

Abstract
The unique photochemical properties of Ru(II)-diimine complexes have helped initiate a series of seminal electron transfer studies in metalloenzymes. It has thus been possible to experimentally determine rate constants for long-range electron transfers. These studies have laid the foundation for the investigation of reactive intermediates in heme proteins and for the design of light-activated biocatalysts. Various metalloenzymes, such as hydrogenase, carbon monoxide dehydrogenase, nitrogenase, laccase and cytochrome P450 BM3 have been functionalized with Ru(II)-diimine complexes. Upon visible light-excitation, these photosensitized metalloproteins are capable of sustaining photocatalytic activity to reduce small molecules such as protons, acetylene, hydrogen cyanide and carbon monoxide or activate molecular dioxygen to produce hydroxylated products. The Ru(II)-diimine photosensitizers are hence able to deliver multiple electrons to metalloenzymes buried active sites circumventing the need for the natural redox partners. In this review, we will highlight the key achievements of the light-driven biocatalysts, which stem from the extensive electron transfer investigations.
1. Introduction
Considerable effort is currently devoted to the development of visible light-driven approaches to produce biofuels or synthetic chemical reactions as sustainable alternatives to meet the global energy needs.[1, 2] In this context, photosynthesis has provided a valuable platform for understanding the underlying mechanisms of light harnessing and efficient light-to-chemical energy conversion.[3] Following light absorption by the chlorophyll antenna, rapid charge separation occurs and a cascade of proton coupled electron transfer steps is promoted by a network of metalloenzymes and redox centers. Eventually, molecular dioxygen is produced at a unique metal cluster active site with the release of electrons and protons to reduce carbon dioxide.[4, 5]
Electron transfers are at the essence of many important biological transformations and have hence attracted a lot of interest from both experimental and theoretical perspectives. The seminal work by Marcus has provided the theoretical framework to consider electrons flowing between distant redox centers.[6] The semiclassical Marcus equation established the critical parameters governing the rates of electron transfer between a donor and an acceptor held at fixed distances and orientation. These parameters include the reaction driving force, a nuclear reorganization factor, and the strength of the electronic coupling between the reactants and products at the transition state. This electronic coupling term depends on the distance between the redox centers and the nature of the medium separating them.[6]
Experimentally, the Gray group pioneered the use of ruthenium complexes to study electron transfers in modified proteins.[7-10] The unique photochemical properties of the Ru(bpy)32+ prototype and its derivatives[11] have rendered these complexes valuable at triggering electron transfer processes.[12] Due to their intense metal-to-ligand charge transfer band and long lived excited state, the Ru(II)-diimine complexes are amenable to flash photolysis studies. Rapid charge separation and minimization of charge recombination via flash quench techniques, generate powerful reductant or oxidant species to initiate redox reactions. Moreover, the functionalization of the ancillary ligands on the Ru(II) complexes has enabled its covalent attachment to macromolecules and permitted intramolecular electron transfers in proteins.
Expanding the application of Ru(II)-diimine modified proteins to the field of light-driven biocatalysis has been achieved in several catalytically competent metalloproteins. Early successes with the reduction and activation of small molecules demonstrated the advantages of harnessing the catalytic potential of enzymes. Indeed, metalloenzymes present unique architectural and catalytic efficiency rarely matched by small molecule models. While there have been many important contributions from light-driven small molecule models,[13-18] this review focuses on the light-driven metalloenzymes functionalized with ruthenium-based photosensitizers.
First, a brief background on Ru(II)-diimine complexes and their excited state properties will be presented. The following sections will introduce the various synthetic strategies to covalently attach these complexes to proteins and the techniques to characterize their attachment. Then, the significant contributions and conclusions from the extensive studies on electron transfer in metalloproteins conducted by the Gray group [7-10] and others [19-21] will be highlighted. These studies have paved the way for the detection of elusive reaction intermediates in heme proteins and eventually to the development of light-driven biocatalysis. The last part of the review will illustrate examples of light-driven metalloproteins where Ru(II)-diimine complexes deliver the necessary electrons to the enzyme active sites for the efficient reduction and activation of small molecules toward the generation of biofuels and hydroxylated products.
2. Background on Ru(II)-diimine complexes
The d6 [Ru(LL)3]2+ complexes where LL = diimine ligands such as bipyridine (bpy), phenanthroline (phen) and their derivatives have been the class of metal complexes most deeply investigated from a photochemical point of view. This is due to their unique combination of chemical stability, luminescence, excited-state lifetime and reactivity. As these topics have been extensively reviewed and are well documented,[11, 22, 23] this section will highlight the key properties of these photosensitizers relevant to electron transfer processes and photocatalytic activity in metalloenzymes.
The most well-studied metal complex in this series, Ru(bpy)32+ (bpy = 2,2′-bipyridine), exhibits a typical strong, broad absorbance in the visible range centered at 450 nm (ε = 15,000 M−1cm−1) assigned to a metal-to-ligand charge transfer (MLCT) transition.[11] Excitation into this band followed by rapid inter-system crossing results in the production of a long-lived triplet excited state. This excited state can decay back to the ground state via a radiative process or encounter other solute molecules called quenchers. The participation in these bimolecular processes results in energy transfer or both reductive and oxidative electron transfers (Figure 1). Depending on the nature of the quencher, a highly reductive, Ru(bpy)3+, or oxidative, Ru(bpy)33+, species can be photogenerated. Some of the most commonly used reductive quenchers are ascorbic acid, dithionite, dithiocarbamate or p-methoxy-dimethylaniline while the oxidative quenchers typically comprise methyl viologen, [Ru(NH3)6]3+ and [Co(NH3)5Cl]2+. Both oxidative and reductive routes have been used to promote electron transfers in metalloproteins and generate reactive intermediates in heme proteins as described in later sections. In addition, modification of the ancillary ligands in the Ru(II)-diimine complexes has permitted the tuning of their photophysical properties[11] and their covalent attachment to various macromolecules.
Fig. 1.

Photocatalytic cycle of a typical [Ru(bpy)3]2+ complex. Encountering of its excited state with a solute molecule, electron donor D or acceptor A, generates a highly reductive ([Ru(bpy)3]+, left) or oxidative ([Ru(bpy)3]3+, right) species, respectively.
3. Covalent attachment of Ru(II)-diimine complexes to proteins
3.A. Synthetic strategies
Early attempts at electron transfer studies or light-driven biocatalysis have used Ru(bpy)32+ complex in solution with the metalloprotein of interest. This bimolecular process often resulted in marginal results requiring the need for efficient electron relay systems as discussed below. Hence, strong efforts have been devoted to covalently attach the photosensitizer to metalloproteins in order to promote unimolecular electron transfer reactions. Various covalent strategies have been undertaken for the selective attachment of Ru(II) complexes by taking advantage of the respective reactivity of amino acid side chains as summarized in Table 1.
Table 1.
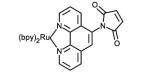
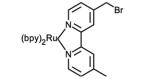
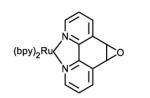
Structures of ruthenium complexes used in the covalent attachment to protein amino acid side chain or cofactors (bpy = 2,2′-bipyridine).
