

Abstract
Thirty-two congeneric rigid adenine nucleoside derivatives containing a North (N)-methanocarba ribose substitution and a 2-arylethynyl group either enhanced (up to 760% of control) or inhibited [125I] methyl (1R,2S,3S)-3-(4-iodophenyl)-8-methyl-8-azabicyclo[3.2.1]octane-2-carboxylate (RTI-55) binding at the human dopamine (DA) transporter (DAT) and inhibited DA uptake. Several nucleosides also enhanced [3H]mazindol [(±)-5-(4-chlorophenyl)-3,5-dihydro-2H-imidazo[2,1-a]isoindol-5-ol] binding to the DAT. The combination of binding enhancement and functional inhibition suggests possible allosteric interaction with the tropanes. The structure-activity relationship of this novel class of DAT ligands was explored: small N6-substition (methyl or ethyl) was favored, while the N1 of the adenine ring was essential. Effective terminal aryl groups include thien-2-yl (compounds 9 and 16), with EC50 values of 35.1 and 9.1 nM, respectively, in [125I]RTI-55 binding enhancement, and 3,4-difluorophenyl as in the most potent DA uptake inhibitor (compound 6) with an IC50 value of 92 nM (3-fold more potent than cocaine), but not nitrogen heterocycles. Several compounds inhibited or enhanced binding at the norepinephrine transporter (NET) and serotonin transporter (SERT) and inhibited function in the micromolar range; truncation at the 4′-position in compound 23 allowed for weak inhibition of the SERT. We have not yet eliminated adenosine receptor affinity from this class of DAT modulators, but we identified modifications that remove DAT inhibition as an off-target effect of potent adenosine receptor agonists. Thus, we have identified a new class of allosteric DAT ligands, rigidified adenosine derivatives, and explored their initial structural requirements. They display a very atypical pharmacological profile, i.e., either enhancement by increasing affinity or inhibition of radioligand binding at the DAT, and in some cases the NET and SERT, and inhibition of neurotransmitter uptake.
Introduction
The dopamine (DA) transporter (DAT) (i.e., SLC6A3) is a sodium-coupled symporter that clears, and thereby inactivates, an extracellular neurotransmitter after its release (Kilty et al., 1991; Torres et al., 2003; Rice and Cragg, 2008). Defects in the function, regulation, or expression of the DAT are implicated in various psychiatric disorders, including attention deficit hyperactivity disorder, depression, mood disorders, and addiction (Laasko et al., 2000, Hansen et al., 2014). Blockade of DA uptake is one of the main mechanisms of action of drugs of abuse such as cocaine (Fig. 1A) (Sekine et al., 2003). A subjective cocaine-induced high is directly proportional to the occupancy of the DAT (Volkow et al., 1997). Attention deficit hyperactivity disorder–associated sequence variants and early onset Parkinson’s disease-associated mutations of the DAT have been identified (Koldsø et al., 2013).
Fig. 1.

Chemical structures of the DAT (A) and A3AR (B) ligands referred to in the text.
The structure of the integral membrane protein DAT was initially deduced by analysis of its amino acid sequence, and by analogy to other proteins in the large family of solute carrier (SLC) transporters, such as the γ-aminobutyric acid transporter (Vaughan and Kuhar, 1996). The neurotransmitters—the norepinephrine transporter (NET) (i.e., SLC6A2) (Axelrod et al., 1961; Wang et al., 2012) and the serotonin transporter (SERT) (i.e., SLC6A4) (Felts et al., 2014)—belong to the same structural family (also known as neurotransmitter sodium symporters) and have much commonality of ligand structures with DAT. Recently, the structure of the Drosophila melanogaster DAT was determined by X-ray crystallography (Penmatsa et al., 2013), and interactions between specific amino acid residues and the substituents of psychostimulants were described (Wang et al., 2015). Having 12 transmembrane domains, the DAT first binds sodium ions at the extracellular side before binding DA, and then both are internalized and released on the cytosolic side.
