

Abstract
We describe in detail a method to introduce optogenetic actuation tools, a mutant version of channelrhodopsin- 2, ChR2(H134R), and archaerhodopsin (ArchT), into primary cardiac fibroblasts (cFB) in vitro by adenoviral infection to yield quick, robust, and consistent expression. Instructions on adjusting infection parameters such as the multiplicity of infection and virus incubation duration are provided to generalize the method for different lab settings or cell types. Specific conditions are discussed to create hybrid co-cultures of the optogenetically modified cFB and non-transformed cardiomyocytes to obtain light- sensitive excitable cardiac syncytium, including stencil-patterned cell growth. We also describe an all-optical framework for the functional testing of responsiveness of these opsins in cFB. The presented methodology provides cell-specific tools for the mechanistic investigation of the functional bioelectric contribution of different non-excitable cells in the heart and their electrical coupling to cardiomyocytes under different conditions.
Keywords: Optogenetics, Cardiac, Non-excitable cells, Fibroblasts, ChR2, ArchT
1 Introduction
Non-excitable cells in the heart (fibroblasts, endothelial cells, smooth muscle cells, pericytes, and macrophages) are an under-investigated mystery compared to the excitable muscle cells. This is particularly true for the largest non-myocyte cell population, the cardiac fibroblasts, cFB [1]. There are no unique markers to identify them [2, 3], and hence no robust cell-specific promoters to genetically target them [4–6] for sensing or manipulation purposes.
Recent evidence has accumulated pointing to quite important and “exciting” roles of non-excitable cells in the heart [7–10], including their key contributions to cardiac electrical activity. For instance, fluctuations in the cFB membrane potential can modulate the operation of the sinoatrial node cells and hence can influence pacing frequency [11]. Through paracrine signaling, cFB can also influence important ion channel expression levels, such as SCN5A and KCNJ2, and hence can actively remodel myocyte electrophysiology [12]. Following cardiac injury, cFB undergo electrical phenotypic transformation to myofibroblasts [11]. This can lead to altered action potential characteristics of co-cultured cardiomyocytes through both paracrine signaling [12] and increased direct electrical coupling via gap junctional (Cx43) channels between the cFB and the cardiomyocytes [13]. Pathological responses of cFB, triggered by cardiac injury, affect their interactions with cardiomyocytes and can become the culprit of a range of pro-arrhythmic consequences, including conduction slowing or block, and enhanced automaticity in the myocytes bordering the injury zone [14–17]. Overall, the lack of investigational research tools to specifically manipulate cFB has hampered the study and understanding of their functional contributions to cardiac activity.
Optogenetic actuation tools (light-sensitive microbial ion channels), when genetically expressed in mammalian cells, can mediate fast and cell-specific optically induced changes in their membrane potential, i.e., can cause depolarization or hyperpolarization [18–20]. Along with optogenetic sensors [21], they can facilitate the mechanistic study not only of excitable (action potential-generating) cells but also of non-excitable cells in the complex multicellular environment of the heart [22, 23]. Pending the identification and development of better cFB-specific promoters or other ways to target cFB [6], optogenetic actuators can allow for unique interrogation of these cells to quantify their contribution to cardiac electrical activity during normal function and during injury response and remodeling in vivo [2]. Separately, in vitro high-throughput coupling assays can be developed [24] in conjunction with micro-patterning or other ways to produce controlled heterotypic cell pairs or hybrid tissue equivalents [25, 26] that can be useful in evaluating stem cell-derived biologics for cardiac tissue repair.
So far, optogenetic actuation tools have found limited use in manipulating excitable cardiac myocytes [27–32]. Furthermore, we have shown that optical manipulation of cardiac syncytium can also be achieved through dedicated donor cells as light-responsive units that are not excitable themselves, i.e., “tandem-cell-unit” approach, demonstrated using different generic somatic cells [33, 34]. This idea can be extended to primary cardiac fibroblasts, as demonstrated in a preliminary study [24] and described here in detail for in vitro application.
2 Materials
2.1 Culturing Non-excitable Cardiac Cells
Neonatal rat ventricular tissue, dissected from 2–3-day-old Sprague-Dawley rats, temporarily submerged in Hanks’ Balanced Salt Solution (HBSS).
Tissue digestion enzymes: Trypsin dissolved in HBSS at 1 mg/mL, and collagenase also dissolved in HBSS at 1 mg/mL. Both enzyme solutions are prepared right before usage, short-term storage at 4 °C.
Cell culture media M199 (500 mL bottle), supplemented with 12 μM l -glutamine, 3.5 μg/mL glucose, 0.05 μg/mL penicillin- streptomycin, 200 μg/mL vitamin B12, 10 mM HEPES, and 2 or 10 % fetal bovine serum (FBS).
175 cm2 Flasks.
2.2 Optogenetic Transgene Delivery by Adenovirus Infection and Optimization
For opsin delivery, we used custom-generated adenovirus vectors (Ad-): Ad-CAG-ChR2-eYFP from the plasmid pcDNA3.1/hChR2(H134R)-EYFP [32] and Ad-CAG-ArchT-eGFP from the plasmid pAAV-CAG-ArchT-GFP. The plasmids are purchased from Addgene (Cambridge, MA) and deposited by K. Deisseroth’s lab (plasmid 20940) and Ed Boyden’s lab (plasmid 29777). Our stock viruses with titer concentration at 1012 units/mL are stored at −20 °C.
