

Abstract
The B. burgdorferi spirochete is the causative agent of Lyme disease, the most common tick-borne disease in the United States. The low abundance of bacterial proteins in human serum during infection imposes a challenge for early proteomic detection of Lyme disease. To address this challenge, we propose to detect membrane proteins released from bacteria due to disruption of their plasma membrane triggered by the innate immune system. These membrane proteins can be separated from the bulk of serum proteins by high-speed centrifugation causing substantial sample enrichment prior to targeted protein quantification using multiple reaction monitoring mass spectrometry. This new approach was first applied to detection of B. burgdorferi membrane proteins supplemented in human serum. Our results indicated that detection of B. burgdorferi membrane proteins, which are ≈ 107 lower in abundance than major serum proteins, is feasible. Therefore quantitative analysis was also carried out for serum samples from three patients with acute Lyme disease. We were able to demonstrate the detection of ospA, the major B. burgdorferi lipoprotein at the level of 4.0 fmol of ospA/mg of serum protein. The results confirm the concept and suggest that the proposed approach can be expanded to detect other bacterial infections in humans, particularly where existing diagnostics are unreliable.
Graphical Abstract
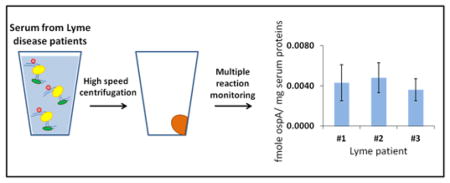
Lyme disease is a multi-organ tick-borne disease caused by spirochetes of the genus Borrelia. Borrelia burgdorferi (B. burgdorferi) is the prototypical Lyme disease spirochete in North America.1, 2 If left untreated, Lyme disease may lead to neurological and rheumatic manifestations that may last for years and adversely affect health-related functioning. Various post-treatment Lyme disease symptoms that may be severe and chronic have been described as well.3 Overall, Lyme disease and post-treatment Lyme disease symptoms are associated with significant health care costs.4
Unfortunately, the diagnosis of Lyme disease at it earliest stage is most often based upon clinical manifestations only including presence of the primary skin lesion, called erythema migrants. Confirmatory laboratory testing is limited to serological tests for the presence of antibodies that react to B. burgdorferi antigens. However serology is hampered by the long time of analysis. It generally takes 3 to 6 weeks before Borrelia-specific antibodies can be detected. In addition, various species-specific factors likely lower the sensitivity and specificity of serological tests, which may be misinterpreted and have false negatives or positives.3 Molecular assays to detect B. burgdorferi based on polymerase chain reaction (PCR) on DNA extracted from tissue or fluid specimens have also been described,3, 5–7 but are currently performed only for confirmation and research purposes. A drawback of using PCR is that B. burgdorferi DNA can be detected in samples long after spirochetes are no longer viable.8 Thus, a positive PCR result can be valuable for early detection, but needs to be interpreted with caution when efficiency of treatment and post-treatment symptoms are evaluated.
In addition to DNA, unique B. burgdorferi proteins can be targets of molecular assays. We have previously used a well-established mass spectrometry-based assay, multiple reaction monitoring (MRM), to detect and quantify target proteins.9–12 MRM assay relies on stable isotope-labeled internal standards added to the biological sample and is typically performed on a triple quadrupole mass spectrometer. Current instrumentation allows for the measurement of many proteins in a single sample, making MRM an ideal assay to perform high-throughput measurements on a panel of target proteins.13–15 Successful application of the MRM assay for detection of Borrelia proteins in human skin biopsies has been recently reported,16 however direct MRM assay in human blood or serum for early detection of Lyme disease poses additional challenges due to the extremely low abundance of total circulating Borrelia proteins.
In the present study, we capitalize on the fact that human serum is a cell-free substance which does not have membrane vesicles under normal conditions. Bacterial infection triggers various defense mechanisms including those of the innate immune system, which can disrupt the integrity of the bacterial wall.17 Broken B. burgdorferi cells18, 19 can release membrane vesicles populated with membrane proteins into the serum. These membrane vesicles can be separated from the bulk of soluble serum proteins by high-speed centrifugation and can be a ready source of unique membrane proteins for MRM detection of B. burgdorferi infection. As an initial proof of concept, we have been able to quantify the B. burgdorferi membrane protein ospA in the serum of Lyme disease patients. We believe this approach can be expanded to detection of many bacterial infections in humans, particularly where existing diagnostics are unreliable.
