 1 mm. (K, M) Scale bar = 300 μm. Higher power images of K and M in Supplemental F1.
MLII feline bones exhibit similar alterations in chondrocyte and osteoblast differentiation
In zebrafish, maturation of dermal bones occurs earlier (3 to 5 dpf) than endochondral bones (6 to 17 dpf). Because MLII embryos die by 6 dpf, analyses of endochondral osteogenesis in zebrafish was limited. Therefore, to both extend these analyses and confirm key findings in a mammalian model, we analyzed growth plates of MLII kittens.(9,11) Together with 6 unaffected WT littermates, the hind legs of three affected kittens (newborn, aged 15 days, and aged 2 months) were assessed by radiography and micro-computed tomography (micro–CT). In all cases, MLII tibia and femurs were smaller (Supplemental Fig. S1), with delayed formation of secondary ossification centers (Fig. 2I and data not shown). The relative trabecular bone volume (BV/TV) was also reduced, confirming osteopenia previously noted in MLII cats and mice.(31,32) Longitudinal bone development depends on the coordinated differentiation of growth plate chondrocytes and osteoclasts and osteoblasts of the primary spongiosia (Fig. 2H). Alcian blue–stained femur sections showed MLII growth plates were 2× wider than WT littermates, with both proliferating and hypertrophic chondrocytes altered (Fig. 2J). As with zebrafish, Sox9 expression was enhanced in MLII felines, especially within hypertrophic chondrocytes (Fig. 2K; Supplemental Fig. S1). Furthermore, Masson trichrome staining of the primary spongiosa showed that bony trabecular collagen content was reduced (Fig. 2L, stained green) and glycosaminoglycan content increased in MLII (Fig. 2J, stained blue). These data indicate a more cartilaginous immature bone, a conclusion supported by the fact that osteoblasts in the primary spongiosa express less Osx and more Sox9 (Fig. 2M, K; Supplemental Fig. S1). These findings, noted in two vertebrate MLII models, confirm that reducing M6P levels overstimulates chondrogenesis and reduces osteoblastogenesis, likely because of imbalanced Sox9 and Osx.
Reducing sox9a levels restores MLII cartilage morphology
To determine if increased sox9a expression actively promotes MLII cartilage defects, gnptab was inhibited within the sox9ahi1134Tg/+ zebrafish line.(33) These animals carry an nLacz-GT viral insertion that disrupts the sox9a gene. Embryos homozygous for the insertion lack sox9a activity and do not develop craniofacial cartilage. Embryos heterozygous for the insertion (+/−) are phenotypically normal but expected to have reduced Sox9a levels compared with homozygote WT (+/+) embryos. We hypothesized that if injected with the gnptab MO, heterozygote carriers (50%) would have improved craniofacial morphology, whereas homozygote WT (25%) embryos would be ML-like (Fig. 3A, a and b). When embryos from uninjected crosses were stained with Alcian blue, 74% were phenotypically WT and 26% lacked staining (Fig. 3B). In contrast, 28% of the embryos from MO-injected clutches contained ML-like cartilage (Class A, predicted to be sox9a +/+), 50% contained “WT-appearing” cartilage (Class B, predicted to be sox9a +/−), and 22% of the embryos lacked Alcian blue staining altogether (Class C, predicted to be sox9a −/−) (Fig. 3C; see Fig. 3D for rescue parameters).(14,34) The expected genotypes of individual Alcian blue–stained animals were confirmed by PCR (Fig. 3E).(33,35)
Fig. 3.
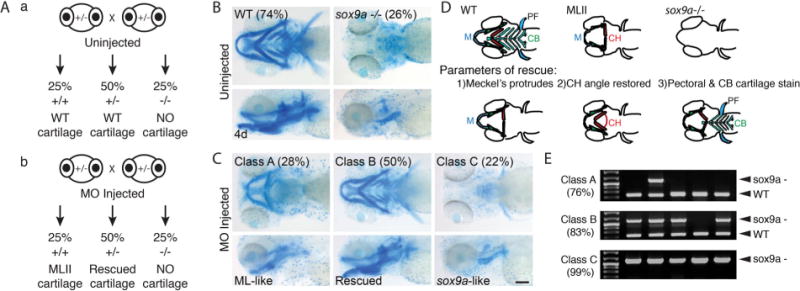
Reduced sox9a improves MLII phenotypes in zebrafish. (A) Schematic outlines the expected craniofacial phenotype in: (a) uninjected and (b) gnptab MO-injected sox9a (+/−) incross progeny. (B) Alcian blue–stained uninjected sox9a (+/−) incross progeny. (C) Alcian blue–stained MO-injected progeny, where Class A embryos (predicted to be sox9a +/+) contain ML-like cartilage, Class B embryos (predicted to be sox9a +/−) contain WT-like “rescued” cartilage, and Class C embryos (predicted to be sox9a−/−) resemble sox9a mutants. Percent values equal the number of embryos within each phenotypic class; n > 300; scale bar = 100 μm. (D) Schematic summarizing cartilage phenotypes in WT, MLII, and sox9a/embryos (top row) and the 3 parameters used to assess phenotypic rescue (bottom row).(15) M = Meckel’s; CH = ceratohyal; CB = ceratobrachials; PF = pectoral fin. (E) Representative PCR of single embryo genotyping for animals within each phenotypic class. Percent value represents number of embryos within the class positive for the expected genotype. n = 50–75 embryos/condition; scale bar = 100 μm.
Unlike sox9a inhibiting col2a1a, an important Sox9a target upregulated in MLII did not improve cartilage phenotypes. For these analyses, a previously validated col2a1a splice blocking MO was co-injected with the gnptab MO (Supplemental Fig. S2).(21) Alcian blue stains showed that gnptab/col2a1a double morphants retained ML-like phenotypes ~94% of the time. Further, confocal analyses of immunohistochemically stained fli1a:EGFP confirmed that although type II collagen expression was reduced, MLII chondrocyte organization was not improved (Supplemental Fig. S2).(14) These findings suggest that although type II collagen accumulation is a consequence of compromised M6P biosynthesis, it does not drive cartilage dysmorphia.