1× Phosphate-buffered saline (PBS).
0.05 % Trypsin-EDTA in HBSS.
10 % Bleach.
2.3 Quantification of Gene Expression and Cell Viability
Prepare a 5× Tyrode’s solution for long- term storage at 4 °C by dissolving the following compounds in 1 L of diH2 O: 1.02 g MgCl2, 2.01 g KCl, 39.45 g NaCl, 0.2 g NaH2PO4, and 5.96 g HEPES. Adjust the pH to 7.4 with 1 M HCl or 0.1 N NaOH at room temperature. For usage, dilute 100 mL 5× stock Tyrode’s solution with 400 mL diH2O to make a total of 500 mL working solution. Add 0.46 g d-glucose, 0.75 mL of 1 M CaCl2 (for 1.5 mM Ca2+ in final volume), and pH the solution to 7.4 at the temperature at which experiments will be done.
Prepare propidium iodide (PI) solution at 2 μg/mL in 1× Tyrode’s solution. Dye solution should be prepared immediately before usage/imaging, and wrapped in aluminum foil.
2.4 Examining Opsin Functionality
Glass-bottom dishes with glass number 1.
Human fibronectin is prepared at a stock concentration of 5 mg/mL in filtered diH2O, which could be stored at −20 °C for long term. We recommend making aliquots of the stock fibronectin. For usage, dilute stock aliquots with 1× PBS to final concentration of 50 μg/mL. Diluted fibronectin could be stored at 4 °C and should be used within 2 weeks.
Sylgard® 184 Silicone Elastomer Kit.
Voltage-sensitive fluorescent dyes: FluoVolt™ Membrane Potential Kit; Di-4ANBDQBS (obtained from Dr. L. Loew, University of Connecticut), dissolved in ethanol at stock concentration of 10 mg/mL. Stock dye is stored at −20 °C. For usage, dilute 2 μL stock dye in 1 mL of Tyrode’s solution to make 35 μM to stain one glass-bottom sample in the dark.
Calcium-sensitive fluorescent dye Quest Rhod-4 AM is dissolved in DMSO (with 20 % Pluronic F17) to make 0.5 mM stock dye concentration. Typically, aliquots of dye are made at 20 μL tubes (each is enough for a 35 mm glass-bottom dish) and stored at −20 °C for long term. For usage, dilute stock aliquots with 1 mL Tyrode’s solution to final concentration of 10 μM immediately before staining.
3 Methods
The overall process from extracting primary cFB through their transduction with opsins to their functional testing requires proper time management (Fig. 1).
Fig. 1.
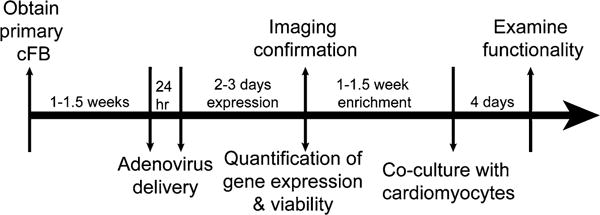
Summary time line. Each study point listed in Subheading 3 is labeled at the arrows, and optimized time is listed
3.1 Culturing Non-excitable Cardiac Cells
Dissection of cardiac tissue and cell isolation steps have been reported in detail previously [34, 35] and are briefly summarized in this section.
Remove neonatal rat ventricular tissue from HBSS and put into 30 mL trypsin solution (1 mg/mL) to digest away extracellular matrix for 12–14 h at 4 °C on a shaker at 75 rpm.
Remove trypsin solution using pipette and avoid picking up tissue. Wash tissue with 20 mL M199 with 10 % FBS at 37 °C for 4 min with swirling motion. Remove M199 and start isolating cardiac cells with collagenase solutions at 37 °C five times, each time adding 10 mL and swirling at 75 rpm for 2 min. Throw away the first 10 mL collagenase solution and collect the later four repetitions.
-
Spin down cells at 300 × g for 8 min, remove collagenase, and resuspend cells in HBSS. Filter out remaining tissue chunks and change HBSS to M199 with 10 % FBS.
Separation of non-myocyte cells (see Note 1) from cardiomyocytes is done using their ability to rapidly adhere to culturing substrates.
Plate the heterogeneous cell mixture from the previous step in a 175 cm2 flask; the typical time frame to avoid cardiomyocyte adhesion but ensure non-myocyte cell attachment is between 45 min and 1 h.
Repeat once by gently transferring floating cells into a second flask for a total of 90 min to 2 h. The plated non-myocyte cells can be maintained in M199 with 2 % FBS until confluent or ready for adenovirus infection; ensure media change every 3–4 days; re-plating should be considered when stored for over 2 weeks.
3.2 Optogenetic Transgene Delivery and Optimization
The following steps specifically focus on transducing cFB with ChR2 and ArchT using adenoviral vectors starting with cell suspension to increase efficiency of infection [32]. This protocol can be scaled up or down by cell number based on experimental design. All solutions should be pre-warmed to 37 °C.
Wash cells with 1× PBS twice.