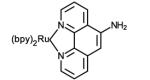



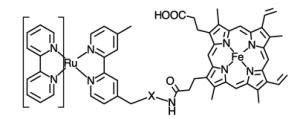
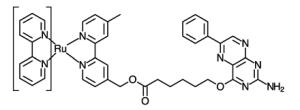
Initial reports involved the direct attachment to histidine residues using aquo complexes such as Ru(NH3)5(H2O),[24, 25] Ru(bpy)2(Im)(H2O) or Ru(tpy)(bpy)(H2O) complexes (bpy = 2,2′-bipyridine, Im = imidazole, tpy = 2,2′:6′,2″-terpyridine) (Table 1, entry 1).[26] Peptide coupling strategies have resulted in the attachment of Ru(II) complexes bearing amino or carboxylic acid moieties to proteins (Table 1, entries 2 and 3). On one hand, the amino group of Ru(bpy)2PhenA (PhenA = 5-amino-1,10-phenthroline) can be coupled with the carboxylic acid side chains of aspartic or glutamic acids in the presence of crosslinking carboxydiimide reagents.[27] On the other hand, the amino side chain of lysine residues can react with N-hydrosuccinimide derivatives of the Ru(bpy)2(dcbpy) complex (dcbpy = 4,4′-dicarboxy-2,2′-bipyridine)[28] or the isocyanate moiety introduced on a phenanthroline ligand (Table 1, entry 4). [29, 30]
Ultimately, the nucleophilicity of cysteine residues and their lower natural abundance [31] have been very advantageous in the selective covalent attachment of various photosensitizers (Table 1, entries 5-8). Sulfhydryl-specific labeling has been achieved via the introduction of reactive maleimide,[32, 33] bromoalkyl,[34] and iodoacetamide[35] substituents onto various luminescent complexes. We have also recently reported the selective sulfhydryl ring opening of an epoxide moiety in several d6 metal complexes containing the 5,6-epoxy-5,6-dihydro-1,10-phenanthroline ligand.[36]
Alternatively, several groups have investigated the attachment of Ru(II) photosensitizers directly to protein cofactors (entries 9 and 10) such as the prosthetic heme group [37, 38] in reconstituted heme proteins or the pterin cofactor of the inducible nitric oxide synthase.[39]
3.B. Characterization of the covalent attachment
A combination of spectrophysical and biological techniques have been utilized to confirm the selective attachment of the photosensitizer to macromolecules by taking advantage of the unique photophysical properties of ruthenium-based photosensitizers. The strong MLCT band can be detected in covalent adducts of metalloproteins even in the presence of the strong absorbing Soret band of the heme cofactor.[38, 40, 41] Measurements of the excited state lifetime can also provide valuable information regarding the local environment around the photosensitizer.[28, 42] Using mass spectrometry, a characteristic shift in the mass of the protein is observed upon covalent attachment of the photosensitizer.[40] Recently, the use of inductively coupled optical emission spectroscopy (ICP-OES) was valuable in establishing the presence of a Ru(II) complex in photosensitized nitrogenase mutants.[43]
X-ray crystallography has remained a unique tool to probe the local environment around the covalently attached Ru(II) photosensitizer and to determine the distances between redox cofactors.[26, 40, 44] However, despite recent progress, crystallization of macromolecules still remains a laborious process. Information on the selective covalent attachment can also be obtained from tryptic digest of the labeled metalloenzymes and analysis of the peptidic fragments.[25, 45] This method was initially used to establish the labeling of ferricytochrome c with pentaamineruthenium(III) complexes. We recently used a similar digestion to unambiguously confirm the position of covalent attachment of the photosensitizer to cytochrome P450 BM3 heme domain mutants.[41] High sequence coverage of the protein was obtained and the labeled peptide fragment could be easily detected and identified using the unique isotopic distribution of the ruthenium atom as fingerprint.
4. Phototriggered electron transfer processes in metalloproteins
4.A. Intramolecular electron transfer studies
First evidence of long range electron tunneling was reported in a Ru(NH3)53+ functionalized ferricytochrome c in 1982,[24] paving the way for the pioneering work of the Gray group. The Ru(NH3)53+ complex was rapidly replaced by Ru(II)-diimine complexes as their unique photophysical properties enabled a wider range of measurements not possible with the nonluminescent amino-based complexes.[9, 12] More than 30 proteins have been functionalized with ruthenium complexes, leading to the systematic investigation of parameters governing electron transfer steps in these models.[7, 8, 10, 46] The proteins comprise the blue copper proteins azurin, plastocyanin and stellacyanin, the high potential iron-sulfur protein (HiPIP) as well as heme proteins such as myoglobin and the cytochromes c, b5 and b562. The measured rates of electron transfers were found to span 7 orders of magnitude between redox centers separated by distances ranging from 12 to 26 Å.[8] In a tabulated timetable for long-range electron transfers, most of the determined rate constants in proteins show exponential distance dependence and are dispersed around a decay constant, β, of 11 nm−1.[7-10] This electron tunneling coupling efficiency is between the efficiency across glassy toluene (β = 12 nm−1) and an all-covalent alkane bridge (β = 10 nm−1), which reflects the heterogeneity and complexity of the protein tertiary structures in mediating electron transfers. These extensive investigations and their implication to the Marcus theory have been well documented in several reviews.[7-10]
In certain cases, electrons have been found to travel distances greater than 25 Å in a timescale not expected for single electron transfer steps. This process would then require multi step tunneling or “hopping”. Gray and coworkers recently demonstrated such feat in azurin where an engineered tryptophan is in van der Waals contact with the photosensitizer. Oxidation of the azurin Cu(I) center occurred two orders of magnitude faster than expected for electron tunneling over the same distance.[44]
4.B. Intermolecular electron transfer and proton coupled electron transfer studies
The phototriggering of electron transfer in metalloproteins has also been expanded to the study of intermolecular processes in protein complexes, notably, the cytochrome c oxidase assembly.[20, 46, 47] Cytochrome c oxidase is the vital protein in the respiratory system of mitochondria. It performs the four-electron reduction of molecular dioxygen to water accompanied with the pumping of protons across the membrane.[48] The electrons are provided from the redox partner cytochrome c and enters the oxidase at the dinuclear copper site, CuA. They are then transferred to the catalytic hemeA-CuB site where the dioxygen reduction takes place. Millet, Durham and coworkers have used a cascade of electron transfer steps initiated at the ruthenium modified cytochrome c to eventually promote the reduction of cytochrome c oxidase.[19, 20, 47] This approach has also enabled the study of proton pumping across the membrane accompanying the oxygen reduction.[19]
An extension of the single electron transfer step is the concomitant motion of a proton summarized as proton coupled electron transfer or PCET. Several reviews have elegantly summarized the experimental and theoretical implication of the PCET mechanism.[49, 50] Worth highlighting are the studies on deciphering the long-range electron transfer pathway in the ribonucleotase reductase (RNR).[21, 51] RNR catalyzes the conversion of nucleotide to deoxynucleotide via hydrogen atom abstraction to provide the precursors required for DNA replication and repair in all organisms. Radical transport occurs over 35 Å and involves the side chains of conserved aromatic amino acids.[51] The electron transport mechanism is thought to be closely coupled to proton motion based on a combination of site-directed mutagenesis and photoinitiated radical transport. [21, 51]
5. Study of elusive reactive intermediates in Ru(II)-diimine modified proteins
The ultrafast techniques developed for the study of electron transfer steps has also allowed the investigation of high-valent reactive intermediates in heme proteins. In a flash quench oxidative route, the excited state of the Ru(bpy)32+ complex is quenched by an electron acceptor solute to generate a Ru(III) species with an estimated potential of 1.26 V vs NHE (Figure 1).[11] Such photogenerated species has been used to oxidize heme cofactors in various proteins.[52, 53] In both horseradish peroxidase and microperoxidase, initial oxidation of the heme group occurred at the porphyrin ring followed by rapid conversion to generate the high valent ferryl Compound II species. A second round of photoexcitation leads to the formation of Compound I, a ferryl porphyrin π cation radical. Oxidation of the heme center was monitored by transient absorption and global fitting of the single wavelength kinetic traces enabled the determination of rate constants for the individual processes.[52, 53] In these proteins, it is worth noting that the heme cofactor is rather surface exposed, and thus prone to oxidation by the photogenerated Ru(III) species in solution. However, no photooxidation could be observed in proteins with more buried active sites.
