

Adapting artificially engineered protein polymers from consensus repeats of natural proteins is an attractive approach to mimic the unprecedented performance of natural materials.[1-9] Tough silk-like polypeptides,[1] thermoresponsive elastin-like polypeptides,[3, 6, 9] and resilient and elastic resilin-like polypeptides[5, 7] have been synthesized to mimic the functions of natural materials. Important design principles have been developed for these artificial biopolymers that enable rational control over their thermodynamic, structural, and mechanical properties. The simplified repeat allows for a detailed understanding of sequence-structure-property relationships to be developed,[8] and these tailor-made materials open up opportunities for applications such as drug delivery, tissue engineering, photonic films and smart responsive devices.[1, 3, 4, 10, 11]
An additional natural material that has interesting engineering properties is the protein matrix which fills the nuclear pore complex (NPC) in the nuclear envelope and controls transport into the nucleus. It allows passage of less than 0.1% of all proteins while translocating over 1,000 molecules per pore per second.[12, 13] The protein matrix is composed of nucleoporins, proteins containing Phe-Gly (FG) repeat sequences which contribute to specific binding of the nuclear transport receptors (NTRs)[14-17] that facilitate transport of a specific subset of biological molecules into the matrix, and the mechanism of molecular transport into the NPC has been actively investigated.[15, 18-22] Individual nucleoporins can form hydrogels in vitro that recapitulate the enhanced permeability of selectively-labelled macromolecules into the gel, similar to the intact NPC, with varying degrees of passive diffusion of inert molecules.[18, 23] This selectivity is rare in synthetic polymer hydrogels, making these natural materials an intriguing model for new filtration technologies. In spite of the advanced filtering function of natural nucleoporin hydrogels, a fundamental understanding of the sequence-structure-property relationships needed for materials engineering is lacking, due to the complex sequence of the proteins and the inability to synthesize them recombinantly in high yields.
To adapt the function of nucleoporin hydrogels in a biosynthetic material, we designed artificially engineered protein polymers that can replicate the biological selective transport of the hydrogel in a synthetic mimic using a consensus repeat adapted from a well-investigated nucleoporin, Nsp1.[15, 24, 25] Recent in vitro results[24] indicate that the recombinant Nsp12-601 can be divided into an N-terminal sequence Nsp12-277 and a C-terminal sequence Nsp1274-601. In Nsp1, the C-terminal sequence contributes to selective transport of NTR-cargo complexes and less non-specific binding of inert molecules, core functions for selective transport. However, the C-terminal sequence alone forms a liquid that cannot restrict the passage of inert molecules. The N-terminal sequence is critical for gelation, suggesting that network formation is required for a fully functional selective transport system. To prepare synthetic gels, we replaced the N-terminal sequence of Nsp1, which gels slowly over a period of hours,[15, 24, 25] with well-investigated pentameric (P) coiled-coil domains[2, 26, 27] flanking the C-terminal sequence (cNsp1, Figure S1). This triblock protein construct, P-cNsp1-P, gels in minutes, and the transient interactions of the P domains allow network relaxation that is thought to be critical to transport.[12]
Analysis of the cNsp1 consensus sequence allows reduction of the protein to a polymer of short repeating segments. Nsp1 is composed of 16 repeats of a 19 amino acid sequence, with a high consensus at each position except position 15, where equal numbers of Asp and Ser are observed (Figure 1a). Therefore, to capture the highest frequency of occurrence in all positions of cNsp1, two separate repeat units were designed: one where position 15 was Asp, and another where position 15 was Ser. These sequences were cloned to form an artificial protein polymer of 16 such units, producing two nucleoporin-like polypeptides (NLPs) denoted 1NLP and 2NLP, respectively (Figure 1b). Both NLPs were genetically fused with P domain endblocks (P-1NLP-P and P-2NLP-P, Figure 1c) to construct polymers that form gels due to coiled-coil physical association (Figure 1d). If simplified NLP polymers can mimic the properties of natural cNsp1, the polymers will provide a valuable tool for material engineering and an opportunity to tune the selectivity, transport rates and barrier function of nucleoporin-inspired materials through rational repeat sequence design.
Figure 1.
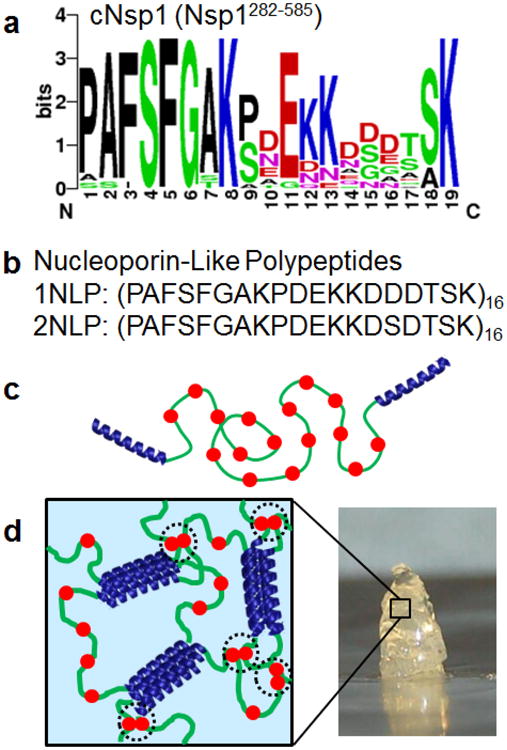
Design of synthetic nucleoporin-like polypeptide hydrogels. a) A sequence logo diagram of the natural nucleoporin, cNsp1 (Nsp1282-585). Color codes represent amino acids with hydrophobic side chains (black), polar side chain (green), negatively charged side chain (red) and positively charged side chain (blue). b) Primary structure for 1NLP and 2NLP, consensus sequences of cNsp1. c) Design of synthetic protein polymers, P-cNsp1-P and P-NLPs-P, which gel by the association of pentameric (P) coiled-coil endblock domains. Red filled circles represent FG sequences. d) 3D gel network formed from assembly of designed artificially engineered proteins. Black dotted circles highlight Phe-mediated interactions within synthetic hydrogels.
