

Abstract
Previous preclinical work has demonstrated the therapeutic potential of antagonists of the group II metabotropic glutamate receptors (mGlus). Still, compounds that are selective for the individual group II mGlus (mGlu2 and mGlu3) have been scarce. There remains a need for such compounds with the balance of properties suitable for convenient use in a wide array of rodent behavioral studies. We describe here the discovery of a selective mGlu3 NAM 106 (VU0650786) suitable for in vivo work. Compound 106 is a member of a series of 5-aryl-6,7-dihydropyrazolo[1,5-a]pyrazine-4(5H)-one compounds originally identified as a mGlu5 positive allosteric modulator (PAM) chemotype. Its suitability for use in rodent behavioral models has been established by extensive in vivo PK studies, and the behavioral experiments presented here with compound 106 represent the first examples in which an mGlu3 NAM has demonstrated efficacy in models where prior efficacy had previously been noted with nonselective group II antagonists.

INTRODUCTION
Glutamate (l-glutamic acid) is the major excitatory neuro-transmitter in the mammalian central nervous system (CNS) and acts on both ionotropic and metabotropic glutamate receptors (mGlus). While ionotropic glutamate receptors are ligand-gated ion channels, mGlus are a family of eight G-protein coupled receptors (GPCRs). Belonging to family C of the GPCRs, the mGlus possess a seven transmembrane (7TM) α-helical domain connected via a cysteine-rich region to a large bilobed extracellular amino-terminal domain containing the orthosteric binding site. The mGlus have been further categorized into three groups according to their homology, preferred signal transduction mechanisms, and pharmacology: group I (mGlu1 and mGlu5), group II (mGlu2 and mGlu3), and group III (mGlu4, mGlu6, mGlu7, and mGlu8).1–3 Both mGlu2 and mGlu3 are primarily located presynaptically in neurons and coupled to Gi/o and the inhibition of adenylyl cyclase activity; in addition, mGlu3 is also expressed in glial cells. The group II mGlus are widely expressed throughout the CNS, including regions of the brain associated with emotional states such as the amygdala, hippocampus, and prefrontal cortex.4,5
Researchers have been successful in designing both orthosteric antagonists and negative allosteric modulators (NAMs), also known as noncompetitive antagonists, of the group II mGlus. Although such compounds are generally selective versus the other six members of the mGlu family, selectivity between mGlu2 and mGlu3 is negligible.6 Still, the use of such compounds in animal models has established a potential role for mGlu2/3 antagonists in a variety of CNS disorders. Many of these studies have been carried out with two orthosteric antagonists, both of which are highly functionalized glutamate analogues, 1 (LY341495)7 and 2 (MGS0039)8 (Figure 1). For example, work with these compounds has helped establish mGlu2/3 inhibition as a potential therapeutic application for obsessive-compulsive disorder (OCD),9,10 anxiety,11 cognition,12 and Alzheimer's disease.13–15 Additionally, antidepressant efficacy has been demonstrated in numerous rodent models of depression with these compounds,8,9,11,16–19 including those meant to assess treatment-resistant depression (TRD),20 anhedonia,21 and depression associated with withdrawal from addictive substances.22,23 Compound 1 has also been used as a tool to establish a potential utility for mGlu2/3 inhibition in the treatment of glioma.24–27
Figure 1.

mGlu2/3 orthosteric antagonist tools 1 and 2, mGlu2/3 NAM tools 3, 4, and 6, and Roche mGlu2/3 NAM clinical compound 5.
Research with mGlu2/3 NAMs has been less extensive than with orthosteric antagonists; however, closely related tools 3 (RO4491533)28 and 4 (RO4432717)29,30 (Figure 1) have been employed in multiple in vivo assays. In particular, these compounds have demonstrated efficacy in multiple models of depression31 and cognition.30,32,33 Furthermore, recent studies in genetically modified mice with 134 and mGlu2/3 NAM 635 (Figure 1) point toward a potential application for group II antagonists in the treatment of certain autism spectrum disorders. Finally, one mGlu2/3 NAM, 5 (decoglurant, RO4995819)36 (Figure 1), has advanced into human clinical trials, including a phase II trial in patients with major depressive disorder (MDD) and resistant to ongoing treatment with antidepressants (NCT01457677).37 In spite of the wealth of preclinical evidence for the potential utility of mGlu2/3 antagonists, identification of compounds with ample selectivity between the two group II mGlus and the balance of pharmacology and drug metabolism and pharmacokinetics (DMPK) properties required for use in vivo has been elusive. Such tools are essential for further validation of the precise roles of each receptor in the etiology of disease.
Our first foray into this arena of research began by an observation of occasional weak mGlu3 NAM activity in a series of 1,2-diphenylethyne mGlu5 positive allosteric modulators (PAMs).38 An optimization plan for mGlu3 activity that centered on modification of the functional groups appended to the two phenyl rings within this scaffold delivered first-generation tool 7 (VU0463597, ML289)39 (Figure 2). Importantly, not only was 7 selective versus mGlu5 but selectivity versus mGlu2 was also notable (>15-fold). Further optimization within this chemotype identified a second-generation compound 8 (VU0469942, ML337)40 (Figure 2) that was devoid of both mGlu2 and mGlu5 activity. While 8 has proven quite useful as an in vitro tool compound and can be used in mice at high doses (100 mg/kg),41 lower CNS penetration and higher protein binding in rats prevent its utility in that species. Furthermore, 1,2-diarylethyne chemotypes similar to this one are prone to bioactivation at the alkyne moiety and subsequent formation of reactive metabolites that can lead to toxicity.42,43 Thus, the discovery of a superior mGlu3 NAM from outside the 1,2-diphenylethyne chemotype, with the balance of pharmacology and DMPK properties for convenient use in both rats and mice, remained a worthy goal and is the subject of this manuscript.
Figure 2.

mGlu3 NAMs from the 1,2-diphenylethyne chemotype.
RESULTS AND DISCUSSION
Lead Identification
The previous success in identification of an mGlu3 NAM lead with inherently good selectivity versus mGlu2 from a mGlu5 PAM chemotype39,40 prompted the mining of our internal collection of mGlu5 PAM chemotypes lacking an alkyne moiety. Utilization of this resource provided some examples of compounds with evidence of mGlu3 NAM activity amid the available associated cross-screening data versus the mGlu family.44,45 We selected many of these interesting compounds as well as additional closely related analogues to arrive at approximately 160 compounds for full concentration response curve (CRC) measurements in our cell-based functional assay for mGlu3. This fluorescence-based assay measures calcium mobilization induced by mGlu3 activation in a cell line stably expressing rat mGlu3 and the promiscuous G-protein Gα15 and is capable of detecting agonists, PAMs, and NAMs of mGlu3. We have developed similar assays and cell lines for rat mGlu2 and rat mGlu5, and both were used throughout this project for assessing selectivity.
One of the most interesting series to emerge from the full CRC mGlu3 screen outlined above is exemplified by compounds 9–11 (Figure 3). These compounds are from within a series of 5-aryl-6,7-dihydropyrazolo[1,5-a]pyrazine-4(5H)-ones that includes many potent mGlu5 PAMs.46 An interesting piece of SAR was noted with respect to the presence of a chiral methyl group at the seven position of the pyrazine-4(5H)-one ring in the (R)-configuration (10). Whereas the potency of (R)-methyl analogue 10 at mGlu5 was only 2-fold less than unsubstituted analogue 9, the efficacy of 10 (Glu Max = 26.5%) was much weaker than that observed with 9 (Glu Max = 93.0%). The (S)-methyl analogue 11 proved highly preferential for mGlu5 PAM activity and was only weakly active at mGlu3, inhibiting the glutamate response only at the top concentration tested (30 μM). Gratifyingly, compound 10 was also inactive up to the top concentration tested (30 μM) in our mGlu2 calcium mobilization assay. Given the potential benefits engendered by this (R)-methyl group, we incorporated this functional group into the design of future compounds.
Figure 3.

Representative compounds from a new mGlu3 lead series.
Development of SAR in the Western Region
Our initial plan centered on the development of structure–activity relationships (SAR) in the area occupied by the 2-pyridyl ether of 10, termed the western region of the scaffold. Intermediate primary alcohol 20 was envisioned as a valuable intermediate for late stage diversification of this area (Scheme 1). The synthesis of 20 began with commercially available phenoxyacetone 12. The sodium enolate of 12 was prepared and treated in situ with diethyl carbonate to afford 2,4-diketone intermediate 13. Reaction of 13 with hydrazine readily provided 14, which could be N-alkylated via a Mitsunobu reaction47 with commercially available chiral alcohol 15. This Mitsunobu reaction was carried out with microwave heating and resulted in inversion of stereochemistry as expected. Treatment of intermediate 16 with acid resulted in cleavage of the tert-butylcarbamate protecting group, and subsequent exposure to aqueous base provided the lactam 17. Chiral HPLC analysis of 17 showed 98.6% ee for this key intermediate. N-Arylation of 17 was accomplished by a copper mediated coupling48 with 4-fluorobromobenzene to yield 18. Treatment of 18 with boron tribromide cleaved the phenoxy ether, affording the corresponding primary bromide, which was then reacted with potassium acetate under mild heating to afford acetate ester 19. Hydrolysis of the acetate group was accomplished with aqueous lithium hydroxide to give the desired intermediate alcohol 20.
Scheme 1.

Synthesis of Primary Alcohol Intermediate 20a
Preliminary mGlu3 NAM SAR obtained from the testing of analogues of lead 10 that were included in the original set of 160 compounds indicated a preference for phenyl and 2-pyridyl ethers in the western portion of the scaffold. Late stage conversion of intermediate alcohol 20 to new analogues was accomplished through one of three methods (Scheme 2). For the synthesis of substituted phenyl ether analogues 22–39, alcohol 20 was converted to the corresponding mesylate, which was then reacted with the desired phenols 21 and cesium carbonate to afford 22–39. Alternatively, the phenols 21 and alcohol 20 were coupled directly via a Mitsunobu reaction.47 For synthesis of 2-pyridyl ether targets 41–51, the sodium alkoxide of 20 was prepared in situ and treated with substituted 2-fluoropyridines 40 to facilitate a SNAr reaction and provide 41–51.
Scheme 2.

Synthesis of New Western Ether Analogues 22–39 and 41–51a
The results obtained from preparing and testing unsubstituted phenyl ether intermediate 18 revealed a near 6-fold preference for mGlu5 vs mGlu3 activity, which ultimately proved a substantial hurdle with this subset of compounds (Table 1). To determine if substitution of the phenyl group could engender preference for mGlu3 activity, we systematically installed a variety of functional groups at all positions (22–34). Most substituents at the 2-position (22, 26, 29) only modestly affected mGlu3 NAM activity relative to 18; however, 2-methoxy analogue 32 was nearly 10-fold less potent than 18. The mGlu5 PAM activity was improved relative to 18 but remained suboptimal with these 2-substituted analogues. 2,4-Difluoro analogue 25 demonstrated modestly enhanced selectivity versus mGlu5 relative to its monosubstituted comparators 22 and 24. Although substitution of the 3-position with fluorine (23) provided little impact on activity at either receptor, installation of larger substituents (27, 30, 33) resulted in reductions in potency at both receptors and two instances of pharmacology mode switching at mGlu5 (27 and 30). Such “molecular switches” have been noted previously in other mGlu5 chemotypes.49,50 Substitution of the 4-position with fluorine (24) only minimally impacted potency at either receptor; larger groups (28, 31, 34) reduced potency at both receptors, although the effect on mGlu5 was more pronounced. Thus, desiring to further examine the effects of substitution at the 4-position, additional analogues were prepared (35–39). The trend toward mGlu3-preferring compounds continued with the 4-ethoxyphenyl analogue 38 and 4-trifluoromethoxy analogue 39, demonstrating no activity versus mGlu5 up to the highest concentration tested (30 μM).
Table 1.
mGlu3 NAM and mGlu5 SAR of Western Phenyl Ethers 18, 22–39