Various ligand tools are available for the study of the DAT, such as the tropane radioligands methyl (1R,2S,3S)-3-(4-fluorophenyl)-8-methyl-8-azabicyclo[3.2.1]octane-2-carboxylate (WIN 35,487) and methyl (1R,2S,3S)-3-(4-iodophenyl)-8-methyl-8-azabicyclo[3.2.1]octane-2-carboxylate (RTI-55), which are analogs of cocaine (Little et al., 1993; Carroll et al., 2004; Schmitt et al., 2013). Mazindol [(±)-5-(4-chlorophenyl)-3,5-dihydro-2H-imidazo[2,1-a]isoindol-5-ol] is another ligand in a different structural class that binds to the DAT and other transporters and blocks neurotransmitter uptake (Severinsen et al., 2014).
Adenosine analogs are under development as potential therapeutic agents for treating chronic neuropathic pain and other diseases (Tosh et al., 2012a; Borea et al., 2015; Little et al., 2015). Among these potent adenosine receptor (AR) agonists are the 9-riboside N6-(3-iodobenzyl)adenosine-5′-N-methylcarboxamide (IB-MECA; Stoilov et al., 2014) and the carbocyclic (1′S,2′R,3′S,4′R,5′S)-4′-{2-chloro-6-[(3-iodophenylmethyl)amino]purin-9-yl}-1 (methylaminocarbonyl)-bicyclo[3.1.0]hexane-2,3-diol MRS1898 (Fig. 1B), which bind selectively to the A3AR subtype and reduce pain in the chronic constriction injury model and other models of prolonged pain (Chen et al., 2012). Recently, we enlarged the set of conformationally constrained A3AR agonists like (1′S,2′R,3′S,4′R,5′S)-4′-{2-chloro-6-[(3-chlorophenylmethyl)amino]purin-9-yl}-1 (methylaminocarbonyl)-bicyclo[3.1.0]hexane-2,3-diol that contain, in place of the natural D-ribose, a sterically rigidified bicyclic bicyclo[3.1.0]hexane (methanocarba) ring system, which maintains a receptor-preferred North (N)-methanocarba conformation (Tosh et al., 2012a). These adenosine derivatives optionally contain a rigid extension at the C2 position, consisting of an arylethynyl group, which enhances A3AR selectivity. In the process of derisking these compounds for possible development as clinical candidates, the National Institute of Mental Health Psychoactive Drug Screening Program (PDSP) (Besnard et al., 2012) conducted broad screening at receptors, ion channels, and transporters. It was noted that some of the analogs that were potent A3AR agonists bound to off-target receptors, such as serotonergic (5HT2B and 5HT2C) and α2- or β3-adrenergic receptors (Paoletta et al., 2014). Furthermore, members of the series were found to modulate radioligand binding to the human DAT (hDAT), although these results were not included in our previous report. Here, we have characterized at the DAT, NET, and SERT the activity of (N)-methanocarba adenosine derivatives and correlated these activities with structure. Both binding and functional activities of this new class of DAT ligands have been characterized.
Materials and Methods
Materials
FetalClone and bovine calf serum were purchased from HyClone (Logan, UT). Most other chemicals were purchased from Sigma-Aldrich (St. Louis, MO). All growth media included 100 units/ml penicillin and 100 mg/ml streptomycin. Most Food and Drug Administration–scheduled substances were obtained from the National Institute on Drug Abuse drug supply program. All radioligands and [3H]neurotransmitters were purchased from Perkin Elmer Life and Analytical Sciences (Boston, MA) except [3H]dihydrotetrabenezine (DHTB), which was purchased from American Radiolabeled Chemicals (St. Louis, MO).
Biogenic Amine Transporters
Inhibition of [125I]RTI-55 binding to—and [3H]neurotransmitter uptake by—the hDAT, human SERT (hSERT), or human NET (hNET) in clonal cells have been described previously (Eshleman et al., 1999). Briefly, human embryonic kidney (HEK)-293 cells expressing the recombinant hDAT (HEK-hDAT), hSERT (HEK-hSERT), or hNET (HEK-hNET) were used. Cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 5% FetalClone, 5% bovine calf serum, and 2 µg puromycin/ml (HEK-hDAT and HEK-hSERT) or 10% FetalClone and 300 µg G418/ml (HEK-hNET). The [125I]RTI-55 (40–80 pM final concentration) and [3H]mazindol binding assays were conducted with duplicate determinations using a total particulate membrane preparation. The [3H]mazindol assay was conducted with modification of the [125I]RTI-55 binding methods, including 1–3 nM [3H]mazindol for the hDAT and hNET assays and 10–13 nM [3H]mazindol in 0.5 ml volume in the hSERT assays. RTI-55 was used to define nonspecific [3H]mazindol binding. The uptake assays were conducted with duplicate determinations and initiated by the addition of [3H]DA, [3H]5-hydroxytryptamine, or [3H]norepinephrine (20 nM final concentration) to intact detached cells. Assays were terminated by filtration using a Wallac 96-well harvester through Perkin Elmer filtermat A filters presoaked in 0.05% polyethylenimine ([3H]neurotransmitter uptake assays). Scintillation fluid was added to the filters, and radioactivity retained on the filters was determined using a Perkin Elmer microbeta plate counter.