Engineered proteins with P domain blocks— P-cNsp1-P, P-1NLP-P and P-2NLP-P— are easily synthesized in much higher yield than recombinant nucleoporin Nsp1. After protein expression and chromatographic purification, the yield of high purity protein is 20-70 fold higher than the recombinant Nsp1 protein (Figure 2 and Figure S2). NLPs without the coiled-coil domain are also isolated at 10 times greater yield than their parent sequence, cNsp1, after the same procedure (Figure S3). Interestingly, when the cNsp1 is fused to the P domain endblocks (P-cNsp1-P), the construct is expressed at a similar yield as the NLP constructs. Based on this observation, a single P domain together with an intein self-cleavage domain[28] was subcloned into the N-terminal cNsp1 gene (P-intein-cNsp1). After the self-cleavage of P-intein domains, cNsp1 was obtained at a similar yield as the NLPs (Figure S3). Significantly improved biosynthetic yields of these artificially engineered proteins enable detailed characterization of their material properties and engineering to control their performance.
Figure 2.
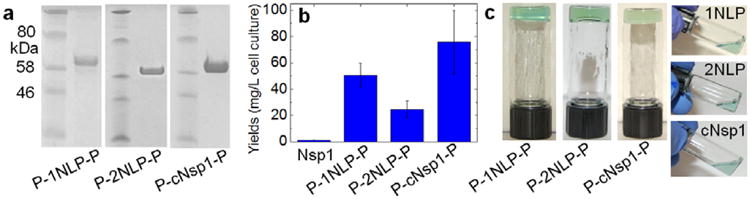
Biosynthesis and hydrogel inversion tests of artificially engineered proteins. a) SDS-PAGE of lyophilized protein samples. b) Yields of designed proteins flanked by P domains. c) Hydrogel inversion tests performed on all artificially engineered proteins with a 20 w/v% concentration in the buffer containing 50 mM Tris/HCl (pH 7.5) and 200 mM NaCl. All proteins with P domains formed gels, while cNsp1 and NLP sequences without P domains did not form gels. All protein sequences and their measured molar masses by MALDI can be found in Figure S1 and Table S1.
Engineered proteins with P domain endblocks rapidly form hydrogels, while NLP midblocks alone fail inversion tests, clearly indicating that structure beyond the FG repeat is necessary to give elastic mechanical properties. Consistent with a previous study on recombinant cNsp1,[24] the NLPs without associating coiled-coil domains do not pass hydrogel inversion tests, while the designed proteins with the P domains form hydrogels within a few minutes, mainly limited by the time required for the lyophilized sample to swell in a buffer (Figure 2c). The engineered proteins gel in all the buffer conditions commonly used for the recombinant Nsp1 (Figure S4), demonstrating that P domain endblocks successfully replace the role of the N-terminal sequences of Nsp1 as a gel crosslinker.
Rheology shows that the midblock sequence has a significant impact on the low frequency viscoelastic properties of the triblock protein gels without affecting the high frequency elastic plateau modulus. Although the midblocks cNsp1, 1NLP and 2NLP are insufficient to cause gelation without the P domain, all three proteins with P domains form gels with a comparable modulus (on the order of 10 kPa) with the crossover between G′ and G′′ occurring below 0.1 rad/s (Figure 3a-c). In all three artificially engineered hydrogels, the addition of 10% hexanediol to a 20 w/v% gel leads to a decrease in the gel relaxation time, increasing the crossover frequency of the gel by approximately a factor of 5, while the stiffness of hydrogels (the plateau modulus G′) changes very little (Figure 3a-c). Aliphatic alcohols, such as 1,6 hexanediol, are known to weaken FG associations, leading to a loss of selective permeability in vivo[29] and in vitro.[17] Comparison to a control hydrogel of similar molar mass but without FG repeats in the midblock[27] shows no effect on the crossover frequency and the high frequency plateau modulus after the addition of hexanediol (Figure 3d), indicating that the endblock P domains are unaffected by hexanediol. Therefore, the changes in crossover frequency in nucleoporin-mimetic gels, characteristic of changes in network relaxation rate, originate from differences in the state of the midblock domain. It has been shown that interchain β-sheets in some nucleoporin FG repeat hydrogels contribute to crosslinking and enhance the FG hydrogel stability,[18, 24, 30] and removing these crosslinks enhances permeability and reduces selectivity.[18] It is expected that the choice of crosslinking group in the hydrogel may affect biomolecular transport and mechanical properties, as crosslinking controls the mesh size of the gel and the relaxation dynamics of the junction points. These properties can influence the transport of macromolecules interacting with the network chains.
Figure 3.
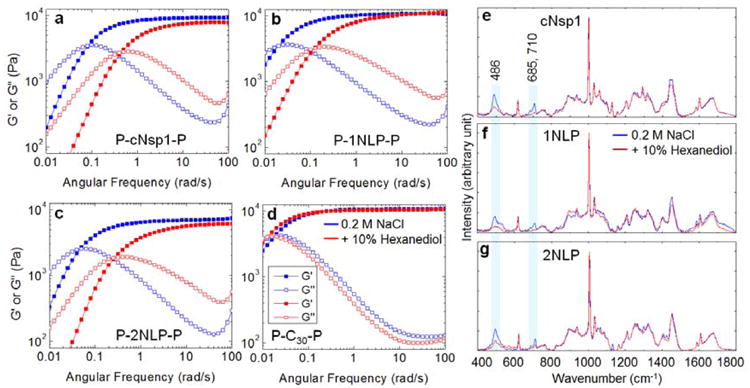
Effect of midblock interactions on hydrogel mechanics. a)-c) Frequency sweep, linear oscillatory shear rheology of 20 w/v% hydrogels in the absence (blue curves) or presence (red curves) of 10 % 1,6 hexanediol. The gel modulus and the crossover frequency in the absence of hexanediol are 9.3 kPa and 0.08 rad/s (a), 10.7 kPa and 0.02 rad/s (b) and 7.5 kPa and 0.04 rad/s (c). Measurements were performed at 25°C with a strain amplitude of 1%, within the linear viscoelastic range (Figure S5). d) Under the same conditions, 20 w/v% P-C30-P gel, which lacks FG repeats in its midblock,27 does not show any effect of the hexanediol (Figure S6). C is the peptide sequence AGAGAGPEG. e)-g) Raman spectra of 20 w/v% midblocks, cNsp1, 1NLP and 2NLP, measured in buffer containing 50 mM Tris/HCl (pH 7.5) and 200 mM NaCl (blue curve) and with the addition of 10% hexanediol (red curve). The shaded boxes highlight Raman bands of 486, 685 and 710 cm-1 that decrease in intensity for all protein midblock polymers with the addition of hexanediol. Assignments of other Raman bands for cNsp1 can be found in Figure S7.