| ||||||||
|---|---|---|---|---|---|---|---|---|
| no. | R | mGlu3 pIC50 (± SEM)a | mGlu3 IC50 (nM)a | % Glu Max (± SEM)a,b | mGlu5 activityc | mGlu5 pEC50 (± SEM)c | mGlu5 EC50 (nM)c | % Glu Max (± SEM)b,c |
| 18 | H | 6.57 ± 0.15 | 267 | 2.21 ± 0.80 | PAM | 7.34 ± 0.02 | 46 | 87.7 ± 1.4 |
| 22 | 2-F | 6.28 ± 0.20 | 530 | 2.90 ± 1.05 | PAM | 6.71 ± 0.04 | 195 | 89.3 ± 3.0 |
| 23 | 3-F | 6.11 ± 0.03 | 773 | 1.19 ± 0.75 | PAM | 7.14 ± 0.02 | 73 | 86.1 ± 2.9 |
| 24 | 4-F | 6.34 ± 0.04 | 462 | 2.09 ± 0.57 | PAM | 7.03 ± 0.05 | 92 | 87.0 ± 1.0 |
| 25 | 2,4-di-F | 6.38 ± 0.22 | 417 | 1.70 ± 0.35 | PAM | 6.51 ± 0.05 | 307 | 82.3 ± 2.7 |
| 26 | 2-Me | 6.14 ± 0.14 | 721 | 1.56 ± 0.37 | PAM | 5.83 ± 0.04 | 1500 | 83.7 ± 2.5 |
| 27 | 3-Me | 5.84 ± 0.05 | 1440 | 2.35 ± 0.18 | NAMd | <5.0 | >10000 | 39.7 ± 7.7 |
| 28 | 4-Me | 5.92 ± 0.02 | 1190 | 1.72 ± 0.55 | PAM | 5.97 ± 0.04 | 1080 | 92.5 ± 2.6 |
| 29 | 2-Cl | 6.27 ± 0.18 | 532 | 1.63 ± 0.33 | PAM | 6.38 ± 0.01 | 419 | 84.4 ± 2.1 |
| 30 | 3-Cl | 5.79 ± 0.02 | 1620 | 1.96 ± 0.11 | NAMd | <5.0 | >10000 | 32.8 ± 6.1 |
| 31 | 4-Cl | 5.93 ± 0.01 | 1170 | 1.86 ± 0.52 | PAM | 6.22 ± 0.04 | 604 | 81.8 ± 0.3 |
| 32 | 2-OMe | 5.59 ± 0.18 | 2540 | 2.32 ± 1.39 | PAMd | <5.0 | >10000 | 63.7 ± 3.1 |
| 33 | 3-OMe | 5.89 ± 0.02 | 1290 | 1.78 ± 0.53 | PAM | 5.74 ± 0.04 | 1840 | 32.4 ± 3.0 |
| 34 | 4-OMe | 5.98 ± 0.02 | 1040 | 1.76 ± 0.39 | PAM | 5.87 ± 0.16 | 1340 | 40.2 ± 4.8 |
| 35 | 4-CF3 | 5.73 ± 0.01 | 1850 | 4.13 ± 0.09 | <4.5 | >30000 | ||
| 36 | 4-Et | 6.08 ± 0.05 | 830 | 2.29 ± 0.84 | PAM | 5.49 ± 0.03 | 3210 | 50.8 ± 3.7 |
| 37 | 4-CN | 6.11 ± 0.12 | 784 | 2.53 ± 0.70 | PAM | 5.71 ± 0.06 | 1940 | 38.6 ± 4.8 |
| 38 | 4-OEt | 5.94 ± 0.06 | 1160 | 3.39 ± 0.87 | <4.5 | >30000 | ||
| 39 | 4-OCF3 | 5.39 ± 0.04 | 4070 | 2.66 ± 0.34 | <4.5 | >30000 | ||
Calcium mobilization mGlu3 assay; values are average of n ≥ 3.
Amplitude of response in the presence of 30 μM test compound as a percentage of maximal response (100 μM glutamate); average of n ≥ 3.
Calcium mobilization mGlu5 assay; values are average of n ≥ 3.
Weak activity; concentration response curve (CRC) does not plateau.
Given the promising profile of initial lead 10, our hope for identifying more attractive compounds within the 2-pyridyl analogues (41–51) remained high; however, this region proved relatively intolerant of substitution (Table 2). Although a 3-fluoro substituent (41) only modestly impacted potency, larger substituents (42, 43) had more deleterious effects. Similarly modest results were observed with 4-position (44–46) and 6-position (49–51) analogues. Still, it was encouraging to identify additional compounds (42, 43, and 50) that displayed no activity versus mGlu5 up to the highest concentration tested (30 μM) as well as several analogues with only weak activity at mGlu5. Fortunately, 5-halo analogues 47 and 48 proved an exception to the general trend of modest mGlu3 potency with these analogues. Furthermore, 5-fluoro analogue 47 demonstrated enhanced selectivity approaching 20-fold versus mGlu5, while 5-chloro analogue 48 was more modest with regard to selectivity at approximately 5-fold.
Table 2.
mGlu3 NAM and mGlu5 SAR of Western 2-Pyridyl Ethers 41–51

| ||||||||
|---|---|---|---|---|---|---|---|---|
| no. | R | mGlu3 pIC50 (± SEM)a | mGlu3 IC50 (nM)a | % Glu Max (± SEM)a,b | mGlu5 activityc | mGlu5 pEC50 (± SEM)c | mGlu5 EC50 (nM)c | % Glu Max (± SEM)b,c |
| 41 | 3-F | 5.96 ± 0.02 | 1100 | 2.22 ± 0.84 | PAM | 5.37 ± 0.06 | 4300 | 78.5 ± 6.1 |
| 42 | 3-CF3 | 5.53 ± 0.02 | 2940 | 2.11 ± 0.96 | <4.5 | >30000 | ||
| 43 | 3-OMe | 5.17 ± 0.02 | 6760 | –1.14 ± 2.54 | <4.5 | >30000 | ||
| 44 | 4-Me | 5.68 ± 0.01 | 2090 | 0.86 ± 0.47 | NAMd | <5.0 | >10000 | 46.9 ± 5.9 |
| 45 | 4-CF3 | 5.78 ± 0.01 | 1670 | 1.62 ± 0.42 | NAMd | <5.0 | >10000 | 12.9 ± 2.9 |
| 46 | 4-OMe | 5.43 ± 0.00 | 3690 | –0.18 ± 1.39 | NAMd | <5.0 | >10000 | 54.7 ± 5.8 |
| 47 | 5-F | 6.27 ± 0.02 | 539 | 1.71 ± 0.55 | NAMd | <5.0 | >10000 | 46.5 ± 3.4 |
| 48 | 5-Cl | 6.22 ± 0.04 | 605 | 1.84 ± 0.51 | PAM | 5.53 ± 0.06 | 2920 | 81.4 ± 2.7 |
| 49 | 6-Me | 5.62 ± 0.02 | 2370 | 1.12 ± 1.08 | NAM | 5.65 ± 0.07 | 2250 | 2.47 ± 0.17 |
| 50 | 6-CF3 | 5.63 ± 0.02 | 2360 | 2.06 ± 0.79 | <4.5 | >30000 | ||
| 51 | 6-OMe | 5.80 ± 0.02 | 1590 | 1.94 ± 0.62 | PAMd | <5.0 | >10000 | 31.2 ± 3.1 |
Calcium mobilization mGlu3 assay; values are average of n ≥ 3.
Amplitude of response in the presence of 30 μM test compound as a percentage of maximal response (100 μM glutamate); average of n ≥ 3.
Calcium mobilization mGlu5 assay; values are average of n ≥ 3.
Weak activity; CRC does not plateau.
With some initial SAR in hand, we further profiled some of the more promising early analogues (38, 47, 48) to assess other properties important for the development of a useful in vivo tool compound (Table 3). In addition to assessing the degree to which the compounds were bound to rat plasma,51 the compounds were evaluated in a rat cassette pharmacokinetics (PK) study using intravenous (IV) dosing52 to assess their metabolic stability in vivo. In spite of its poor selectivity profile versus mGlu5, 4-fluorophenyl ether 24 was also included with these compounds as a comparator compound. In fact, analogue 24 exhibited moderate clearance, with a half-life of over 1.5 h. Not surprisingly, replacement of the fluorine atom (24) with the more metabolically labile ethoxy group (38) increased clearance to approximately hepatic blood flow and reduced half-life. The results obtained with the 2-pyridyl analogues 47 and 48 revealed a profound metabolic difference. Whereas 5-fluoro analogue 47 was rapidly cleared, 5-chloro analogue 48 had a moderate clearance and a half-life of approximately 2 h. As indicated by their ligand-lipophilicity efficiency (LLE) values,53 the 2-pyridyl ethers possessed a better balance of potency and lipophilicity than the phenyl ether compounds. Notably, 4-ethoxyphenyl ether 38 also exhibited exceedingly high protein binding to rat plasma. These factors led to the conclusion that optimization should continue in the context of the 2-pyridyl ethers. Superior LLE, selectivity versus mGlu5, and plasma unbound fraction relative to 48, made 5-fluoro analogue 47 an attractive starting point for further optimization; however, its high plasma clearance remained an issue. To avoid focusing future optimization efforts exclusively in the context of a potential PK liability, the decision was made to prepare analogues of both 47 and 48 in parallel.
Table 3.
DMPK Profiling of Early Analogues

| ||||||||
|---|---|---|---|---|---|---|---|---|
| no. | cLogPa | LLEb | mGlu3 IC50 (nM) | fold vs mGlu5 | rat plasma Fuc | CLplasma (mL/min/kg)d | VSS (L/kg)d | |
| 24 | 3.96 | 2.38 | 462 | 0.20 | 0.060 | 95 | 40 | 3.5 |
| 38 | 4.24 | 1.70 | 1160 | >25 | 0.005 | 32 | 82 | 2.2 |
| 47 | 3.06 | 3.21 | 539 | >18 | 0.135 | 88 | 82 | 6.6 |
| 48 | 3.57 | 2.65 | 605 | 4.8 | 0.034 | 141 | 29 | 5.2 |
Calculated using Dotmatics Elemental (www.dotmatics.com/products/elemental/).
LLE (ligand-lipophilicity efficiency) = pIC50 – cLogP.
Fu = fraction unbound.
Rat IV PK results (n = 2); dose = 0.2 mg/kg; solution in 9% EtOH, 37% PEG 400, 54% DMSO (2 mg/mL).
Development of SAR in the Eastern Region
To enable final step diversification and the rapid synthesis of new analogues, modified syntheses were required for development of SAR in the eastern portion of the chemotype. Synthesis of new 5-fluoropyridin-2-yl ether analogues 55–72 was accomplished via a reordering of the previously outlined reactions (Scheme 3). Intermediate 17 was treated with boron tribromide and subsequently potassium acetate to afford acetate ester 52. Hydrolysis of 52 was accomplished with lithium hydroxide; however, the yield of alcohol 53 suffered due to difficulty in isolation of this polar and water-soluble intermediate. Still, preparation of the 5-fluoropyridin-2-yl ether 54 via SNAr chemistry was readily accomplished, which enabled a final stage installation of the aryl or heteroaryl eastern ring according to methods described previously to yield the desired products 55–72.
Scheme 3.

Synthesis of 5-Fluoropyridin-2-yl Ether Analogues 55–72a
Because of the poor yield encountered in the synthesis of intermediate 53, we employed what proved to be a more scalable and shorter synthesis in the preparation of the 5-chloropyridin-2-yl ether analogues (Scheme 4). Synthesis of intermediate 75 from commercially available 73 was accomplished via an analogous two-step Mitsunobu coupling47 and deprotection/cyclization sequence as described previously. The lactam nitrogen was protected with a 4-methoxybenzyl group to afford 76. Reduction of the ethyl ester was accomplished with sodium borohydride to yield 77. Formation of the 5-chloropyridin-2-yl ether 78 was carried out via SNAr chemistry as before. Oxidative removal of the 4-methoxybenzyl protecting group was carried out with ceric ammonium nitrate to provide penultimate intermediate 79. Conversion of 79 into analogues 80–110 utilized the copper mediate N-arylation methods employed previously.
Scheme 4.