In order to relay electrons to these buried sites, Ru(II)-diimine based molecular wires were developed to promote non-covalent binding to metalloproteins using their recognition binding sites. A variety of molecular wires showed tight binding to the active site of P450 cam,[54, 55] amine oxidase[56] and nitric oxide synthase.[39] Only a few cases showed heme reduction by the ruthenium-based molecular wires. [54, 55] Additional approaches have emerged to covalently attach the Ru(II) photosensitizer to heme prosthetic group or pterin cofactor (Table 1, entries 9 and 10). Several synthetic steps are required to assemble such systems and only marginal results have been observed. In the case of myoglobin where the original cofactor could be replaced by a Ru(II)-modified heme, a porphyrin radical has been observed in two separate studies followed by formation of Compound II (Fe(IV)=O) in one case [37] or an amino acid radical in the other.[38] For the covalently modified pterin cofactor, rapid heme reduction of the inducible nitric oxide synthase protein was observed[39] but more work is still needed to form valuable reactive intermediates.
Eventually, photoxidation of the heme cofactor in cytochromes P450 was achieved using a Ru(II) photosensitizer covalently attached at an engineered non-native single cysteine mutant of the P450 BM3 heme domain.[40] Upon photoexcitation and quenching with the oxidative quencher, Ru(NH3)63+, rapid oxidation of the porphyrin ring was observed which rearranged to the ferryl Compound II species. The pH dependence study further established the protonated form of the ferryl species that is characteristic of cys-coordinated heme proteins.[57]
6. Light-driven biocatalytic processes
Since Ru(II) complexes could promote electron transfers in metalloproteins upon excitation, there has been a strong motivation to drive catalytically competent enzymes with visible light and capitalize on their unique synthetic ability. Early evidence of photocatalytic activity was provided using a derivative of sperm whale myoglobin functionalized with pentaamineruthenium(III) complexes. This synthetic “oxidation-reduction” enzyme catalyzes the reduction of dioxygen coupled with oxidation of ascorbate and durohydroquinone with kcat of 0.30 to 0.60 s−1.[58] From their strong absorption in the visible range and unique photophysical properties highlighted in the previous sections, Ru(II)-diimine photosensitizers have become functional redox partners to activate metalloproteins upon visible light excitation. Often, a reductive photocatalytic cycle, highlighted in Figure 1, is used to deliver multiple electrons to their unique active sites.
This section will illustrate the various enzymes powered by Ru(II) photosensitizers that resulted in the photogeneration of valuable products. Dihydrogen production was established in light-driven hydrogenase systems. Reduction of small molecules such as carbon dioxide or protons, acetylene and hydrogen cyanide have been achieved with photosensitized carbon monoxide dehydrogenase and nitrogenase, respectively. Meanwhile, under aerobic conditions, reduction of molecular dioxygen was obtained with a light-driven laccase and efficient O2 activation in Ru(II)-diimine functionalized P450 BM3 enzymes has led to high photocatalytic production of hydroxylated fatty acids.
6.A. Photoproduction of H2 from photosensitized hydrogenases
Hydrogenases are a large class of metalloenzymes that catalyze the conversion of dihydrogen into protons and electrons as well as the reverse reaction. There are three main types of hydrogenases, depending on the metal present in their active site: an all iron active site, the [FeFe] hydrogenase, a mixed metal [NiFe] hydrogenase and an [Fe] only hydrogenase.[59] Hydrogenases are typically O2 sensitive, but a more O2 tolerant subclass, the [NiFeSe] hydrogenase, was identified where the coordinating cysteine group is replaced by a seleno cysteine. The redox partners are either NAD(P)+ or a cytochrome of b- or c-type.
Various contributions to powering hydrogenases with visible light have focused on the interactions between the light harvesting units and the enzyme.[60] The first account of H2 photoproduction using a Ru(II) photosensitizer was reported more than 30 years ago by Okura et al. using a [NiFe] hydrogenase.[61] Electron relays such as methyl viologen and cytochrome c3 were necessary for activity (Figure 2A).[62] Nevertheless, the system suffers from low efficiency due to high oxygen sensitivity. Improvements have followed using photosystem I as the light harvester, leading to the H2 production at a rate of 0.58 umol/mg of enzyme/h.[63] More recently, entrapping the hydrogenase into a nanoporous glass plate in the presence of Ru(bpy)32+ and methyl viologen has resulted in the production of hydrogen with an efficiency of 3.7 μmol H2 m−2 s−1 under external aerobic conditions.[64]
Fig. 2.

Strategies for the light-driven production of dihydrogen using photosensitized hydrogenases (PDB ID: 3ZE9). A) The use of methyl viologen (MV) or a methyl viologen-cytochrome c assembly (MV1) with Ru(bpy)32+ resulted in minimal H2 production; B) Covalent attachment of the photosensitizer to a [NiFe] hydrogenase in the presence of both methyl viologen and EDTA improved the rate of hydrogen production; C) Higher H2 production rate was obtained with a more O2 tolerant [NiFeSe] hydrogenase sensitized with a TiO2 nanoparticle containing a phosphonated Ru(II)-diimine complex. Adapted with permission from [67]. Copyright 2009 American Chemical Society.