Prominent changes in Raman bands upon the addition of hexanediol confirm that these changes in gel mechanics are caused by disruption of FG repeats involved in molecular interactions within the midblocks, indicating that naturally observed FG interactions have been successfully adapted into the biosynthetic hydrogels. Upon the addition of hexanediol, a significant decrease is observed in the Raman band at 486 cm-1 corresponding to a Phe vibrational mode[31] for cNsp1 and both consensus repeat NLP midblocks. Other Phe Raman bands (band assignments in Figure S7) are similar for all midblock polymers in the presence and absence of the hexanediol (Figure 3e-g), indicating that natural cNsp1 and synthetic NLPs have a similar physicochemical environment for the Phe residues. Other common changes upon the addition of hexanediol are observed in Raman bands at 685 and 710 cm-1. These bands do not appear in lyophilized cNsp1 or NLP (Figure S8) or in individual amino acids included in NLPs in water solution from a previous study.[32] This suggests that the bands are a result of the association between midblocks in water. Molecular interactions between Phe and CH3 and Pro and Lys have been suggested in cNsp1 based upon Nuclear Overhauser Exchange Spectroscopy NMR spectra by Ader et al.[24] The addition of 10% hexanediol suppresses Raman bands responsible for Phe (486 cm-1), Pro (856 and 1097 cm-1), Lys (1442 cm-1) and CH3 (1452 cm-1) in cNsp1 (Figure 3e), consistent with the NMR result,[24] and therefore, the observed Raman bands at 685 and 710 cm-1 may also relate to those residues. The similar shifts in the crossover frequency in all designed hydrogels (Figure 3a-c) and large intensity differences of the 486, 685 and 710 cm-1 (Figure 3e-g) by the addition of hexanediol suggest that hydrophobic interactions, including Phe-mediated associations, between the midblocks exist, similar to the natural Nsp1 hydrogel. These interactions can influence the gel relaxation without contributing significantly to the plateau modulus G′ (Figure 3a-c).
Engineered protein gels containing cNsp1, 1NLP, and 2NLP midblocks selectively enhanced transport of specific biomolecules into the hydrogels, mimicking the property of natural Nsp1 gels. A fluorescence assay originally established to test recombinant nucleoporin hydrogels[17, 25] was performed in a capillary geometry (Figure 4a) to test whether cargo-NTR complexes can permeate through the engineered biosynthetic gels with enhanced transport accumulation, while other molecules and cargo without the NTR are significantly retarded. For the assay, importin β (95 kg/mol; blue filled circles in Figure 4a) was chosen as a NTR due to its well-known binding to cargo with an importin β binding (IBB) domain and to the FG repeat on nucleoporin hydrogels.[14, 33, 34] To reduce the passage of cargo without the NTR and easily quantify the transport of selected cargo, recombinant IBB - maltose binding protein (MBP) - enhanced green fluorescent protein (EGFP) protein fusions were prepared as a model cargo protein[16] (75 kg/ mol; green filled circles in Figure 4a; Stokes' radii of MBP and GFP are reported as 2.85 nm and 2.42 nm, respectively.[35]). Based on the widely applied dextran diffusion method,[21] it is expected that cargo diffusion into the gel will be significantly reduced since the pore size of the gels is smaller than non-interacting 40 kg/mol dextran probe (4.5 nm of Stokes' radius; Figure S9). When importin β and the cargo were physically mixed and added to the capillary prepared with the engineered hydrogels, selective partitioning into the hydrogel occurs over time, while a slab diffusion profile is detected in the absence of importin β (Figure 4b-e and Figure S10). The enhanced transport into the gel occurs due to a combination of diffusion and convection caused by gel swelling with buffer and cargo complexes (Figure S11).[10, 19]
Figure 4.