Synthesis of 5-Chloropyridin-2-yl Ether Analogues 80–110a
Testing of new 5-fluoropyridin-2-yl ether analogues 55–72 showed eastern ring modification was a useful strategy for enhancing mGlu3 NAM activity (Table 4). In the case of monosubstituted phenyl ethers (55–65), several analogues exhibited mGlu3 IC50 values less than 200 nM. Specifically, at the 2-position, fluorine (55) and chlorine (57) substitution was preferred to methyl (60) and methoxy (63) substituents, while little difference in mGlu3 activity was observed with variation of the same substituents at the 3-position (56, 58, 61, 64). Substitution at the 4-position (59, 62, 65) was slightly less favorable. Encouragingly, several of these analogues also demonstrated only weak activity at mGlu5. Some difluorophenyl analogues (66–68) were also prepared and found to exhibit good potency. Simple pyridyl analogues (69–71) were less potent versus mGlu3 than the majority of the phenyl analogues; however, selectivity versus mGlu5 was notable in the case of 70 and 71. Finally, simple fluorine substitution of the pyridine ring (72) enhanced mGlu3 activity relative to unsubstituted comparator 69.
Table 4.
mGlu3 NAM and mGlu5 SAR of 5-Fluoropyridin-2-yl Ether Analogues 55–72

| ||||||||
|---|---|---|---|---|---|---|---|---|
| no. | R | mGlu3 pIC50 (± SEM)a | mGlu3 IC50 (nM)a | % Glu Max (± SEM)a,b | mGlu5 activityc | mGlu5 pEC50 (± SEM)c | mGlu5 EC50 (nM)c | % Glu Max (± SEM)b,c |
| 55 | 2-fluorophenyl | 6.79 ± 0.17 | 162 | 1.53 ± 0.12 | NAMf | <5.0 | >10000 | 29.1 ± 7.3 |
| 56 | 3-fluorophenyl | 6.72 ± 0.08 | 192 | 1.44 ± 0.38 | NAM | 5.56 ± 0.24 | 2720 | 9.71 ± 5.78 |
| 57 | 2-chlorophenyl | 6.85 ± 0.16 | 141 | 1.37 ± 0.72 | NAM | 6.00 ± 0.25 | 1010 | 8.78 ± 5.29 |
| 58 | 3-chlorophenyl | 6.84 ± 0.10 | 145 | 2.30 ± 0.17 | NAMe | 5.73 ± 0.24 | 1850 | 19.2 ± 10.0 |
| 59 | 4-chlorophenyl | 6.71 ± 0.13 | 197 | 2.17 ± 0.40 | PAM | 6.44 ± 0.03 | 364 | 51.6 ± 7.7 |
| 60 | 2-methylphenyl | 6.46 ± 0.06 | 346 | 1.72 ± 0.16 | NAMf | <5.0d | >10000d | 35.8d |
| 61 | 3-methylphenyl | 6.67 ± 0.15 | 216 | 1.93 ± 0.56 | NAMf | <5.0 | >10000 | 36.6 ± 8.7 |
| 62 | 4-methylphenyl | 6.57 ± 0.03 | 269 | 1.04 ± 0.45 | NAMf | <5.0 | >10000 | 54.8 ± 9.4 |
| 63 | 2-methoxyphenyl | 6.47 ± 0.10 | 339 | 1.49 ± 0.60 | NAM | 6.52 ± 0.17 | 301 | 3.28 ± 0.45 |
| 64 | 3-methoxyphenyl | 6.70 ± 0.06 | 198 | 1.02 ± 0.12 | NAMf | <5.0 | >10000 | 59.7 ± 6.1 |
| 65 | 4-methoxyphenyl | 6.49 ± 0.10 | 320 | 1.23 ± 0.38 | PAM | 5.86 ± 0.05 | 1380 | 32.3 ± 5.2 |
| 66 | 2,3-difluorophenyl | 6.73 ± 0.16 | 184 | 2.15 ± 0.33 | NAMe | 5.68 ± 0.20 | 2110 | 21.0 ± 10.6 |
| 67 | 2,5-difluorophenyl | 6.91 ± 0.11 | 123 | 2.62 ± 0.47 | NAMf | <5.0 | >10000 | 18.8 ± 6.1 |
| 68 | 2,6-difluorophenyl | 6.74 ± 0.08 | 181 | 1.49 ± 0.17 | NAMe | 5.91d | 1230d | 50.8d |
| 69 | pyridin-2-yl | 6.04 ± 0.05 | 915 | 0.96 ± 0.63 | NAM | 5.45 ± 0.04 | 3520 | 3.71 ± 0.47 |
| 70 | pyridin-3-yl | 6.36 ± 0.13 | 436 | 0.64 ± 0.70 | NAMf | <5.0 | >10000 | 46.3 ± 2.9 |
| 71 | pyridin-4-yl | 6.05 ± 0.04 | 881 | 0.77 ± 0.44 | <4.5 | >30000 | ||
| 72 | 5-fluoropyridin-2-yl | 6.46 ± 0.07 | 349 | 1.31 ± 0.11 | NAM | 5.01 ± 0.13 | 9740 | 7.0 ± 2.5 |
Calcium mobilization mGlu3 assay; values are average of n ≥ 3.
Amplitude of response in the presence of 30 μM test compound as a percentage of maximal response (100 μM glutamate); average of n ≥ 3.
Calcium mobilization mGlu5 assay; values are average of n ≥ 3.
Average of n = 2.
Partial NAM; CRC plateaus above 10% glutamate maximum.
Weak activity; CRC does not plateau.
Although several of these new 5-fluoropyridin-2-yl ether analogues demonstrated improved mGlu3 NAM potency, modest levels of mGlu5 selectivity, and improved LLE values53 (Table 5), their DMPK profiles remained a critical unanswered question. Thus, selected analogues were profiled in our aforementioned rat protein binding assay,51 and metabolic stability was also assessed in vitro by measuring the intrinsic clearance of the compound when incubated with rat liver microsomes (RLM).54 While the fraction unbound in rat plasma was encouraging for most compounds, metabolic stability was uniformly poor. Unfortunately, on the basis of their intrinsic clearance in RLM, the compounds were predicted to exhibit hepatic clearance near blood flow.55
Table 5.
In Vitro DMPK Profiling of Select 5-Fluoropyridin-2-yl Ether Analogues

| |||||||
|---|---|---|---|---|---|---|---|
| no. | R | cLogPa | LLEb | mGlu3 IC50 (nM) | fold vs mGlu5 | rat plasma Fuc | rat CLhep (mL/min/kg)d |
| 55 | 2-fluorophenyl | 3.06 | 3.73 | 162 | >61 | 0.098 | 64.7 |
| 56 | 3-fluorophenyl | 3.06 | 3.66 | 192 | 14 | 0.094 | 53.0 |
| 57 | 2-chlorophenyl | 3.57 | 3.28 | 141 | 7.2 | 0.054 | 66.5 |
| 58 | 3-chlorophenyl | 3.57 | 3.27 | 145 | 13 | 0.034 | 51.9 |
| 61 | 3-methylphenyl | 3.23 | 3.44 | 216 | 17 | 0.051 | 58.4 |
| 66 | 2,3-difluorophenyl | 3.16 | 3.57 | 184 | 11 | 0.092 | 63.6 |
| 67 | 2,5-difluorophenyl | 3.16 | 3.75 | 123 | >81 | 0.089 | 49.6 |
| 68 | 2,6-difluorophenyl | 3.16 | 3.58 | 181 | 6.8 | 0.064 | 64.6 |
Calculated using Dotmatics Elemental (www.dotmatics.com/products/elemental/).
LLE (ligand-lipophilicity efficiency) = pIC50 – cLogP.
Fu = fraction unbound.
Predicted hepatic clearance based on intrinsic clearance in rat liver microsomes.
Fortunately, testing of new 5-chloropyridin-2-yl ether analogues 80–101 ultimately provided another path forward for the design of both potent and highly selective mGlu3 NAMs (Table 6). Monosubstituted phenyl analogues (80–93) generally exhibited similar activity at mGlu3 (IC50 = 200–600 nM) regardless of the position of the substituent on the ring. 4-Chlorophenyl analogue 84, 2-methoxyphenyl analogue 88, and 3-cyanophenyl analogue 92 were exceptions to this trend with each exhibiting reduced activity at mGlu3. Although most of these monosubstituted phenyl analogues (80–93) demonstrated only modest selectivity versus mGlu5, 4-methylphenyl analogue 87, 2-methoxyphenyl analogue 88, and 3-cyanophenyl analogue 92 exhibited weak activity at that receptor. Preparation of disubstituted phenyl analogues (94–99) yielded additional compounds with improved selectivity versus mGlu5. 3,5-Difluorophenyl analogue 97 was a weak mGlu5 PAM, and 2-cyano-5-fluorophenyl analogue 98 was inactive versus mGlu5 up to the highest concentration tested (30 μM). Finally, encouraging results were observed with unsubstituted pyridyl analogues 100 and 101, where both proved inactive versus mGlu5 while maintaining good mGlu3 NAM activity.
Table 6.
mGlu3 NAM and mGlu5 SAR of 5-Chloropyridin-2-yl Ether Analogues 80–101

| ||||||||
|---|---|---|---|---|---|---|---|---|
| no. | R | mGlu3 pIC50 (± SEM)a | mGlu3 IC50 (nM)a | % Glu Max (± SEM)a,b | mGlu5 activityc | mGlu5 pEC50 (± SEM)c | mGlu5 EC50 (nM)c | % Glu Max (± SEM)b,c |
| 80 | 2-fluorophenyl | 6.65 ± 0.04 | 226 | 1.74 ± 0.10 | PAM | 5.49 ± 0.06 | 3240 | 57.3 ± 4.6 |
| 81 | 3-fluorophenyl | 6.69 ± 0.04 | 206 | 1.38 ± 0.47 | PAM | 5.22 ± 0.05 | 5970 | 63.5 ± 0.8 |
| 82 | 2-chlorophenyl | 6.48 ± 0.08 | 328 | 1.87 ± 0.29 | PAM | 5.26 ± 0.03 | 5450 | 51.0 ± 3.9 |
| 83 | 3-chlorophenyl | 6.25 ± 0.07 | 558 | 1.63 ± 0.20 | PAM | 5.54 ± 0.02 | 2870 | 65.8 ± 8.8 |
| 84 | 4-chlorophenyl | 5.91 ± 0.25 | 1230 | 0.94 ± 1.46 | PAM | 5.48 ± 0.15 | 3310 | 51.2 ± 10.3 |
| 85 | 2-methylphenyl | 6.24 ± 0.18 | 571 | 1.95 ± 0.70 | PAM | 5.64 ± 0.03 | 2280 | 80.7 ± 0.5 |
| 86 | 3-methylphenyl | 6.41 ± 0.05 | 392 | 1.71 ± 0.44 | PAM | 5.44 ± 0.11 | 3600 | 69.9 ± 6.5 |
| 87 | 4-methylphenyl | 6.39 ± 0.21 | 410 | 1.39 ± 0.25 | PAMe | <5.0 | >10000 | 70.8 ± 3.1 |
| 88 | 2-methoxyphenyl | 6.16 ± 0.15 | 690 | 1.29 ± 0.18 | NAMe | <5.0 | >10000 | 58.5 ± 1.2 |
| 89 | 3-methoxyphenyl | 6.50 ± 0.05 | 313 | 2.00 ± 0.32 | PAM | 5.29 ± 0.08 | 5070 | 78.0 ± 1.8 |
| 90 | 4-methoxyphenyl | 6.36 ± 0.11 | 439 | 2.07 ± 0.68 | PAM | 5.36 ± 0.06 | 4330 | 59.4 ± 7.3 |
| 91 | 2-cyanophenyl | 6.37 ± 0.05 | 429 | 1.64 ± 0.04 | PAM | 5.30 ± 0.10 | 4970 | 38.1 ± 5.0 |
| 92 | 3-cyanophenyl | 6.08 ± 0.26 | 825 | 1.75 ± 0.41 | PAMe | <5.0 | >10000 | 32.2 ± 3.7 |
| 93 | 4-cyanophenyl | 6.27 ± 0.05 | 539 | 1.84 ± 0.19 | PAM | 5.48 ± 0.08 | 3320 | 23.1 ± 6.8 |
| 94 | 2,3-difluorophenyl | 6.07 ± 0.23 | 852 | 0.33 ± 0.71 | PAM | 5.17 ± 0.29 | 6680 | 36.1 ± 8.4 |
| 95 | 2,4-difluorophenyl | 6.55 ± 0.08 | 280 | 1.34 ± 0.25 | PAM | 5.59 ± 0.07 | 2580 | 70.8 ± 0.8 |
| 96 | 2,6-difluorophenyl | 6.65 ± 0.04 | 225 | 2.06 ± 0.31 | PAM | 5.85 ± 0.09 | 1420 | 40.3 ± 5.6 |
| 97 | 3,5-difluorophenyl | 6.38 ± 0.07 | 420 | 2.18 ± 0.19 | PAMe | <5.0 | >10000 | 33.1 ± 2.3 |
| 98 | 2-cyano-5-fluorophenyl | 6.28 ± 0.06 | 529 | 1.55 ± 0.31 | <4.5 | >30000 | ||
| 99 | 3-cyano-5-fluorophenyl | 6.50 ± 0.07 | 315 | 1.92 ± 0.12 | PAM | 5.98d | 1044d | 28.0d |
| 100 | pyridin-2-yl | 6.48 ± 0.07 | 328 | 1.07 ± 0.04 | <4.5 | >30000 | ||
| 101 | pyridin-3-yl | 6.22 ± 0.10 | 608 | 1.73 ± 0.53 | <4.5 | >30000 | ||
Calcium mobilization mGlu3 assay; values are average of n ≥ 3.
Amplitude of response in the presence of 30 μM test compound as a percentage of maximal response (100 μM glutamate); average of n ≥ 3.
Calcium mobilization mGlu5 assay; values are average of n ≥ 3.
Average of n = 2.
Weak activity; CRC does not plateau.
Encouraged by the selectivity profiles seen with pyridyl analogues 100 and 101 but suspecting that these unsubstituted compounds may be prone to rapid metabolism, we immediately prepared several substituted analogues of each (Table 7). Unfortunately, in the pyridin-2-yl set (102–105), the majority of these modifications enhanced mGlu5 activity, albeit only slightly. Cyano analogue 103 was the lone exception; however, this compound was approximately 2-fold less potent than unsubstituted analogue 100. Results with the pyridin-3-yl set (106–110) were more encouraging as all new compounds except 6-fluoro analogue 110 maintained the excellent selectivity profile versus mGlu5 observed with 101 without a loss of activity at mGlu3. At this point, several compounds with good mGlu3 NAM activity and devoid of mGlu5 activity were in hand, which set the stage for further profiling in pursuit of compounds meriting extensive in vivo evaluation.
Table 7.
mGlu3 NAM and mGlu5 SAR of Additional 5-Chloropyridin-2-yl Ether Analogues 102–110

| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| no. | series | R | mGlu3 pIC50 (± SEM)a | mGlu3 IC50 (nM)a | % Glu Max (± SEM)a,b | mGlu5 activityc | mGlu5 pEC50 (± SEM)c | mGlu5 EC50 (nM)c | % Glu Max (± SEM)b,c |
| 102 | I | 3-F | 6.44 ± 0.03 | 362 | 1.64 ± 0.36 | PAMd | <5.0 | >10000 | 32.4 ± 4.4 |
| 103 | I | 3-CN | 6.17 ± 0.06 | 677 | 1.35 ± 0.55 | <4.5 | >30000 | ||
| 104 | I | 5-F | 6.59 ± 0.09 | 260 | 2.44 ± 0.32 | PAMd | <5.0 | >10000 | 47.9 ± 5.1 |
| 105 | I | 6-F | 6.53 ± 0.08 | 294 | 1.75 ± 0.19 | NAMd | <5.0 | >10000 | 24.8 ± 5.5 |
| 106 | II | 2-F | 6.41 ± 0.05 | 392 | 1.71 ± 0.41 | <4.5 | >30000 | ||
| 107 | II | 4-F | 6.32 ± 0.03 | 482 | 1.45 ± 0.22 | <4.5 | >30000 | ||
| 108 | II | 5-F | 6.32 ± 0.03 | 481 | 1.85 ± 0.32 | <4.5 | >30000 | ||
| 109 | II | 5-CN | 6.22 ± 0.01 | 605 | 1.08 ± 0.50 | <4.5 | >30000 | ||
| 110 | II | 6-F | 6.39 ± 0.06 | 408 | 1.45 ± 0.29 | PAMd | <5.0 | >10000 | 30.6 ± 2.4 |
Calcium mobilization mGlu3 assay; values are average of n ≥ 3.
Amplitude of response in the presence of 30 μM test compound as a percentage of maximal response (100 μM glutamate); average of n ≥ 3.
Calcium mobilization mGlu5 assay; values are average of n ≥ 3.
Weak activity; CRC does not plateau.
As before, we moved several promising compounds into assays to assess protein binding51 in rat plasma as well as metabolic stability in RLM54 (Table 8). The lone eastern phenyl analogue 99 was slightly more protein bound than its eastern pyridyl comparators, which was not surprising given its higher lipophilicity. A range of predicted hepatic clearance values based on the intrinsic clearance of the compound in RLM were observed including one analogue of less than one-third hepatic blood flow (109) and one analogue near hepatic blood flow (100). The remaining analogues were predicted to have moderate clearance in vivo. Somewhat surprisingly, unsubstituted pyridine-3-yl analogue 101 was more metabolically stable than its regioisomeric comparator 100. Still, substituted versions of 101 (106–109) did exhibit increased stability relative to 101.
Table 8.
In Vitro DMPK Profiling of Select 5-Chloropyridin-2-yl Ether Analogues

| |||||||
|---|---|---|---|---|---|---|---|
| no. | R | cLogPa | LLEb | mGlu3 IC50 (nM) | fold vs mGlu5 | rat plasma Fuc | rat CLhep (mL/min/kg)d |
| 99 | 2-cyano-5-fluorophenyl | 3.29 | 2.99 | 529 | >56 | 0.045 | 47.7 |
| 100 | pyridin-2-yl | 2.57 | 3.91 | 328 | >91 | 0.051 | 69.5 |
| 101 | pyridin-3-yl | 2.16 | 4.06 | 608 | >49 | 0.118 | 42.0 |
| 106 | 2-fluoropyridin-3-yl | 2.67 | 3.74 | 392 | >76 | 0.083 | 36.9 |
| 107 | 4-fluoropyridin-3-yl | 2.26 | 4.06 | 482 | >62 | 0.085 | 36.0 |
| 108 | 5-fluoropyridin-3-yl | 2.26 | 4.06 | 481 | >62 | 0.092 | 26.6 |
| 109 | 5-cyanopyridin-3-yl | 1.88 | 4.34 | 605 | >49 | 0.078 | 22.8 |
Calculated using Dotmatics Elemental (www.dotmatics.com/products/elemental/).
LLE (ligand-lipophilicity efficiency) = pIC50 – cLogP.
Fu = fraction unbound.
Predicted hepatic clearance based on intrinsic clearance in rat liver microsomes.
Several interesting analogues were next advanced into rat cassette PK studies using IV dosing52 to assess their metabolic stability in vivo (Table 9). As the most promising analogue with an eastern phenyl group, 99 was selected for these studies. Unsubstituted eastern pyridyl analogue 101 and several substituted comparators (106, 108, and 109) were also chosen based on the totality of data collected to that point. Distinguishing between analogues 108 and 107 was difficult; however, compound 108 was ultimately selected as it had a marginally better in vitro DMPK profile. 5-Cyanopyridin-3-yl analogue 109 was selected as it was predicted to have the lowest clearance, and 2-fluoropyridin-3-yl analogue 106 was chosen as it was the most potent analogue with a moderate predicted clearance. Compound 99 exhibited a lower clearance in vivo than expected and had a long half-life in excess of 3 h. The in vivo clearance for the pyridine-3-yl analogues was generally well predicted by the RLM experiments, with analogue 108 being the lone exception. Analogues 99, 106, and 109 were thus selected for further study in single time point (15 min) tissue distribution studies at a higher dose (10 mg/kg).56 Intraperitoneal (IP) dosing was chosen for these studies as this route is convenient for use in our planned behavioral studies. The same three compounds were also examined in protein binding assays with rat brain homogenates.51 CNS penetration with each compound was excellent, with compound 106 exhibiting the highest plasma and brain levels. Both 99 and 106 exhibited unbound brain to unbound plasma ratios (Kp,uu) of one, indicating distribution equilibrium between the compartments and a low probability of the compounds being substrates for transporters.57 Compound 109 had a Kp,uu value of 0.49, indicating possible efflux; however, additional experiments would be required to determine such conclusively. Compound 106 (VU0650786) was deemed the most attractive and targeted for extensive profiling.
Table 9.
In Vivo DMPK Profiling of Select 5-Chloropyridin-2-yl Ether Analogues

| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| rat IV PK resultsb |
rat IP tissue distribution resultsc,d,e |
||||||||
| no. | rat plasma Fua | rat brain Fua | t1/2 (min) | CLplasma (mL/min/kg) | VSS (L/kg) | plasma conc (μM) | brain conc (μM) | K p f | K p,uu g |
| 99 | 0.045 | 0.032 | 194 | 13 | 2.2 | 1.68 | 2.41 | 1.4 | 1.0 |
| 101 | 0.118 | 21 | 50 | 1.2 | |||||
| 106 | 0.083 | 0.052 | 42 | 37 | 1.6 | 9.49 | 15.85 | 1.7 | 1.0 |
| 108 | 0.092 | 30 | 50 | 1.9 | |||||
| 109 | 0.078 | 0.033 | 59 | 22 | 1.4 | 4.52 | 5.28 | 1.2 | 0.49 |
Fu = fraction unbound.
n = 2; dose = 0.2 mg/kg; solution in 10% EtOH, 38–40% PEG 400, 50–52% DMSO (2 mg/mL).
n = 2; time point = 15 min; dose = 10 mg/kg.
For 99 and 109, fine homogeneous suspension in 0.1% Tween 80 and 0.5% methyl cellulose in H2O (4 mg/mL).
For 106, fine homogeneous suspension in 10% EtOH and 90% PEG400 (4 mg/mL).
Kp = total brain to total plasma ratio.
Kp,uu = unbound brain (brain Fu × total brain) to unbound plasma (plasma Fu × total plasma) ratio
Profiling of Compound 106
Profiling of compound 106 began with determination of its full selectivity versus other members of the mGlu family. In addition to mGlu5, selectivity versus fellow group II receptor subtype, mGlu2, was critical to assess. We evaluated the selectivity of 106 versus rat mGlu2 using full CRC analysis, and the compound was inactive up to the highest concentration tested (30 μM). Thus, compound 106 has been established to have no functional activity at either mGlu2 or mGlu5 up to a concentration that is more than 75-fold over the functional potency at mGlu3. The effect of 10 μM 106 on the orthosteric agonist CRC was measured in fold-shift experiments to evaluate selectivity versus the other mGlus.44,45 No significant effect was found, indicating the compound was inactive at those receptors as well. We further evaluated the nature of the interaction between 106 and mGlu3 by examining the effects of increasing concentrations of 106 on the glutamate CRC in a progressive fold-shift experiment. If an antagonist acts via a noncompetitive mechanism, we anticipate increasing concentrations of antagonist would shift the glutamate curve to the right and decrease the maximal signal of glutamate. Figure 4 depicts the effects of multiple concentrations of 106 and known mGlu2/3 NAM 111 (MNI-137)58 on the glutamate CRC. As expected, both compounds exhibited the characteristic rightward shift and depressed glutamate maximum typically observed with NAMs, suggesting that 106 does not bind to the orthosteric glutamate binding site but instead acts via an allosteric mechanism.
Figure 4.

Progressive fold-shift of the glutamate CRC by mGlu3 NAM 106 (A) and mGlu2/3 NAM 111 (B); compound concentrations shown in M.
To evaluate the ancillary pharmacology of the compound, a commercially available radioligand binding assay panel of 68 clinically relevant GPCRs, ion channels, kinases, and transporters was employed,59 and only a single significant response (5-HT2B, 65% inhibition) was found at 10 μM 106.60 Because 5-hydroxytryptamine receptor 2B (5-HT2B) agonists are associated with cardiotoxicity,61 we followed this result up with a functional cell-based assay to assess potential agonist or antagonist activity of the compound at 5-HT2B.62 Fortunately, testing up to 10 μM in this assay revealed no functional activity for 106 at 5-HT2B. Potential for drug–drug interactions was negligible as assessed in a human liver microsomes (HLM) cocktail assay with probe substrates for four common P450s (Table 10).63 Finally, permeability and potential for P-glycoprotein (P-gp) mediated efflux was assessed in Madin–Darby canine kidney (MDCK) cells transfected with the human MDR1 gene.64 Not surprisingly, compound 106 was highly permeable with no evidence of efflux in this assay.
Table 10.
P450 Inhibition and Permeability Profile of Compound 106

| |||
|---|---|---|---|
| P450 inhibitiona |
permeabilityb |
||
| CYP1A2 IC50 | >30 μM | A–B Papp | 45.0 × 10–6 cm/s |
| CYP2C9 IC50 | 25.6 μM | B–A Papp | 49.9 × 10–6 cm/s |
| CYP2D6 IC50 | >30 μM | efflux ratio | 1.1 |
| CYP3A4 IC50 | >30 μM | ||
Cocktail assay in HLM.
MDR1-MDCK cells.
Confident that 106 possessed a favorable profile with respect to its pharmacology, in vitro DMPK properties, and preliminary in vivo DMPK properties, we moved the compound into several definitive in vivo DMPK studies in rats and mice (Table 11).56 Time course studies using IP dosing revealed nearly identical and rapidly reached Cmax values in both rats and mice. A single time point (30 min) tissue distribution study in mice analogous to the one previously conducted in rats showed excellent CNS penetration in that species as well. Again, the Kp,uu value was near one as would be expected for a highly permeable compound devoid of efflux issues.57 A definitive rat IV PK study (1.0 mg/kg) was conducted, and results were essentially identical to those collected with the prior cassette study (0.2 mg/kg). Finally, a rat oral PK study (3.0 mg/kg) was carried out to assess bioavailability, which proved quite good (60%). Taking into account the data summarized herein, it was estimated that the unbound Cmax in the CNS following the 10 mg/kg IP studies in both rats and mice was at or beyond the measured functional mGlu3 NAM activity. With that in mind, evaluation of compound 106 in rodent behavioral models known to be sensitive to mGlu2/3 antagonists was initiated.
Table 11.
Rodent PK Profile of Compound 106