Dyer et al. confirmed minimal H2 production with a Ru(bpy)32+/ascorbic acid / [NiFe] hydrogenase.[65] This system was found to be 100 times less efficient compared to a quantum dot system. The difference was mainly attributed to a difference in binding of the photosensitizer between the positively charged Ru(II) based photosensitizer and the negatively capped quantum dot.[65] The slow kinetics with the Ru(II) system conveniently enabled the observation and characterization of most intermediates in the hydrogenase catalytic cycle. The direct attachment of a ruthenium photosensitizer to a [NiFe]-hydrogenase (Figure 2B) led to the production of dihydrogen with a rate of 16 nmol of H2/min/mg of protein, which corresponds to 592 units/uM of ruthenium photosensitizer.[27] Armstrong et al. took on a different approach using a TiO2 nanoparticle sensitized with a Ru(II) complex bearing phosphonate groups (Figure 2C).[66, 67] In this case, the TiO2 nanoparticle acts not only as anchor for both the enzyme and the light-harvesting compound but also as an electron mediator in the process.[67] A first report established the photoproduction of dihydrogen from the sensitized nanoparticle assemblies.[66] A more systematic investigation of several hydrogenases and photosensitizers led to H2 production at a rate of 50s−1 with the more dioxygen tolerant [NiFeSe]-hydrogenase.
6.B. Photoreduction of CO2 using a sensitized carbon monoxide dehydrogenase
The carbon monoxide dehydrogenase, CODH I, has an unusual [Ni-4Fe4S] active site that catalyzes the reversible oxidation of CO to CO2. Following a similar system developed for the hydrogenase, Armstrong and coworkers turned their attention to the two electron reduction of CO2 to carbon monoxide using TiO2 nanoparticles sensitized with the phosphonated Ru(II) complex.[68] In controlling the absorption of the CODH I enzyme onto the nanoparticle, 5 μmol of CO was produced after four hours of irradiation, corresponding to an average turnover number of 250 μmol of CO/gram of TiO2/hour. Successive improvements in the use of anatase/rutile TiO2 and in the control of enzyme binding onto the surface led to enhancements of the overall activity and tolerance to dioxygen.[69]
6.C. Photoreduction of small molecules in Ru(II)-modified nitrogenase
Dinitrogen reduction to two molecules of ammonia is only achieved industrially through the costly and high energy demanding Haber-Bosch process. Meanwhile, nitrogenase achieves the biological eight-electron reduction of dinitrogen at its unique FeMoCo active site under ambient conditions.[70, 71] The strictly required FeP protein couples the hydrolysis of ATP molecules with the delivery of electrons, via the P cluster, to the catalytic FeMoCo cofactor of the iron-molybdenum protein (MoFeP), where dinitrogen reduction occurs (Figure 3). Recently, Tezcan and coworkers have covalently attached a Ru(II) photosensitizer (Table 1, entry 7) onto the MoFeP protein and demonstrated the two electron reduction of protons and acetylene to their corresponding products H2 and C2H4, respectively.[43] Three positions of covalent attachment were investigated. While two of the positions did not lead to any product formation, the third position (Ru-C158 in close proximity to the P cluster) lead to the evolution of both products in equal quantities (Figure 3), with the average velocities of 16 nmol C2H4/min and 14 nmol H2/min per mg of MoFeP. Photodriven C2H4 and H2 production reached a plateau after 50 min despite the presence of excess reductive quencher, dithionite, yielding a turnover number of ~110 per active site for both products. This light-driven generation of products established that nitrogenase could be activated without the use of its redox partner, FeP, protein and the high consumption of ATP (Figure 3). Later on, with a similar MoFeP construct, the six-electron reduction of hydrogen cyanide to methane was also established with an initial velocity of 0.4 nmol of CH4/min per mg of MoFeP, indicating the ability of the photosensitizer to deliver multiple electrons to the active site.[72]
Fig. 3.

Light-driven reduction of small molecules (acetylene, protons, hydrogen cyanide) using a photosensitized component, FeMoP (dark gray), of nitrogenase. The covalently attached Ru(II)-diimine complex (red) is able to deliver the necessary electrons to the FeMoCo active site and thus bypasses the need for the ATP dependent FeP protein (light gray) (PDB ID: 4WZB).
While the aforementioned light-driven biocatalysis was obtained under reduced oxygen atmosphere, a challenging task has been to carry photocatalysis under aerobic conditions with Ru(II)-diimine functionalized metalloenzymes. Indeed, dioxygen is known to react with the Ru(II) excited state and the photogenerated reductive species to yield detrimental reactive oxygen species.[73] Singlet dioxygen is produced from energy transfer between dioxygen and the Ru(II) excited state while formation of superoxide or hydrogen peroxide is due to reductive electron transfers from the photosensitizer. Nevertheless, recent examples using laccase and the monooxygenase cytochrome P450 BM3 demonstrated the feasibility of light-driven biocatalysis under aerobic conditions.
6.D. Photosensitized Laccase
Laccases belong to a class of blue copper enzymes found in fungi and plants that have the ability to efficiently reduce molecular dioxygen at a trinuclear copper center.[74] The four-electron reduction is usually coupled to oxidation of phenolic compounds, aromatic amines and ascorbate. These enzymes have found potential use in diverse biotechnological and environmental applications.[75] In biofuel cells, laccases have been immobilized on carbon based polypyrrole film where various embedded Ru(II) and Os(II) complexes act as redox mediators to create biocathodes.[76, 77] Such redox hydrogel design has led to efficient dioxygen electroreduction outperforming platinum-based catalysts.[76-78]
Toward light-driven biocatalytic applications, Tron et al. have utilized photocatalytic reductive cycles to power laccase with various light-harvesting units.[79, 80] The first report included Ru(bpy)32+ as the photosensitizer with EDTA as the sacrificial electron donor.[80] While modest O2 consumption was observed with this system, higher rates (1.7 μmolL−1s−1) were achieved when switching to a soluble zinc porphyrin photosensitizer.[79]
6.E. Light-driven P450 BM3 biocatalysts
One attractive target for biocatalysis has been the superfamily of cytochrome P450 enzymes due to their high regio- and stereoselective synthetic potential.[81] This superfamily of enzymes catalyzes the insertion of an oxygen atom into unactivated C-H bonds of a wide range of organic substrates using molecular dioxygen and two electrons.[82] The reducing equivalents are delivered by the electron providing reductase from the NAD(P)H cofactor.
Our laboratory has initially focused on the soluble fatty acid hydroxylase, P450 BM3.[83] Our earlier work on the photooxidization of the heme cofactor in a Ru(II)-diimine functionalized P450 BM3 mutant[40] established the electronic coupling between the two redox centers. These findings prompted the investigation of a photoreductive route to activate the P450 enzyme upon visible light irradiation and generate hydroxylated products. Low turnover numbers in the hydroxylation of the model substrate, lauric acid, were obtained with the initial mutants.[84] However, varying the position and nature of the photosensitizer to optimize the electron transfer pathway[85] resulted in a mutant, sL407C-2, containing a Ru(II) photosensitizer with electron donating groups covalently attached to an engineered cysteine residue at position L407C (Figure 4). This hybrid P450 BM3 mutant showed an enhanced activity in the light-driven hydroxylation of the C12 fatty acid. More than 900 total turnover numbers were obtained with a fast initial reaction rate of 120 eq/min.[41] It is worth noting that the ratio of hydroxylated products in the photocatalytic enzyme reaction is identical to that observed with the natural system. This light-driven P450 biocatalyst was also used in the one-step synthesis of a valuable synthon in route to the synthesis of several natural products.[86] The high photocatalytic activity establishes that the photogenerated reductive species is able to inject electrons to the heme active site and sustain photocatalytic activity. In addition, this approach circumvents the use of the natural electron transfer pathway, reductase and NAD(P)H cofactor, (Figure 4) and could therefore be more applicable to other members of the large family of cytochrome P450 enzymes.