[125I]RTI-55 Saturation Binding Assays.
Saturation binding assays were conducted to determine whether the nucleoside derivative–induced enhancement of [125I]RTI-55 binding was due to an increase in affinity or an increase in the Bmax value. The methods for [125I]RTI-55 saturation binding to HEK-hDAT membranes have been reported previously (Eshleman et al., 1999). Briefly, saturation binding experiments were conducted in triplicate by diluting the specific activity of [125I]RTI-55 with unlabeled RTI-55 ranging in concentration from 0.036 to 16.6 nM. Buffer or compounds 6, 16, or mazindol at the indicated concentrations was added prior to the addition of HEK-hDAT membranes. Protein concentrations ranged from 2.7 to 7.8 µg. GraphPad Prism software (San Diego, CA) was used to analyze the saturation curves to yield the Kd and Bmax values.
Cocaine Antagonism Assay.
A cocaine antagonist is expected to shift the dose-response curve for cocaine in the [3H]DA uptake assay to the right without having an effect on uptake by itself. Control cocaine curves, in the presence of 0.1% dimethylsulfoxide and three-to-six cocaine dose-response curves in the presence of selected concentrations of the test compound, were conducted. Nine concentrations of cocaine ranging from 21.6 nM to 10 μM were used. Cocaine IC50 values were calculated using GraphPAD Prism.
HEK-hDAT [3H]DA Release Assay.
The methods for characterizing drug-induced release of preloaded [3H]DA from HEK-hDAT cells have been described previously (Eshleman et al., 2013). Drugs were perfused for 22 minutes during the assays.
Vesicular Monoamine Transporter 2 (VMAT2)
Inhibition of [3H]DHTB binding to human VMAT2 in clonal cells has been previously described (Eshleman et al., 1999). Inhibition of [3H]ketanserin binding to human VMAT2 was conducted in an identical fashion except that DHTB (10 µM) was used to define nonspecific binding.
Data and Statistical Analyses
At least three independent competition experiments were conducted. GraphPAD Prism was used to analyze the data, with the IC50 values converted to Ki values using the equation Ki = IC50/{1+ [(drug*)/Kd drug*]}, where (drug*) is the concentration of the labeled ligand used in the binding assays (Cheng and Prusoff, 1973). The Kd values used in the equation for [125I]RTI are as previously reported (Eshleman et al., 1999), and the Kd values for [3H]mazindol were 54, 82, and 3.9 nM for the hDAT, hSERT, and hNET, respectively. When a drug enhanced the radioligand binding, an EC50 value was determined. Differences in affinities, potencies, or Bmax values were assessed by one-way analysis of variance using the logarithms of the Ki values for the test compounds. Dunnett’s multiple comparisons test was used to compare the effects of the test compounds with the control values.