Remove the second PBS wash, and add 5 mL of 0.05 % trypsin in HBSS to the flasks or enough to make sure that the entire monolayer is submerged. Immediately put the cell and trypsin mixture in a 37 °C incubator and wait for 5 min. At the end of 5 min, agitate the cell culture container (swirl and tap on the sides) and observe under a light microscope for floating cells in a quick manner. Stop trypsin digestion by adding equal volume of M199 with 2 % FBS and collect the cells in a conical. Wash the culture substrate once with M199 with 2 % FBS.
Perform cell counting. Optional: Add trypan blue to the cell sample to label nonviable cells and subtract from the total count.
Spin cells down at 180 × g for 3 min, remove the supernatant mixture of trypsin, and add fresh M199 with 2 % FBS (see Note 2) to keep concentration between 7.2 and 12 × 104 for plating on glass-bottom dishes or enough volume to keep density between 1 and 2 × 104 cells/cm2 for enrichment (see Note 3).
Remove stock virus from −20 °C freezer and put on ice. Invert or tap virus stock, but avoid vortexing.
- Add virus to cFB solution at multiplicity of infection (MOI) of 2000 viral particles per cell. Primary cFB are much more resistant to adenoviral infection compared to cardiomyocytes and require higher MOIs [23]; for both ChR2 and ArchT infection, we recommend a starting MOI 2000. An optimized MOI is determined based on expression efficiency and cell viability post-infection. For first-time infection experiments, we recommend to test a large range of MOI covering 3 orders of magnitude from several viral particles per cell to several 103 viral particles per cell (see Note 4) using the appropriate virus solution volume:
Mix cells and virus by pipetting up and down (see Note 5) before plating the mixture. If samples are to be used for imaging, plate cFB on glass-bottom dishes (pre-coated with fibronectin for 2 h at 37 °C) at 3–5×104 cells/cm2. Let samples sit in the laminar flow hood for 15 min to allow cells to settle to the bottom. Then move the dishes into 37 °C incubator for 2 h before filling up the entire dish with additional 2 mL M199 with 2 % FBS. If samples are to be used for flow cytometry/sorting, plate cells in flasks or tissue culture-treated dishes at similar density. Add more media if necessary and swirl to distribute cells evenly.
Keep virally infected cFB in a 37 °C incubator for 24 h. Incubation duration is strongly dependent on cell type; we recommend testing a wide range from brief exposure (2 h) up to 2 days.
At the end of the incubation period, remove virus by simply aspirating off the media from cFB monolayer and adding fresh M199 with 2 % FBS (10 % FBS could be used to stimulate growth). Wait for 2 days before examining expression.
3.3 Quantification of Gene Expression and Cell Viability
-
Initial confirmation of ChR2 or ArchT expression in cFB can come from reporter gene fluorescence, eYFP or eGFP, respectively, under fluorescence microscopy (see Note 6).
To quantify viability by flow cytometry, we recommend applying PI stain before harvesting the cells with trypsin to avoid unnecessary excessive uptake of the dye and raised background from partially compromised cell membranes (see Note 7).
Aspirate cell culture media from the 35 mm dish sample, add 1 mL of 2 μg/mL PI stain, and wait for 2 min.
Remove PI stain, add 0.05 % trypsin-EDTA in HBSS, and repeat the same harvesting procedure described in step 2 of Subheading 3.2.
Optional: Count cells and adjust concentration in the next step by replacing trypsin solution with cell culture media. The optimal cell concentration should be based on the specific flow cytometer. Typically, 0.5 × 106 cells in 0.5 mL media are needed for BD Calibur flow cytometer.
Spin cells down, aspirate trypsin mixture, and add M199 with 2 % FBS to dispense the cell pellet. Make sure to pipette the cells up and down multiple times to break any clusters that can clog the flow cytometer tube. Run the cells through a filter with appropriate pore size. For cFBs, a filter with 40 μm diameter is recommended.
Transfer cFBs into flow cytometry tubes and perform analysis (see Note 8).
If ChR2 or ArchT expression is low, especially in difficult-to-transduce cells, enrichment by flow-cytometry-assisted cell sorting (FACS) can be applied. However, this step adds an additional cycle of lifting and plating, which may be undesirable for primary cells.
This infection protocol, based on high virus dose and longer virus incubation (24 h), has provided consistent and improved expression efficiency for both ChR2 and ArchT in cFB, without detrimental effects on cell viability (Fig. 2).
Fig. 2.