Smad2,3 activation is increased in MLII chondrocytes
Work in several systems suggests that type II collagen expression is regulated by the cooperative action of Sox9 and canonical TGFß effectors Smad2,3 (Fig. 4A).(36) Because both sox9a and col2a1a are increased in MLII, we asked if TGFß-like Smad2,3 signaling was also increased in affected chondrocytes. Cartilage sections from WT and MLII fli1a:EGFP embryos were stained immunohistochemically for either Smad2,3 or the transcription-ally active phospho-Smad2 (pSmad2) (Fig. 4B). Confocal analyses revealed high levels of Smad2,3 in MLII chondrocytes with very little staining evident in WT. Co-stains with the nuclear marker TOPRO showed significant Smad2,3 localization within MLII nuclei. pSmad2 staining was also higher in MLII cartilages with the majority present within TOPRO-positive nuclei. Nuclear localized pSmad2 was noted in >80% of MLII chondrocytes, whereas only 2% to 3% of WT nuclei were pSmad2 positive. This phenotype was noted in 95% of MLII embryos assayed. qRT-PCR of RNA isolated from WT and MLII heads confirmed that MLII tissues expressed more smad2 (56%) and smad3a (36%) than WT tissues (Supplemental Fig. S3). An increase in productive Smad2,3 signaling was confirmed using a transgenic reporter line, Tg(col2a1aBAC:mCherry), in which the TGFß-responsive col2a1a promoter controls mCherry expression (Fig. 4B).(19) These data demonstrate that loss of M6P biosynthesis increases Smad2,3 signaling within MLII chondrocytes, providing a mechanism to explain arrested chondrogenesis and high type II collagen levels.
Fig. 4.
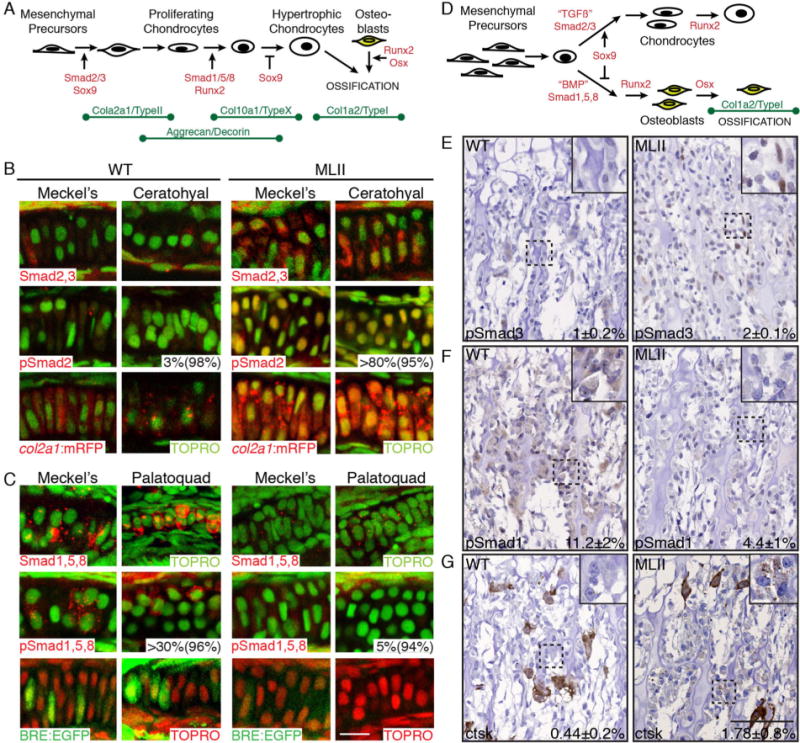
TGFß- and BMP-related Smad activation is imbalanced in MLII chondrocytes. (A) Schematic of chondrogenesis summarizing transcriptional regulators and ECM protein expression patterns. (B) Immunohistochemistry of either Smad2,3 (red) or pSmad2 (red) in cartilage sections of WT and MLII fli1a:EGFP embryos (top two rows). WT and MLII col2a1a:mRFP (red) transgenic embryos (third row). TOPRO+ nuclei (green). First percent value equals the number of phospho-Smad-positive cells per structure; the second equals the number of experimental animals similar to that pictured. (C) Immunohistochemistry of either Smad1,5,8 (red) or pSmad1,5,8 (red) in cartilage sections of WT and MLII fli1a:EGFP embryos (top two rows). WT and MLII embryos in BRE:EGFP (green) BMP reporter line (third row). TOPRO+ nuclei green in upper panels, red in lower EGFP panels. (D) Schematic summarizing reported regulation of long bone osteogenesis. (E, F) pSmad3 and pSmad1 immunohistochemistry in WT and MLII kitten femur. (G) Increased ctsk reactivity in MLII feline osteoblasts compared with WT. Ctsk expression levels in feline osteoclasts (large brown cells) are similar between WT and MLII samples. The percent values listed represent the number of cells staining positively for the given marker. For G, this number excludes the larger osteoclast cells. For E–G, WT n = 4–7 and MLII n = 2–3. Scale bar = 100 μm. Higher power images of E and F in Supplement F1.
Smad1,5,8 activation is reduced in zebrafish MLII chondrocytes
As chondrocytes mature, TGFß-propagated Smad2,3 signals are thought to wane in exchange for increasing BMP-regulated Smad1,5,8 signals (Fig. 4A).(26) Similarly, osteoblast maturation has also been shown to coincide with increasing BMP signals that drive expression of osteogenic factors like Osx. Because chondrocytic expression of Smad2,3-regulated col2a1a is high in MLII whereas osteoblast-specific expression of Smad1,5,8-regulated col10a1 is low, we asked if heightened Smad2,3 activation was also matched in chondrocytes by reduced Smad1,5,8 signals. Cartilage sections from WT and MLII fli1a: EGFP embryos were stained immunohistochemically for either Smad1,5,8 or pSmad1,5,8 (Fig. 4C). Confocal analyses showed lower Smad1,5,8 levels in MLII chondrocytes than in WT. Further, pSmad1,5,8, abundant in several WT regions, was barely detected in MLII structures. Nuclear localized pSmad1,5,8 was noted in 40% to 50% of WT cells from 96% of the embryos. In contrast, pSmad1,5,8 staining was only detected in ~5% of MLII chondrocytes. Reduced Smad expression was confirmed by qRT-PCR, where analyses of RNA from WT and MLII heads demonstrated 60% and 57% decreases in Smad5 and 8 (Supplemental Fig. S3). Reduced BMP-like Smad1,5,8 signaling was also assessed with a transgenic reporter line, Tg(BRE-AAVmlp: EGFP), in which a BMP-responsive element (BRE) regulates EGFP expression (Fig. 4C).(18) Using this line, we found that >50% of WT chondrocytes exhibited robust EGFP expression that was rarely detected (<10% of the cells) in MLII structures, a phenotype noted in 92% of MLII embryos.
MLII kittens also exhibit altered Smad signaling
In long bones where chondrocytes and osteoblasts arise from a common progenitor, cell fate is controlled by the ratio of Sox9/Osx and TGFß/BMP (Fig. 4D). TGFß-activated Smads and Sox9 are thought to favor a chondrogenic fate, whereas BMP-activated Smads and their gene target Osx favor the osteogenic fate. Because Sox9 and Osx levels were altered in both zebrafish and kitten cells, we asked if altered TGFß/BMP signaling was also a general feature of MLII cartilage and bones. Femur sections of MLII kittens were stained for either pSmad3 or pSmad1. Although we could not detect significant pSmad staining in growth plate chondrocytes, osteoblasts in the primary spongiosa expressed more nuclear pSmad3 and less pSmad1 (Fig. 4E, F; Supplemental Fig. S1). Thus, imbalanced TGFß/BMP activity also correlated with altered chondrogenesis and osteoblastogenesis in primary spongiosa of MLII kittens.