EXPERIMENTAL SECTION
Borrelia Extracts
Low-passage-number, B. burgdorferi strain B31 was grown in modified Barbour-Stoenner-Kelly II medium at 34 °C. Bacteria are harvested from log phase cultures, washed in PBS, sonicated and filtered as described.20 Protein concentration was adjusted to 1mg/ml and frozen at −80 °C.
Lyme disease patient serum
Serum samples were obtained from three patients with early, untreated Lyme disease at the time of diagnosis (1st visit) and two months later on the 3rd visit. All patients had a physician-observed erythema migrans rash at time of the 1st visit, as well as a recent history of flu-like symptoms, including fever, fatigue, and/or musculoskeletal pain. Two of the three patients were positive on standard, commercial two-tier serologic testing at the time of enrollment, and the third seroconverted during treatment and was positive at repeat testing three weeks later. Control samples were obtained from non-hospitalized individuals with no prior history of Lyme disease. Handling of serum for sample processing conformed to University of Maryland regulations. All data were analyzed anonymously. Demographic information on the de-identified donors is summarized in the Supporting Information Table S1.
Two-dimensional PAGE
The first dimension of separation was performed on 7-cm strips with 3–10 immobilized pH gradient using a PROTEAN IEF cell (Bio-Rad Laboratories, Hercules, CA). The strips were rehydrated with 125 μl of a protein solution in 2 mol/L thiourea, 7 mol/L urea, 4 % CHAPS, 0.5 % ASB-14, 0.2 % of (3–10) Biolytes, and bromphenol blue. Isoelectric focusing was conducted at 50 V for 12 hr, linearly increased over 2 h to a maximum of 4000 V, and then run to accumulate a total of 20000 V/h. For the second dimension, the immobilized pH gradient strips were equilibrated for 15 min in 50 mmol/L Tris-HCl (pH 8.8), 6 mol/L urea, 30 % glycerol, 2 % SDS, and bromophenol blue. The strips were then embedded in 0.7 % (w/v) agarose on the top of 9 % home-made polyacrylamide gels and proteins were separated by SDS-PAGE. In some experiments, 8–16 % mini-PROTEAN TGX gels from Bio-Rad were used as well. All gels were stained with a Pierce silver stain kit for mass spectrometry.
In-gel protein identification
Silver-stained gel pieces were excised and destained in accordance with the manufacturer’s protocol. In-gel digestion was then carried out with a sequencing grade modified trypsin (Promega, Madison, WI) in 25 mmol/L NH4HCO3 (pH 7.9) for 15 h at 37 °C. After digestion, samples were dried, and dissolved in 5 mg/mL α-cyano-4-hydroxycinnamic acid in 50 % acetonitrile containing 0.1 % trifluoroacetic acid (TFA). After spotting onto an ABI 01-192-6-AB target plate, the MS analysis was performed using an AB4700 Proteomics Analyzer (Applied Biosystems, Framingham, MA). All MALDI mass spectra were internally calibrated using the monoisotopic masses of the autolysis peptides of trypsin at 842.51 and 2211.10. Automated combined acquisition of MS and MS/MS data was controlled with 4000 Series Explorer software 3.0. Data analysis was performed with GPS Explorer software 3.5 utilizing Mascot 2.0 (MatrixScience, London, UK) as the search engine. During searching, the mass tolerance was 0.08 Da for the precursor ions and 0.2 Da for the fragment ions. A protein was listed as identified protein when the MOWSE score was higher than a MOWSE score at which statistical significance (p < 0.05) occurred for that particular search.
15N-Labeled QconCAT expression, purification, and characterization
The quantification concatamer (QconCAT) is an artificial protein composed of concatenated tryptic peptides from targeted proteins and used as an internal standard for quantification of these targeted proteins by MRM. The amino acid sequence of a QconCAT designed for quantification of B. burgdorferi proteins was coded into the corresponding DNA sequence and incorporated into the pET21a expression vector, with codon optimization for E. coli (Biomatik, Cambridge, Ontario). The plasmid was transformed into One Shot BL21 (DE3) competent E. coli cells (Invitrogen, Grand Island, NY) and grown in M9 minimal media with 1 g/L 15NH4Cl (Cambridge Isotope Laboratories, Andover, MA) as the sole nitrogen source at 37 °C until the optical density reached 0.6 to 0.8 at 600 nm. Protein expression was induced by 0.5 mmol/L isopropyl β-D-1-thiogalactopyranoside. After 3 h of growth, the cells were harvested by centrifugation at 5000 g for 10 min and resuspended in 0.1 mmol/L dithiothreitol (DTT) and sonicated. Following centrifugation at 35000g for 30 min, the supernatant was discarded and the pellet which contained the QconCAT was resuspended in 100 mmol/L Na2HPO4/10 mmol/L Tris-HCl (pH 8.0) containing 8 mol/L urea and 10 mmol/L imidazole. The suspension was centrifuged at 20000g for 10 min and the supernatant was used for QconCAT purification.