| ||||
|---|---|---|---|---|
| Protein Binding (Fu)a | Rat IP PKb | |||
|
rat plasma | 0.083 | dose | 10 mg/kg |
| rat brain homogenates | 0.052 | plasma Cmax | 4.57 ± 0.67 μM | |
| mouse plasma | 0.163 | plasma Tmax | 12.3 ± 2.7 minutes | |
| mouse brain homogenates | 0.035 | plasma AUC0-∞ | 11.5 ± 0.4 μM·h | |
| Rat IVc and POd PK | Mouse IP Tissue Distributione | Mouse IP PKh | |||||
|---|---|---|---|---|---|---|---|
| t1/2 | 49 minutes | dose | 10 mg/kg | dose | 10 mg/kg | ||
| CLplasma | 30 mL/min/kg | plasma concentration | 2.59 ± 0.29 μM | plasma Cmax | 4.48 ± 0.56 μM | ||
| VSS | 1.4 L/kg | brain concentration | 9.36 ± 0.61 μM | plasma Tmax | 30 ± 15 minutes | ||
| F | 60% | Kpf | Kp,uug | 3.6 | 0.78 | plasma AUC0-∞ | 7.26 ± 0.24 μM·h |
Fu = fraction unbound.
n = 3; fine microsuspension in 0.1% Tween 80 and 0.5% methyl cellulose in H2O (4 mg/mL).
n = 2; dose =1.0 mg/kg; solution in 10% EtOH, 50% PEG 400, 40% saline (1 mg/mL).
n = 2; dose = 3.0 mg/kg; fine microsuspension in 0.1% Tween 80 and 0.5% methyl cellulose in H2O (0.3 mg/mL).
n = 3; time point = 30 min post dose; fine microsuspension in 0.1% Tween 80 and 0.5% methyl cellulose in H2O (1 mg/mL).
Kp = total brain to total plasma ratio.
Kp,uu = unbound brain (brain Fu × total brain) to unbound plasma (plasma Fu × total plasma) ratio.
n = 3 per time point; fine microsuspension in 0.1% Tween 80 and 0.5% methyl cellulose in H2O (1 mg/mL).
Behavioral Pharmacology of Compound 106
It is well-known that naïve mice will bury foreign objects, such as glass marbles, in deep bedding. This behavior can be inhibited by pretreatment with low doses of certain benzodiazepines, such as diazepam,65 as well as certain selective serotonin reuptake inhibitors (SSRIs), such as fluvoxamine.66 Likewise, this behavior has proven sensitive to a variety of mGlu5 NAM compounds from diverse chemotypes.38,67,68 As such, the marble burying assay has often been used as a convenient method for assessing anxiolytic activity. It is worth noting that recent reports have argued that the assay reflects a repetitive and perseverative behavior such as OCD as opposed to novelty-induced anxiety.69 For our purposes, the most important fact was that the mGlu2/3 orthosteric antagonists 1 and 2 have both been previously shown to inhibit marble burying.9,10 Thus, examination of the selective mGlu3 NAM 106 in this assay was warranted to determine the contribution of mGlu2 versus mGlu3 to this effect (Figure 5). Gratifyingly, dose dependent efficacy was observed in this assay, with statistically significant effects at all three doses and essentially complete inhibition at the highest dose (56.6 mg/kg). The positive control for this assay was the well characterized mGlu5 NAM 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]-pyridine (112).70 On the basis of the available DMPK data, it is estimated that CNS unbound exposure of compound 106 at the 10 mg/kg dose in this study reached a peak of 564 nM or approximately 1.5-fold the functional IC50.
Figure 5.

Inhibition of marble burying in mice by compound 106. n = 8–10 male CD-1 mice per treatment group; vehicle = 10% Tween 80 in H2O; 15 min pretreatment with compound or vehicle; 30 min burying time; *, p < 0.05 vs vehicle control group. Compound 112 is the mGlu5 NAM 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]-pyridine.
Having established its efficacy in an anxiolytic/OCD mouse model, profiling of compound 106 in a rat model of depression was considered valuable for illustrating the utility of the compound. The forced swim test (FST) measures immobility time in rats placed in a tank of water from which they cannot escape and is sensitive to many antidepressants including several SSRIs.71 Importantly, efficacy with the mGlu2/3 orthosteric antagonists 1 and 2 has been demonstrated in this assay,8 making the examination of 106 compelling (Figure 6). In this case, significant effects in decreasing immobility time were observed at the highest dose (56.6 mg/kg). On the basis of the available DMPK data and an assumption of dose linearity, it is estimated that CNS unbound exposure of 106 at the high dose in these studies reached a peak of 2.3 μM or approximately 6-fold over the functional IC50. The positive control in this assay was the N-methyl-d-aspartate (NMDA) receptor antagonist drug ketamine,72 which was introduced to the market over 50 years ago.73 Ketamine has recently demonstrated rapid acting antidepressant efficacy in TRD patients.74–76 Unfortunately, ketamine produces several undesirable side effects including psychotomimetic effects.77 Preclinical studies implicate common downstream signaling pathways in the antidepressant effects of ketamine and mGlu2/3 antagonists, including activation of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor and the mammalian target of rapamycin (mTOR) pathway.18,19,78–82 Such studies have raised the possibility that mGlu2/3 antagonists or selective antagonists of each individual group II mGlu might represent novel approaches to rapid acting antidepressants without the side effect profile of ketamine. A selective and CNS penetrant mGlu3 NAM compound such as 106 will be a valuable tool in shedding light on such questions.
Figure 6.