Fig. 4.

Light-driven hydroxylation of long chain fatty acids using Ru(II)-diimine functionalized P450 BM3 biocatalysts. The necessary electrons for the activation of molecular dioxygen at the heme active site (blue) are delivered by the Ru(II) photosensitizer covalently attached to a non-native single cysteine (L407C) in place of the reductase cofactor (FMN domain shown in light gray, PDB ID: 1BVY).
7. Conclusion
For more than three decades, the post-modification of proteins with Ru(II)-diimine photosensitizers has enabled the systematic study of intra- and intermolecular electron transfers and the deciphering of parameters governing the electron transfer rates. These investigations have also highlighted the importance of the protein tertiary structures in mediating electron transfer steps. Building on this work, the field of Ru(II)-diimine functionalized metalloproteins has been expanded to catalytically competent enzymes. The growing examples of light-driven systems establish that Ru(II)-diimine photosensitizers are amenable to deliver multiple electrons to buried active sites and sustain photocatalytic activity upon visible light excitation under both anaerobic and aerobic conditions. Moreover, the need for redox partner enzymes or expensive cofactors is often alleviated. While a number of alternative photosensitizers such as nanoparticles, quantum dots, metal complexes or organic dyes are emerging,[87-91] Ru(II)-diimine photosensitizers still remain a premier choice in light driven processes thanks to their unique tunable photochemical properties combined with an ease of synthesis and derivatization.
Acknowledgements
This work was supported by the National Institute of General Medical Science of the National Institutes of Health under award number SC3 GM095415.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Gray HB. Powering the planet with solar fuel. Nat Chem. 2009;1:7–7. doi: 10.1038/nchem.141. [DOI] [PubMed] [Google Scholar]
- [2].Lewis NS, Nocera DG. Powering the planet: Chemical challenges in solar energy utilization. P Natl Acad Sci USA. 2007;104:20142–20142. doi: 10.1073/pnas.0603395103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Nelson N, Ben-Shem A. The complex architecture of oxygenic photosynthesis. Nat Rev Mol Cell Bio. 2004;5:971–982. doi: 10.1038/nrm1525. [DOI] [PubMed] [Google Scholar]
- [4].Tommos C, Babcock GT. Oxygen production in nature: A light-driven metalloradical enzyme process. Accounts Chem Res. 1998;31:18–25. [Google Scholar]
- [5].Styring S, Sjoholm J, Mamedov F. Two tyrosines that changed the world: Interfacing the oxidizing power of photochemistry to water splitting in photosystem II. Bba-Bioenergetics. 2012;1817:76–87. doi: 10.1016/j.bbabio.2011.03.016. [DOI] [PubMed] [Google Scholar]
- [6].Marcus RA, Sutin N. Electron Transfers in Chemistry and Biology. Biochim Biophys Acta. 1985;811:265–322. [Google Scholar]
- [7].Gray HB, Winkler JR. Electron flow through metalloproteins. Bba-Bioenergetics. 2010;1797:1563–1572. doi: 10.1016/j.bbabio.2010.05.001. [DOI] [PubMed] [Google Scholar]
- [8].Winkler JR, Gray HB. Electron Flow through Metalloproteins. Chem Rev. 2014;114:3369–3380. doi: 10.1021/cr4004715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gray HB, Winkler JR. Electron transfer in proteins. Annu Rev Biochem. 1996;65:537–561. doi: 10.1146/annurev.bi.65.070196.002541. [DOI] [PubMed] [Google Scholar]
- [10].Winkler JR, Gray HB. Long-range electron tunneling. J Am Chem Soc. 2014;136:2930–2939. doi: 10.1021/ja500215j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kalyanasundaram K. Photophysics, Photochemistry and Solar-Energy Conversion with Tris(Bipyridyl)Ruthenium(Ii) and Its Analogs. Coordin Chem Rev. 1982;46:159–244. [Google Scholar]
- [12].Chang IJ, Gray HB, Winkler JR. High-Driving-Force Electron-Transfer in Metalloproteins - Intramolecular Oxidation of Ferrocytochrome-C by Ru(2,2′-Bpy)2(Im)(His-33)3+ J Am Chem Soc. 1991;113:7056–7057. [Google Scholar]
- [13].Kotani H, Suenobu T, Lee YM, Nam W, Fukuzumi S. Photocatalytic Generation of a Non-Heme Oxoiron(IV) Complex with Water as an Oxygen Source. J Am Chem Soc. 2011;133:3249–3251. doi: 10.1021/ja109794p. [DOI] [PubMed] [Google Scholar]
- [14].Berardi S, Drouet S, Francas L, Gimbert-Surinach C, Guttentag M, Richmond C, Stoll T, Llobet A. Molecular artificial photosynthesis. Chem Soc Rev. 2014;43:7501–7519. doi: 10.1039/c3cs60405e. [DOI] [PubMed] [Google Scholar]
- [15].Frischmann PD, Mahata K, Wurthner F. Powering the future of molecular artificial photosynthesis with light-harvesting metallosupramolecular dye assemblies. Chem Soc Rev. 2013;42:1847–1870. doi: 10.1039/c2cs35223k. [DOI] [PubMed] [Google Scholar]
- [16].Karkas MD, Verho O, Johnston EV, Akermark B. Artificial Photosynthesis: Molecular Systems for Catalytic Water Oxidation. Chem Rev. 2014;114:11863–12001. doi: 10.1021/cr400572f. [DOI] [PubMed] [Google Scholar]
- [17].Fukuzumi S. Artificial photosynthetic systems for production of hydrogen. Current Opinion in Chemical Biology. 2015;25:18–26. doi: 10.1016/j.cbpa.2014.12.008. [DOI] [PubMed] [Google Scholar]
- [18].Simmons TR, Berggren G, Bacchi M, Fontecave M, Artero V. Mimicking hydrogenases: From biomimetics to artificial enzymes. Coordin Chem Rev. 2014;270:127–150. [Google Scholar]
- [19].Durham B, Millett F. Design of photoactive ruthenium complexes to study electron transfer and proton pumping in cytochrome oxidase. Bba-Bioenergetics. 2012;1817:567–574. doi: 10.1016/j.bbabio.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Millett F, Durham B. Design of photoactive ruthenium complexes to study interprotein electron transfer. Biochemistry-Us. 2002;41:11315–11324. doi: 10.1021/bi0262956. [DOI] [PubMed] [Google Scholar]