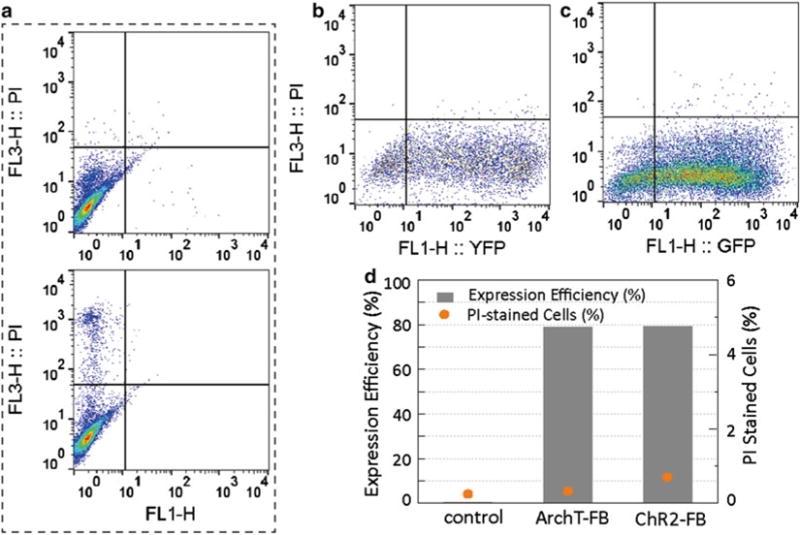
Cell viability and expression efficiency. Flow cytometry analysis indicates that cells subjected to 24-h infection incubation show high expression efficiency and low toxicity. (a) Negative (top) and positive (bottom) control for PI stain on non-transduced cFB (positive control of PI stain was done with prolonged trypsin digestion to break cell membrane). (b) ChR2-cFB analysis (5000 events) and (c) ArchT-FB analysis (20,000 events) were acquired based on gates determined in control condition. (d) Both ChR2 and ArchT delivery at MOI2000 and 24-h incubation achieved over 70 % efficiency
3.4 Opsin Functionality Testing
The most direct way to ensure opsin functionality is to measure light-evoked ChR2- or ArchT-photocurrents in single cells using patch clamp methods [30]. Alternatively, opsin functionality in cFBs can be tested within multicellular preparations. We use a co-culture of opsin-transformed primary cFBs and cardiomyocytes, and probe cFB responsiveness to light by measuring the cardiomyocyte activity, based on the “tandem-cell-unit” concept [34].
3.4.1 Co-culture with Cardiomyocytes
Different patterns of co-culture of ChR2-cFB and cardiomyocytes can be created, e.g., diffuse uniform co-culture or spatially localized ChR2-cFB cluster surrounded by cardiomyocytes. Typically, the clustered pattern of opsin-expressing non-myocytes yields better optical excitability. Cell patterning can be done using polydimethylsiloxane (PDMS) stencils. The thickness of the stencil determines the volume of cells that can be deposited, and it can be easily adjusted by the amount of elastomer cured in a fixed area.
The stencil stiffness can be varied by the ratio of elastomer to curing agent; here 10:1 ratio is used. A combined weight of 9.5 g makes approximately 1 mm thick stencils in a 100 mm wide Petri dish. In a plastic cup, weigh out elastomer and curing agent at the desired volume and ratio. Mix thoroughly.
Pour the elastomer mixture into 100 mm Petri dish and swirl the dish to cover the entire bottom surface.
Put the Petri dish in a desiccator and turn on vacuum to remove bubbles for 60 min. Occasional de-pressure helps draw bubbles out. However, when opening up the chamber, slowly turn the air valve to open to avoid disturbing the sample by strong flow of air.
Put de-bubbled elastomer mixture on a leveled surface in oven, and bake at 50–60 °C for 2 h. Paper towel can be put below the petri dish to ensure even heating.
The stencils will be applied to the glass area of glass-bottom dishes (14 or 20 mm). Cut out 1 cm × 1 cm squares (or desired size) using a pattern printout placed below the petri dish (Fig. 3a). Puncture a circle Ø = 0.4 cm in the middle of the square with a glass puncher.
-
Sterilize the PDMS stencils by submerging them in ethanol. Depending on the specific cell type and their ability to couple with cardiomyocytes and to proliferate, the ratio for co-culturing optically sensitized cells with cardiomyocytes needs to be calculated. Overloading the co-culture system with cFB can cause overcrowding and tissue culture peeling, since their proliferation potentials are reinforced by high serum (10 %) media required by cardiomyocytes during the first days of culturing. Vice versa, a low ratio of cFB to cardiomyocytes may make it difficult to optically excite (despite the presence of functional opsins) due to source-load mismatch. A ratio of 1 opsin-expressing cFB to 16 cardiomyocytes in co-cultures has been reliable in our experiences in testing optical excitability of ChR2-cFB with relatively low light levels.
When ChR2-cFB or ArchT-cFB come to confluence and are ready to be experimented on (see Note 9), they should be collected in parallel with the cardiomyocyte dissection/isolation procedure (Fig. 1).
Coat 14 mm glass-bottom dishes with 0.25 mL fibronectin, and incubate dishes at 37 °C for at least 2 h. If PDMS pattern stencil is used, rinse the ethanol off them by submerging them in PBS twice. Put one pattern on the fibronectin coating.
Isolate neonatal cardiac tissue using previously described methods (Subheading 3.1). After obtaining purified cardiomyocytes, determine cell count.
-
During the step of separating cardiomyocytes from non-myocytes (usually 90 min to 2 h), harvest ChR2-cFB or ArchT-cFB using previously described trypsin digestion steps (Subheading 3.2, steps 1 and 2) and count cell number.
For a diffused pattern (Fig. 4a): we typically use plating density of 0.35 × 106 cells/cm2 on a 14 mm glass-bottom dish (area = 1.43 cm2) that holds 0.6 mL in the well, leading to mixed cell concentration of 0.83 × 106 cells/mL.