Decrease in immobility in the FST in rats by compound 106. n = 8–10 male Sprague–Dawley rats per treatment group; 106 vehicle = 10% Tween 80 in H2O; ketamine vehicle = saline; 30 min pretreatment with compound or vehicle; 6 min testing session; *, p < 0.05 vs vehicle control group.
CONCLUSION
A cross screening hit from a nonalkyne mGlu5 PAM chemotype served as a successful launching point for the discovery of compound 106, a highly selective mGlu3 NAM with DMPK properties that enable its convenient use in rodent models of psychiatric disorders. In addition to these features, the compound displays moderate clearance and good bioavailability in rats. Furthermore, the compound is highly permeable, not a substrate for P-gp mediated efflux, and possesses an attractive P450-inhibition profile. The compound has demonstrated efficacy in two rodent models previously shown to be sensitive to mGlu2/3 inhibition. This highly selective mGlu3 NAM can thus serve as a useful tool for elucidating the role of selective inhibition of mGlu3 and its potential utility as a novel therapeutic target. Such studies will constitute the subject of future communications.
EXPERIMENTAL SECTION
Diethyl (R)-1-(1-((tert-Butoxycarbonyl)amino)propan-2-yl)-1H-pyrazole-3,5-dicarboxylate (74)
Diethyl 3,5-pyrazoledicarboxylate 73 (4.24 g, 20 mmol, 1.0 equiv) and tert-butyl (S)-(2-hydroxypropyl) carbamate 15 (7.01 g, 40 mmol, 2.0 equiv) were dissolved in THF (100 mL. 0.2 M), and triphenyl phosphine (9.44 g, 36 mmol, 1.8 equiv) was added. After 5 min, the mixture was cooled to 0 °C and di-tert-butyl azodicarboxylate (8.29 g, 36 mmol, 1.8 equiv) was added. The reaction mixture was then subjected to microwave irradiation for 25 min at 120 °C. The mixture was cooled to room temperature, and the solvent was removed in vacuo. Purification via flash chromatography on silica gel provided the title compound as a semisolid (8.2 g, yield was not determined due to contamination of DtBAD byproduct, di-tert-butyl hydrazine-1,2-dicarboxylate). 1H NMR (400 MHz, MeOD) δ 7.29 (s, 1H), 5.69–5.01 (m, 1H), 4.41–4.35 (m, 4H), 3.51–3.39 (m, 2H), 1.51 (d, J = 6.8 Hz, 3H), 1.41–1.38 (m, 15H). 13C NMR (100 MHz, CDCl3) δ 161.7, 159.0, 155.8, 142.6, 134.2, 114.0, 79.4, 61.4, 61.1, 53.4, 45.1, 28.2 (3C), 18.5, 14.3, 14.2. LCMS (method A): RT = 1.052 min, m/z = 314.2 [M + H]+. HRMS, calcd for C17H27N3O6 [M], 369.1900; found, 369.1899.
Ethyl (R)-7-Methyl-4-oxo-4,5,6,7-tetrahydropyrazolo[1,5-a]-pyrazine-2-carboxylate (75)
Compound 74 (8.2 g, 22.2 mmol, 1.0 equiv) was treated with a solution of 4 N HCl in 1,4-dioxane (78 mL). Deprotection of the tert-butyl carbamate protecting group was monitored by LCMS. Once deprotection was complete, the reaction mixture was carefully basified with saturated aqueous NaHCO3 (verified by pH paper) and was allowed to stir at room temperature overnight. The mixture was diluted with dichloromethane, and the aqueous layer was extracted with dichloromethane (3×). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated in vacuo to provide the title compound as a white solid (4.4 g, 89% yield over two steps), which was used without further purification. 1H NMR (400 MHz, CDCl3) δ 7.35 (s, 1H), 7.13 (bs, 1H), 4.68–4.62 (m, 1H), 4.41 (q, J = 7.1 Hz, 2H), 3.86 (ddd, J = 15.8, 10.2, 1.4 Hz, 1H), 3.51 (ddd, J = 9.4, 6.4, 3.0 Hz, 1H), 1.65 (d, J = 6.6 Hz, 3H), 1.39 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 161.7, 159.3, 143.8, 134.4, 110.7, 61.3, 53.2, 46.0, 17.4, 14.3. LCMS (method A): RT = 0.546 min, m/z = 224.2 [M + H]. HRMS, calcd for C10H13N3O3 [M], 223.0957; found, 223.0957. [α]25D = −29.9° (c 0.500, CHCl3).
Ethyl (R)-5-(4-Methoxybenzyl)-7-methyl-4-oxo-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrazine-2-carboxylate (76)
Compound 75 (2.23 g, 10 mmol, 1.0 equiv) was dissolved in DMF (50 mL, 0.2 M), cooled to 0 °C, and treated with 60% sodium hydride in mineral oil (480 mg, 12 mmol, 1.2 equiv) in five portions. The reaction mixture was stirred for 15 min, and 4-methoxybenzyl chloride (1.63 mL, 12 mmol, 1.2 equiv) was added. After 16 h, the reaction mixture was diluted with water and extracted with EtOAc (3×). The combined extracts were washed with water and brine, dried over Na2SO4, filtered, and concentrated in vacuo. The crude material was purified by flash chromatography on silica gel to provide the title compound (2.51 g, 73% yield) as a pale-yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.37 (s, 1H), 7.23 (d, J = 14.1, 2H), 6.87 (d, J = 14.1, 2H), 4.76 (d, J = 14.5, 1H), 4.59–4.50 (m, 2H), 4.44–4.36 (m, 2H), 3.79 (s, 3H), 3.69 (dd, J = 13.1, 4.6 Hz, 1H), 3.35 (dd, J = 13.1, 6.3 Hz, 1H), 1.47 (d, J = 6.6 Hz, 3H), 1.38 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 161.7, 159.5, 137.1, 143.9, 134.8, 129.9 (2C), 127.9, 114.3, 110.9 (2C), 61.2, 55.3, 52.9, 50.4, 48.8, 17.5, 14.3. LCMS (method A): RT = 0.955 min, m/z = 344.2 [M + H]+. HRMS, calcd for C18H21N3O4 [M], 343.1532; found, 343.1533. [α]25D = −8.1° (c 0.157, CHCl3).
(R)-2-(Hydroxymethyl)-5-(4-methoxybenzyl)-7-methyl-6,7-dihydropyrazolo[1,5-a]pyrazin-4(5H)-one (77)
Sodium borohydride (1.16 g, 30.6 mmol, 5.0 equiv) was added slowly to a solution of compound 76 (2.1 g, 6.11 mmol, 1.0 equiv) in THF (20 mL) and MeOH (5.0 mL) at 0 °C. The reaction was heated to 60 °C, and after 30 min at that temperature, the reaction mixture was diluted with water and extracted with dichloromethane. The aqueous layer was acidified with a 1 M aqueous HCl solution and extracted with dichloromethane (2×). The combined extracts were dried over Na2SO4 and concentrated in vacuo. Purification by flash chromatography on silica gel provided the title compound as a viscous oil (1.55 g, 84% yield). 1H NMR (400 MHz, CDCl3) δ 7.23–7.21 (m, 2H), 6.87–6.84 (m, 3H), 4.72 (d, J = 14.5 Hz, 1H), 4.68 (s, 2H), 4.59 (d, J = 14.4 Hz, 1H), 4.43–4.35 (m, 1H), 3.78 (s, 3H), 3.60 (dd, J = 13.0, 4.6 Hz, 1H), 3.31 (dd, J = 13.0, 7.4 Hz, 1H), 1.42 (d, J = 6.5 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 159.4, 157.8, 152.6, 134.5, 129.8 (2C), 128.2, 114.3 (2C), 106.5, 58.7, 55.3, 52.0, 50.8, 48.7, 17.2. LCMS (method A): RT = 0.680 min, m/z = 302.2 [M + H]+. HRMS, calcd for C16H19N3O3 [M], 301.1426; found, 301.1428. [α]25D = −5.3° (c 0.98, CHCl3).
(R)-2-(((5-Chloropyridin-2-yl)oxy)methyl)-5-(4-methoxybenzyl)-7-methyl-6,7-dihydropyrazolo[1,5-a]pyrazin-4(5H)-one(78)
To a solution of compound 77 (1.5 g, 4.98 mmol, 1.0 equiv) in DMF (25 mL, 0.2 M) at 0 °C was added NaH (300 mg, 12.44 mmol, 2.5 equiv). The resulting mixture was stirred for 15 min, and 5-chloro-2-fluoropyridine (1.25 mL, 12.44 mmol, 2.5 equiv) was added. The mixture was stirred overnight and extracted with EtOAc (3×). The combined extracts were concentrated in vacuo. Purification by flash chromatography on silica gel afforded the title compound (1.72 g, 84% yield) as a viscous oil. 1H NMR (400 MHz, CDCl3) δ 8.13 (d, J = 2.6 Hz, 1H), 7.54 (dd, J = 8.8, 2.6 Hz, 1H), 7.25 (d, J = 8.5 Hz, 2H), 6.98 (s, 1H), 6.88 (d, J = 8.5 Hz, 2H), 6.75 (d, J = 8.8 Hz, 1H), 5.39 (s, 2H), 4.75 (d, J = 14.5 Hz, 1H), 4.63 (d, J = 14.5 Hz, 1H), 4.50–4.42 (m, 1H), 3.81 (s, 3H), 3.64 (dd, J = 13.0, 4.6 Hz, 1H), 3.35 (dd, J = 13.0, 7.4 Hz, 1H), 1.48 (d, J = 6.5 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 161.6, 159.4, 157.7, 148.9, 145.1, 138.6, 134.5, 129.8 (2C), 128.2, 124.4, 114.2 (2C), 112.3, 108.2, 61.6, 55.3, 52.1, 50.8, 48.8, 17.2. LCMS (method A): RT = 1.080 min, m/z = 413.2 [M + H]+. HRMS, calcd for C21H21ClN4O3 [M], 412.1302; found, 412.1305. [α]25D = −10.3° (c 1.512, CHCl3).
(R)-2-(((5-Chloropyridin-2-yl)oxy)methyl)-7-methyl-6,7-dihydropyrazolo[1,5-a]pyrazin-4(5H)-one (79)
Compound 78 (1.65 mg, 4.0 mmol, 1.0 equiv) was dissolved in MeCN (40 mL, 0.1 M). and a solution of ceric ammonium nitrate (6.57 g, 12 mmol, 4.0 equiv) in water (12 mL) was added. After 30 min at room temperature, solvents were removed in vacuo. Purification using flash chromatography on silica gel provided the title compound (764 mg, 65% yield) as a pale-yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.26 (d, J = 2.7 Hz, 1H), 8.21 (s, 1H), 7.83 (dd, J = 8.8, 2.7 Hz, 1H), 6.92 (dd, J = 8.8, 0.5 Hz, 1H), 6.77 (s, 1H), 5.29 (s, 2H), 4.51–4.46 (m, 1H), 3.66 (ddd, J = 13.0, 8.7, 8.7 Hz, 1H), 3.34 (ddd, J = 13.1, 7.9, 2.2 Hz, 1H), 1.45 (d, J = 6.5 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 161.9, 158.7, 147.7, 145.3, 139.7, 135.1, 124.1, 112.9, 107.4, 61.7, 52.3, 45.6, 17.0. LCMS (method A): RT = 0.804 min, m/z = 293.2 [M + H]+. HRMS, calcd for C13H13ClN4O2 [M], 292.0727; found, 292.0727. [α]25D = −38.3° (c 0.442, CHCl3).
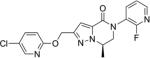
(R)-2-(((5-Chloropyridin-2-yl)oxy)methyl)-5-(2-fluoropyridin-3-yl)-7-methyl-6,7-dihydropyrazolo[1,5-a]pyrazin-4(5H)-one(106)
Copper(I) iodide (13.7 mg, 0.072 mmol, 2.1 equiv) was added to a suspension of compound 79 (10 mg, 0.035 mmol, 1.0 equiv), 3-bromo-2-fluoropyridine (7.40 μL, 0.072 mmol, 2.1 equiv), potassium carbonate (10 mg, 0.072 mmol, 2.1 equiv), and N,N′-dimethylethylenediamine (20.7 μL, 0.19 mmol, 5.5 equiv) in toluene (0.44 mL) in a sealed reaction vial. The reaction mixture was stirred at 120 °C. After 16 h, the mixture was diluted with EtOAc, filtered through a Celite pad which was rinsed with EtOAc (2×), and concentrated in vacuo. Purification using reserve phase HPLC method 1 with 39–71% CH3CN in H2O (0.1% TFA) over 4 min provided the title compound (7.2 mg, 53% yield) as a white powder. 1H NMR (400 MHz, DMSO-d6) δ 8.27 (dd, J = 2.7, 0.5 Hz, 1H), 8.23 (ddd, J = 4.8, 2.7, 2.7 Hz, 1H), 8.09 (ddd, J = 9.6, 7.7, 1.8 Hz, 1H), 7.84 (dd, J = 8.8, 2.7 Hz, 1H), 7.50 (ddd, J = 9.0, 4.9, 1.3 Hz, 1H) 6.95 (s, 1H), 6.92 (d, J = 0.5 Hz, 1H), 5.35 (s, 2H), 4.82–4.75 (m, 1H), 4.27 (dd, J = 12.8, 4.3 Hz, 1H), 3.98 (dd, J = 12.8, 7.2 Hz, 1H) 1.55 (d, J = 6.5 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 161.9, 158.2 (d, JC,F = 239 Hz), 156.6, 148.4, 146.4 (d, JC,F = 14 Hz), 145.3, 133.9 (d, JC,F = 13 Hz), 139.8, 134.0, 124.4 (d, JC,F = 28 Hz), 124.2, 123.2 (d, JC,F = 4 Hz), 112.9, 108.7, 61.6, 54.0, 52.7, 17.1. LCMS (method A): RT = 0.944 min, m/z = 388.2 [M + H]+. HRMS, calcd for C18H15ClFN5O2 [M], 387.0898; found, 387.0899. [α]25D = −23.6° (c 0.100, DMSO).
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge the generous support of the National Institute of Mental Health for the funding of this work, NIMH R01MH099269 (K.A.E) and U54MH084659 (C.W.L.).
ABBREVIATIONS USED
- Ac
acetate
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- AUC
area under the curve
- C
concentration
- CL
clearance
- CNS
central nervous system
- CRC
concentration response curve
- DIAD
di-iso-propyl azodicarboxylate
- DMF
N,N-dimethylformamide
- DMPK
drug metabolism and pharmacokinetics
- DMSO
dimethyl sulfoxide
- DtBAD
di-tert-butyl azodicarboxylate
- Et
ethyl
- FST
forced swim test
- F
bioavailability
- Fu
fraction unbound
- GPCR
G-protein-coupled receptors
- HLM
human liver microsomes
- IP
intraperitoneal
- IV
intravenous
- Kp
brain to plasma ratio
- Kp,uu
unbound brain to unbound plasma ratio
- LLE
ligand-lipophilicity efficiency
- max
maximum
- MDCK
Madin–Darby canine kidney
- MDD
major depressive disorder
- Me
methyl
- mGlu
metabotropic glutamate receptor
- Ms
methanesulfonyl
- mTOR
mammalian target of rapamycin
- NAM
negative allosteric modulator
- NMDA
N-methyl-d-aspartate
- OCD
obsessive-compulsive disorder
- PAM
positive allosteric modulator
- PEG
polyethylene glycol
- Ph
phenyl
- PK
pharmacokinetics
- PO
oral
- PS
polystyrene
- RLM
rat liver microsomes
- SAR
structure–activity relationships
- THF
tetrahydrofuran
- TRD
treatment-resistant depression
- T
time
- t1/2
half-life
- VSS
volume of distribution at steady-state
- 5-HT2B
5-hydroxytryptamine receptor 2B
- 7TM
seven transmembrane
Footnotes
Author Contributions
Drs. Emmitte and Lindsley directed and designed the chemistry. Drs. Engers and Konkol performed the medicinal chemistry. Drs. Conn and Niswender directed and designed the molecular pharmacology experiments. Dr. Rodriguez directed and performed molecular pharmacology experiments. Mr. Venable and Mr. Loch performed molecular pharmacology experiments. Dr. Daniels directed and designed the DMPK experiments. Dr. Blobaum directed DMPK experiments and performed bioanalytical work. Mr. Morrison performed bioanalytical work. Mr. Chang performed in vitro DMPK work. Mr. Byers performed in vivo DMPK work. Dr. Jones directed and designed the behavioral experiments. Dr. Thompson performed the behavioral experiments.
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.5b01005.