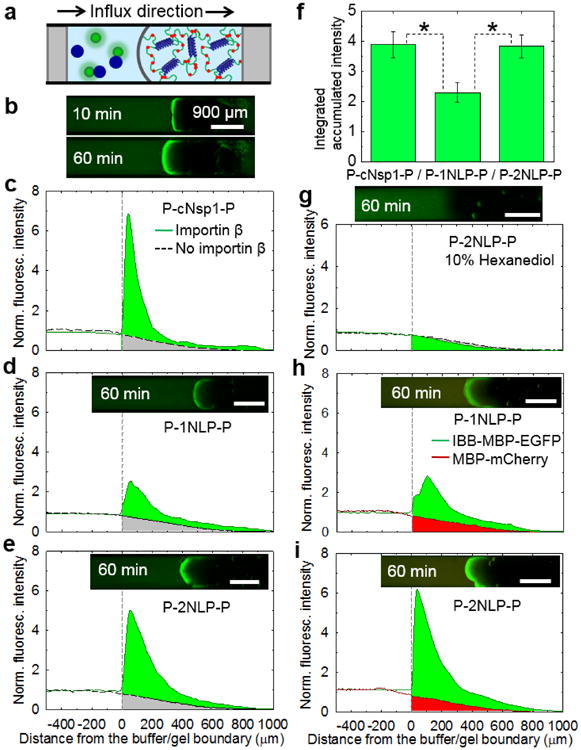
Artificially engineered protein hydrogels can mimic the enhanced selective transport of natural Nsp1. a) A schematic of capillary transport assay set-up. Blue and green circles are representing importin β and IBB-MBP-EGFP, respectively. All tests were measured with 5 μM IBB-MBP-EGFP in the presence or absence of 5 μM importin β. b) A time course transport measurement of 20 w/v% P-cNsp1-P hydrogel with importin β. c)-e) 20 w/v% hydrogels in the presence (solid line) or absence (dotted line) of importin β. f) P-cNsp1-P, P-1NLP-P and P-2NLP-P hydrogels absorbed 3.9±0.4 (mean±S.D., n = 3), 2.3±0.1 (n = 3) and 3.8±0.3 (n = 6) times more cargo-importin β complexes than inert molecules in an hour (Figure S10). * denotes p < 0.05. g) Fluorescence intensity measurements on P-2NLP-P (P-cNsp1-P in Figure S13) hydrogels (20 w/v%) with 10% 1,6 hexanediol show that FG interactions are critical for transport. h)-i). Selective permeability test performed on P-1NLP-P and P-2NLP-P biosynthetic hydrogels (20 w/v%) with the addition of 5 μM MBP-mCherry, a model inert molecule, into 5 μM IBB-MBP-EGFP/ importin β cargo complex mixtures. Over an hour, the cargo-carrier complexes accumulate 3.0 and 5.3 times more than MBP-mCherry (without the IBB domain) in P-1NLP-P and P-2NLP-P hydrogels, respectively. Time lapse measurement of P-2NLP-P gel is included in Figure S14. All scale bars are 900 μm.
Because the length scale of the measurement is much larger than the molecular size and gel mesh size, the gel can be considered as a uniform, semi-infinite slab. The gels can also be treated as macroscopically homogeneous since they are optically clear (absence of inhomgeneity that would scatter on the length scale of visible light) and did not phase separate upon centrifugation (Figure 2c and Figure S4). Under these conditions, the permeability coefficient is the product of the diffusivity and solubility coefficients. The discontinuous concentration profile at the interface during the capillary experiments suggests that the cargo-importin β complexes are more soluble in the gel phase than the cargo alone because of the physical association between importin β and FG repeats.[14] This increase in solubility enhances the permeability of the cargo complexes.
The two simplified NLP midblocks, both consensus sequences of cNsp1 but differing by a single amino acid in the repeating peptide, showed quantitative differences in transport properties. When accumulated green fluorescence intensities are calculated compared to the intensities without importin β (Figure 4f), the P-2NLP-P gel shows almost twice the intensity of the P-1NLP-P gel. Both NLPs have equal numbers of FG repeats (Figure 1), the same P domain crosslinkers for gelation (Figure 2), similar secondary structure as determined by circular dichroism (Figure S12), and similar passive diffusion profiles for inert molecules over time (dotted black curves in Figure 4d-e). Therefore, this quantitative change in permeability must originate from the change in the consensus repeat sequence. Since the single amino acid change Asp in 1NLP to Ser in 2NLP (Figure 1b) occurred in the middle of the peptide between FG repeats that are known to bind importin β, the change from an anionic to neutral residue (change from a formal charge of -16 to 0 for the entire midblock of 16 repeats) suggests that electrostatic effects may affect molecular transport. It is interesting to note that the hydrogel made from P-cNsp1-P, where the midblock has a formal charge of + 6, shows higher cargo-carrier accumulation on the gel interface than the P-2NLP-P hydrogel (max. fluorescence intensity: 7.6 ± 0.7 and 5.0 ± 0.9 for P-cNsp1-P and P-2NLP-P, respectively). However, the depth-integrated accumulation in an hour is the same for both materials (Figure 4f), indicating that the P-2NLP-P gel has better permeability to the cargo-carrier than P-cNsp1-P gel. Changes in the charge of the protein based on a single substitution per repeat unit between 1NLP and 2NLP impact on biomolecular transport through the designed hydrogels, despite the use of high ionic strength buffers that screen charge as under physiological conditions. Recent studies of related biological hydrogels such as mucus and cartilage have similarly observed that electrostatic effects influence molecular transport at the physiologically relevant ionic strength conditions.[36, 37] Selective binding can be added to synthetic systems by conjugating FG peptide onto polymer gels.[38] The results presented here on NLP hydrogels indicate that not only FG sequences but also residues far from the FG repeat can play an important role in the performance of nuclear pore-mimetic synthetic hydrogels.
After identifying P-2NLP-P as the top performing construct, additional experiments were performed to explore its performance. The addition of 10% hexanediol to disrupt FG interactions eliminates selective accumulation of cargo complexes in P-2NLP-P gels (Figure 4g), showing that FG repeat interactions are essential for enhanced selective transport in the biosynthetic hydrogels. This indicates that the engineered hydrogels have a filtering mechanism similar to the natural nucleoporin system[17, 29] Using blends of model proteins with and without the IBB domain establishes the ability of the biosynthetic NLP gels to actively transport cargo proteins compared to the inert proteins. A model cargo protein incapable of binding importin β, MBP-mCherry (67 kg/mol), has smaller size than IBB-MBP-EGFP (75 kg/mol) but shows retarded transport through the biosynthetic NLP hydrogels even in the presence of importin β (Figure 4h-i, 3 hours assay in 10 w/v% and 20 w/v% of P-2NLP-P gel results in Figure S15). The result clearly illustrates that the designed hydrogels can mimic both the selectivity and enhanced transport of natural nucleoporin hydrogels.[16, 18]
The successful development of new functional, biosynthetic hydrogels that mimic selectively enhanced transport biomolecules by the nuclear pore system using artificial protein engineering is a significant advance in adapting biological mechanisms of separation to synthetic systems. Due to decoupled gel stiffness by endblock gel crosslinkers and selective transport property by the midblock NLPs, stiffer or softer selective filtering hydrogels than those investigated here can be constructed as fusions of NLP consensus repeats with other types of gel forming domains[39] or by changing crosslink density,[27, 40] enabling the selective separation and gel stiffness to be independently tuned. There is also the potential to develop sequence libraries based on simplified NLP systems to both understand sequence-structure-property relationships and tune the selective transporting rates of these artificial protein polymer gels. Therefore, these scalable and easily processable engineered hydrogels have great potential for use in a wide variety of novel filtering, separation, and barrier technologies.