Ctsk inhibition restores normal col2a1 and col10a1 levels to MLII chondrocytes
Prior work on MLII zebrafish demonstrated a significant increase in activity of the cysteine protease, Ctsk.(15) Importantly, immunohistochemical analyses of MLII kitten long bones also showed increased Ctsk reactivity. This was particularly prominent within osteoblasts of the primary spongiosa (boxed regions, Fig. 4G), the same cells exhibiting altered TGFß/BMP signaling and differentiation. Notably, osteoclasts also expressed Ctsk, but this expression was overtly normal in MLII kittens compared with WT littermates (large brown cells, Fig. 4G). In zebrafish, inhibition of Ctsk ameliorated several aspects of MLII cartilage pathology, including restoration of craniofacial morphology and type II collagen immunohistochemistry.(15) Reduced type II collagen staining upon ctsk inhibition was surprising because Ctsk is a known collagenase.(37) To further address this, we asked if ctsk inhibition also restored the molecular alterations associated with the cartilage and bone defects. MLII embryos were co-injected with 0.1 μM of a ctsk splice blocking MO as previously described(15) and ISH of col2a1a, col10a1, and sox9a performed. Inhibition of ctsk substantially improved col2a1a and col10a1 expression patterns (Fig. 5), which were altered in 93% to 94% of MLII embryos. Co-injection with the ctsk MO restored WT col2a1a expression levels in 64.5% of the embryos (white arrows, Fig. 5A) and increased col10a1 expression in 62.5% of the embryos (boxed area ventral views, Fig. 5B). Importantly, reduced sox9a transcript levels were also noted (Fig. 5C) with ctsk inhibition.
Fig. 5.
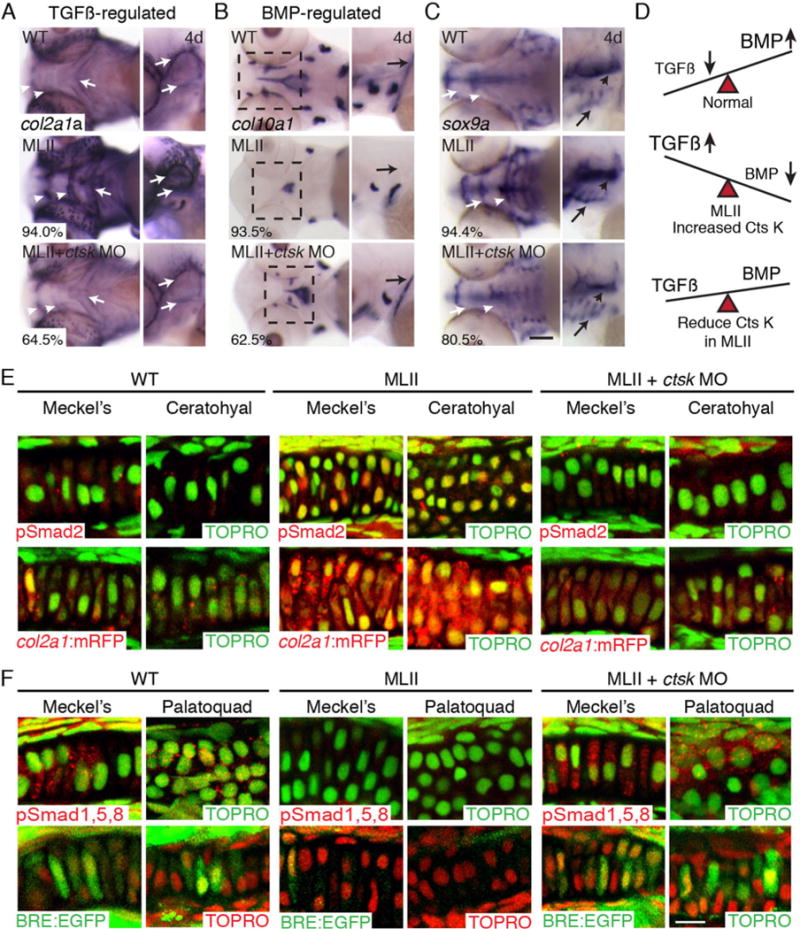
ctsk inhibition improves altered TGFß and BMP Smad activation in zebrafish. ISH of (A) TGFß gene target col2a1a, (B) BMP target gene col10a1a, and (C) sox9a. The percent values shown indicate the number of embryos with the phenotype pictured. n = 100 embryos. (D) Schematic showing idea that TGFß signals decline and BMP signals increase as normal cartilage matures. This balance is disrupted in MII but restored with ctsk reduction. (E) Immunohistochemistry of pSmad2 (red) in WT, MLII, and ctsk-inhibited MLII (upper row). Analyses in col2a1a:mRFP (red) reporter line (lower row). TOPRO+ nuclei green. (F) Immunohistochemistry of pSmad1,5,8 (red) in WT, MLII, and ctsk-inhibited MLII embryos (upper row). Analyses in BMP reporter line BRE:EGFP (green; lower row). TOPRO+ nuclei are green in upper panels and red in lower panels. Ctsk inhibition restored WT levels of pSmad2 and pSmad1,5,8 activation in >60% and >50% of embryos, respectively. n = 30–50 embryos, 3 experiments. Scale bar = 10 μm.
Ctsk inhibition improved imbalanced Smad levels within MLII chondrocytes
As illustrated in Fig. 5D, chondrogenic maturation in many systems is characterized by decreasing TGFß (Smad2,3) and increasing BMP (Smad1,5,8) signal propagation. Because col2a1a expression is thought to be regulated by TGFßs and col10a1 expression by BMPs, we asked whether ctsk inhibition also restored normal TGFß-like and BMP-like Smad activation to MLII chondrocytes. fli1a:EGFP embryos were injected with either the gnptab MO alone or co-injected with the ctsk MO. Immunohistochemically stained cartilage sections showed substantial improvement in MLII Smad activation after ctsk inhibition (Fig. 5E). Both nuclear-localized pSmad2 levels and col2a1a-driven mCherry expression were reduced in >60% of ctsk-inhibited MLII chondrocytes (Fig. 5E). This was matched by increased pSmad1,5,8 staining and BMP-responsive EGFP expression in >50% of the embryos (Fig. 5F).