Combine the two cell type s into one conical tube at a ratio of 1 opsin-expressing cFB:16 CM. Adjust the concentration of the cell mixture to 0.83 × 106 cells/mL by spinning the cells down at 1000 rpm for 4 min and remove/add M199 with 10 % FBS.
-
Remove fibronectin coating from the glass-bottom dishes and deposit 0.6 mL of well-mixed cells in the glass well.
For a localized/confined cluster pattern (Fig. 4b): We typically pipette 30 μL of opsin-expressing cFB in the circular cutout space of the PDMS stencil. At the 1 opsin-expressing cFB:16 CM ratio and plating density of 0.35 × 106 cells/cm2, each glass-bottom dish will have 0.03 × 106 opsin-expressing cFB and 0.471 × 106 cardiomyocytes, necessitating concentration of the opsin-expressing cFBs at 0.98 × 106/mL and cardiomyocyte concentration at 0.785 × 106/mL.
Remove the fibronectin coating from the glass-bottom dishes. Leave the dishes uncovered to allow the glass to become completely dry so that the PDMS stencil seals to the glass. Keep the side of the stencil touching the fibronetin on the glass (Fig. 3b).
Deposit the 30 μL of opsin-expressing cFB at 0.98 × 106 cells/mL into the PDMS stencil and let it sit in the humidified 37 °C incubator for at least 1 h for cell attachment.
After incubation, in a quick manner, gently aspirate off the cell media from the surface of the PDMS stencil, remove the PDMS stencil, and deposit 0.6 mL cardiomyocytes at 0.785×106/mL to fill up the glass-bottom well.
Leave the samples in the hood still for 15 min to avoid spillage of cells from the glass to the plastic region. Then move the samples to the humidified 37 °C incubator and wait for 2 h before filling up the entire glass-bottom dish with 2 mL of M199 with 10 % FBS.
On the next day approximately 24 h after plating, remove cell culture media from the samples and wash in PBS with shaking for 5 min to remove loosely attached dead cells. Remove PBS, fill glass-bottom dishes with M199 with 10 % FBS, and keep samples incubated in humidified 37 °C incubator for another 48 h. At this point, cardiomyocytes no longer need high-serum supplement and samples will be switched to M199 with 2 % serum for the rest of their maintenance.
Fig. 3.
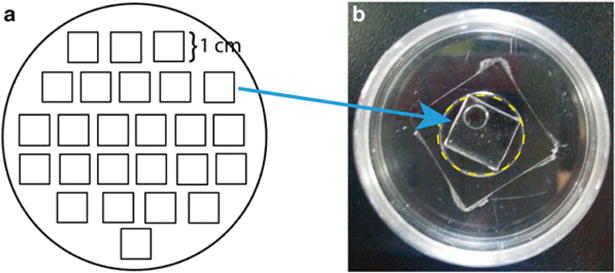
PDMS stencil for cell patterning. (a) Template printout for trace-cut cured PDMS in a 100 mm petri dish. (b) Each PDMS square dimension is 1 cm by 1 cm to fit into the glass well, indicated by yellow dash line, of a 14 mm glass-bottom dish. A circle with Ø=0.4 cm is punctured through the PDMS square to create the stencil space for cFB cluster
Fig. 4.
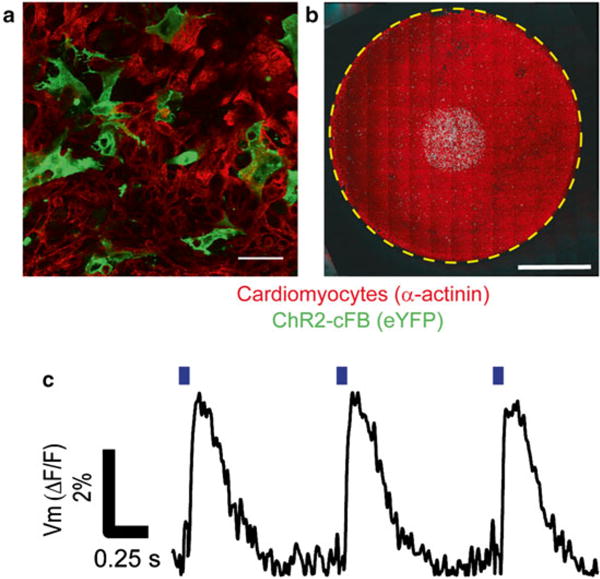
Co-culture of ChR2-cFB and cardiomyocytes and confirmation of opsin functionality. (a) Fluorescent images of diffuse plating. Scale bar is 50 μm. (b) Panoramic macroscopic fluorescent image of patterned ChR-cFB cluster surrounded and covered by cardiomyocytes. Scale bar is 4 mm. (c) Optical activation of ChR2-cFB leads to excitation in cardiomyocytes. Blue bars indicate time of 470 nm light pulse. Optical stimulation was 10-ms pulse at 0.31 mW/mm2. Action potentials were acquired via Di4-ANBDQBS optically at 390 frames/s
3.4.2 Optical Mapping
Using the 1 opsin-expressing cFB:16 CM conditions, experiments can be conducted on the 4th day after plating. Remove sample from the incubator and aspirate culture media. Stain ChR2-cFB with 1 mL of 10 μM of the calcium-sensitive dye Quest Rhod4-AM for 20 min and wash the sample once with 20-min incubation of Tyrode’s solution. Stain ArchT-cFB samples with the voltage-sensitive dye FluoVolt™ following the manufacturer’s manual. Incubate sample in dye (and PowerLoad™) for 15 min and wash sample twice with Tyrode’s solution. Alternatively, both opsin’s activation spectra are compatible with that of the voltage-sensitive dye Di4-ANBDQBS. Incubate samples in 35 μM Di4-ANBDQBS in 1 mL Tyrode’s for 5 min. Wash samples with 5-min incubation in Tyrode’s solution. Remove the wash and add fresh Tyrode’s solution.