Purification of 6xHis-tagged QconCAT was performed using Ni-NTA agarose resin by the gravity-flow method (Qiagen, Valencia, CA). The binding, washing, and eluting buffers were 100 mmol/L Na2HPO4/10 mmol/L Tris-HCl (pH 8.0) containing 8 mol/L urea and 10 mmol/L, 30 mmol/L and 100 mmol/L imidazole, respectively. The eluted fraction was concentrated and buffer-exchanged to 100 mmol/L Na2HPO4/10 mmol/L Tris-HCl (pH 8.0) containing 6 mol/L urea using an Amicon filter (30 kDa MWCO, Millipore, Billerica, MA). The QconCAT concentration was subsequently determined by a BCA protein assay with bovine serum albumin as a standard (Thermo Scientific, Waltham, MA). QconCAT expression and purification were evaluated with SDS-PAGE and mass spectrometry analysis on a 4700 Proteomics Analyzer. The isotope incorporation was determined at the peptide level after digestion of the purified QconCAT with trypsin. MALDI spectra of three representative peptides were imported to Isotopic Enrichment Calculator (http://www.nist.gov/mml/bmd/bioanalytical/isoenrichcalc.cfm)21 and the mean value was higher than 99 % of 15N incorporation. This was accepted as a complete labeling and no correction was applied to the data.
Processing of human serum samples
To prove the concept, 1 mL samples of normal human serum (cat. # S-7023, Sigma-Aldrich, St. Louis, MO) were supplemented with 3.0 μg, 1.0 μg, 0.3 μg, 0.1 μg, 0.03 μg, or 0.01 μg of total B. burgdorferi protein. The membrane proteins were pulled down by high-speed centrifugation at 106000 g for 60 min at 4 °C. The membrane pellet was then resuspended in 1 mL of 0.1 mol/L Na2CO3 and centrifuged at 179000 g for 60 min at 4 °C. The Na2CO3-washed membrane pellet was resuspended in 150 μL of 25 mmol/L NH4HCO3 /1 % SDS/10 mmol/L DTT and supplemented with 15 pmoles of QconCAT. The mixture was incubated at room temperature for 60 min to allow reduction of cysteines and then treated with 55 mmol/L iodoacetamide for another 60 min to alkylate the reduced cysteines. Alkylated samples were precipitated with chloroform/methanol22 to deplete salts, urea, and SDS from the samples. Protein pellets were then sonicated in 100 μL of 25 mmol/L NH4HCO3/0.1 % RapiGest SF surfactant (Waters, Milford, MA) and treated with trypsin for 15 h at 37 °C. The substrate/trypsin ratio was 10:1 (w/w). After trypsin digestion, the samples were treated with 0.5 % TFA for 30 min at 37 °C to break down acid-cleavable RapiGest. The insoluble by-product of RapiGest was then removed by centrifugation at 106000 g for 30 min. After centrifugation, the supernatants were dried using a vacuum centrifuge (Vacufuge, Eppendorf AG, Hamburg, Germany).
Human serum samples from control and Lyme disease patients (1 mL each) were processed as described above without supplementation with spirochete proteins, but supplemented with 3 pmoles of QconCAT.
LC-MS/MS analysis
Dried peptides were reconstituted in 30 μL of 3 % acetonitrile/97 % water containing 0.1 % formic acid and 5 μL were used for each LC-MS/MS run. Instrumental analyses were performed on an Agilent Zorbax Eclipse Plus C18 RRHD column (2.1 mm × 50 mm, 1.8 μm particle) coupled to an Agilent 6490 Triple Quadrupole LC/MS system (Santa Clara, CA). Peptides were eluted over a 35-min gradient from 5 % to 80 % acetonitrile containing 0.1 % formic acid at a flow rate of 200 μL/min. The gradient settings were: 5 % to 10 % solvent B in 5 min, 10 % to 30 % solvent B in 25 min, 30 % to 80 % solvent B in 5 min, then returned to 5 % solvent B in 5 min. Solvent A was water containing 0.1 % formic acid and solvent B was 100 % acetonitrile with 0.1 % formic acid. The acquisition method used the following parameters in positive mode: fragmentor 380 V, electron multiplier 500 V, and capillary voltage 3500 V. Collision energy was optimized for each peptide using the default equation from Agilent, CE = 0.036 m/z - 4.8. Dwell time for all transitions was set at 120 ms.