Experimental procedures and spectroscopic data for additional compounds, molecular pharmacology methods, DMPK methods, behavioral pharmacology methods, and the ancillary pharmacology profile details of 106 (PDF)
Molecular formula strings (CSV)
REFERENCES
- 1.Niswender CM, Conn PJ. Metabotropic glutamate receptors: Physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 2010;50:295–322. doi: 10.1146/annurev.pharmtox.011008.145533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schoepp DD, Jane DE, Monn JA. Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuro-pharmacology. 1999;38:1431–1476. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]
- 3.Conn PJ, Pin J-P. Pharmacology and functions of metabotropic glutamate receptors. Annu. Rev. Pharmacol. Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 4.Chaki S, Ago Y, Palucha-Paniewiera A, Matrisciano F, Pilc A. mGlu2/3 and mGlu5 receptors: Potential targets for novel antidepressants. Neuropharmacology. 2013;66:40–52. doi: 10.1016/j.neuropharm.2012.05.022. [DOI] [PubMed] [Google Scholar]
- 5.Palucha A, Pilc A. Metabotropic glutamate receptor ligands as possible anxiolytic and antidepressant drugs. Pharmacol. Ther. 2007;115:116–147. doi: 10.1016/j.pharmthera.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 6.Célanire S, Sebhat I, Wichmann J, Mayer S, Schann S, Gatti S. Novel metabotropic glutamate receptor 2/3 antagonists and their therapeutic applications: a patent review (2005 − present). Expert Opin. Ther. Patents. 2015;25:69–90. doi: 10.1517/13543776.2014.983899. [DOI] [PubMed] [Google Scholar]
- 7.Ornstein PL, Bleisch TJ, Arnold MB, Kennedy JH, Wright RA, Johnson BG, Tizzano JP, Helton DR, Kallman MJ, Schoepp DD, Hérin M. 2-Substituted (2SR)-2-amino-2-((1SR,2SR)-2-carboxycycloprop-1-yl)glycines as potent and selective antagonists of group II metabotropic glutamate receptors. 2. Effects of aromatic substitution, pharmacological characterization, and bioavailability. J. Med. Chem. 1998;41:358–378. doi: 10.1021/jm970498o. [DOI] [PubMed] [Google Scholar]
- 8.Chaki S, Yoshikawa R, Hirota S, Shimazaki T, Maeda M, Kawashima N, Yoshimizu T, Yasuhara A, Sakagami K, Okuyama S, Nakanishi S, Nakazato A. MGS0039: a potent and selective group II metabotropic glutamate receptor antagonist with antidepressant-like activity. Neuropharmacology. 2004;46:457–467. doi: 10.1016/j.neuropharm.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 9.Bespalov AY, van Gaalen MM, Sukhotina IA, Wicke K, Mezler M, Schoemaker H, Gross G. Behavioral characterization of the mGlu group II/III receptor antagonist, LY-341495, in animal models of anxiety and depression. Eur. J. Pharmacol. 2008;592:96–102. doi: 10.1016/j.ejphar.2008.06.089. [DOI] [PubMed] [Google Scholar]
- 10.Shimazaki T, Iijima M, Chaki S. Anxiolytic-like activity of MGS0039, a potent group II metabotropic glutamate receptor antagonist, in a marble-burying behavior test. Eur. J. Pharmacol. 2004;501:121–125. doi: 10.1016/j.ejphar.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 11.Yoshimizu T, Shimazaki T, Ito A, Chaki S. An mGluR2/3 antagonist, MGS0039, exerts antidepressant and anxiolytic effects in behavioral models in rats. Psychopharmacology. 2006;186:587–593. doi: 10.1007/s00213-006-0390-7. [DOI] [PubMed] [Google Scholar]
- 12.Higgins GA, Ballard TM, Kew JN, Richards JG, Kemp JA, Adam G, Woltering T, Nakanishi S, Mutel V. Pharmacological manipulation of mGlu2 receptors influences cognitive performance in the rodent. Neuropharmacology. 2004;46:907–917. doi: 10.1016/j.neuropharm.2004.01.018. [DOI] [PubMed] [Google Scholar]
- 13.Kim SH, Steele JW, Lee SW, Clemenson GD, Carter TA, Treuner K, Gadient R, Wedel P, Glabe C, Barlow C, Ehrlich ME, Gage FH, Gandy S. Proneurogenic group II mGluR antagonist improves learning and reduces anxiety in Alzheimer Aβ oligomer mouse. Mol. Psychiatry. 2014;19:1235–1242. doi: 10.1038/mp.2014.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim SH, Fraser PE, Westaway D, St. George-Hyslop PH, Ehrlich ME, Gandy S. Group II metabotropic glutamate receptor stimulation triggers production and release of Alzheimer's amyloid β42 from isolated intact nerve terminals. J. Neurosci. 2010;30:3870–3875. doi: 10.1523/JNEUROSCI.4717-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoshimizu T, Chaki S. Increased cell proliferation in the adult mouse hippocampus following chronic administration of group II metabotropic glutamate receptor antagonist, MGS0039. Biochem. Biophys. Res. Commun. 2004;315:493–496. doi: 10.1016/j.bbrc.2004.01.073. [DOI] [PubMed] [Google Scholar]
- 16.Gleason SD, Li X, Smith IA, Ephlin JD, Wang XS, Heinz BA, Carter JH, Baez M, Yu J, Bender DM, Witkin JM. mGlu2/3 agonist-induced hyperthermia: An in vivo assay for detection of mGlu2/3 receptor antagonism and its relation to antidepressant-like efficacy in mice. CNS Neurol. Disord.: Drug Targets. 2013;12:554–566. doi: 10.2174/18715273113129990079. [DOI] [PubMed] [Google Scholar]
- 17.Koike H, Fukumoto K, Iijima M, Chaki S. Role of BDNF/TrkB signaling in antidepressant-like effects of a group II metabotropic glutamate receptor antagonist in animal models of depression. Behav. Brain Res. 2013;238:48–52. doi: 10.1016/j.bbr.2012.10.023. [DOI] [PubMed] [Google Scholar]
- 18.Koike H, Iijima M, Chaki S. Involvement of the mammalian target of rapamycin signaling in the antidepressant-like effect of group II metabotropic glutamate receptor antagonists. Neuropharmacology. 2011;61:1419–1423. doi: 10.1016/j.neuropharm.2011.08.034. [DOI] [PubMed] [Google Scholar]
- 19.Karasawa J, Shimazaki T, Kawashima N, Chaki S. AMPA receptor stimulation mediates the antidepressant-like effect of a group II metabotropic glutamate receptor antagonist. Brain Res. 2005;1042:92–98. doi: 10.1016/j.brainres.2005.02.032. [DOI] [PubMed] [Google Scholar]
- 20.Ago Y, Yano K, Araki R, Hiramatsu N, Kita Y, Kawasaki T, Onoe H, Chaki S, Nakazato A, Hashimoto H, Baba A, Takuma K, Matsuda T. Metabotropic glutamate 2/3 receptor antagonists improve behavioral and prefrontal dopaminergic alterations in the chronic corticosterone-induced depression model in mice. Neuropharmacology. 2013;65:29–38. doi: 10.1016/j.neuropharm.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 21.Dwyer JM, Lepack AE, Duman RS. mGluR2/3 blockade produces rapid and long-lasting reversal of anhedonia caused by chronic stress exposure. J. Mol. Psychiatry. 2013;1:15. doi: 10.1186/2049-9256-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iijima M, Koike H, Chaki S. Effect of an mGlu2/3 receptor antagonist on depressive behavior induced by withdrawal from chronic treatment with methamphetamine. Behav. Brain Res. 2013;246:24–28. doi: 10.1016/j.bbr.2013.02.039. [DOI] [PubMed] [Google Scholar]
- 23.Markou A. Metabotropic glutamate receptor antagonists: Novel therapeutics for nicotine dependence and depression? Biol. Psychiatry. 2007;61:17–22. doi: 10.1016/j.biopsych.2006.03.053. [DOI] [PubMed] [Google Scholar]
- 24.Ciceroni C, Bonelli M, Mastrantoni E, Niccolini C, Laurenza M, Larocca LM, Pallini R, Traficante A, Spinsanti P, Ricci-Vitiani L, Arcella A, De Maria R, Nicoletti F, Battaglia G, Melchiorri D. Type-3 metabotropic glutamate receptors regulate chemoresistance in glioma stem cells, and their levels are inversely related to survival in patients with malignant gliomas. Cell Death Differ. 2013;20:396–407. doi: 10.1038/cdd.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ciceroni C, Arcella A, Mosillo P, Battaglia G, Mastrantoni E, Oliva MA, Carpinelli G, Santoro F, Sale P, Ricci-Vitiani L, De Maria R, Pallini R, Giangaspero F, Nicoletti F, Melchiorri D. Type-3 metabotropic glutamate receptors negatively modulate bone morphogenetic protein receptor signaling and support the tumourigenic potential of glioma-initiating cells. Neuropharmacology. 2008;55:568–576. doi: 10.1016/j.neuropharm.2008.06.064. [DOI] [PubMed] [Google Scholar]
- 26.Arcella A, Carpinelli G, Battaglia G, D’Onofrio M, Santoro F, Ngomba RT, Bruno V, Casolini P, Giangaspero F, Nicoletti F. Pharmacological blockade of group II metabotropic glutamate receptors reduces the growth of glioma cells in vivo. Neuro-Oncology. 2005;7:236–245. doi: 10.1215/S1152851704000961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.D'Onofrio M, Arcella A, Bruno V, Ngomba RT, Battaglia G, Lombari V, Ragona G, Calogero A, Nicoletti F. Pharmacological blockade of mGlu2/3 metabotropic glutamate receptors reduces cell proliferation in cultured human glioma cells. J. Neurochem. 2003;84:1288–1295. doi: 10.1046/j.1471-4159.2003.01633.x. [DOI] [PubMed] [Google Scholar]
- 28.Campo B, Kalinichev M, Lambeng N, Yacoubi ME, Royer-Urios I, Schneider M, Legrand C, Parron D, Girard F, Bessif A, Poli S, Vaugeois J-M, Le Poul E, Celanire S. Characterization of an mGluR2/3 negative allosteric modulator in rodent models of depression. J. Neurogenet. 2011;25:152–166. doi: 10.3109/01677063.2011.627485. [DOI] [PubMed] [Google Scholar]
- 29.Pritchett D, Jagannath A, Brown LA, Tam SKE, Hasan S, Gatti S, Harrison PJ, Bannerman DM, Foster RG, Peirson SN. Deletion of metabotropic glutamate receptors 2 and 3 (mGlu2 & mGlu3) in mice disrupts sleep and wheel-running activity, and increases the sensitivity of the circadian system to light. PLoS One. 2015;10:e0125523. doi: 10.1371/journal.pone.0125523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Woltering TJ, Wichmann J, Goetschi E, Knoflach F, Ballard TM, Huwyler J, Gatti S. Synthesis and characterization of 1,3-dihydro-benzo[b][1,4]diazepin-2-one derivatives: Part 4. In vivo active potent and selective non-competitive metabotropic glutamate receptor 2/3 antagonists. Bioorg. Med. Chem. Lett. 2010;20:6969–6974. doi: 10.1016/j.bmcl.2010.09.125. [DOI] [PubMed] [Google Scholar]
- 31.Yacoubi ME, Vaugeois J-M, Kalinichev M, Célanire S, Parron D, Le Poul E, Campo B. Effects of a mGluR2/3 negative allosteric modulator and a reference mGluR2/3 orthosteric antagonist in a genetic mouse model of depression.. Behavioral Studies of Mood Disorders; Proceedings of the 40th Annual Meeting of the Society for Neuroscience; San Diego, CA. Nov 13−17, 2010; Washington, DC: Society for Neuroscience; 2010. 886.14/VV7. [Google Scholar]
- 32.Goeldner C, Ballard TM, Knoflach F, Wichmann J, Gatti S, Umbricht D. Cognitive impairment in major depression and the mGlu2 receptor as a therapeutic target. Neuropharmacology. 2013;64:337–346. doi: 10.1016/j.neuropharm.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 33.Kalinichev M, Campo B, Lambeng N, Célanire S, Schneider M, Bessif A, Royer-Urios I, Parron D, Legrand C, Mahious N, Girard F, Le Poul E. An mGluR2/3 negative allosteric modulator improves recognition memory assessed by natural forgetting in the novel object recognition test in rats.. Memory Consolidation and Reconsolidation: Molecular Mechanisms II; Proceedings of the 40th Annual Meeting of the Society for Neuroscience; San Diego, CA. Nov 13−17, 2010; Washington, DC: Society for Neuroscience; 2010. 406.9/MMM57. [Google Scholar]
- 34.Choi CH, Schoenfeld BP, Bell AJ, Hinchey P, Kollaros M, Gertner MJ, Woo NH, Tranfaglia MR, Bear MF, Zukin RS, McDonald TV, Jongens TA, McBride SM. Pharmacological reversal of synaptic plasticity deficits in the mouse model of fragile X syndrome by group II mGluR antagonist or lithium treatment. Brain Res. 2011;1380:106–119. doi: 10.1016/j.brainres.2010.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gatti McArthur S, Saxe M, Wichmann J, Woltering T. mGlu2/3 antagonists for the treatment of autistic disorders. 2014 May 1; PCT Int. Pat. Appl. WO 2014/064028 A1.
- 36.Structure of decoglurant disclosed in Recommended International Nonproprietary Names (INN) WHO Drug Information. 3. Vol. 27. World Health Organization; Geneva, Switzerland: 2013. p. 150. [Google Scholar]
- 37.ARTDeCo Study: A study of RO4995819 in patients with major depressive disorder and inadequate response to ongoing antidepressant treatment. U.S. National Institutes of Health; Bethesda, MD: 2011. ClinicalTrials.gov https://www.clinicaltrials.gov/ct2/show/NCT01457677 (accessed August 12, 2015) [Google Scholar]