Ctsk is mistargeted outside of ML-II chondrocytes
Because loss of M6P can cause selective enzyme hypersecretion, we hypothesized that if mislocalized outside MLII chondrocytes Ctsk may directly affect TGFß or BMP growth factors. To address this possibility, we examined how the M6P tag is affected in MLII embryos and whether its reduction altered Ctsk’s localization. To assess the M6P levels on Ctsk, WT and MLII embryonic lysates were passed over a cation-independent M6P receptor (CI-MPR) affinity column and Ctsk activity from the bound and unbound fractions assessed.(38) As shown in Supplemental Table S1, 86% of the Ctsk activity and 16% of the Ctsd activity from WT lysates bound the CI-MPR column. This indicates that at least one of two N-linked sugar chains on zebrafish Ctsk normally bears M6P, whereas the single sugar chain on Ctsd is minimally modified. Analyses of WT adult brains confirmed that in zebrafish, Ctsk (93.1%) but not Ctsd (24.2%) is normally highly M6P-modified (Supplemental Table S1). In contrast, only 17.2% and 5.0% of the Ctsk and Ctsd activities from MLII lysates bound the column, reflecting the substantial loss of M6P tags in ML-II embryos.
Previous analyses showed that MLII whole embryo lysates contain 3 to 5 times more Ctsk activity than WT lysates.(15) To ask whether this increased “activity” was primarily intracellular or extracellular, WT and MLII fli1a:EGFP embryos were dissociated into single cells and the GFP (+) and (−) populations FACS-sorted and collected. Despite the fact that total MLII lysates contain more Ctsk activity,(15) the level of activity within dissociated MLII cells was 2 to 5 times lower than within dissociated WT cells (Fig. 6A). In contrast, Ctsd activity levels were similar within WT and MLII sorted cells (Fig. 6B). These data suggest that the majority of Ctsk (but not Ctsd) activity in MLII embryos is in the extracellular space and therefore removed when the embryos are dissociated into single cells. Indeed, Western blot analyses only detected enzymatically active mature Ctsk (~30 kDa) within WT sorted cells (Fig. 6C). Enzyme hypersecretion from MLII cartilage was confirmed by immunostaining and confocal microscopy. For these analyses, the intracellular compartment of chondrocytes was marked by cytosolic EGFP and the extracellular space with Alexa fluor 647-conjugated wheat germ agglutinin (WGA), which binds ECM. Although intracellular Ctsk (red) was detected in both WT and MLII chondrocytes, extracellular staining was only ever noted in MLII samples (Fig. 6D). True overlap with the extracellular marker WGA was determined by rotating overlap regions 90°, such that z axes were visible from the side. Lateral views show that although Ctsk and WGA staining were distinct in WT cartilages, regions of overlap occur in MLII elements. WT gnptab mRNA restored intracellular Ctsk localization (n = 20).
Fig. 6.
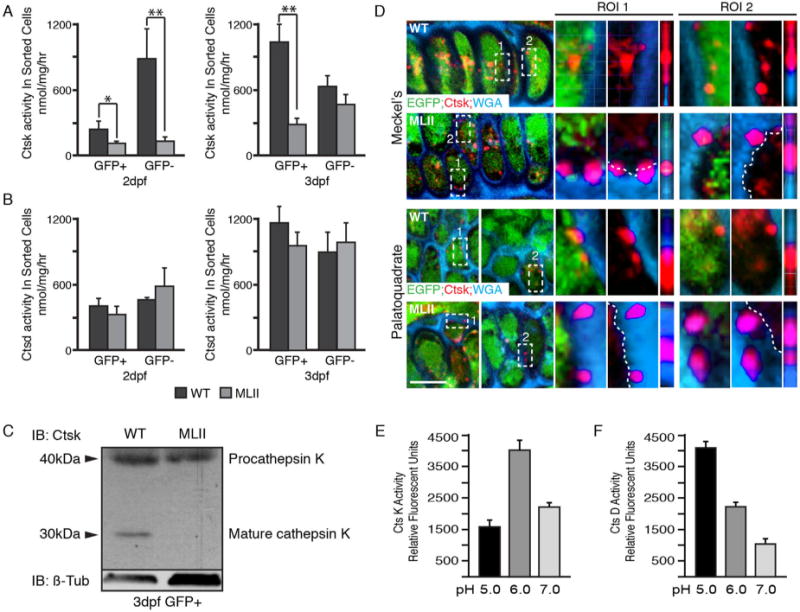
Ctsk is secreted outside MLII chondrocytes. Ctsk (A) and Ctsd (B) enzyme activities in GFP(+) and (−) cells isolated from 2 and 3 dpf WT (black bars) and MLII (gray bars) fli1a:EGFP embryos show that the intracellular levels of Ctsk (A) but not Ctsd (B) are reduced in MLII cells; n = 3 experiments. GFP+ cells are enriched for chondrocytes. (C) Western blot of Ctsk on 3 dpf WT and MLII chondrocyte-enriched GFP+ cells shows the 30 kDa mature form only present inside WT cells. n = 2 independent experiments with cells isolated from 300 embryos each time. (D) Confocal analyses of WT and MLII fli1a:EGFP cartilage sections stained for Ctsk (red) and WGA (blue). Two regions of interest (ROI) shown for each set. The 90° rotations are the third view under each region. n = 20–25 embryos/condition. (E) CtsK activity at multiple pHs (n = 3 experiments). (F) CtsD activity at multiple pHs (n = 3 experiments). Error values = SD.
For Ctsk to act on ECM-deposited proteins, it should retain activity at or near neutral pH. Activity assays run at multiple pHs (5.0, 6.0, and 7.0) showed that Ctsk had the highest relative activity at pH 6.0. Further, Ctsk retained ~50% of its maximal activity at pH 7.0 (Fig. 6E). In contrast, Ctsd was most active at pH 5.0 only retaining ~10% of its maximal activity at pH 7.0 (Fig. 6F). To further confirm that ML craniofacial phenotypes do not actually result from intracellular Ctsk deficiency, its expression was suppressed in WT embryos using a previously validated MO.(15) Alcian blue staining and immunohistochemical studies showed that inhibiting intracellular ctsk does not recapitulate either the MLII morphological or molecular phenotypes (Supplemental Fig. S4). Together these data suggest that Ctsk mediates its effect instead by a gain of extracellular function.