Data analysis
MRM peak area integration was performed using Agilent MassHunter Qualitative Analysis B.06. Excel was used to calculate peak area ratios. Peak integration was manually inspected and adjusted if necessary. The peak ratios from transitions were averaged to yield the peptide ratios. All experiments were performed in duplicate with three replicate injections to assess error and reproducibility. Data is represented as the mean ± SD.
RESULTS AND DISCUSSION
Selection of target B. burgdorferi proteins and design of a QconCAT
Our primary focus was on those B. burgdorferi proteins that demonstrate the highest abundance and possess unique sequence. We have used a cultured B. burgdorferi B31 isolate A3. Figure 1 shows a two-dimensional gel pattern for the whole homogenate of this prototypical B. burgdorferi. Based on silver staining, the list of most abundant proteins includes integral outer membrane protein P66 (p66), aminopeptidase 1 (apeA), basic membrane protein A (bmpA), outer surface protein A (ospA), chaperone protein Dnak (dnaK), 60 kDa chaperonin (groL), enolase (eno), and glyceraldehyde 3-phosphate dehydrogenase (gap). The identification statistic of these proteins is summarized in the Supporting Information Table S2. For all of these identifications, the MOWSE score from Mascot software (http://www.matrixscience.com/search_form_select.html) search was higher than the score at which statistical significance (p<0.05) occurred for that particular search. Identified proteins also correspond well to the expected molecular mass and PI values.
Figure 1.

Two-dimensional PAGE pattern of B. burgdorferi proteins. The first and second dimensions were performed on 7-cm pH 3–10 immobilized pH gradient strips and 9 % polyacrylamide slab gel, respectively. After separation, proteins were detected by silver staining. The proteins identified in the numbered spots are: integral outer membrane protein P66 (#1), aminopeptidase 1 (#2), basic membrane protein A (#3), outer surface protein A (#4), chaperone protein Dnak (#5), 60 kDa chaperonin (#6), enolase (#7), and glyceraldehyde 3-phosphate dehydrogenase (#8).
To be selected as a target protein for B. burgdorferi detection in human serum, the protein amino acid sequence has to be distinguishable from any human protein(s). BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) and LALIGN (http://www.ch.embnet.org/software/LALIGN_form.html) searches revealed that dnaK, groL, eno, and gap proteins from B. burgdorferi have high level of homology with corresponding human proteins and cannot be used for selective B. burgdorferi detection. Four other proteins, p66, apeA, bmpA, and ospA, have amino acid sequences that can generate multiple tryptic peptides, which are unique and will identify B. burgdorferi unambiguously. Although not observed in our two-dimensional gel, three more B. burgdorferi proteins, namely outer surface protein C (ospC), flagellar filament 41 kDa core protein (fla), and DNA-binding protein HU (hup) have also attracted our attention. These proteins were previously reported as abundant B. burgdorferi proteins16, 23–26 and have unique tryptic peptides for selective detection of B. burgdorferi. Consequently, the list of tryptic peptides for the seven B. burgdorferi proteins was generated in silico. Based on published rules,9 peptides acceptable for MRM analysis were selected and compiled into a QconCAT sequence (Supporting Information Figure S1). The 15N-labeled QconCAT was expressed, purified, and characterized (Supporting Information Figure S2).
While this QconCAT was designed to quantify B. burgdorferi only, it is important to note that the target proteins possess many tryptic peptides which are not only species-selective, but are also strain-selective. This means it is possible to design QconCATs, which will allow selective quantification of specific Borrelia species (such as B. burgdorferi, B. garinii, B. afzelli, etc) in one LC-MS/MS run. Selective quantification of specific strains of B. burgdorferi is possible as well.
