

Introduction
Mutations in the SAMHD1 (SAM domain and HD domain–containing protein 1) gene are implicated in SAMS (stroke, aneurysm, moyamoya, and stenosis) association,1, 2 also described as Aicardi-Goutieres syndrome type 5 (AGS-5).3 We present a patient with homozygous germline mutations in SAMHD1 who subsequently had a CD8+ aggressive epidermotropic cutaneous T-cell lymphoma (CTCL).
Case report
A 29-year-old Amish woman with intellectual disability and arthritis caused by the homozygous mutation c.1411-2A > G in the SAMHD1 gene (identified by Xin et al1), presented with fever, cervical lymphadenopathy, and a diffuse rash. The rash began 8 weeks before presentation and consisted of reddish-brown scaly plaques on the chest, abdomen, and back, which were treated with topical steroids by her primary physician. She then became acutely ill and was admitted to the medical intensive care unit of our institution for circulatory shock and hypoxemic respiratory failure.
On examination by the dermatology service, the patient had diffuse, red-brown, circinate, coalescing, scaly plaques involving the trunk, proximal extremities, and face (Fig 1, A). A 3-cm × 3-cm red-purple, exophytic tumor was noted on the right posterior thigh (Fig 1, B). Joint examination found deformities of the bilateral hands without active synovitis. Laboratory evaluation was notable for normocytic anemia, thrombocytopenia, elevated erythrocyte sedimentation rate, elevated C-reactive protein, positive antinuclear antibodies with 1:10,240 titer, positive anti–double-stranded DNA antibodies, and positive antichromatin antibodies. Blood, lower respiratory, and urine cultures were negative. Computed tomography of the neck, chest, abdomen, and pelvis showed bilateral pulmonary infiltrates and cervical, axillary, retroperitoneal, and gastrohepatic lymphadenopathy.
Fig 1.
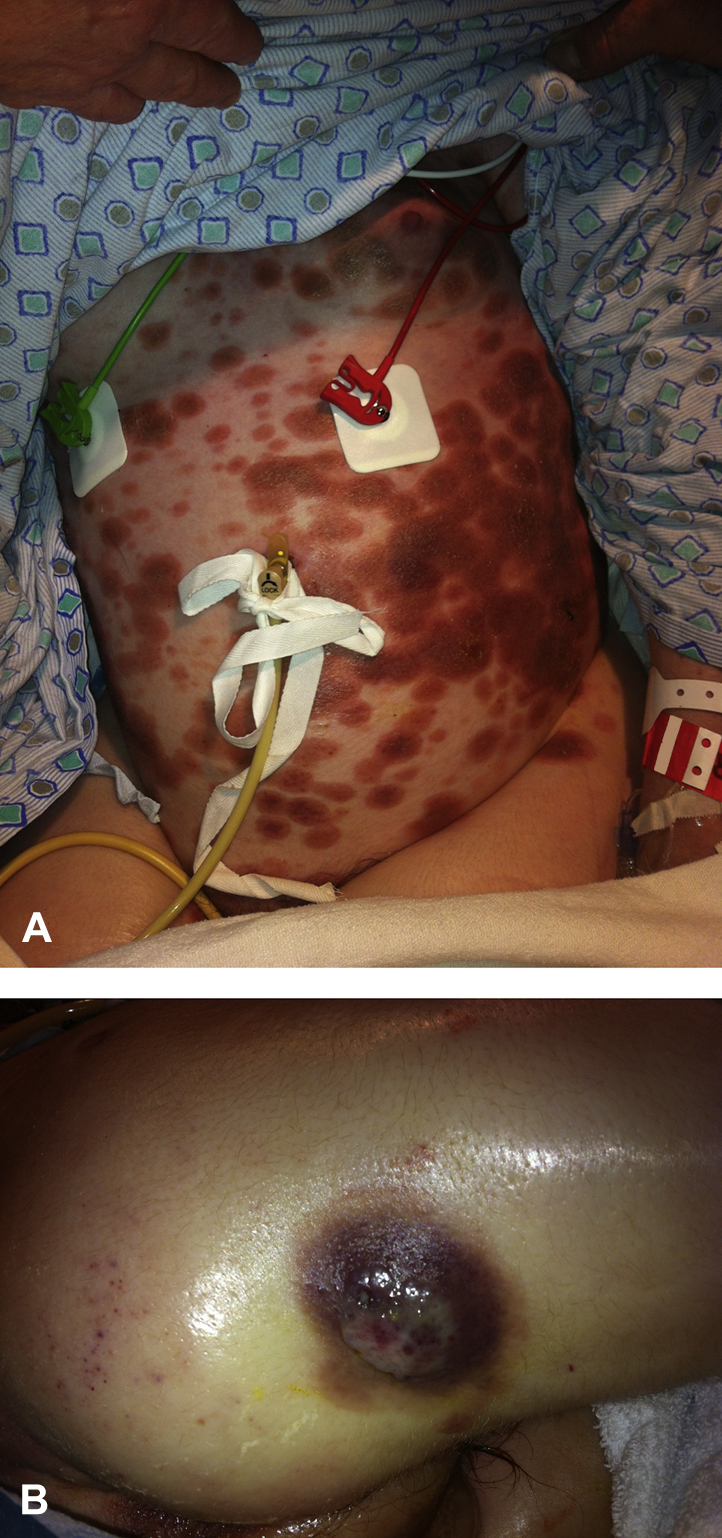
Aggressive CD8+ epidermotropic CTCL. Cutaneous findings on initial presentation from the abdomen (A) and right posterior thigh (B).