CTSK increases in vitro activation of latent TGFß complexes and reduces BMP2 activity
Analyses of multiple TGFß and BMP isoforms showed no major differences in relative transcript abundance between WT and MLII samples (Supplemental Fig. S5). Therefore, the imbalanced Smad signals and persistent type II collagen expression support a post-translational mechanism whereby latent TGFß may undergo sustained activation in MLII cartilage. Sustained TGFß activity could arise if extracellular Ctsk excessively liberates TGFß ligand from latent complexes, which form upon noncovalent association of the latency-associated peptide (LAP) and mature ligand. In most tissues, this small latent complex (SLC) binds latent TGFß binding proteins (LTBPs) to form a large latent complex (LLC), which incorporates into ECM via interactions between LTBP and ECM proteins (reviewed in Robertson and Rifkin(39)). A direct cathepsin-mediated activation mechanism relies on the protease’s ability to preferentially cleave either the LAP or LTBP, while sparing the mature TGFß ligand. Analysis of amino acid sequences of human and zebrafish TGFß1 precursors identified multiple potential Ctsk cleavage sites within the LAP portion of both molecules but not in any of the three mature TGFß ligands. To confirm this specificity, in vitro digestions using recombinant human CTSK and purified components of the SLC were performed at multiple pHs. Human LAP dimer was efficiently degraded at low pH but cleaved into distinct polypeptide fragments at neutral pH (Fig. 7A). Over the same pH and CTSK activity range, mature TGFß ligand was unaffected (Fig. 7B). The lack of an effect on TGFß activity after CTSK digestion was confirmed using a TGFß reporter cell line expressing firefly luciferase under the control of the plasminogen activator inhibitor-1 (PAI-1) promoter (Fig. 7C).(40) These data provide evidence that SLC components are differentially sensitive to CTSK cleavage, in a manner that favors activation of latent growth factor complexes.
To ask whether CTSK can also act on the large latent complex (LLC) to release a signaling-competent form of ligand, concentrated media from PMA-stimulated human erythroleukemia (HEL) cells was used as a source of LLC.(41) Liberation of bioactive ligand was again assessed with the TGFß reporter cells. When media-derived LLC was subjected to temperature-dependent activation, 80 °C yielded maximal release of ligand (Fig. 7D). In contrast exposure to 75 °C was suboptimal, giving 35% of the luciferase induction noted at 80 °C. To compare a known latent TGFß activator, LLC concentrates were digested with plasmin, which cleaves LTBP, before incubation on the reporter cells. Although no induction was found with plasmin digestion alone, plasmin followed by exposure to suboptimal temperature (75 °C) synergistically increased luciferase expression. This increase was equivalent to the activation found at 80 °C. Similarly, treatment of LLC with CTSK alone at pH 7 did not induce luciferase, but digestion followed by 75 °C yielded a significant increase in luciferase induction. These data suggest that at neutral pH CTSK digestion compromises LLC stability, lowering the barrier for heat-induced activation. To determine the respective effects on LTBP and LAP stability, plasmin and CTSK-digested LLC were analyzed by Western blot (Fig. 7E). Although plasmin completely degraded LTBP, CTSK led to protein fragmentation. Under these conditions, neither protease affected LAP stability, indicating that the physiological structure of the LLC may protect it from proteolysis.
In light of reduced BMP-related signals noted in MLII cartilage, we also asked whether CTSK could directly influence the activity of BMP2. Here, a C2C12 myoblast BMP-reporter line in which a BMP-responsive element (BRE) drives luciferase expression was used (Fig. 7F).(42) After incubation with CTSK, the activity of recombinant human BMP2 was reduced 50% within this reporter assay. Western blot analyses showed that the loss of BMP2 activity likely results from its degradation (Fig. 7G). These data indicate that although CTSK promotes latent TGFß activation, under the same conditions it inactivates BMP ligand.
Discussion
Cartilage and bone development depend on the coordinated action of multiple growth factor pathways,(26) with a transition between TGFß- and BMP-activated signals thought to play a central role. Here we report a novel pathogenic mechanism by which secreted Ctsk disrupts this transition, causing sustained Smad2,3 and suppressed Smad1,5,8 signaling (Fig. 8). This imbalance alters chondrocyte and osteoblast differentiation, adversely affecting cartilage and bone development. These findings provide a direct link between the loss of carbohydrate-dependent lysosomal targeting and MLII pathogenesis and point to secreted proteases as key mediators of tissue dysfunction.
Fig. 8.
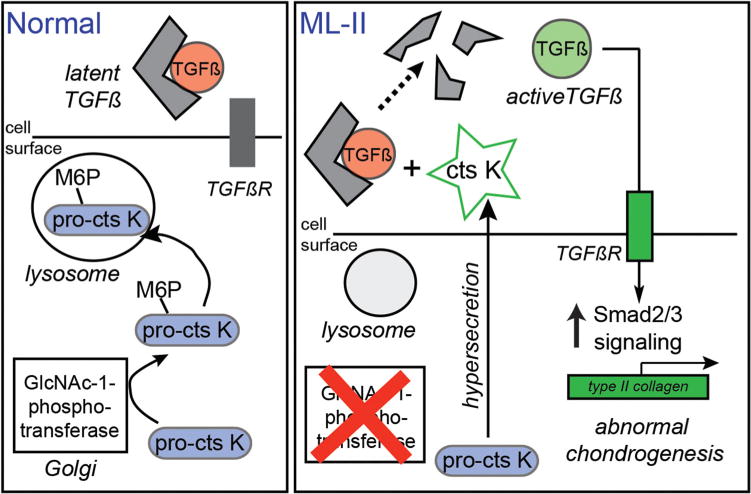
Model for how Ctsk alters growth factor signaling and chondrocyte differentiation in MLII cartilage. In MLII chondrocytes, the hypersecretion and extracellular action of Ctsk causes sustained activation of latent TGFß and expression of type II collagen, disrupting chondrogenesis. In vitro data also support the possibility that CtsK degrades BMPs, which may contribute to the imbalance in TGFß/BMP signaling during tissue development.