Supplementary Material
Figure S1. Protein sequences used in this study. Calculated and measured molar masses of proteins are in Table S1.
Figure S2. Protein expression levels by SDS-PAGE after the first step of protein purification. To reduce protein loss during washing step in Ni-NTA chromatography denaturing purification, the washing buffer did not contain imidazole and was prepared at pH 8, causing increased impurity during elution. After the elution, eluted proteins were run on the denaturing gels shown here (L: protein ladder; E: elution fraction). While Nsp1 (60 kg/mol), one of nuclear pore proteins, was expressed at low levels and difficult to identify on the gel, engineered proteins (P-cNsp1-P and P-NLPs-P) were highly expressed. For better purity, anion exchange chromatography was performed, with final purified products shown in Figure 2a. Note that all proteins ran slower in SDS-PAGE (Figure 2a, Figure S2-S3a) than their calculated and measured molar masses (Table S1), similar to previous reports for P-C30-P protein.[2]
Figure S3. Biosynthesis of NLPs and cNsp1. Similar to Nsp1 expression (Figure S2), cNsp1 (Nsp1282-585) was difficult to produce. On the other hand, both NLPs and cNsp1, as a product from P-intein-cNsp1, were synthesized in higher yields. The purity of lyophilized samples was confirmed by SDS-PAGE.
Figure S4. Hydrogel inversion tests of P-cNsp1-P in multiple buffer conditions. While cNsp1 is known not to form hydrogels,[7, 10, 11] P-cNsp1-P formed gels in all commonly used buffer conditions for Nsp1at a protein concentration of 200 mg/mL. 1: water; 2: 0.1% TFA (in water), followed by neutralization with 1/4 volume of the buffer (400 mM Tris-base, 100 mM Tris/HCl pH 7.5, 1 M NaCl) [5]; 3: 0.1% TFA (in water) and neutralized with 1/5 volume of 200 mM Tris-HCl (pH 8.5) [4, 6]; 4: 0.2% TFA (in water) and neutralized with 1/5 volume of 200 mM Tris-HCl (pH 9); 5: 100 mM phosphate buffer[7].
Figure S5. Strain sweep oscillatory shear rheology of 20 w/v% hydrogels at 100 rad/s and 25°C. Frequency sweeps were performed within the linear viscoelastic range at a strain amplitude of 1%.
Figure S6. The 1,6-hexanediol effect on other types of physically crosslinked hydrogels with P coiled coil domains. P-C30-P (37 kg/mol)[2] was synthesized and hydrogels were prepared by hydrating lyophilized samples (20 w/v%) with the buffer containing 50 mM Tris/HCl (pH 7.5) and 200 mM NaCl in the presence or absence of 10% 1,6-hexanediol. Frequency sweeps were measured at 37°C with a strain amplitude of 1%. Hexanediol did not affect the mechanical properties of P-C30-P hydrogels. Note that P-C30-P protein is known to run significantly slower than its calculated and measured molar masses in SDS-PAGE.[2]
Figure S7. Raman spectra of cNsp1 (20 w/v%) with selected band assignments. [12-16] The buffer contained 50 mM Tris/HCl (pH 7.5) and 200 mM NaCl (∆ = deformation and σ = stretching).
Figure S8. Raman spectra of cNsp1 and 1NLP lyophilized samples with selected band assignments. [17] The bands representing interacting moieties that were disrupted by hexanediol in solution (486 cm-1, 685 cm-1 and 710 cm-1 in Figure 3e-g) were not observed in lyophilized samples.
Figure S9. The pore size of designed hydrogels was measured by the dextran diffusion method. Various sizes of FITC–dextran (4, 10, 40, 70 and 150 kg/mol) were used for the fluorescence capillary assay under the same conditions as the transport measurements in Figure 4. Similar to the dextran diffusion result by Bestembayeva et al., [18] non-interacting 40 kg/mol, 70 kg/mol and 150 kg/mol dextran probes almost remained excluded while 4 and 10 kDa pass through the hydrogels, suggesting that the pore sizes of all designed hydrogels were nearly identical. Based on Stokes' radii of the FITC-dextran polymers in the SI Methods section, the gel pore radii can be estimated between 2.3 nm and 4.5 nm. White dotted curves were drawn on the gel/buffer interface for easy visualization.
Figure S10. Examples of engineered hydrogel permeability profiles. The florescence intensity profiles on the gel at 1 hour are shown. Areas under the solid curves (> 0 μm) were calculated (Figure 4) and compared to the passive diffusion by inert molecules (black dashed curve).
Figure S11. Gel and buffer interface changes by gel swelling during time lapse measurement of the capillary assay. The gel interface changes in Figure S10 and Figure 4c-e were measured and plotted as a function of time. Gel swelling was expected due to differences in chemical potential between the gel and the water bath, resulting in net flow into the gel (initial 20 w/v% concentration). Larger gel swelling was observed when importin β was presented in the capillary due to the additional effect of the cargo complex in increasing gel swelling. The enhanced transport rate of cargo-carrier complexes (IBB-MBP-GFP + importin β; green area in Figure 4c-e) compared to the cargo alone (gray area in Figure 4c-e) added more mass into the gel, causing larger gel swelling.
Figure S12. CD analysis of cNsp1 and NLPs. 5μM of the lyophilized proteins were hydrated in the buffer containing 50 mM Tris/HCl (pH 7.5) and 200 mM NaCl. By the CDSSTR method, [9] it was found that approximately 50% structures of all proteins were disordered and others were β-strand and β-turns.