Skin biopsy from the abdomen showed significant exocytosis of lymphocytes with mildly enlarged hyperchromatic nuclei without significant cytoplasm. Some lymphocytes were surrounded by clear halos. There was also a moderate superficial and mid-dermal interstitial and periadnexal lymphocytic infiltrate (Fig 2). Flow cytometry from affected skin found a population of atypical T cells (32% of all events) that were CD2+, CD3+, CD4−, CD7+, CD8+, CD25dim, CD26dim, CD56dim, CD30−, HLA-DR−, CD45RO−, and CD45RA−. The cells were T-cell receptor (TCR)-α/β positive and did not react with any antibodies used in the TCR-Vβ analysis, consistent with an abnormal clonal T-cell population. Peripheral blood flow cytometry found no evidence of a peripheral lymphoproliferative disorder. TCR-γ chain gene rearrangement from affected skin was negative.
Fig 2.
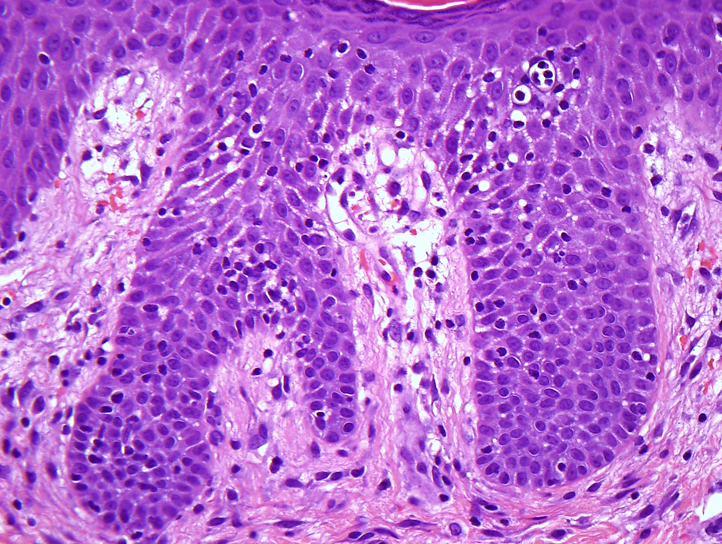
Aggressive CD8+ epidermotropic CTCL. Histopathology from lesional skin shows significant exocytosis of normal-size atypical lymphocytes. The lymphocytes stain with antibodies against CD3 and CD45RB, without significant CD4 or CD8 staining. (Hematoxylin-eosin stain; original magnification: ×40.)
Based on these results, an aggressive CD8+ epidermotropic CTCL was diagnosed. Lymph node biopsy was declined, as it would not change management, and because of severe thrombocytopenia, it represented an unnecessary risk to the patient. The patient's family declined treatment with systemic chemotherapy, and she subsequently died under hospice care.
Discussion
SAMS association was first described in the Old Order Amish in 2011. The clinical phenotype is highly heterogeneous, ranging from normal cognitive development to severe intellectual impairment and early death from recurrent strokes.1 It is likely that AGS-5 and SAMS association might be the same entity,3 although most patients from the Amish cohort, particularly the mildly affected patients, do not fit the clinical spectrum of AGS-5.2 The clinical features of AGS-5 include progressive encephalopathy, basal ganglia calcification, white matter changes, and cerebrospinal fluid abnormalities of lymphocytosis and elevated interferon alfa levels.4, 5, 6
The SAMHD1 protein degrades the intracellular pool of deoxynucleoside triphosphates, maintaining genomic stability and the innate immune system.7 SAMHD1 is upregulated in response to viral infections and is a restriction factor for HIV-1 infection.8 Absence of SAMHD1 causes an imbalance in DNA precursors, which acts as a danger signal to the innate immune system, triggering a type I interferon immune response.7
Activation of the innate immune system plays a central role in SAMS association/AGS-5 and may lead to autoimmunity. Many of the symptoms of SAMS association overlap with autoimmune conditions such as vasculitis, systemic lupus erythematosus (SLE), rheumatoid arthritis, and mixed connective tissue disease, and AGS is associated with systemic autoimmunity and SLE.1, 6 Our patient met 4 of 11 American College of Rheumatology classification criteria required for the diagnosis of SLE (arthritis, thrombocytopenia, positive antinuclear antibodies and anti–double-stranded DNA antibodies).