Importantly, the alterations noted in MLII zebrafish cartilage and bone was mirrored within the growth plates of the MLII kittens. In both cases, downstream effectors of the canonical TGFß pathway (Smad2/3) and its targets (type II collagen in zebrafish, GAGs in feline growth plates) were increased, whereas Smad1,5,8 signaling and expression of BMP targets (collagen type X and I) were decreased. In both organisms, altered growth factor-driven ECM expression also corresponded with increased Sox9 and decreased Runx and Osx expression. Although Sox9 is essential for early chondrogenesis, when sustained its expression can inhibit later stages of maturation.(30) An inhibitory role for Sox9 in MLII pathogenesis is supported by the observation that reducing Sox9a (but not its gene target col2a1) in MLII zebrafish improves craniofacial dysmorphia. In long bones where chondrocytes and osteoblasts arise from a common progenitor, high Sox9 expression favors the chondrogenic over the osteogenic fate. The increased size of MLII feline growth plates, delayed formation of secondary ossification centers, and reduced trabecular bone volume is consistent with the ability of Sox9 to delay osteogenic maturation. In both systems, the mechanism underlying increased Sox9 expression remains unclear. However, several reports demonstrate the ability of TGFß signals to promote Sox9 expression, suggesting that in MLII, increased Smad2,3 activation may contribute to its sustained activity.(43,44)
The finding that ctsk inhibition restores normal levels of both TGFß-related Smad activation and col2a1, col10a1, and sox9a transcripts support it as a key mediator of altered differentiation in MLII chondrocytes. Ctsk’s effect on MLII chondrogenesis appears to be driven by its increased extracellular activity, as opposed to loss of its intracellular activity. Its clear presence outside MLII chondrocytes (Fig. 6D) places Ctsk in proximity to both the growth factor ligands and their ECM-deposited latent complexes. Its loss from chondrocytes parallels observations in cultured murine MLII osteoclasts, where Ctsk was shown to be missing from secretory lysosomes.(45) In both cases, this hypersecretion is selective because Ctsd localization is unaltered. The facts that reducing Ctsk expression in WT zebrafish does not generate ML-like morphological or molecular phenotypes (Supplemental Fig. S4) support a deleterious gain of its extracellular function during MLII cartilage and bone development. Although our data suggest this as the primary disease mechanism in cartilage, intracellular hydrolase deficiency may contribute to pathogenesis in other tissues. In particular, cathepsin deficiency was recently reported to impair antigen processing in B cells.(46)
In vitro analyses with recombinant CTSK and components of the SLC showed that LAP but not mature ligand was digested at neutral pH. Moreover, digestion of HEL-derived LLC indicated that LTBP is the component of latent complexes most sensitive to CTSK proteolysis (Fig. 7E). This differential sensitivity supports a mechanism where secreted Ctsk disrupts the latent complex and liberates ligand to promote receptor-mediated activation. Protease-mediated activation of the latent complex resembles the effects of mutations in LTBP or LAP also known to cause excessive TGFß activity.(47–49) Although the details by which Ctsk mediates activation of latent TGFß in zebrafish cartilage are not yet known, in vitro experiments suggest that cathepsin proteolysis may prime complexes for heightened activation by intrinsic modes such as interaction with integrins.(50) In vitro data also show that unlike TGFß, Ctsk readily degrades BMP ligands, reducing their activity. Although compelling, these findings do not address whether Ctsk directly interacts with either the TGFß or BMP growth factor ligands in vivo. Other indirect mechanisms, including cross-talk between the two pathways, may also contribute to altered activation of Smad effectors. Such mechanisms may be particularly relevant for reduced BMP signaling in vivo.(51) Future work in the fish and feline systems will necessarily address the relationship between Ctsk and the TGFß-related ligands and their signaling cascades.
Imbalances in TGFß and BMP signaling have been reported in the context of other cartilage and bone disorders as well as in various genetic mutants. In many cases, the imbalance arises from ECM alterations that indirectly affect the incorporation, release, or activation of latent growth factors (reviewed in Robertson and Rifkin(39) and Olivieri and colleagues(52)). Although our data show cathepsin proteases can directly impact latent growth factors, we cannot rule out the possibility that cathepsin-mediated degradation of ECM proteins also contributes to pathogenesis. Interestingly, defects in GAG sulfation arising from mutations in the chondroitin 4-O sulfyltransferase also affect the balance of TGFß/BMP signals in murine skeletal tissue.(53) This is compelling because GAGs have been shown to directly modulate CTSK,(37,54) with certain structures stimulating its activity and others inhibiting it. This is intriguing in the context of MLII, where GAG expression is not only affected, but secretion of M6P modified sulfatases may also alter their composition.
This work highlights the ability of cathepsin proteases to influence growth factor signaling, either through enhanced activation of latent complexes or degradation of mature ligands. The finding that Smad2,3 signaling is increased in MLII animals may be significant clinically, as MLII joins a growing list of skeletal disorders characterized by excessive TGFß activity.(55,56) For diseases like Marfan syndrome, the development of anti-TGFß therapies has been promising,(57–60) raising the possibility that similar strategies may be leveraged for MLII. Considering other novel interventions such as cathepsin protease inhibition may be warranted because enzyme replacement is not feasible and other treatment modes have proven unsuccessful.(61)
Supplementary Material
Acknowledgments
We thank Dr Daniel Rifkin for providing the TGFß and BMP reporter cell lines, Dr Brian Link (MCW, Milwaukee, WI, USA) for the BRE-EGFP reporter line, Dr Chrissy Hammond for the col2a1a:mRFP line, and Dr Peter Lobel (CABM-Rutgers) for the CI-MPR affinity matrix. We also recognize Jérôme Amiaud, Celine Charrier, and Sevine Battaglia (Inserm UMR957, Nantes) for their expert help in histology and micro-CT. This work was supported by grants from NIGMS (GM-086524 and P40 OD-101939) and the National ISMRD/MPS Society.
Footnotes
Additional Supporting Information may be found in the online version of this article.
Disclosures
All authors state that they have no conflicts of interest.
Authors’ roles: HFS and RS conceived ideas, performed experiments, interpreted data, generated figures, and wrote the manuscript. MA performed experiments and assessed data. BK performed experiments and managed zebrafish care. PG performed feline immune-histochemistry. AP performed experiments. MH provided feline samples, interpreted data, and edited the manuscript. FB oversaw and interpreted feline experiments, generated figures, and wrote the manuscript.