Place stained sample on optical mapping setup, which has been described in detail previously [34] (Fig. 5).
Set up appropriate filters for the fluorescent dye used. We used 525 nm excitation light sheet and 605/70 nm emission filter behind a macro-lens (50 mm, f/1.0) to collect Quest Rhod4- AM signals. Similarly, 475 nm excitation light sheet and 535/50 nm emission filter to collect FluoVolt™ signals. For Di 4-ANBDQBS, the excitation and emission filters are 655/40 nm and 700 nm long pass, respectively.
Apply optical stimulation of ChR2-cFB from underneath the glass-bottom dish using a pulsed 470 nm laser or LED (Thorlabs) coupled to an optical fiber (core Ø = 1 mm). The pulse duration and the needed irradiance are inversely related; hence more light is needed if pulse duration is shortened. Typically a spot size covering 75–80 % of the plated glass bottom with irradiance of 0.5 mW/mm2 is enough for pulses longer than 10 ms. Successful excitation of cardiomyocytes by light via optically stimulated ChR2-cFB indicates functional opsins.
-
ArchT is an inhibitory opsin that can be used to suppress spontaneous or electrode-triggered activity. Optical stimulation of ArchT-cFB may require co-stimulation from electrodes in quiescent cells to check opsin functionality. Apply electrical stimulation (10 V and 5 ms pulses) to tissue. When the co-cultured tissue follows pacing reliably, introduce a 10-s pulse of 590 nm light to activate ArchT. Successful suppression of electrically evoked excitation or shortening of the action potentials indicates functional opsins.
Optical excitation or suppression of electrical activity in cardiomyocytes can be applied to extract different electrophysiology-relevant parameters, such as action potential duration or conduction velocity (Fig. 4c).
Fig. 5.
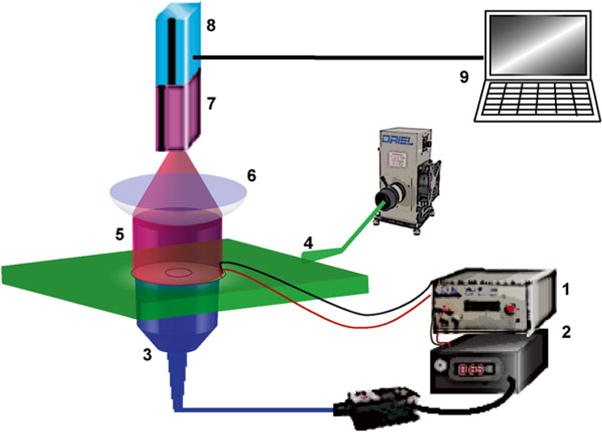
Ultrahigh-resolution high-speed optical imaging system was used to capture electrically and optically induced excitation waves, reported by calcium-sensitive fluorescent dye Rhod-4 or voltage-sensitive fluorescent dye FluoVolt™: (1) Pulse generator for electrical pacing (analog modulation) and optical pacing (TTL modulation). (2) Fiber optic-coupled 470 nm laser as activation light source for ChR2 or 590 nm LED as activation light source for ArchT. (3) Global illumination of co-cultured ChR2-cFB/cardiomyocytes or ArchT-cFB/cardiomyocytes was delivered from below the sample. (4) Excitation for Rhod-4 or FluoVolt was delivered to the sample as a light sheet from a filtered arc lamp. (5 and 6) Rhod-4 and FluoVolt emission fluorescence was collected through appropriate filters for high-resolution macroscopic lens. (7) Gen III MCP intensifier. (8) Pco 1200hs CMOS camera. (9) Computer system and software for image acquisition
Acknowledgments
This work was supported by NIH-NHLBI grant R01-HL-111649 and NSF-Biophotonics grant 1511353 (to E.E.), and partially by a NYSTEM grant C026716 to the Stony Brook Stem Cell Center. We thank Christina Ambrosi and Aleks Klimas for helpful discussions.
Footnotes
The majority of non-myocytes are cardiac fibroblasts and small percentage of endothelial cells, pericytes, smooth muscle cells, and macrophages [7–9]. Hence we treat the non-myocyte population as a clean fibroblast culture. Small amount of residual cardiomyocytes from the separation step eventually lose functionality and get washed away during media change in the duration of subsequent maintenance and infection process.
Virus entry can be less effective in high-serum condition.
This is a good opportunity to set some cells aside as a control group without infection for viability testing. The control group should go through same infection process except without the actual virus.
For low MOI using high-virus titers, a dilution in PBS could be done.