Genomic instability caused by SAMHD1 mutations may lead to increased mutagenesis and cancer development. SAMHD1 somatic mutations have been identified in 11% of refractory cases of chronic lymphocytic leukemia and in lung adenocarcinoma; glioblastoma; and breast, pancreatic, and colorectal cancers. Recently, Clifford et al8 and de Silva et al9 found decreased SAMHD1 expression in 8 patients with Sezary syndrome and 1 with advanced-stage mycosis fungoides.
Aggressive CD8+ epidermotropic CTCL is a rare subtype associated with a poor prognosis. Median expected lifespan from diagnosis is 22.5 to 33 months, with an 18% chance of 5-year survival. Tumor markers CD2− and CD7+ appear to predict a more aggressive clinical course.10 Our patient's CTCL was CD2+ and CD7+. Although CD8+ CTCL is often clonal,10 our patient did not have TCR-γ gene rearrangement. Potential explanations include insufficient representation of the clone, genetic instability leading to deletion of portions of the TCR-γ gene, or other unknown factors.
The absence of prior reports of CTCL in patients with homozygous mutations of SAMHD1 could relate to the rarity of AGS-5 or SAMS association and the reduced lifespan caused by recurrent cerebrovascular accidents seen in these patients. Along with de Silva et al,9 our case provides further evidence for a possible link between SAMHD1 deficiency and CTCL. Additional studies to further characterize the role of SAMHD1 in the pathogenesis of CTCL are indicated.
Acknowledgments
Author Contributions: Drs Gerstenblith and Merati, had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Study concept and design: Drs Merati, Buethe, Cooper, Honda, Wang, Gerstenblith
Acquisition, analysis, and interpretation of data: Drs Merati, Buethe, Cooper, Honda, Wang, Gerstenblith
Drafting of the manuscript: Drs Merati, Buethe, Cooper, Honda, Wang, Gerstenblith
Critical revision of the manuscript for important intellectual content: Drs Merati, Buethe, Cooper, Honda, Wang, Gerstenblith
Footnotes
Funding sources: None.
Conflicts of interest: None declared.
References
- 1.Xin B., Jones S., Puffenberger E.G. Homozygous mutation in SAMHD1 gene causes cerebral vasculopathy and early onset stroke. Proc Natl Acad Sci. 2011;108(13):5372–5377. doi: 10.1073/pnas.1014265108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xin B., Li W., Bright A., Hinze C., Wang H., Reply to du Moulin, et al Cerebral vasculopathy is a common hallmark in individuals with SAMHD1 mutations. Proc Natl Acad Sci. 2011;108(26):E233. doi: 10.1073/pnas.1104699108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rice G.I., Bond J., Asipu A. Mutations involved in Aicardi-Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet. 2009;41(7):829–832. doi: 10.1038/ng.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thiele H., du Moulin M., Barczyk K. Cerebral Arterial Stenoses and Stroke: Novel Features of Aicardi-Goutières Syndrome Caused by the Arg164X Mutation in SAMHD1 Are Associated with Altered Cytokine Expression. Hum Mutat. 2010;31(11):E1836–E1850. doi: 10.1002/humu.21357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramesh V., Bernardi B., Garone C. Intracerebral large artery disease in Aicardi-Goutières syndrome implicates SAMHD1 in vascular homeostasis. Dev Med Child Neurol. 2010;52(8):725–732. doi: 10.1111/j.1469-8749.2010.03727.x. [DOI] [PubMed] [Google Scholar]
- 6.Ramantani G., Kohlhase J., Hertzberg C. Expanding the phenotypic spectrum of lupus erythematosus in Aicardi-Goutières syndrome. Arthritis Rheum. 2010;62:1469–1477. doi: 10.1002/art.27367. [DOI] [PubMed] [Google Scholar]
- 7.Kretschmer S., Wolf C., König N. SAMHD1 prevents autoimmunity by maintaining genome stability. Ann Rheum Dis. 2015;74(3):e17. doi: 10.1136/annrheumdis-2013-204845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clifford R., Louis T., Robbe P. SAMHD1 is mutated recurrently in chronic lymphocytic leukemia and is involved in response to DNA damage. Blood. 2014;123(7):1021–1031. doi: 10.1182/blood-2013-04-490847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Silva S., Wang F., Hake T.S., Porcu P., Wong H.K., Wu L. Downregulation of SAMHD1 expression correlates with promoter DNA methylation in Sézary syndrome patients. J Invest Dermatol. 2014;134(2):562–565. doi: 10.1038/jid.2013.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berti E., Tomasini D., Vermeer M.H., Meijer C.J., Alessi E., Willemze R. Primary cutaneous CD8-positive epidermotropic cytotoxic T cell lymphomas: a distinct clinicopathological entity with an aggressive clinical behavior. Am J Pathol. 1999;155(2):483–492. doi: 10.1016/S0002-9440(10)65144-9. [DOI] [PMC free article] [PubMed] [Google Scholar]