Figure S13. Fluorescence intensity measurements on P-cNsp1-P hydrogel (20 w/v%) with 10% 1,6 hexanediol in the buffer containing 50 mM Tris/HCl (pH 7.5) and 200 mM NaCl. The selective permeability function of the hydrogel was lost due to disruption of hydrophobic interactions in the gel midblock, cNsp1.
Figure S14. Time lapse measurements of P-2NLP-P gel (20 w/v%). a) Fluorescence profiles were measured to investigate the selective permeability of the gel in the mixture of IBB-MBP-EGFP, MBP-mCherry and importin β. Solid curves represent green fluorescence intensity observed in the gel. b) Gel swelling in the presence of Importin β can be inferred from the movement of the gel/buffer boundary over the course of the selective permeability tests. For the control experiment, only MBP-mCherry was also tested and the gel boundary was measured as a function of time.
Figure S15. Selective permeability tests of P-2NLP-P gel in 20 w/v% and 10 w/v%. After finding that gels made with 2NLP have better selective accumulation than those made with1NLP and better permeability than those made with cNsp1 (Figure 4), we further investigated whether P-2NLP-P can show selective biomolecule transport at lower gel concentration, where natural FG repeat nucleoporins form inhomogeneous hydrogels with significantly reduced selective permeability. [5, 6] When hydrated in buffer at 20% and 10%, the proteins formed optically-clear gels that did not phase separate upon centrifugation, suggesting they formed macroscopically homogeneous networks under these conditions. Over three hours, the cargo-carrier complexes accumulate in the gel 10.2 and 5.2 times more than MBP-mCherry (no IBB domain), indicating that the 10 w/v% gel still shows enhanced transport of the selected biomolecules and the total accumulation depends on the number of FG sequences in the hydrogels (i.e. there are twice as many FG sequences in the 20 w/v% gel compared to the 10 w/v% gel).
Table S1. Protein molar masses measured by MALDI
Table S2. DNA plasmids that were used in this study
Acknowledgments
We are grateful to Dr. B. T. Chait (The Rockefeller University, USA) for the Nsp1 gene and Dr. Dirk Görlich (Max-Planck-Gesellschaft, Germany) for pSF851, pSF844, pSF856 and pQE30-scImpβ. M.K. thanks Carley M. Allen (Wellesley College, USA) for assisting with protein synthesis. This work was supported by the Defense Threat Reduction Agency under contract HDTRA1-13-1-0038 (MK, WGC, MJG, KR and BDO), NIH 5-T32-GM008834 (fellowships for W.G.C.,M.J.G.), the NIH NIBIB (P41EB015871-28; J.W.K.) and MIT WILtY-VUH SkolTech initiative (J.W.K.). The NIH (R01-EB017755) supported the development of methods for transport assays. The MRSEC Program of the National Science Foundation supported the infrastructure in KR's lab under award number DMR-0819762.
Footnotes
Supporting Information: Materials and methods are described in the Supporting Information. Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Minkyu Kim, Department of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA.
Wesley G. Chen, Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
Jeon Woong Kang, Laser Biomedical Research Center, G. R. Harrison Spectroscopy Laboratory, Massachusetts, Institute of Technology, Cambridge, MA 02139, USA.
Matthew J. Glassman, Department of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139
Katharina Ribbeck, Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA.
Bradley D. Olsen, Email: bdolsen@mit.edu, Department of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139.
References
- 1.Omenetto FG, Kaplan DL. Science. 2010;329:528. doi: 10.1126/science.1188936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petka WA, Harden JL, McGrath KP, Wirtz D, Tirrell DA. Science. 1998;281:389. doi: 10.1126/science.281.5375.389. [DOI] [PubMed] [Google Scholar]
- 3.MacKay JA, Chen MN, McDaniel JR, Liu WG, Simnick AJ, Chilkoti A. Nature Materials. 2009;8:993. doi: 10.1038/nmat2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hubbell JA. Nat Biotech. 1995;13:565. [Google Scholar]
- 5.Elvin CM, Carr AG, Huson MG, Maxwell JM, Pearson RD, Vuocolo T, Liyou NE, Wong DCC, Merritt DJ, Dixon NE. Nature. 2005;437:999. doi: 10.1038/nature04085. [DOI] [PubMed] [Google Scholar]
- 6.Lee TAT, Cooper A, Apkarian RP, Conticello VP. Adv Mater. 2000;12:1105. [Google Scholar]
- 7.Li L, Charati MB, Kiick KL. J Polym Sci A Polym Chem. 2010;1:1160. doi: 10.1039/b9py00346k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.García Quiroz F, Chilkoti A. Sequence-Controlled Polymers: Synthesis, Self-Assembly and Properties, Vol 1170, American Chemical Society. 2014:15. [Google Scholar]
- 9.Urry DW. Journal of Physical Chemistry B. 1997;101:11007. [Google Scholar]
- 10.Peppas NA, Hilt JZ, Khademhosseini A, Langer R. Adv Mater. 2006;18:1345. [Google Scholar]
- 11.Nath N, Chilkoti A. Adv Mater. 2002;14:1243. [Google Scholar]
- 12.Ribbeck K, Görlich D. Embo J. 2001;20:1320. doi: 10.1093/emboj/20.6.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang WD, Gelles J, Musser SM. P Natl Acad Sci USA. 2004;101:12887. doi: 10.1073/pnas.0403675101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bayliss R, Littlewood T, Stewart M. Cell. 2000;102:99. doi: 10.1016/s0092-8674(00)00014-3. [DOI] [PubMed] [Google Scholar]