Detecting B. burgdorferi membrane proteins supplemented in the human serum
We hypothesized that upon bacterial infection, the broken bacterial cells would generate outer membrane vesicles populated with membrane proteins and become the source of membrane proteins in the serum. Five B. burgdorferi proteins included in the QconCAT are membrane proteins: p66 is a single pass transmembrane protein; bmpA, ospA, and ospC have an N-terminal lipid anchor; apeA was recovered from a membrane fraction.24 Previously, we have demonstrated that washing the membrane pellet with 0.1 mol/L Na2CO3 can enrich the sample with those membrane proteins which were not detectable in membrane pellet after high-speed centrifugation.10 However, prior to applying 0.1 mol/L Na2CO3 washing to our samples, we confirmed that this washing step does not cause loss of the target proteins. Visual comparison of p66, apeA, bmpA, and ospA staining on the two-dimensional gels shows no changes in the amount of these proteins before and after 0.1 mol/L Na2CO3 washing (Supporting Information Figure S3). At the same time, measurement of the total protein in these samples shows that washing with 0.1 mol/L Na2CO3 removed 57 % of the total protein, resulting in approximately 2-fold sample enrichment with target proteins. Such a step is important for detection of low-abundance proteins.
To prove the proposed concept, we supplemented 1 mL of normal human serum with various amount of total bacterial protein obtained from B. burgdorferi B31 isolate A3. The membrane proteins were pulled down by high-speed centrifugation and washed with 0.1 mol/L Na2CO3. The washed membrane pellet was supplemented with 15N-labeled QconCAT and mass spectrometry analysis was focused on detecting B. burgdorferi proteins included in the QconCAT. The quantitative data are summarized in Table 1. Two proteins, apeA and hup, were not detected in these experiments and are not included in Table 1. Quantification of five other proteins for different added amount of total B. burgdorferi protein was consistently reproducible with the SD generally under 20 % of the mean value. The lowest detection for these proteins concurs well with their relative abundance in B. burgdorferi. For example, bmpA with an average concentration 79 ± 7 pmol/mg of total B. burgdorferi protein was detected at 1 μg supplementation while ospA with an average concentration 4220 ± 349 pmol/mg of total B. burgdorferi protein was detected at 0.03 μg supplementation. None of the proteins was detected for 0.01 μg supplementation of total B. burgdorferi protein. The data in Table 1 allow two simple calculations. First, the molecular mass of ospA is 29.4 kDa and the concentration 4220 pmol of ospA per mg of total B. burgdorferi protein means that ospA represents approximately 12 % of total protein in B. burgdorferi B31 isolate A3. Second, 0.03 μg supplementation represents approximately 4 ng of ospA. This amount was supplemented into 1 mL of human serum, which has 60 mg of total protein. The resulting dynamic range between 60 mg/mL and 4 ng/mL is 1.5 × 107. We believe this would be the largest dynamic range reported for quantitative measurements of a target protein in the human serum. Overall, the developed protocol for quantitative analysis of bacterial membrane proteins in human serum encouraged us to proceed to measurements of serum samples from control and Lyme disease-diagnosed patients.
Table 1.
Quantification of B. burgdorferi proteins supplemented in the 1 mL of human serum
| Supplementation, μg of B. burgdorferi protein/mL of human serum | Average concentration | ||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| 3.0 | 1.0 | 0.3 | 0.1 | 0.03 | 0.01 | ||
|
| |||||||
| Protein/peptide | Concentration, pmol of a protein/mg of B. burgdorferi proteina | ||||||
|
| |||||||
| ospA | 4220 ± 349 | ||||||
| YDLIATVDK | 4073 ± 437 | 4974 ± 1040 | 3874 ± 559 | 3862 ± 1548 | ndb | nd | |
| EGTVTLSK | 4093 ± 258 | 4749 ± 82 | 3994 ± 278 | nd | nd | nd | |
| GYVLEGTLTAEK | 4345 ± 187 | 4419 ± 514 | 3983 ± 492 | 4014 ± 483 | 4254 ± 1166 | nd | |
|
| |||||||
| fla | 987 ± 125 | ||||||
| NNGINAANLSK | 843 ± 128 | 1056 ± 49 | 1062 ± 136 | nd | nd | nd | |
|
| |||||||
| p66 | 302 ± 60 | ||||||
| STYYGFPSNDR | 293 ± 53 | 311 ± 79 | 237 ± 58 | nd | nd | nd | |
| LDLTFAIGGTGTGNR | 270 ± 17 | 296 ± 13 | 354 ± 22 | nd | nd | nd | |
| NLLDQNEDTK | 237 ± 14 | 294 ±20 | 430 ± 53 | nd | nd | nd | |
|
| |||||||
| ospC | 83 ± 6 | ||||||
| EVEALLSSIDEIAAK | 77 ± 10 | 85 ± 21 | 88 ± 16 | nd | nd | nd | |
|
| |||||||
| bmpA | 79 ± 7 | ||||||
| ALNIFTSNHLK | 81 ± 39 | 77 ± 21 | nd | nd | nd | nd | |
All experiments were performed in duplicate with three replicate injections. Data is presented as the mean ± SD.
nd means not detected.