- [21].Reece SY, Nocera DG. Proton-Coupled Electron Transfer in Biology: Results from Synergistic Studies in Natural and Model Systems. Annu Rev Biochem. 2009;78:673–699. doi: 10.1146/annurev.biochem.78.080207.092132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Balzani V, Credi A, Venturi M. Photochemistry and photophysics of coordination compounds: An extended view. Coordin Chem Rev. 1998;171:3–16. [Google Scholar]
- [23].Campagna S, Puntoriero F, Nastasi F, Bergamini G, Balzani V. Photochemistry and photophysics of coordination compounds: Ruthenium. Top Curr Chem. 2007;280:117–214. [Google Scholar]
- [24].Winkler JR, Nocera DG, Yocom KM, Bordignon E, Gray HB. Electron-Transfer Kinetics of Pentaammineruthenium(Iii)(Histidine-33)-Ferricytochrome-C - Measurement of the Rate of Intramolecular Electron-Transfer between Redox Centers Separated by 15-a in a Protein. J Am Chem Soc. 1982;104:5798–5800. [Google Scholar]
- [25].Yocom KM, Shelton JB, Shelton JR, Schroeder WA, Worosila G, Isied SS, Bordignon E, Gray HB. Preparation and Characterization of a Pentaammineruthenium(Iii) Derivative of Horse Heart Ferricytochrome-C. Proceedings of the National Academy of Sciences of the United States of America-Physical Sciences. 1982;79:7052–7055. doi: 10.1073/pnas.79.22.7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Crane BR, Di Bilio AJ, Winkler JR, Gray HB. Electron tunneling in single crystals of Pseudomonas aeruginosa azurins. J Am Chem Soc. 2001;123:11623–11631. doi: 10.1021/ja0115870. [DOI] [PubMed] [Google Scholar]
- [27].Zadvornyy OA, Lucon JE, Gerlach R, Zorin NA, Douglas T, Elgren TE, Peters JW. Photo-induced H-2 production by [NiFe]-hydrogenase from T. roseopersicina covalently linked to a Ru(II) photosensitizer. J Inorg Biochem. 2012;106:151–155. doi: 10.1016/j.jinorgbio.2011.09.012. [DOI] [PubMed] [Google Scholar]
- [28].Terpetschnig E, Szmacinski H, Malak H, Lakowicz JR. Metal-Ligand Complexes as a New Class of Long-Lived Fluorophores for Protein Hydrodynamics. Biophys J. 1995;68:342–350. doi: 10.1016/S0006-3495(95)80193-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ryan EM, Okennedy R, Feeney MM, Kelly JM, Vos JG. Covalent Linkage of Ruthenium Polypyridyl Compounds to Poly(L-Lysine), Albumins, and Immunoglobulin-G. Bioconjugate Chem. 1992;3:285–290. doi: 10.1021/bc00016a005. [DOI] [PubMed] [Google Scholar]
- [30].Youn HJ, Terpetschnig E, Szmacinski H, Lakowicz JR. Fluorescence Energy-Transfer Immunoassay Based on a Long-Lifetime Luminescent Metal-Ligand Complex. Anal Biochem. 1995;232:24–30. doi: 10.1006/abio.1995.9966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chalker JM, Bernardes GJL, Lin YA, Davis BG. Chemical Modification of Proteins at Cysteine: Opportunities in Chemistry and Biology. Chem-Asian J. 4 doi: 10.1002/asia.200800427. [DOI] [PubMed] [Google Scholar]
- [32].Terpetschnig E, Dattelbaum JD, Szmacinski H, Lakowicz JR. Synthesis and spectral characterization of a thiol-reactive long-lifetime Ru(II) complex. Anal Biochem. 1997;251:241–245. doi: 10.1006/abio.1997.2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Weh J, Duerkop A, Wolfbeis OS. A resonance energy transfer immunoassay based on a thiol-reactive ruthenium donor dye and a longwave-emitting acceptor. Chembiochem. 2007;8:122–128. doi: 10.1002/cbic.200600316. [DOI] [PubMed] [Google Scholar]
- [34].Geren L, Durham B, Millett F. Use of Ruthenium Photoreduction Techniques to Study Electron Transfer in Cytochrome Oxidase. Methods in Enzymology. 2009;456:507–520. doi: 10.1016/S0076-6879(08)04428-5. Vol 456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Castellano FN, Dattelbaum JD, Lakowicz JR. Long-lifetime Ru(II) complexes as labeling reagents for sulfhydryl groups. Anal Biochem. 1998;255:165–170. doi: 10.1006/abio.1997.2468. [DOI] [PubMed] [Google Scholar]
- [36].Dwaraknath S, Tran NH, Dao T, Colbert A, Mullen S, Nguyen A, Cortez A, Cheruzel L. A facile and versatile methodology for cysteine specific labeling of proteins with octahedral polypyridyl d(6) metal complexes. J Inorg Biochem. 2014;136:154–160. doi: 10.1016/j.jinorgbio.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hamachi I, Tsukiji S, Shinkai S, Oishi S. Direct observation of the ferricporphyrin cation radical as an intermediate in the phototriggered oxidation of ferric- to ferryl-heme tethered to Ru(bpy)(3) in reconstituted myoglobin. J Am Chem Soc. 1999;121:5500–5506. [Google Scholar]
- [38].Immoos CE, Di Bilio AJ, Cohen MS, Van der Veer W, Gray HB, Farmer PJ. Electron-transfer chemistry of Ru-linker-(Heme)-modified myoglobin: Rapid intraprotein reduction of a photogenerated porphyrin cation radical. Inorg Chem. 2004;43:3593–3596. doi: 10.1021/ic049741h. [DOI] [PubMed] [Google Scholar]
- [39].Glazer EC, Nguyen YHL, Gray HB, Goodin DB. Probing inducible nitric oxide synthase with a pterin-ruthenium(II) sensitizer wire. Angew Chem Int Edit. 2008;47:898–901. doi: 10.1002/anie.200703743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ener ME, Lee YT, Winkler JR, Gray HB, Cheruzel L. Photooxidation of cytochrome P450-BM3. P Natl Acad Sci USA. 2010;107:18783–18786. doi: 10.1073/pnas.1012381107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Tran NH, Nguyen D, Dwaraknath S, Mahadevan S, Chavez G, Nguyen A, Dao T, Mullen S, Nguyen TA, Cheruzel LE. An Efficient Light-Driven P450 BM3 Biocatalyst. J Am Chem Soc. 2013;135:14484–14487. doi: 10.1021/ja409337v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Szmacinski H, Castellano FN, Terpetschnig E, Dattelbaum JD, Lakowicz JR, Meyer GJ. Long-lifetime Ru(II) complexes for the measurement of high molecular weight protein hydrodynamics. Bba-Protein Struct M. 1998;1383:151–159. doi: 10.1016/s0167-4838(97)00196-9. [DOI] [PubMed] [Google Scholar]