- 38.Rodriguez AL, Grier MD, Jones CK, Herman EJ, Kane AS, Smith RL, Williams R, Zhou Y, Marlo JE, Days EL, Blatt TN, Jadhav S, Menon UN, Vinson PN, Rook JM, Stauffer SR, Niswender CM, Lindsley CW, Weaver CD, Conn PJ. Discovery of novel allosteric modulators of metabotropic glutamate receptor subtype 5 reveals chemical and functional diversity and in vivo activity in rat behavioral models of anxiolytic and antipsychotic activity. Mol. Pharmacol. 2010;78:1105–1123. doi: 10.1124/mol.110.067207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sheffler DJ, Wenthur CJ, Bruner JA, Carrington SJS, Vinson PN, Gogi KK, Blobaum AL, Morrison RD, Vamos M, Cosford NDP, Stauffer SR, Daniels JS, Niswender CM, Conn PJ, Lindsley CW. Development of a novel, CNS-penetrant, metabotropic glutamate receptor 3 (mGlu3) NAM probe (ML289) derived from a closely related mGlu5 PAM. Bioorg. Med. Chem. Lett. 2012;22:3921–3925. doi: 10.1016/j.bmcl.2012.04.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wenthur CJ, Morrison R, Felts AS, Smith KA, Engers JL, Byers FW, Daniels JS, Emmitte KA, Conn PJ, Lindsley CW. Discovery of (R)−−(2-fluoro-4-((−4-methoxyphenyl)ethynyl)-phenyl(3-hydroxypiperidin-1-yl)methanone (ML337), An mGlu3 selective and CNS penetrant negative allosteric modulator (NAM). J. Med. Chem. 2013;56:5208–5212. doi: 10.1021/jm400439t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walker AG, Wenthur CJ, Xiang Z, Rook JM, Emmitte KA, Niswender CM, Lindsley CW, Conn PJ. Metabotropic glutamate receptor 3 activation is required for long-term depression in medial prefrontal cortex and fear extinction. Proc. Natl. Acad. Sci. U. S. A. 2015;112:1196–1201. doi: 10.1073/pnas.1416196112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang L, Balan G, Barreiro G, Boscoe BP, Chenard LK, Cianfrogna J, Claffey MM, Chen L, Coffman KJ, Drozda SE, Dunetz JR, Fonseca KR, Galatsis P, Grimwood S, Lazzaro JT, Mancuso JY, Miller EL, Reese MR, Rogers BN, Sakurada I, Skaddan M, Smith DL, Stepan AF, Trapa P, Tuttle JB, Verhoest PR, Walker DP, Wright AS, Zaleska MM, Zasadny K, Shaffer CL. Discovery and preclinical characterization of 1-methyl-3-(4-methylpyridin-3-yl)-6-(pyridin-2-ylmethoxy)-1H-pyrazolo-[3,4-b]pyrazine (PF470): a highly potent, selective, and efficacious metabotropic glutamate receptor 5 (mGluR5) negative allosteric modulator. J. Med. Chem. 2014;57:861–877. doi: 10.1021/jm401622k. [DOI] [PubMed] [Google Scholar]
- 43.Zhuo X, Huang XS, Degnan AP, Snyder LB, Yang F, Huang H, Shu Y-Z, Johnson BM. Identification of glutathione conjugates of acetylene-containing positive allosteric modulators of metabotropic glutamate receptor subtype 5. Drug Metab. Dispos. 2015;43:578–589. doi: 10.1124/dmd.114.061879. [DOI] [PubMed] [Google Scholar]
- 44.Noetzel MJ, Rook JM, Vinson PN, Cho H, Days E, Zhou Y, Rodriguez AL, Lavreysen H, Stauffer SR, Niswender CM, Xiang Z, Daniels JS, Jones CK, Lindsley CW, Weaver CD, Conn PJ. Functional impact of allosteric agonist activity of selective positive allosteric modulators of metabotropic glutamate receptor subtype 5 in regulating central nervous system function. Mol. Pharmacol. 2012;81:120–133. doi: 10.1124/mol.111.075184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Niswender CM, Johnson KA, Luo Q, Ayala JE, Kim C, Conn PJ, Weaver CD. A novel assay of Gi/o-linked G protein-coupled receptor coupling to potassium channels provides new insights into the pharmacology of the group III metabotropic glutamate receptors. Mol. Pharmacol. 2008;73:1213–1224. doi: 10.1124/mol.107.041053. [DOI] [PubMed] [Google Scholar]
- 46.Conn PJ, Lindsley CW, Stauffer SL, Bartolome-Nebreda JM, Conde-Ceide S, MacDonald GJ, Tong HM, Jones CK, Alcazar-Vaca JM, Andres-Gil JI, Malosh C. Bicyclic triazole and pyrazole lactams as allosteric modulators of mGluR5 receptors. 2012 Jun 21; PCT Int. Pat. Appl. WO 2012/083224 A1.
- 47.But TYS, Toy PH. The Mitsunobu reaction: Origin, mechanism, improvements, and applications. Chem. - Asian J. 2007;2:1340–1355. doi: 10.1002/asia.200700182. [DOI] [PubMed] [Google Scholar]
- 48.Klapars A, Huang X, Buchwald SL. A general and efficient copper catalyst for the amidation of aryl halides. J. Am. Chem. Soc. 2002;124:7421–7428. doi: 10.1021/ja0260465. [DOI] [PubMed] [Google Scholar]
- 49.Gregory KJ, Nguyen ED, Reiff SD, Squire EF, Stauffer SR, Lindsley CW, Meiler J, Conn PJ. Probing the metabotropic glutamate receptor 5 (mGlu5) positive allosteric modulator (PAM) binding pocket: discovery of point mutations that engender a “molecular switch” in PAM pharmacology. Mol. Pharmacol. 2013;83:991–1006. doi: 10.1124/mol.112.083949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lamb JP, Engers DW, Niswender CM, Rodriguez AL, Venable DF, Conn PJ, Lindsley CW. Discovery of molecular switches within the ADX-47273 mGlu5 PAM scaffold that modulate modes of pharmacology to afford potent mGlu5 NAMs, PAMs and partial antagonists. Bioorg. Med. Chem. Lett. 2011;21:2711–2714. doi: 10.1016/j.bmcl.2010.11.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kalvass JC, Maurer TS. Influence of nonspecific brain and plasma binding on CNS exposure: implications for rational drug discovery. Biopharm. Drug Dispos. 2002;23:327–338. doi: 10.1002/bdd.325. [DOI] [PubMed] [Google Scholar]
- 52.Bridges TM, Morrison RD, Byers FW, Luo S, Daniels JS. Use of a novel rapid and resource-efficient cassette dosing approach to determine the pharmacokinetics and CNS distribution of small molecule 7-transmembrane receptor allosteric modulators in rat. Pharmacol. Res. Perspect. 2014;2:e00077. doi: 10.1002/prp2.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leeson PD, Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discovery. 2007;6:881–890. doi: 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- 54.Obach RS. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: An examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab. Dispos. 1999;27:1350–1359. [PubMed] [Google Scholar]
- 55.Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm. Res. 1993;10:1093–1095. doi: 10.1023/a:1018943613122. [DOI] [PubMed] [Google Scholar]
- 56.Bridges TM, Rook JM, Noetzel MJ, Morrison RD, Zhou Y, Gogliotti RD, Vinson PN, Xiang Z, Jones CK, Niswender CM, Lindsley CW, Stauffer SL, Conn PJ, Daniels JS. Biotransformation of a novel positive allosteric modulator of metabotropic glutamate receptor subtype 5 contributes to seizure-like adverse events in rats involving a receptor agonism-dependent mechanism. Drug Metab. Dispos. 2013;41:1703–1714. doi: 10.1124/dmd.113.052084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Di L, Rong H, Feng B. Demystifying brain penetration in central nervous system drug discovery. J. Med. Chem. 2013;56:2–12. doi: 10.1021/jm301297f. [DOI] [PubMed] [Google Scholar]
- 58.Hemstapat K, Da Costa H, Nong Y, Brady AE, Luo Q, Niswender CM, Tamagnan GD, Conn PJ. A novel family of potent negative allosteric modulators of group II metabotropic glutamate receptors. J. Pharmacol. Exp. Ther. 2007;322:254–264. doi: 10.1124/jpet.106.117093. [DOI] [PubMed] [Google Scholar]
- 59.LeadProfilingScreen; (catalogue no. 68) Eurofins Panlabs, Inc.; Redmond, WA: ( www.eurofinspanlabs.com) [Google Scholar]
- 60.Significant responses are defined as those that inhibited more than 50% of radioligand binding.
- 61.Fitzgerald LW, Burn TC, Brown BS, Patterson JP, Corjay MH, Valentine PA, Sun JH, Link JR, Abbaszade I, Hollis JM, Largent BL, Hartig PR, Hollis GF, Meunier PC, Robichaud AJ, Robertson DW. Possible role of valvular serotonin 5-HT(2B) receptors in the cardiopathy associated with fenfluramine. Mol. Pharmacol. 2000;57:75–81. [PubMed] [Google Scholar]
- 62.Serotonin (5-Hydroxytryptamine) 5-HT2B, IP1; (catalogue no. 355260) Eurofins Panlabs, Inc.; Redmond, WA: ( www.eurofinspanlabs.com) [Google Scholar]
- 63.Zientek M, Miller H, Smith D, Dunklee MB, Heinle L, Thurston A, Lee C, Hyland R, Fahmi O, Burdette D. Development of an in vitro drug-drug interaction assay to simultaneously monitor five cytochrome P450 isoforms and performance assessment using drug library compounds. J. Pharmacol. Toxicol. Methods. 2008;58:206–214. doi: 10.1016/j.vascn.2008.05.131. [DOI] [PubMed] [Google Scholar]
- 64.Wang Q, Rager JD, Weinstein K, Kardos PS, Dobson GL, Li J, Hidalgo IJ. Evaluation of the MDR-MDCK cell line as a permeability screen for the blood−brain barrier. Int. J. Pharm. 2005;288:349–359. doi: 10.1016/j.ijpharm.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 65.Njung'e K, Handley SL. Evaluation of marble-burying behavior as a model of anxiety. Pharmacol., Biochem. Behav. 1991;38:63–67. doi: 10.1016/0091-3057(91)90590-x. [DOI] [PubMed] [Google Scholar]
- 66.Njung'e K, Handley SL. Effects of 5-HT uptake inhibitors, agonists and antagonists on the burying of harmless objects by mice; a putative test for anxiolytic agents. Br. J. Pharmacol. 1991;104:105–112. doi: 10.1111/j.1476-5381.1991.tb12392.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nicolas LB, Kolb Y, Prinssen EP. A combined marble burying-locomotor activity test in mice: a practical screening test with sensitivity to different classes of anxiolytics and antidepressants. Eur. J. Pharmacol. 2006;547:106–115. doi: 10.1016/j.ejphar.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 68.Felts AS, Rodriguez AL, Morrison RD, Venable DF, Manka JT, Bates BS, Blobaum AL, Byers FW, Daniels JS, Niswender CM, Jones CK, Conn PJ, Lindsley CW, Emmitte KA. Discovery of VU0409106: A negative allosteric modulator of mGlu5 with activity in a mouse model of anxiety. Bioorg. Med. Chem. Lett. 2013;23:5779–5785. doi: 10.1016/j.bmcl.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thomas A, Burant A, Bui N, Graham D, Yuva-Paylor LA, Paylor R. Marble burying reflects a repetitive and perseverative behavior more than novelty-induced anxiety. Psychopharmacology. 2009;204:361–373. doi: 10.1007/s00213-009-1466-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cosford ND, Tehrani L, Roppe J, Schweiger E, Smith ND, Anderson J, Bristow L, Brodkin J, Jiang X, McDonald I, Rao S, Washburn M, Varney MA. 3-[(2-Methyl-1,3-thiazol-4-yl)-ethynyl]-pyridine: a potent and highly selective metabotropic glutamate subtype 5 receptor antagonist with anxiolytic activity. J. Med. Chem. 2003;46:204–206. doi: 10.1021/jm025570j. [DOI] [PubMed] [Google Scholar]
- 71.Detke MJ, Rickels M, Lucki I. Active behaviors in the rat forced swimming test differentially produced by serotonergic and noradrenergic antidepressants. Psychopharmacology (Berl) 1995;121:66–72. doi: 10.1007/BF02245592. [DOI] [PubMed] [Google Scholar]
- 72.Garcia LS, Comim CM, Valvassori SS, Réus GZ, Barbosa LM, Andreazza AC, Stertz L, Fries GR, Gavioli EC, Kapczinski F, Quevedo J. Acute administration of ketamine induces antidepressant-like effects in the forced swimming test and increases BDNF levels in the rat hippocampus. Prog. Neuro-Psychopharmacol. Biol. Psychiatry. 2008;32:140–144. doi: 10.1016/j.pnpbp.2007.07.027. [DOI] [PubMed] [Google Scholar]
- 73.Kohrs R, Durieux ME. Ketamine: Teaching an old drug new tricks. Anesth. Analg. 1998;87:1186–1193. doi: 10.1097/00000539-199811000-00039. [DOI] [PubMed] [Google Scholar]
- 74.Murrough JW, Iosifescu DV, Chang LC, Al Jurdi RK, Green CE, Perez AM, Iqbal S, Pillemer S, Foulkes A, Shah A, Charney DS, Mathew SJ. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am. J. Psychiatry. 2013;170:1134–1142. doi: 10.1176/appi.ajp.2013.13030392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Browne CA, Lucki I. Antidepressant effects of ketamine: mechanisms underlying fast-acting novel antidepressants. Front. Pharmacol. 2013;4 doi: 10.3389/fphar.2013.00161. DOI: 10.3389/fphar.2013.00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.aan het Rot M, Collins KA, Murrough JW, Perez AM, Reich DL, Charney DS, Mathew SJ. Safety and efficacy of repeated-dose intravenous ketamine for treatment-resistant depression. Biol. Psychiatry. 2010;67:139–145. doi: 10.1016/j.biopsych.2009.08.038. [DOI] [PubMed] [Google Scholar]
- 77.Morgan CJA, Curran HV. Ketamine use: A review. Addiction. 2012;107:27–38. doi: 10.1111/j.1360-0443.2011.03576.x. [DOI] [PubMed] [Google Scholar]
- 78.Zhou W, Wang N, Yang C, Li X-M, Zhou Z-Q, Yang J-J. Ketamine-induced antidepressant effects are associated with AMPA receptors-mediated upregulation of mTOR and BDNF in rat hippocampus and prefrontal cortex. Eur. Psychiatry. 2014;29:419–423. doi: 10.1016/j.eurpsy.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 79.Koike H, Chaki S. Requirement of AMPA receptor stimulation for the sustained antidepressant activity of ketamine and LY341495 during the forced swim test in rats. Behav. Brain Res. 2014;271:111–115. doi: 10.1016/j.bbr.2014.05.065. [DOI] [PubMed] [Google Scholar]
- 80.Duman RS, Li N, Liu R-J, Duric V, Aghajanian G. Signaling pathways underlying the rapid antidepressant actions of ketamine. Neuropharmacology. 2012;62:35–41. doi: 10.1016/j.neuropharm.2011.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dwyer JM, Lepack AE, Duman RS. mTOR activation is required for the antidepressant effects of mGluR2/3 blockade. Int. J. Neuropsychopharmacol. 2012;15:429–434. doi: 10.1017/S1461145711001702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, Li XY, Aghajanian G, Duman RS. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329:959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.