References
- 1.Kornfeld S, Reitman ML, Varki A, Goldberg D, Gabel CA. Steps in the phosphorylation of the high mannose oligosaccharides of lysosomal enzymes. Ciba Found Symp. 1982;(92):138–56. doi: 10.1002/9780470720745.ch8. [DOI] [PubMed] [Google Scholar]
- 2.Reitman ML, Kornfeld S. Lysosomal enzyme targeting. N-Acetylglu-cosaminylphosphotransferase selectively phosphorylates native lysosomal enzymes. J Biol Chemi. 1981;256(23):11977–80. [PubMed] [Google Scholar]
- 3.Tiede S, Storch S, Lubke T, et al. Mucolipidosis II is caused by mutations in GNPTA encoding the alpha/beta GlcNAc-1-phospho-transferase. Nat Med. 2005;11(10):1109–12. doi: 10.1038/nm1305. [DOI] [PubMed] [Google Scholar]
- 4.Kudo M, Bao M, D’Souza A, et al. The alpha- and beta-subunits of the human UDP-N-acetylglucosamine:lysosomal enzyme N-acetylglu-cosamine-1-phosphotransferase are encoded by a single cDNA. J Biol Chem. 2005;280(43):36141–9. doi: 10.1074/jbc.M509008200. [DOI] [PubMed] [Google Scholar]
- 5.Qian Y, Lee I, Lee WS, et al. Functions of the alpha, beta, and gamma subunits of UDP-GlcNAc:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase. J Biol Chem. 2010;285(5):3360–70. doi: 10.1074/jbc.M109.068650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marschner K, Kollmann K, Schweizer M, Braulke T, Pohl S. A key enzyme in the biogenesis of lysosomes is a protease that regulates cholesterol metabolism. Science. 2011;333(6038):87–90. doi: 10.1126/science.1205677. [DOI] [PubMed] [Google Scholar]
- 7.Kollmann K, Pohl S, Marschner K, et al. Mannose phosphorylation in health and disease. Eur J Cell Biol. 2010;89(1):117–23. doi: 10.1016/j.ejcb.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 8.Leroy JG, Cathey S, Friez MJ. Mucolipidosis II. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews®. Seattle (WA): University of Washington; 1993. [Internet] [Google Scholar]
- 9.Cathey SS, Leroy JG, Wood T, et al. Phenotype and genotype in mucolipidoses II and III alpha/beta: a study of 61 probands. J Med Genet. 2010;47(1):38–48. doi: 10.1136/jmg.2009.067736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kudo M, Brem MS, Canfield WM. Mucolipidosis II (I-cell disease) and mucolipidosis IIIA (classical pseudo-hurler polydystrophy) are caused by mutations in the GlcNAc-phosphotransferase alpha/beta -subunits precursor gene. Am J Hum Genet. 2006;78(3):451–63. doi: 10.1086/500849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.David-Vizcarra G, Briody J, Ault J, et al. The natural history and osteodystrophy of mucolipidosis types II and III. J Paediatr Child Health. 2010;46(6):316–22. doi: 10.1111/j.1440-1754.2010.01715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herman TE, McAlister WH. Neonatal mucolipidosis II (I-cell disease) with dysharmonic epiphyseal ossification and butterfly vertebral body. J Perinatol. 1996;16(5):400–2. [PubMed] [Google Scholar]
- 13.Sprigz RA, Doughty RA, Spackman TJ, et al. Neonatal presentation of I-cell disease. J Pediatr. 1978;93(6):954–8. doi: 10.1016/s0022-3476(78)81218-9. [DOI] [PubMed] [Google Scholar]
- 14.Flanagan-Steet H, Sias C, Steet R. Altered chondrocyte differentiation and extracellular matrix homeostasis in a zebrafish model for mucolipidosis II. Am J Pathol. 2009;175(5):2063–75. doi: 10.2353/ajpath.2009.090210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petrey AC, Flanagan-Steet H, Johnson S, et al. Excessive activity of cathepsin K is associated with cartilage defects in a zebrafish model of mucolipidosis II. Dis Model Mech. 2012;5(2):177–90. doi: 10.1242/dmm.008219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lawson ND, Weinstein BM. In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev Biol. 2002;248(2):307–18. doi: 10.1006/dbio.2002.0711. [DOI] [PubMed] [Google Scholar]
- 17.DeLaurier A, Eames BF, Blanco-Sanchez B, et al. Zebrafish sp7:EGFP: a transgenic for studying otic vesicle formation, skeletogenesis, and bone regeneration. Genesis. 2010;48(8):505–11. doi: 10.1002/dvg.20639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Collery RF, Link BA. Dynamic smad-mediated BMP signaling revealed through transgenic zebrafish. Dev Dyn. 2011;240(3):712–22. doi: 10.1002/dvdy.22567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitchell RE, Huitema LF, Skinner RE, et al. New tools for studying osteoarthritis genetics in zebrafish. Osteoarthritis Cartilage. 2013;21(2):269–78. doi: 10.1016/j.joca.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Dev Dyn. 1995;203(3):253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- 21.Mangos S, Lam PY, Zhao A, et al. The ADPKD genes pkd1a/b and pkd2 regulate extracellular matrix formation. Dis Model Mech. 2010;3(5–6):354–65. doi: 10.1242/dmm.003194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thisse C, Thisse B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat Protoc. 2008;3(1):59–69. doi: 10.1038/nprot.2007.514. [DOI] [PubMed] [Google Scholar]
- 23.Li N, Felber K, Elks P, Croucher P, Roehl HH. Tracking gene expression during zebrafish osteoblast differentiation. Dev Dyn. 2009;238(2):459–66. doi: 10.1002/dvdy.21838. [DOI] [PubMed] [Google Scholar]
- 24.Kang JS, Oohashi T, Kawakami Y, Bekku Y, Izpisua Belmonte JC, Ninomiya Y. Characterization of dermacan, a novel zebrafish lectican gene, expressed in dermal bones. Mech Dev. 2004;121(3):301–12. doi: 10.1016/j.mod.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 25.Javidan Y, Schilling TF. Development of cartilage and bone. Methods Cell Biol. 2004;76:415–36. doi: 10.1016/s0091-679x(04)76018-5. [DOI] [PubMed] [Google Scholar]
- 26.Goldring MB, Tsuchimochi K, Ijiri K. The control of chondrogenesis. J Cell Biochem. 2006;97(1):33–44. doi: 10.1002/jcb.20652. [DOI] [PubMed] [Google Scholar]
- 27.Bi W, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B. Sox9 is required for cartilage formation. Nat Genet. 1999;22(1):85–9. doi: 10.1038/8792. [DOI] [PubMed] [Google Scholar]
- 28.Bi W, Huang W, Whitworth DJ, et al. Haploinsufficiency of Sox9 results in defective cartilage primordia and premature skeletal mineralization. Proc Natl Acad Sci USA. 2001;98(12):6698–703. doi: 10.1073/pnas.111092198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yan YL, Willoughby J, Liu D, et al. A pair of Sox: distinct and overlapping functions of zebrafish sox9 co-orthologs in craniofacial and pectoral fin development. Development. 2005;132(5):1069–83. doi: 10.1242/dev.01674. [DOI] [PubMed] [Google Scholar]
- 30.Olsen BR, Reginato AM, Wang W. Bone development. Ann Rev Cell Dev Biol. 2000;16:191–220. doi: 10.1146/annurev.cellbio.16.1.191. [DOI] [PubMed] [Google Scholar]
- 31.Kollmann K, Pestka JM, Kuhn SC, et al. Decreased bone formation and increased osteoclastogenesis cause bone loss in mucolipidosis II. EMBO Mol Med. 2013;5(12):1871–86. doi: 10.1002/emmm.201302979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mazrier H, Van Hoeven M, Wang P, et al. Inheritance, biochemical abnormalities, and clinical features of feline mucolipidosis II: the first animal model of human I-cell disease. J Hered. 2003;94(5):363–73. doi: 10.1093/jhered/esg080. [DOI] [PubMed] [Google Scholar]