Results
Various sterically constrained adenine nucleoside derivatives (Supplemental Material; (Supplemental Table 1) (Table 1) have been synthesized and studied for their potent binding to the A1AR (compound 2) (Tosh et al., 2012b) or A3AR (compounds 1, 3–9, 14–19, 21–28, 31, and 32) (Tosh et al., 2012a, 2014, 2015a,b). Many of these A3AR agonists reduce chronic neuropathic pain in a phenotypic screen, and the AR binding affinities of the previously reported nucleosides are provided (Supplemental Material; (Supplemental Table 2). These rigid nucleosides were tested at 10 µM for inhibition or enhancement of radioligand binding at the DAT, NET, and SERT in a preliminary screen by the PDSP (Besnard et al., 2012). A large fraction of those compounds tested either inhibited or enhanced the binding of radioligand at the DAT (Supplemental Table 1), with negligible activity at the NET and SERT. Six compounds were found to enhance DAT binding using radiolabeled methyl (1R,2S,3S)-3-(4-fluorophenyl)-8-methyl-8-azabicyclo[3.2.1]octane-2-carboxylate, which was confirmed in full concentration-response curves performed by the PDSP (Supplemental Material). Based on these findings, additional compounds (10–13, 20, 29, and 30) were synthesized to explore the structure-activity relationship (SAR) at these three transporters. The synthetic procedures and the AR activity of the latter set of compounds are reported elsewhere (Tosh et al., 2015a).
TABLE 1.
Structures and modulation of binding and activity at the hDAT ([125I]RTI-55, unless noted) of (N)-methanocarba adenosine derivatives
The derivatives include simple C2 derivatives (compounds 1, 2 and 21), alkyne compounds 3–20 (5′-amides) and 22–24 (truncated), and triazole derivatives (compounds 25–32). Each compound that produced an effect is associated with data for inhibition or enhancement, in separate columns. —, no effect.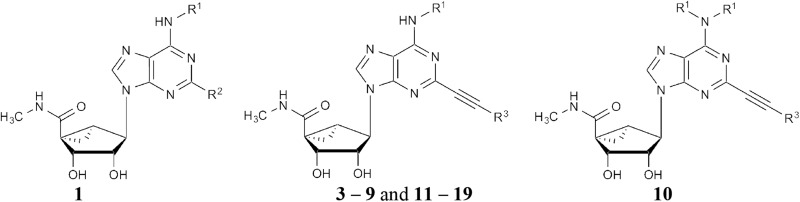 Compounds 1, 3–9 and 11–19, and 10
Compounds 1, 3–9 and 11–19, and 10