- [43].Roth LE, Nguyen JC, Tezcan FA. ATP- and Iron-Protein-Independent Activation of Nitrogenase Catalysis by Light. J Am Chem Soc. 2010;132:13672–13674. doi: 10.1021/ja1071866. [DOI] [PubMed] [Google Scholar]
- [44].Shih C, Museth AK, Abrahamsson M, Blanco-Rodriguez AM, Di Bilio AJ, Sudhamsu J, Crane BR, Ronayne KL, Towrie M, Vlcek A, Richards JH, Winkler JR, Gray HB. Tryptophan-accelerated electron flow through proteins. Science. 2008;320:1760–1762. doi: 10.1126/science.1158241. [DOI] [PubMed] [Google Scholar]
- [45].Chang IJ, Gray HB, Albin M. Identification of Chemical Modification Sites on Metalloproteins by Capillary Electrophoresis. Anal Biochem. 1993;212:24–27. doi: 10.1006/abio.1993.1285. [DOI] [PubMed] [Google Scholar]
- [46].Winkler JR, Malmstrom BG, Gray HB. Rapid Electron Injection into Multisite Metalloproteins - Intramolecular Electron-Transfer in Cytochrome-Oxidase. Biophys Chem. 1995;54:199–209. doi: 10.1016/0301-4622(94)00156-e. [DOI] [PubMed] [Google Scholar]
- [47].Millett F, Havens J, Rajagukguk S, Durham B. Design and use of photoactive ruthenium complexes to study electron transfer within cytochrome bc(1) and from cytochrome bc(1) to cytochrome c. Bba-Bioenergetics. 2013;1827:1309–1319. doi: 10.1016/j.bbabio.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Yoshikawa S, Shimada A. Reaction Mechanism of Cytochrome c Oxidase. Chem Rev. 2015;115:1936–1989. doi: 10.1021/cr500266a. [DOI] [PubMed] [Google Scholar]
- [49].Reece SY, Hodgkiss JM, Stubbe J, Nocera DG. Proton-coupled electron transfer: the mechanistic underpinning for radical transport and catalysis in biology. Philos T R Soc B. 2006;361:1351–1364. doi: 10.1098/rstb.2006.1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Dempsey JL, Winkler JR, Gray HB. Proton-Coupled Electron Flow in Protein Redox Machines. Chem Rev. 2010;110:7024–7039. doi: 10.1021/cr100182b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Stubbe J, Nocera DG, Yee CS, Chang MCY. Radical initiation in the class I ribonucleotide reductase: Long-range proton-coupled electron transfer? Chem Rev. 2003;103:2167–2201. doi: 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]
- [52].Berglund J, Pascher T, Winkler JR, Gray HB. Photoinduced oxidation of horseradish peroxidase. J Am Chem Soc. 1997;119:2464–2469. [Google Scholar]
- [53].Low DW, Winkler JR, Gray HB. Photoinduced oxidation of microperoxidase-8: Generation of ferryl and cation-radical porphyrins. J Am Chem Soc. 1996;118:117–120. [Google Scholar]
- [54].Dunn AR, Dmochowski IJ, Winkler JR, Gray HB. Nanosecond photoreduction of cytochrome P450cam by channel-specific Ru-diimine electron tunneling wires. J Am Chem Soc. 2003;125:12450–12456. doi: 10.1021/ja0294111. [DOI] [PubMed] [Google Scholar]
- [55].Wilker JJ, Dmochowski IJ, Dawson JH, Winkler JR, Gray HB. Substrates for rapid delivery of electrons and holes to buried active sites in proteins. Angew Chem Int Edit. 1999;38:90–92. [Google Scholar]
- [56].Langley DB, Brown DE, Cheruzel LE, Contakes SM, Duff AP, Hilmer KM, Dooley DM, Gray HB, Guss JM, Freeman HC. Enantiomer-specific binding of ruthenium(II) molecular wires by the amine oxidase of Arthrobacter globiformis. J Am Chem Soc. 2008;130:8069–8078. doi: 10.1021/ja801289f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Green MT, Dawson JH, Gray HB. Oxoiron(IV) in chloroperoxidase compound II is basic: Implications for P450 chemistry. Science. 2004;304:1653–1656. doi: 10.1126/science.1096897. [DOI] [PubMed] [Google Scholar]
- [58].Margalit R, Pecht I, Gray HB. Oxidation Reduction Catalytic Activity of a Pentaammineruthenium(Iii) Derivative of Sperm Whale Myoglobin. J Am Chem Soc. 1983;105:301–302. [Google Scholar]
- [59].Lubitz W, Ogata H, Rudiger O, Reijerse E. Hydrogenases. Chem Rev. 2014;114:4081–4148. doi: 10.1021/cr4005814. [DOI] [PubMed] [Google Scholar]
- [60].King PW. Designing interfaces of hydrogenase-nanomaterial hybrids for efficient solar conversion. Bba-Bioenergetics. 2013;1827:949–957. doi: 10.1016/j.bbabio.2013.03.006. [DOI] [PubMed] [Google Scholar]
- [61].Okura I, Kimthuan N. Kinetic-Studies of Photoinduced Methyl Viologen Reduction with Ruthenium Complexes and Hydrogen Evolution from Water by Hydrogenase. J Chem Soc Farad T 1. 1981;77:1411–1415. [Google Scholar]
- [62].Asakura N, Hiraishi T, Kamachi T, Okura I. Photoinduced hydrogen evolution with cytochrome c(3)-viologen-ruthenium(II) triad complex and hydrogenase. J Mol Catal a-Chem. 2001;172:1–7. [Google Scholar]
- [63].Ihara M, Nishihara H, Yoon KS, Lenz O, Friedrich B, Nakamoto H, Kojima K, Honma D, Kamachi T, Okura I. Light-driven hydrogen production by a hybrid complex of a [NiFe]-hydrogenase and the cyanobacterial photosystem I. Photochem Photobiol. 2006;82:676–682. doi: 10.1562/2006-01-16-RA-778. [DOI] [PubMed] [Google Scholar]
- [64].Noji T, Kondo M, Jin T, Yazawa T, Osuka H, Higuchi Y, Nango M, Itoh S, Dewa T. Light-Driven Hydrogen Production by Hydrogenases and a Ru-Complex inside a Nanoporous Glass Plate under Aerobic External Conditions. J Phys Chem Lett. 2014;5:2402–2407. doi: 10.1021/jz5008164. [DOI] [PubMed] [Google Scholar]
- [65].Greene BL, Joseph CA, Maroney MJ, Dyer RB. Direct Evidence of Active-Site Reduction and Photodriven Catalysis in Sensitized Hydrogenase Assemblies. J Am Chem Soc. 2012;134:11108–11111. doi: 10.1021/ja3042367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Reisner E, Fontecilla-Camps JC, Armstrong FA. Catalytic electrochemistry of a [NiFeSe]-hydrogenase on TiO2 and demonstration of its suitability for visible-light driven H-2 production. Chem Commun. 2009:550–552. doi: 10.1039/b817371k. [DOI] [PubMed] [Google Scholar]