- 15.Frey S, Richter RP, Goerlich D. Science. 2006;314:815. doi: 10.1126/science.1132516. [DOI] [PubMed] [Google Scholar]
- 16.Frey S, Görlich D. Embo J. 2009;28:2554. doi: 10.1038/emboj.2009.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patel SS, Belmont BJ, Sante JM, Rexach MF. Cell. 2007;129:83. doi: 10.1016/j.cell.2007.01.044. [DOI] [PubMed] [Google Scholar]
- 18.Labokha AA, Gradmann S, Frey S, Hulsmann BB, Urlaub H, Baldus M, Görlich D. Embo J. 2013;32:204. doi: 10.1038/emboj.2012.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schoch RL, Kapinos LE, Lim RY. Proc Natl Acad Sci U S A. 2012;109:16911. doi: 10.1073/pnas.1208440109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim J, Izadyar A, Nioradze N, Amemiya S. J Am Chem Soc. 2013;135:2321. doi: 10.1021/ja311080j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bestembayeva A, Kramer A, Labokha AA, Osmanovic D, Liashkovich I, Orlova EV, Ford IJ, Charras G, Fassati A, Hoogenboom BW. Nat Nanotechnol. 2015;10:60. doi: 10.1038/nnano.2014.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamada J, Phillips JL, Patel S, Goldfien G, Calestagne-Morelli A, Huang H, Reza R, Acheson J, Krishnan VV, Newsam S, Gopinathan A, Lau EY, Colvin ME, Uversky VN, Rexach MF. Mol Cell Proteomics. 2010;9:2205. doi: 10.1074/mcp.M000035-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jovanovic-Talisman T, Tetenbaum-Novatt J, McKenney AS, Zilman A, Peters R, Rout MP, Chait BT. Nature. 2009;457:1023. doi: 10.1038/nature07600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ader C, Frey S, Maas W, Schmidt HB, Görlich D, Baldus M. P Natl Acad Sci USA. 2010;107:6281. doi: 10.1073/pnas.0910163107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frey S, Görlich D. Cell. 2007;130:512. doi: 10.1016/j.cell.2007.06.024. [DOI] [PubMed] [Google Scholar]
- 26.Shen W, Zhang K, Kornfield JA, Tirrell DA. Nat Mater. 2006;5:153. doi: 10.1038/nmat1573. [DOI] [PubMed] [Google Scholar]
- 27.Olsen BD, Kornfield JA, Tirrell DA. Macromolecules. 2010;43:9094. doi: 10.1021/ma101434a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mathys S, Evans TC, Chute IC, Wu H, Chong S, Benner J, Liu XQ, Xu MQ. Gene. 1999;231:1. doi: 10.1016/s0378-1119(99)00103-1. [DOI] [PubMed] [Google Scholar]
- 29.Ribbeck K, Görlich D. Embo J. 2002;21:2664. doi: 10.1093/emboj/21.11.2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petri M, Frey S, Menzel A, Görlich D, Techert S. Biomacromolecules. 2012;13:1882. doi: 10.1021/bm300412q. [DOI] [PubMed] [Google Scholar]
- 31.Podstawka E, Ozaki Y, Proniewicz LM. Appl Spectrosc. 2004;58:570. doi: 10.1366/000370204774103408. [DOI] [PubMed] [Google Scholar]
- 32.Zhu G, Zhu X, Fan Q, Wan X. Spectrochim Acta A Mol Biomol Spectrosc. 2011;78:1187. doi: 10.1016/j.saa.2010.12.079. [DOI] [PubMed] [Google Scholar]
- 33.Cingolani G, Petosa C, Weis K, Muller CW. Nature. 1999;399:221. doi: 10.1038/20367. [DOI] [PubMed] [Google Scholar]
- 34.Görlich D, Henklein P, Laskey RA, Hartmann E. Embo J. 1996;15:1810. [PMC free article] [PubMed] [Google Scholar]
- 35.Mohr D, Frey S, Fischer T, Guttler T, Görlich D. Embo J. 2009;28:2541. doi: 10.1038/emboj.2009.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li LD, Crouzier T, Sarkar A, Dunphy L, Han J, Ribbeck K. Biophys J. 2013;105:1357. doi: 10.1016/j.bpj.2013.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bajpayee AG, Scheu M, Grodzinsky AJ, Porter RM. J Orthop Res. 2014;32:1044. doi: 10.1002/jor.22630. [DOI] [PubMed] [Google Scholar]
- 38.Bird SP, Baker LA. Biomacromolecules. 2011;12:3119. doi: 10.1021/bm200820x. [DOI] [PubMed] [Google Scholar]
- 39.Wright ER, Conticello VP. Adv Drug Deliv Rev. 2002;54:1057. doi: 10.1016/s0169-409x(02)00059-5. [DOI] [PubMed] [Google Scholar]
- 40.Tanaka F, Edwards SF. Macromolecules. 1992;25:1516. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Protein sequences used in this study. Calculated and measured molar masses of proteins are in Table S1.
Figure S2. Protein expression levels by SDS-PAGE after the first step of protein purification. To reduce protein loss during washing step in Ni-NTA chromatography denaturing purification, the washing buffer did not contain imidazole and was prepared at pH 8, causing increased impurity during elution. After the elution, eluted proteins were run on the denaturing gels shown here (L: protein ladder; E: elution fraction). While Nsp1 (60 kg/mol), one of nuclear pore proteins, was expressed at low levels and difficult to identify on the gel, engineered proteins (P-cNsp1-P and P-NLPs-P) were highly expressed. For better purity, anion exchange chromatography was performed, with final purified products shown in Figure 2a. Note that all proteins ran slower in SDS-PAGE (Figure 2a, Figure S2-S3a) than their calculated and measured molar masses (Table S1), similar to previous reports for P-C30-P protein.[2]
Figure S3. Biosynthesis of NLPs and cNsp1. Similar to Nsp1 expression (Figure S2), cNsp1 (Nsp1282-585) was difficult to produce. On the other hand, both NLPs and cNsp1, as a product from P-intein-cNsp1, were synthesized in higher yields. The purity of lyophilized samples was confirmed by SDS-PAGE.