- 33.Yan YL, Miller CT, Nissen RM, et al. A zebrafish sox9 gene required for cartilage morphogenesis. Development. 2002;129(21):5065–79. doi: 10.1242/dev.129.21.5065. [DOI] [PubMed] [Google Scholar]
- 34.Qian Y, Flanagan-Steet H, van Meel E, Steet R, Kornfeld SA. The DMAP interaction domain of UDP-GlcNAc:lysosomal enzyme N-acetylglu-cosamine-1-phosphotransferase is a substrate recognition module. Proc Natl Acad Sci USA. 2013;110(25):10246–51. doi: 10.1073/pnas.1308453110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walker MB, Kimmel CB. A two-color acid-free cartilage and bone stain for zebrafish larvae. Biotech Histochem. 2007;82(1):23–8. doi: 10.1080/10520290701333558. [DOI] [PubMed] [Google Scholar]
- 36.Furumatsu T, Tsuda M, Taniguchi N, Tajima Y, Asahara H. Smad3 induces chondrogenesis through the activation of SOX9 via CREB-binding protein/p300 recruitment. J Biol Chem. 2005;280(9):8343–50. doi: 10.1074/jbc.M413913200. [DOI] [PubMed] [Google Scholar]
- 37.Lecaille F, Bromme D, Lalmanach G. Biochemical properties and regulation of cathepsin K activity. Biochimie. 2008;90(2):208–26. doi: 10.1016/j.biochi.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 38.Fan X, Klein M, Flanagan-Steet HR, Steet R. Selective yolk deposition and mannose phosphorylation of lysosomal glycosidases in zebra-fish. J Biol Chem. 2010;285(43):32946–53. doi: 10.1074/jbc.M110.158295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robertson IB, Rifkin DB. Unchaining the beast; insights from structural and evolutionary studies on TGFbeta secretion, sequestration, and activation. Cytokine Growth Factor Rev. 2013;24(4):355–72. doi: 10.1016/j.cytogfr.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abe M, Harpel JG, Metz CN, Nunes I, Loskutoff DJ, Rifkin DB. An assay for transforming growth factor-beta using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal Biochem. 1994;216(2):276–84. doi: 10.1006/abio.1994.1042. [DOI] [PubMed] [Google Scholar]
- 41.Miyazono K, Olofsson A, Colosetti P, Heldin CH. A role of the latent TGF-beta 1-binding protein in the assembly and secretion of TGF-beta 1. EMBO J. 1991;10(5):1091–101. doi: 10.1002/j.1460-2075.1991.tb08049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zilberberg L, ten Dijke P, Sakai LY, Rifkin DB. A rapid and sensitive bioassay to measure bone morphogenetic protein activity. BMC Cell Biol. 2007;8:41. doi: 10.1186/1471-2121-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Furumatsu T, Ozaki T, Asahara H. Smad3 activates the Sox9-dependent transcription on chromatin. Int J Biochem Cell Biol. 2009;41(5):1198–204. doi: 10.1016/j.biocel.2008.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chimal-Monroy J, Rodriguez-Leon J, Montero JA, et al. Analysis of the molecular cascade responsible for mesodermal limb chondrogenesis: Sox genes and BMP signaling. Dev Biol. 2003;257(2):292–301. doi: 10.1016/s0012-1606(03)00066-6. [DOI] [PubMed] [Google Scholar]
- 45.van Meel E, Boonen M, Zhao H, et al. Disruption of the Man-6-P targeting pathway in mice impairs osteoclast secretory lysosome biogenesis. Traffic. 2011;12(7):912–24. doi: 10.1111/j.1600-0854.2011.01203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Otomo T, Schweizer M, Kollmann K, et al. Mannose 6 phosphorylation of lysosomal enzymes controls B cell functions. J Cell Biol. 2015;208(2):171–80. doi: 10.1083/jcb.201407077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dietz HC. TGF-beta in the pathogenesis and prevention of disease: a matter of aneurysmic proportions. J Clin Invest. 2010;120(2):403–7. doi: 10.1172/JCI42014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Janssens K, Gershoni-Baruch R, Guanabens N, et al. Mutations in the gene encoding the latency-associated peptide of TGF-beta 1 cause Camurati-Engelmann disease. Nat Genet. 2000;26(3):273–5. doi: 10.1038/81563. [DOI] [PubMed] [Google Scholar]
- 49.Janssens K, ten Dijke P, Ralston SH, Bergmann C, Van Hul W. Transforming growth factor-beta 1 mutations in Camurati-Engel-mann disease lead to increased signaling by altering either activation or secretion of the mutant protein. J Biol Chem. 2003;278(9):7718–24. doi: 10.1074/jbc.M208857200. [DOI] [PubMed] [Google Scholar]
- 50.Annes JP, Chen Y, Munger JS, Rifkin DB. Integrin alphaVbeta6-mediated activation of latent TGF-beta requires the latent TGF-beta binding protein-1. J Cell Biol. 2004;165(5):723–34. doi: 10.1083/jcb.200312172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van der Kraan PM, Blaney Davidson EN, Blom A, van den Berg WB. TGF-beta signaling in chondrocyte terminal differentiation and osteoarthritis: modulation and integration of signaling pathways through receptor-Smads. Osteoarthritis Cartilage. 2009;17(12):1539–45. doi: 10.1016/j.joca.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 52.Olivieri J, Smaldone S, Ramirez F. Fibrillin assemblies: extracellular determinants of tissue formation and fibrosis. Fibrogenesis Tissue Repair. 2010;3:24. doi: 10.1186/1755-1536-3-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kluppel M, Wight TN, Chan C, Hinek A, Wrana JL. Maintenance of chondroitin sulfation balance by chondroitin-4-sulfotransferase 1 is required for chondrocyte development and growth factor signaling during cartilage morphogenesis. Development. 2005;132(17):3989–4003. doi: 10.1242/dev.01948. [DOI] [PubMed] [Google Scholar]
- 54.Li Z, Hou WS, Escalante-Torres CR, Gelb BD, Bromme D. Collagenase activity of cathepsin K depends on complex formation with chondroitin sulfate. J Biol Chem. 2002;277(32):28669–76. doi: 10.1074/jbc.M204004200. [DOI] [PubMed] [Google Scholar]
- 55.Grafe I, Yang T, Alexander S, et al. Excessive transforming growth factor-beta signaling is a common mechanism in osteogenesis imperfecta. Nat Med. 2014;20(6):670–5. doi: 10.1038/nm.3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Neptune ER, Frischmeyer PA, Arking DE, et al. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33(3):407–11. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- 57.Gallo EM, Loch DC, Habashi JP, et al. Angiotensin II-dependent TGF-beta signaling contributes to Loeys-Dietz syndrome vascular pathogenesis. J Clin Invest. 2014;124(1):448–60. doi: 10.1172/JCI69666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Habashi JP, Judge DP, Holm TM, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312(5770):117–21. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Holm TM, Habashi JP, Doyle JJ, et al. Noncanonical TGFbeta signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science. 2011;332(6027):358–61. doi: 10.1126/science.1192149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kalluri R, Han Y. Targeting TGF-beta and the extracellular matrix in Marfan’s syndrome. Dev Cell. 2008;15(1):1–2. doi: 10.1016/j.devcel.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 61.Lund TC, Cathey SS, Miller WP, et al. Outcomes after hematopoietic stem cell transplantation for children with I-cell disease. Biol Blood Marrow Transplant. 2014;20(11):1847–51. doi: 10.1016/j.bbmt.2014.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.