Detecting B. burgdorferi membrane proteins in the serum of Lyme disease-diagnosed patients
There is a well-documented diversity of the spirochetes protein expression pattern in response to changing environmental factors23, 25–29. In the previous section, we have used a clonal and low-passage infectious B. burgdorferi B31 isolate A3 to optimize the serum processing protocol and detecting spirochetes proteins supplemented in human serum. The average protein concentrations presented in Table 1 are correct for this specific isolate and its cultured conditions. For example, average concentration for ospC at 83 pmol/mg of total B. burgdorferi protein is 50-fold lower than concentration for ospA, which is 4220 pmol of ospA per mg of total B. burgdorferi protein. This explains why we do not see ospC in our 2D-PAGE images (Figure 1 and Supporting Information Figure S3). However, previously published 2D-PAGE images of a different isolate of B. burgdorferi B3124 show approximately equal levels of expression for both proteins. Therefore, when approaching measurements in human serum from naturally infected patients, we decided to track all proteins included in QconCAT no matter how abundant they are in the B. burgdorferi B31 isolate A3.
In our measurements, we used 1 mL size serum samples from control patients and from Lyme disease patients, collected at the time of their 1st and 3rd visits (Supporting Information Table S1). We did not detect B. burgdorferi proteins in the control and 3rd visit samples, but we were able to detect ospA in the Lyme disease samples, collected at the time of 1st visit. Detection has been made based on three ospA peptides (Figure 2): YDLIATVDK (two transitions), EGTVTLSK (two transitions), and GYVLEGTLTAEK (three transitions). It is important to emphasize that the relative ratios of these transitions for every pair of labeled and non-labeled peptides were almost identical (Figure 2). This confirms that ospA quantification is not affected by nonspecific interference from the biological sample. In the contrast to supplementation experiments, we cannot normalize these measurements to the amount of the total B. burgdorferi protein. However, taking a value of 60 mg of serum protein/mL, we can normalize ospA measurements to a mg of total serum protein. Based on YDLIATVDK, EGTVTLSK, and GYVLEGTLTAEK peptides, we have detected an average concentration of ospA as 2.9 ± 0.6, 3.5 ± 1.4, and 5.1 ± 1.3 fmol of ospA/mg of serum protein for three Lyme disease patients, respectively (Table 2). As a consensus, it brings us to the detection limit on the level of approximately 4.0 fmol of ospA/mg of serum protein.
Figure 2.
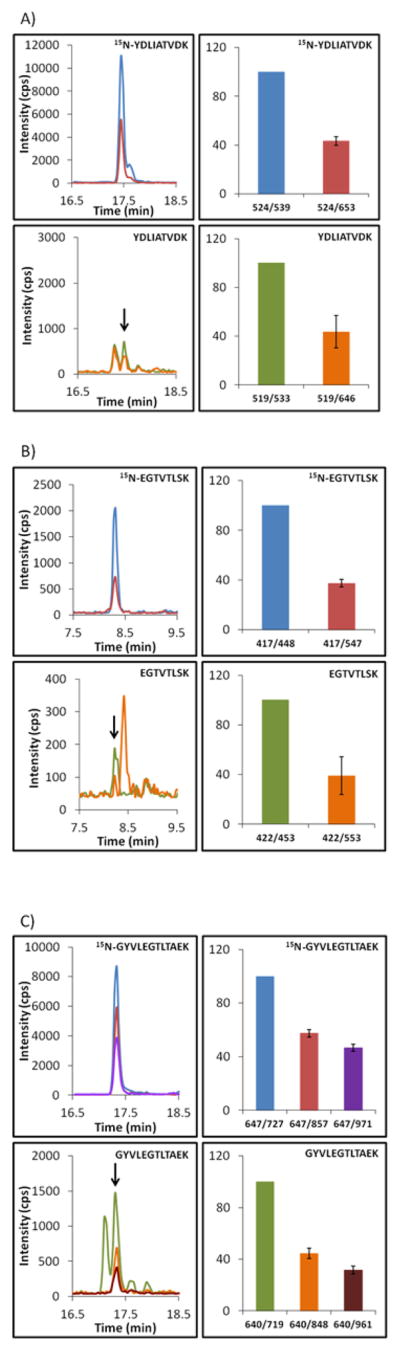
Extracted ion chromatograms and MRM transitions ratio monitored for ospA in the serum from Lyme disease patients, collected at the time of 1st visit. Data are presented for the YDLIATVDK (A), EGTVTLSK (B), and GYVLEGTLTAEK (C) peptides. Heavy and light versions of peptides represent QconCAT and endogenous ospA, respectively. Overlaid extracted ion chromatograms and bars for transitions ratio are color-coordinated. For transitions ratio, the most intensive transition was taken as a 100 and intensities of other transitions were plotted as a portion of a 100. Measurements were performed in duplicate for 3 Lyme disease patients with three analytical replicates.