Figure S4. Hydrogel inversion tests of P-cNsp1-P in multiple buffer conditions. While cNsp1 is known not to form hydrogels,[7, 10, 11] P-cNsp1-P formed gels in all commonly used buffer conditions for Nsp1at a protein concentration of 200 mg/mL. 1: water; 2: 0.1% TFA (in water), followed by neutralization with 1/4 volume of the buffer (400 mM Tris-base, 100 mM Tris/HCl pH 7.5, 1 M NaCl) [5]; 3: 0.1% TFA (in water) and neutralized with 1/5 volume of 200 mM Tris-HCl (pH 8.5) [4, 6]; 4: 0.2% TFA (in water) and neutralized with 1/5 volume of 200 mM Tris-HCl (pH 9); 5: 100 mM phosphate buffer[7].
Figure S5. Strain sweep oscillatory shear rheology of 20 w/v% hydrogels at 100 rad/s and 25°C. Frequency sweeps were performed within the linear viscoelastic range at a strain amplitude of 1%.
Figure S6. The 1,6-hexanediol effect on other types of physically crosslinked hydrogels with P coiled coil domains. P-C30-P (37 kg/mol)[2] was synthesized and hydrogels were prepared by hydrating lyophilized samples (20 w/v%) with the buffer containing 50 mM Tris/HCl (pH 7.5) and 200 mM NaCl in the presence or absence of 10% 1,6-hexanediol. Frequency sweeps were measured at 37°C with a strain amplitude of 1%. Hexanediol did not affect the mechanical properties of P-C30-P hydrogels. Note that P-C30-P protein is known to run significantly slower than its calculated and measured molar masses in SDS-PAGE.[2]
Figure S7. Raman spectra of cNsp1 (20 w/v%) with selected band assignments. [12-16] The buffer contained 50 mM Tris/HCl (pH 7.5) and 200 mM NaCl (∆ = deformation and σ = stretching).
Figure S8. Raman spectra of cNsp1 and 1NLP lyophilized samples with selected band assignments. [17] The bands representing interacting moieties that were disrupted by hexanediol in solution (486 cm-1, 685 cm-1 and 710 cm-1 in Figure 3e-g) were not observed in lyophilized samples.
Figure S9. The pore size of designed hydrogels was measured by the dextran diffusion method. Various sizes of FITC–dextran (4, 10, 40, 70 and 150 kg/mol) were used for the fluorescence capillary assay under the same conditions as the transport measurements in Figure 4. Similar to the dextran diffusion result by Bestembayeva et al., [18] non-interacting 40 kg/mol, 70 kg/mol and 150 kg/mol dextran probes almost remained excluded while 4 and 10 kDa pass through the hydrogels, suggesting that the pore sizes of all designed hydrogels were nearly identical. Based on Stokes' radii of the FITC-dextran polymers in the SI Methods section, the gel pore radii can be estimated between 2.3 nm and 4.5 nm. White dotted curves were drawn on the gel/buffer interface for easy visualization.
Figure S10. Examples of engineered hydrogel permeability profiles. The florescence intensity profiles on the gel at 1 hour are shown. Areas under the solid curves (> 0 μm) were calculated (Figure 4) and compared to the passive diffusion by inert molecules (black dashed curve).
Figure S11. Gel and buffer interface changes by gel swelling during time lapse measurement of the capillary assay. The gel interface changes in Figure S10 and Figure 4c-e were measured and plotted as a function of time. Gel swelling was expected due to differences in chemical potential between the gel and the water bath, resulting in net flow into the gel (initial 20 w/v% concentration). Larger gel swelling was observed when importin β was presented in the capillary due to the additional effect of the cargo complex in increasing gel swelling. The enhanced transport rate of cargo-carrier complexes (IBB-MBP-GFP + importin β; green area in Figure 4c-e) compared to the cargo alone (gray area in Figure 4c-e) added more mass into the gel, causing larger gel swelling.
Figure S12. CD analysis of cNsp1 and NLPs. 5μM of the lyophilized proteins were hydrated in the buffer containing 50 mM Tris/HCl (pH 7.5) and 200 mM NaCl. By the CDSSTR method, [9] it was found that approximately 50% structures of all proteins were disordered and others were β-strand and β-turns.
Figure S13. Fluorescence intensity measurements on P-cNsp1-P hydrogel (20 w/v%) with 10% 1,6 hexanediol in the buffer containing 50 mM Tris/HCl (pH 7.5) and 200 mM NaCl. The selective permeability function of the hydrogel was lost due to disruption of hydrophobic interactions in the gel midblock, cNsp1.
Figure S14. Time lapse measurements of P-2NLP-P gel (20 w/v%). a) Fluorescence profiles were measured to investigate the selective permeability of the gel in the mixture of IBB-MBP-EGFP, MBP-mCherry and importin β. Solid curves represent green fluorescence intensity observed in the gel. b) Gel swelling in the presence of Importin β can be inferred from the movement of the gel/buffer boundary over the course of the selective permeability tests. For the control experiment, only MBP-mCherry was also tested and the gel boundary was measured as a function of time.
Figure S15. Selective permeability tests of P-2NLP-P gel in 20 w/v% and 10 w/v%. After finding that gels made with 2NLP have better selective accumulation than those made with1NLP and better permeability than those made with cNsp1 (Figure 4), we further investigated whether P-2NLP-P can show selective biomolecule transport at lower gel concentration, where natural FG repeat nucleoporins form inhomogeneous hydrogels with significantly reduced selective permeability. [5, 6] When hydrated in buffer at 20% and 10%, the proteins formed optically-clear gels that did not phase separate upon centrifugation, suggesting they formed macroscopically homogeneous networks under these conditions. Over three hours, the cargo-carrier complexes accumulate in the gel 10.2 and 5.2 times more than MBP-mCherry (no IBB domain), indicating that the 10 w/v% gel still shows enhanced transport of the selected biomolecules and the total accumulation depends on the number of FG sequences in the hydrogels (i.e. there are twice as many FG sequences in the 20 w/v% gel compared to the 10 w/v% gel).
Table S1. Protein molar masses measured by MALDI
Table S2. DNA plasmids that were used in this study