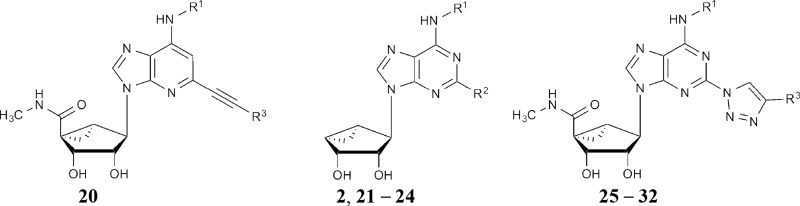 Compounds 20, 2 and 21–24, and 25–32
Compounds 20, 2 and 21–24, and 25–32
| Compound Numbera |
R1 |
R2 or R3 |
DAT Binding Inhibition,b
Ki |
DAT Binding Enhancement,b EC50 |
Dopamine Uptake Assay IC50c |
|---|---|---|---|---|---|
| nM | nM or % of control | nM | |||
| 1 | 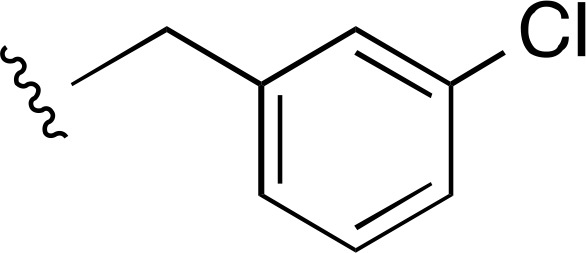 |
Cl | a | a | ND |
| 2 |  |
Cl | >10,000 (4) | — | >10,000 (3) |
| 3 | 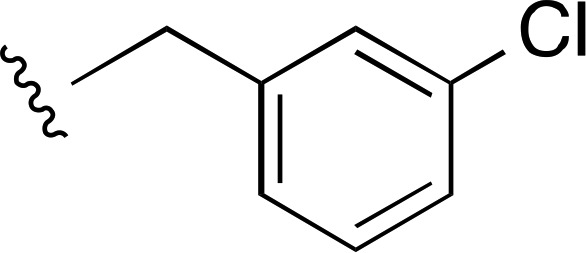 |
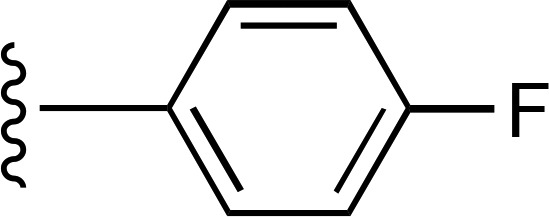 |
a | a | ND |
| 4 | 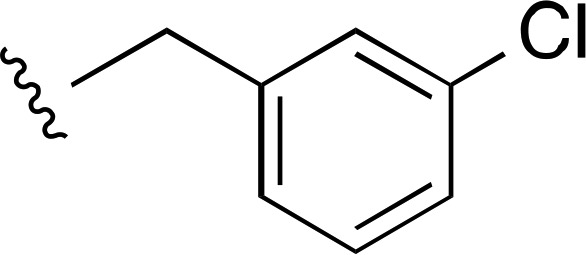 |
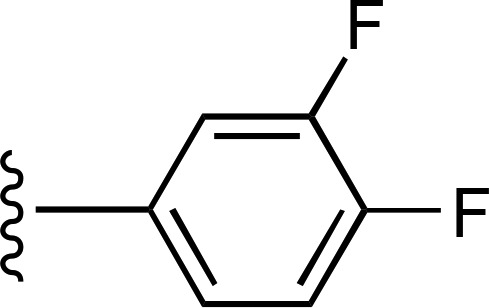 |
>2500a (5) | — | >10,000 (2) |
| 5 | 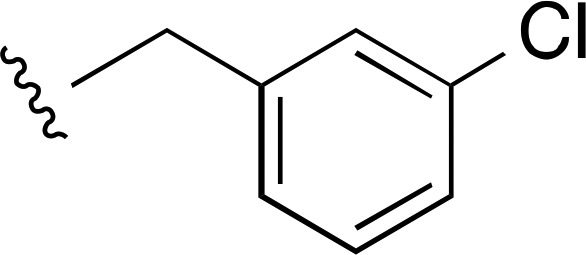 |
 |
2650 ± 300d (6) | — | >10,000 (2) |
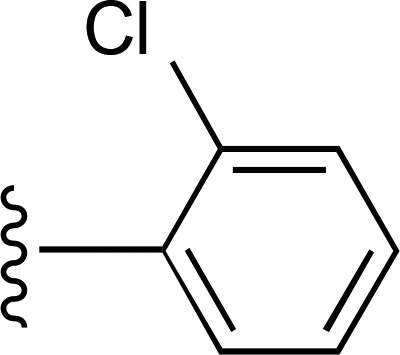
| 6 | CH3 | 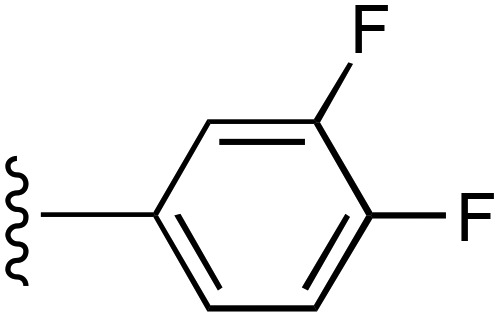 |
— | 70 ± 26e (4), 690% ± 180% | 92 ± 16 (5) |
| 7 | CH3 |  |
— | 540 ± 160e (5), 194% ± 12% | >9500 (3) |
| 8 | CH3 | 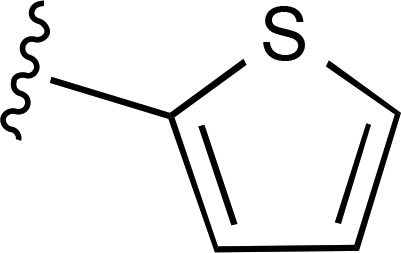 |
— | 446 ± 76 (8), 387% ± 48% | 3170 ± 380 (5) |