Table 2.
Quantification of ospA in the serum from Lyme disease patients collected on their 1st visit.
| 15N/14N ratioa | fmol of ospA/mg serum proteinb | |||||
|---|---|---|---|---|---|---|
| Patient #1 | Patient #2 | Patient #3 | Average | Consensus | ||
| YDLIATVDK | 16.3 ± 4.0 | 2.4 ± 0.5 | 3.3 ± 0.6 | 3.0 ± 0.6 | 2.9 ± 0.6 | 3.8 ± 0.4 |
| EGTVTLSK | 14.7 ± 5.4 | 3.7 ± 1.4 | 4.1 ± 1.6 | 2.7 ± 1.0 | 3.5 ± 1.4 | |
| GYVLEGTLTAEK | 9.4 ± 2.5 | 5.4 ± 1.4 | 5.8 ± 0.8 | 4.2 ± 1.0 | 5.1 ± 1.3 | |
Average ratio shows the proportion between 15N-labeled internal standard and 14N-analyte signal intensities.
All experiments for three patients were performed in duplicate with three replicate injections. Data is presented as the mean ± SD.
CONCLUSIONS
In the summary, broken bacterial cells may be a source of membrane proteins in human serum during early bacterial infection with B. burgdorferi. Targeting these proteins is strongly supported by the fact that a simple experimental step such as high-speed centrifugation allows substantial enrichment of the sample before LC-MS/MS analysis. Therefore detection of proteins, whose abundance is ≈ 107 lower than abundance of major serum proteins, became feasible. We report here a proof of concept based on detecting ospA protein in the serum from patients diagnosed with Lyme disease and we believe that this approach may be universally applicable to detection of other bacterial infections in human serum. We anticipate that future investigations with additional serum samples will further substantiate this approach and expand its potential range of applications.
Supplementary Material
Acknowledgments
This work was supported in part by the Lyme Disease Research Foundation, Inc. (www.lymemd.org), the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under award number P30AR05350, and the Stabler Foundation (www.stablerfoundation.org). Certain commercial materials, instruments, and equipment are identified in this manuscript in order to specify the experimental procedure as completely as possible. In no case does such identification imply a recommendation or endorsement by the National Institute of Standards and Technology nor does it imply that the materials, instruments, or equipment identified are necessarily the best available for the purpose.
Footnotes
ASSOCIATED CONTENT
Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Centers for Disease Control and Prevention. Reported Cases of Lyme Disease by Year, United States 1996–2010. Available: http://www.cdc.gov/lyme/stats/chartstables/casesbyyear.html.