Waste disposal of virus contaminated tools, such as pipette tips and pipettes, should be put in 10 % bleach.
Pattern of expression varies among cell types [23]. cFB tend to show more cytosolic expression of ChR2-eYFP or ArchT-eGFP without strong membrane-bound fluorescence.
PI gets incorporated into the nuclei of cells with compromised membrane in a short time. However, prolonged staining incubation will eventually stain most cells’ nuclei and bias signal-to-background ratio.
Depending on filter settings, eYFP fluorescence can appear stronger than eGFP fluorescence. If both ChR2- and ArchT- infected cells are examined in the same flow cytometry experiment, extra attention should be paid in setting the compensation for bleed thru. In addition, if PI stain for viability is done in dual channel with reporter genes, a separate sample with no PI stain is needed to establish baseline.
Cardiac fibroblasts have limited proliferation cycles. Functional examination of opsins via the TCU approach needs coordination between the age of the ChR-cFB and extracting fresh cardiomyocytes. Our experience has been using 2- to 4-week-old fibroblasts since their isolation.
References
- 1.Lajiness JD, Conway SJ. Origin, development, and differentiation of cardiac fibroblasts. J Mol Cell Cardiol. 2013 doi: 10.1016/j.yjmcc.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vasquez C, Benamer N, Morley GE. The cardiac fibroblast: functional and electrophysiological considerations in healthy and diseased hearts. J Cardiovasc Pharmacol. 2011;57(4):380–388. doi: 10.1097/FJC.0b013e31820cda19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bursac N. Cardiac fibroblasts in pressure overload hypertrophy: the enemy within? J Clin Invest. 2014;124(7):2850–2853. doi: 10.1172/JCI76628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doetschman T, Azhar M. Cardiac-specific inducible and conditional gene targeting in mice. Circ Res. 2012;110(11):1498–1512. doi: 10.1161/CIRCRESAHA.112.265066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeisberg EM, Kalluri R. Origins of cardiac fibroblasts. Circ Res. 2010;107(11):1304–1312. doi: 10.1161/CIRCRESAHA.110.231910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krenning G, Zeisberg EM, Kalluri R. The origin of fibroblasts and mechanism of cardiac fibrosis. J Cell Physiol. 2010;225(3):631–637. doi: 10.1002/jcp.22322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baudino TA, Carver W, Giles W, Borg TK. Cardiac fibroblasts: friend or foe? Am J Physiol. 2006;291(3):H1015–H1026. doi: 10.1152/ajpheart.00023.2006. [DOI] [PubMed] [Google Scholar]
- 8.Nag AC. Study of non-muscle cells of the adult mammalian heart: a fine structural analysis and distribution. Cytobios. 1980;28(109):41–61. [PubMed] [Google Scholar]
- 9.Camelliti P, Borg TK, Kohl P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc Res. 2005;65(1):40–51. doi: 10.1016/j.cardiores.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 10.Kohl P, Gourdie RG. Fibroblast-myocyte electrotonic coupling: does it occur in native cardiac tissue? J Mol Cell Cardiol. 2014 doi: 10.1016/j.yjmcc.2013.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rohr S. Arrhythmogenic implications of fibroblast-myocyte interactions. Circ Arrhythm Electrophysiol. 2012;5(2):442–452. doi: 10.1161/circep.110.957647. [DOI] [PubMed] [Google Scholar]
- 12.Pedrotty DM, Klinger RY, Kirkton RD, Bursac N. Cardiac fibroblast paracrine factors alter impulse conduction and ion channel expression of neonatal rat cardiomyocytes. Cardiovasc Res. 2009;83(4):688–697. doi: 10.1093/cvr/cvp164. doi: 10.1093/cvr/cvp164, cvp164 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vasquez C, Mohandas P, Louie KL, Benamer N, Bapat AC, Morley GE. Enhanced fibroblast-myocyte interactions in response to cardiac injury. Circ Res. 2010;107(8):1011–1020. doi: 10.1161/CIRCRESAHA.110.227421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miragoli M, Salvarani N, Rohr S. Myofibroblasts induce ectopic activity in cardiac tissue. Circ Res. 2007;101(8):755–758. doi: 10.1161/CIRCRESAHA.107.160549. [DOI] [PubMed] [Google Scholar]
- 15.Miragoli M, Gaudesius G, Rohr S. Electrotonic modulation of cardiac impulse conduction by myofibroblasts. Circ Res. 2006;98(6):801–810. doi: 10.1161/01.RES.0000214537.44195.a3. [DOI] [PubMed] [Google Scholar]