| 9 | CH3 | 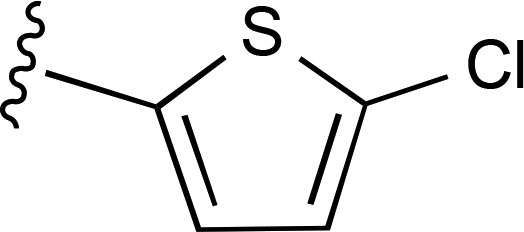 |
— | 35.1 ± 8.4e (5), 550% ± 110% | 253 ± 92 (5) |
| 10 | N,N-di-CH3 | 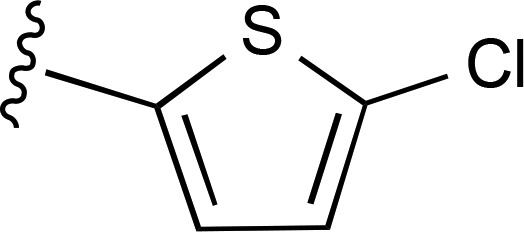 |
a | a | ND |
| 11 | C2H5 | 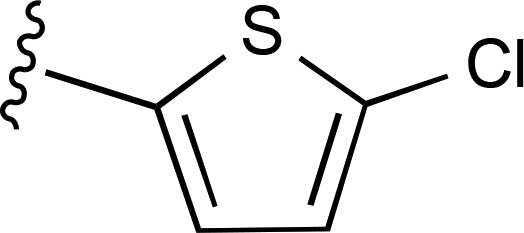 |
— | 300 ± 150e (6), 760% ± 260% | 206 ± 61 (4) |
| 12 | (CH2)2CH3 |  |
— | 1120 ± 220e (4), 268% ± 32% | >8700 (4) |
| 13 |  |
 |
— | 560 ± 190e (6), 251% ± 30% | >6600 (4) |
| 14 |  |
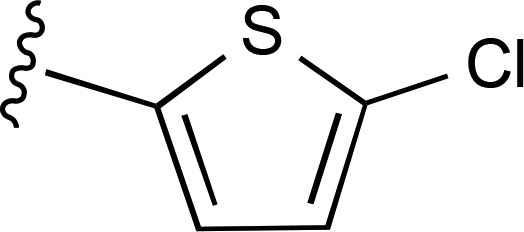 |
a | a | ND |
| 15 | 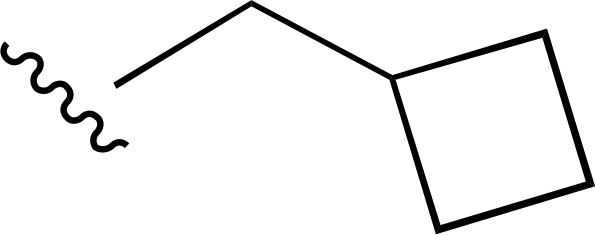 |
 |
a | a | ND |
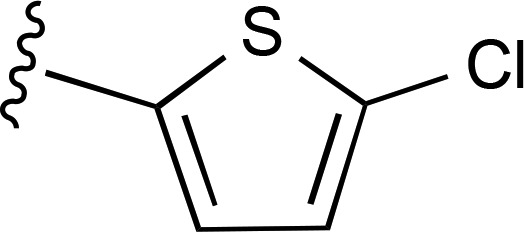
| 16 | CH3 | 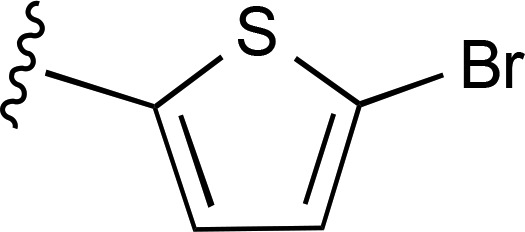 |
— | 9.1 ± 1.7e (4), 217% ± 24% | 229 ± 30 (3) |
| 17 | CH3 | 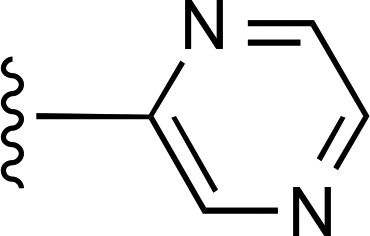 |
>10,000d (2) | >10,000d (2) | >10,000 (2) |
| 18 | CH3 | 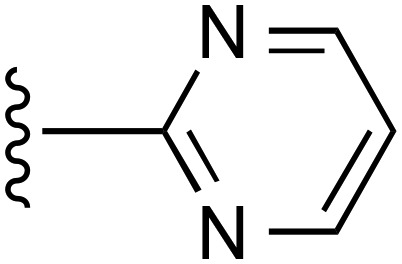 |
a | a | ND |
| 19 | CH3 | 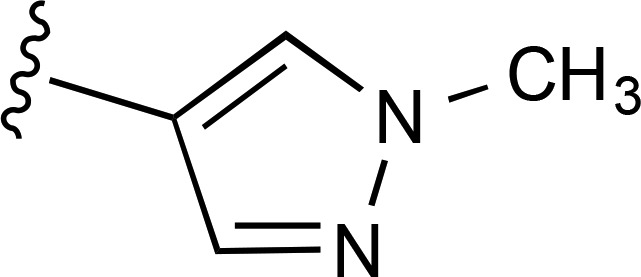 |
— | 3370 ± 820e (3), 163% ± 14% | >10,000 (2) |
| 20 | CH3 | 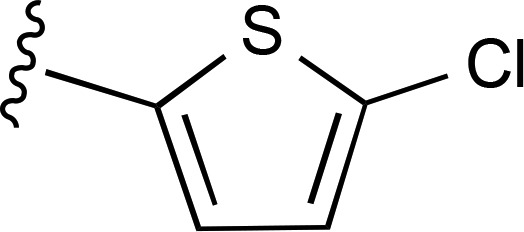 |