- 2.Hinckley AF, Connally NP, Meek JI, Johnson BJ, Kemperman MM, Feldman KA, White JL, Mead PS. Clin Infect Dis. 2014;59:676–681. doi: 10.1093/cid/ciu397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borchers AT, Keen CL, Huntley AC, Gershwin ME. J Autoimmun. 2015;57:82–115. doi: 10.1016/j.jaut.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 4.Adrion ER, Aucott J, Lemke KW, Weiner JP. PLoS ONE. 2015;10:e0116767. doi: 10.1371/journal.pone.0116767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nocton JJ, Dressier F, Rutledge BJ, Rys PN, Persing DH, Steere AC. N Eng J Med. 1994;330:229–234. doi: 10.1056/NEJM199401273300401. [DOI] [PubMed] [Google Scholar]
- 6.Cerar T, Ogrinc K, Cimperman J, Lotric-Furlan S, Strle F, Ruzic-Sabljic E. J Clin Microbiol. 2008;46:3375–3379. doi: 10.1128/JCM.00410-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eshoo MW, Crowder CC, Rebman AW, Rounds MA, Matthews HE, Picuri JM, Soloski MJ, Ecker DJ, Schutzer SE, Aucott JN. PLoS ONE. 2012;7:e36825. doi: 10.1371/journal.pone.0036825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iyer R, Mukherjee P, Wang K, Simons J, Wormser GP, Schwartz I. J Clin Microbiol. 2013;51:857–862. doi: 10.1128/JCM.02785-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liao W-L, Heo G-Y, Dodder NG, Pikuleva IA, Turko IV. Anal Chem. 2010;82:5760–5767. doi: 10.1021/ac100811x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang M, Heo G-Y, Omarova S, Pikuleva IA, Turko IV. Anal Chem. 2012;84:5186–5191. doi: 10.1021/ac300587v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen J, Huang RY-C, Turko IV. Anal Chem. 2013;85:6011–6017. doi: 10.1021/ac400831z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheung CSF, Anderson KW, Wang M, Turko IV. Anal Chem. 2015;87:1097–1102. doi: 10.1021/ac503697j. [DOI] [PubMed] [Google Scholar]
- 13.Liebler DC, Zimmerman LJ. Biochemistry. 2013;52:3797–3806. doi: 10.1021/bi400110b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gillette MA, Carr SA. Nat Methods. 2013;10:28–34. doi: 10.1038/nmeth.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Picotti P, Aebersold R. Nat Methods. 2012;9:555–566. doi: 10.1038/nmeth.2015. [DOI] [PubMed] [Google Scholar]
- 16.Schnell G, Boeuf A, Westermann B, Jaulhac B, Lipsker D, Carapito C, Boulanger N, Ehret-Sabatier L. Mol Cell Proteomics. 2015;14:1254–1264. doi: 10.1074/mcp.M114.046540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rus H, Cudrici C, Niculescu F. Immunol Res. 2005;33:103–112. doi: 10.1385/IR:33:2:103. [DOI] [PubMed] [Google Scholar]
- 18.Kochi SK, Johnson RC, Dalmasso AP. J Immunol. 1991;146:3964–3970. [PubMed] [Google Scholar]
- 19.Breitner-Ruddock S, Wurzner R, Schulze J, Brade V. Med Microbiol Immunol. 1997;185:253–260. doi: 10.1007/s004300050038. [DOI] [PubMed] [Google Scholar]
- 20.Craft JE, Grodzicki RL, Steere AC. J Infect Dis. 1984;149:789–795. doi: 10.1093/infdis/149.5.789. [DOI] [PubMed] [Google Scholar]
- 21.Kilpatrick EL, Liao W-L, Camara JE, Turko IV, Bunk DM. Protein Expr Purif. 2012;85:94–99. doi: 10.1016/j.pep.2012.06.019. [DOI] [PubMed] [Google Scholar]
- 22.Liao W-L, Turko IV. Anal Biochem. 2008;377:55–61. doi: 10.1016/j.ab.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 23.Jacobs JM, Yang X, Luft BJ, Dunn JJ, Camp DG, II, Smith RD. Proteomics. 2005;5:1446–1453. doi: 10.1002/pmic.200401052. [DOI] [PubMed] [Google Scholar]
- 24.Nowalk AJ, Nolder C, Clifton DR, Carroll JA. Proteomics. 2006;6:2121–2134. doi: 10.1002/pmic.200500187. [DOI] [PubMed] [Google Scholar]
- 25.Yang X, Promnares K, Qin J, He M, Shroder DY, Kariu T, Wang Y, Pal U. J Proteome Res. 2011;10:4556–4566. doi: 10.1021/pr200395b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gesslbauer B, Poljak A, Handwerker C, Schuler W, Schwendenwein D, Weber C, Lundberg U, Meinke A, Kungl AJ. Proteomics. 2012;12:845–858. doi: 10.1002/pmic.201100211. [DOI] [PubMed] [Google Scholar]
- 27.Revel AT, Talaat AM, Norgard MV. Proc Natl Acad Sci USA. 2002;99:1562–156.7. doi: 10.1073/pnas.032667699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Angel TE, Luft BJ, Yang X, Nicora CD, Camp DG, II, Jacobs JM, Smith RD. PLoS ONE. 2010;5:e13800. doi: 10.1371/journal.pone.0013800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schnell G, Boeuf A, Jaulhac B, Boulanger N, Collin E, Barthel C, De Martino S, Ehret-Sabatier L. Proteomics. 2015;15:1280–1290. doi: 10.1002/pmic.201400177. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.