- 16.Vasquez C, Morley GE. The origin and arrhythmogenic potential of fibroblasts in cardiac disease. J Cardiovasc Transl Res. 2012;5(6):760–767. doi: 10.1007/s12265-012-9408-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen W, Frangogiannis NG. Fibroblasts in post-infarction inflammation and cardiac repair. Biochim Biophys Acta. 2013;1833(4):945–953. doi: 10.1016/j.bbamcr.2012.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nagel G, Brauner M, Liewald JF, Adeishvili N, Bamberg E, Gottschalk A. Light activation of channelrhodopsin-2 in excitable cells of Caenorhabditis elegans triggers rapid behavioral responses. Curr Biol. 2005;15(24):2279–2284. doi: 10.1016/j.cub.2005.11.032. doi: 10.1016/j.cub.2005.11.032, S0960-9822(05)01407-7 [pii] [DOI] [PubMed] [Google Scholar]
- 19.Han X, Chow BY, Zhou H, Klapoetke NC, Chuong A, Rajimehr R, Yang A, Baratta MV, Winkle J, Desimone R, Boyden ES. A high-light sensitivity optical neural silencer: development and application to optogenetic control of non-human primate cortex. Front Syst Neurosci. 2011;5:18. doi: 10.3389/fnsys.2011.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chow BY, Han X, Dobry AS, Qian X, Chuong AS, Li M, Henninger MA, Belfort GM, Lin Y, Monahan PE, Boyden ES. Highperformance genetically targetable optical neural silencing by light-driven proton pumps. Nature. 2010;463(7277):98–102. doi: 10.1038/nature08652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dugue GP, Akemann W, Knopfel T. A comprehensive concept of optogenetics. Prog Brain Res. 2012;196:1–28. doi: 10.1016/B978-0-444-59426-6.00001-X. [DOI] [PubMed] [Google Scholar]
- 22.Entcheva E. Cardiac optogenetics. Am J Physiol Heart Circ Physiol. 2013;304(9):H1179–H1191. doi: 10.1152/ajpheart.00432.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ambrosi CM, Klimas A, Yu J, Entcheva E. Cardiac applications of optogenetics. Prog Biophys Mol Biol. 2014;115(2–3):294–304. doi: 10.1016/j.pbiomolbio.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu JZ, Boyle PM, Ambrosi CM, Trayanova NA, Entcheva E. High-throughput contactless optogenetic assay for cellular coupling: illustration by Chr2-light-sensitized cardiac fibroblasts and cardiomyocytes. Circulation. 2013;128(22) [Google Scholar]
- 25.Pedrotty DM, Klinger RY, Badie N, Hinds S, Kardashian A, Bursac N. Structural coupling of cardiomyocytes and noncardiomyocytes: quantitative comparisons using a novel micropatterned cell pair assay. Am J Physiol Heart Circ Physiol. 2008;295(1):H390–H400. doi: 10.1152/ajpheart.91531.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nguyen H, Badie N, McSpadden L, Pedrotty D, Bursac N. Quantifying electrical interactions between cardiomyocytes and other cells in micropatterned cell pairs. Methods Mol Biol. 2014;1181:249–262. doi: 10.1007/978-1-4939-1047-2_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arrenberg AB, Stainier DY, Baier H, Huisken J. Optogenetic control of cardiac function. Science. 2010;330(6006):971–974. doi: 10.1126/science.1195929. [DOI] [PubMed] [Google Scholar]
- 28.Bruegmann T, Malan D, Hesse M, Beiert T, Fuegemann CJ, Fleischmann BK, Sasse P. Optogenetic control of heart muscle in vitro and in vivo. Nat Methods. 2010;7(11):897–900. doi: 10.1038/nmeth.1512. [DOI] [PubMed] [Google Scholar]
- 29.Abilez OJ, Wong J, Prakash R, Deisseroth K, Zarins CK, Kuhl E. Multiscale computational models for optogenetic control of cardiac function. Biophys J. 2011;101(6):1326–1334. doi: 10.1016/j.bpj.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Williams JC, Xu J, Lu Z, Klimas A, Chen X, Ambrosi CM, Cohen IS, Entcheva E. Computational optogenetics: empirically- derived voltage- and light-sensitive channelrhodopsin-2 model. PLoS Comput Biol. 2013;9(9):e1003220. doi: 10.1371/journal.pcbi.1003220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vogt CC, Bruegmann T, Malan D, Ottersbach A, Roell W, Fleischmann BK, Sasse P. Systemic gene transfer enables optogenetic pacing of mouse hearts. Cardiovasc Res. 2015 doi: 10.1093/cvr/cvv004. [DOI] [PubMed] [Google Scholar]
- 32.Ambrosi C, Entcheva E. Optogenetic control of cardiomyocytes via viral delivery. In: Radisic M, Black LD Iii, editors. Cardiac tissue engineering, vol 1181, Methods in molecular biology. Springer; New York: 2014. pp. 215–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nussinovitch U, Shinnawi R, Gepstein L. Modulation of cardiac tissue electrophysiological properties with light-sensitive proteins. Cardiovasc Res. 2014;102(1):176–187. doi: 10.1093/cvr/cvu037. [DOI] [PubMed] [Google Scholar]
- 34.Jia Z, Valiunas V, Lu Z, Bien H, Liu H, Wang HZ, Rosati B, Brink PR, Cohen IS, Entcheva E. Stimulating cardiac muscle by light: cardiac optogenetics by cell delivery. Circ Arrhythm Electrophysiol. 2011;4(5):753–760. doi: 10.1161/CIRCEP.111.964247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Entcheva E, Bien H. Mechanical and spatial determinants of cytoskeletal geodesic dome formation in cardiac fibroblasts. Integr Biol. 2009;1(2):212–219. doi: 10.1039/B818874b. [DOI] [PMC free article] [PubMed] [Google Scholar]