- [67].Reisner E, Powell DJ, Cavazza C, Fontecilla-Camps JC, Armstrong FA. Visible Light-Driven H-2 Production by Hydrogenases Attached to Dye-Sensitized TiO2 Nanoparticles. J Am Chem Soc. 2009;131:18457–18466. doi: 10.1021/ja907923r. [DOI] [PubMed] [Google Scholar]
- [68].Woolerton TW, Sheard S, Reisner E, Pierce E, Ragsdale SW, Armstrong FA. Efficient and Clean Photoreduction of CO2 to CO by Enzyme-Modified TiO2 Nanoparticles Using Visible Light. J Am Chem Soc. 2010;132:2132. doi: 10.1021/ja910091z. + [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Woolerton TW, Sheard S, Pierce E, Ragsdale SW, Armstrong FA. CO2 photoreduction at enzyme-modified metal oxide nanoparticles. Energ Environ Sci. 2011;4:2393–2399. [Google Scholar]
- [70].Hoffman BM, Lukoyanov D, Yang ZY, Dean DR, Seefeldt LC. Mechanism of Nitrogen Fixation by Nitrogenase: The Next Stage. Chem Rev. 2014;114:4041–4062. doi: 10.1021/cr400641x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Tezcan FA, Kaiser JT, Mustafi D, Walton MY, Howard JB, Rees DC. Nitrogenase complexes: Multiple docking sites for a nucleotide switch protein. Science. 2005;309:1377–1380. doi: 10.1126/science.1115653. [DOI] [PubMed] [Google Scholar]
- [72].Roth LE, Tezcan FA. ATP-Uncoupled, Six-Electron Photoreduction of Hydrogen Cyanide to Methane by the Molybdenum-Iron Protein. J Am Chem Soc. 2012;134:8416–8419. doi: 10.1021/ja303265m. [DOI] [PubMed] [Google Scholar]
- [73].Ruggi A, van Leeuwen FWB, Velders AH. Interaction of dioxygen with the electronic excited state of Ir(III) and Ru(II) complexes: Principles and biomedical applications. Coordin Chem Rev. 2011;255:2542–2554. [Google Scholar]
- [74].Jones SM, Solomon EI. Electron transfer and reaction mechanism of laccases. Cell Mol Life Sci. 2015;72:869–883. doi: 10.1007/s00018-014-1826-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Kudanga T, Le Roes-Hill M. Laccase applications in biofuels production: current status and future prospects. Appl Microbiol Biot. 2014;98:6525–6542. doi: 10.1007/s00253-014-5810-8. [DOI] [PubMed] [Google Scholar]
- [76].Cardoso FP, Neto SA, Crepaldi LB, Nikolaou S, Barros VP, De Andrade AR. Biocathodes for Enzymatic Biofuel Cells Using Laccase and Different Redox Mediators Entrapped in Polypyrrole Matrix. J Electrochem Soc. 2014;161:F445–F450. [Google Scholar]
- [77].Barton SC, Kim HH, Binyamin G, Zhang YC, Heller A. The “wired” laccase cathode: High current density electroreduction of O-2 to water at+0.7 V (NHE) at pH 5. J Am Chem Soc. 2001;123:5802–5803. doi: 10.1021/ja010408b. [DOI] [PubMed] [Google Scholar]
- [78].Mano N, Soukharev V, Heller A. A laccase-wiring redox hydrogel for efficient catalysis of O-2 electroreduction. J Phys Chem B. 2006;110:11180–11187. doi: 10.1021/jp055654e. [DOI] [PubMed] [Google Scholar]
- [79].Lazarides T, Sazanovich IV, Simaan AJ, Kafentzi MC, Delor M, Mekmouche Y, Faure B, Reglier M, Weinstein JA, Coutsolelos AG, Tron T. Visible Light-Driven O-2 Reduction by a Porphyrin-Laccase System. J Am Chem Soc. 2013;135:3095–3103. doi: 10.1021/ja309969s. [DOI] [PubMed] [Google Scholar]
- [80].Simaan AJ, Mekmouche Y, Herrero C, Moreno P, Aukauloo A, Delaire JA, Reglier M, Tron T. Photoinduced Multielectron Transfer to a Multicopper Oxidase Resulting in Dioxygen Reduction into Water. Chem-Eur J. 2011;17:11743–11746. doi: 10.1002/chem.201101282. [DOI] [PubMed] [Google Scholar]
- [81].Urlacher VB, Girhard M. Cytochrome P450 monooxygenases: an update on perspectives for synthetic application. Trends Biotechnol. 2012;30:26–36. doi: 10.1016/j.tibtech.2011.06.012. [DOI] [PubMed] [Google Scholar]
- [82].Denisov IG, Makris TM, Sligar SG, Schlichting I. Structure and chemistry of cytochrome P450. Chem Rev. 2005;105:2253–2277. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- [83].Whitehouse CJC, Bell SG, Wong LL. P450(BM3) (CYP102A1): connecting the dots. Chem Soc Rev. 2012;41:1218–1260. doi: 10.1039/c1cs15192d. [DOI] [PubMed] [Google Scholar]
- [84].Tran NH, Huynh N, Bui T, Nguyen Y, Huynh P, Cooper ME, Cheruzel LE. Light-initiated hydroxylation of lauric acid using hybrid P450 BM3 enzymes. Chem Commun. 2011;47:11936–11938. doi: 10.1039/c1cc15124j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Tran NH, Huynh N, Chavez G, Nguyen A, Dwaraknath S, Nguyen TA, Nguyen M, Cheruzel L. A series of hybrid P450 BM3 enzymes with different catalytic activity in the light-initiated hydroxylation of lauric acid. J Inorg Biochem. 2012;115:50–56. doi: 10.1016/j.jinorgbio.2012.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Kato M, Nguyen D, Gonzalez M, Cortez A, Mullen SE, Cheruzel LE. Regio- and stereoselective hydroxylation of 10-undecenoic acid with a light-driven P450 BM3 biocatalyst yielding a valuable synthon for natural product synthesis. Bioorgan Med Chem. 2014;22:5687–5691. doi: 10.1016/j.bmc.2014.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Ma Y, Wang XL, Jia YS, Chen XB, Han HX, Li C. Titanium Dioxide-Based Nanomaterials for Photocatalytic Fuel Generations. Chem Rev. 2014;114:9987–10043. doi: 10.1021/cr500008u. [DOI] [PubMed] [Google Scholar]
- [88].Filice M, Palomo JM. Cascade Reactions Catalyzed by Bionanostructures. Acs Catal. 2014;4:1588–1598. [Google Scholar]
- [89].Kim JH, Nam DH, Park CB. Nanobiocatalytic assemblies for artificial photosynthesis. Curr Opin Biotech. 2014;28:1–9. doi: 10.1016/j.copbio.2013.10.008. [DOI] [PubMed] [Google Scholar]
- [90].Mifsud M, Gargiulo S, Iborra S, Arends IWCE, Hollmann F, Corma A. Photobiocatalytic chemistry of oxidoreductases using water as the electron donor. Nat Commun. 2014;5 doi: 10.1038/ncomms4145. [DOI] [PubMed] [Google Scholar]
- [91].Park JH, Lee SH, Cha GS, Choi DS, Nam DH, Lee JH, Lee JK, Yun CH, Jeong KJ, Park CB. Cofactor-Free Light-Driven Whole-Cell Cytochrome P450 Catalysis. Angew Chem Int Edit. 2015;54:969–973. doi: 10.1002/anie.201410059. [DOI] [PubMed] [Google Scholar]