>10,000a (2) | — | >10,000 (2) |
| 21 | 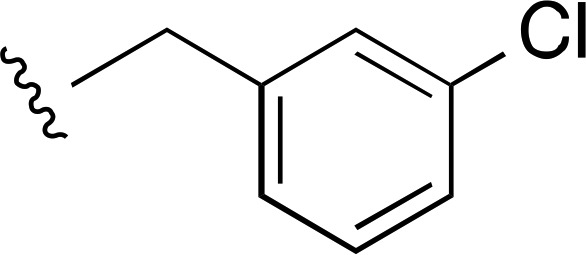 |
H | a | a | ND |
| 22 | 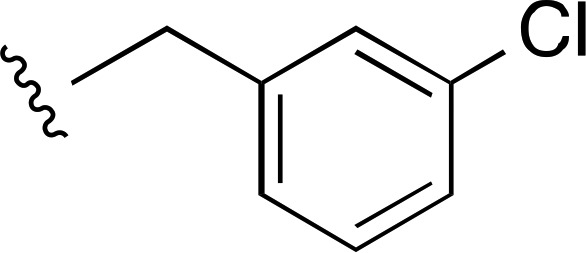 |
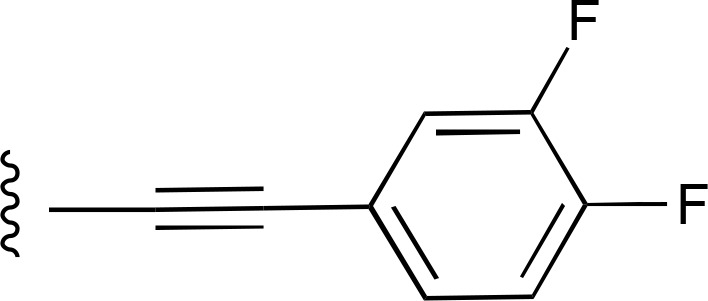 |
3950 ± 900a (3) | — | >10,000 (3) |
| 23 | CH3 | 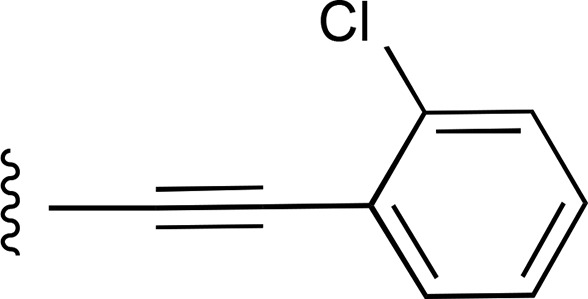 |
— | 4110 ± 540 (3), 277% ± 36% | >10,000 (2) |
| 24 | C2H5 |  |
— | >7700 (9), 163.2% ± 8.6% | >10,000 (2) |
| 25 | CH3 |  |
>10,000 (4) | >10,000 (4) | >10,000 (2) |
| 26 | CH3 |  |
— | >7100 (9), 136.2% ± 8.0% | >10,000 (2) |
| 27 | CH3 |  |
(Ki 4.48 µM)d | — | ND |
| 28 | CH3 | 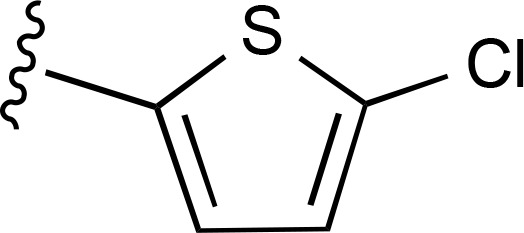 |
a | a | ND |
| 29 | 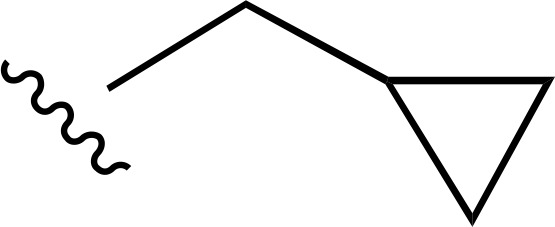 |
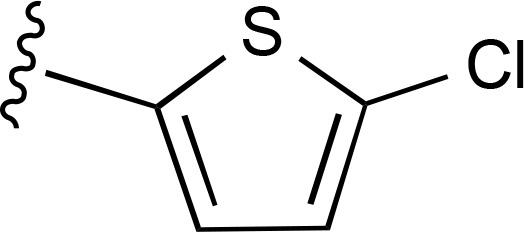 |
a | a | ND |
| 30 | 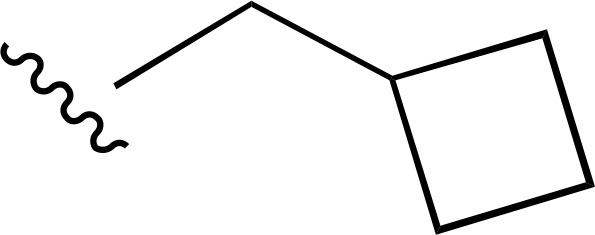 |
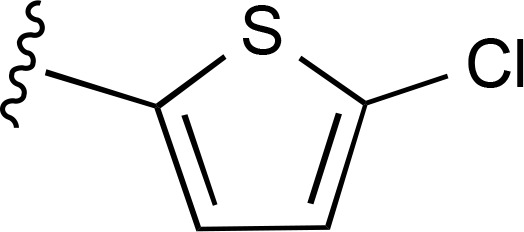 |
a | a | ND |
| 31 | 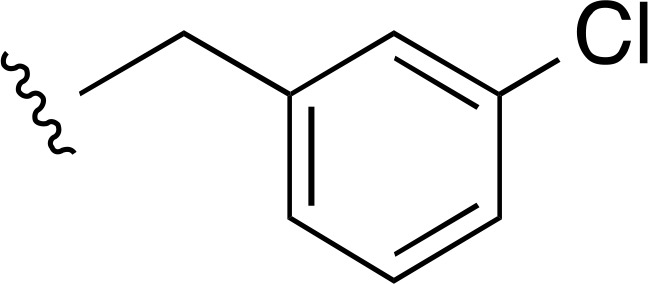 |
 |
7300 ± 1700d (3) | — | >10,000 (2) |
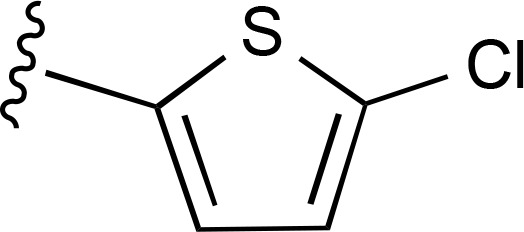
| 32 | CH3 | 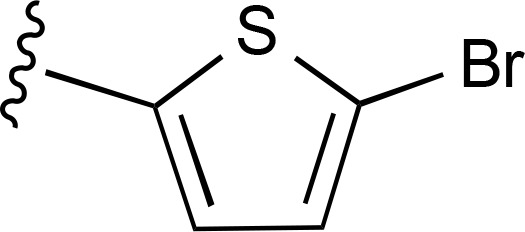 |
>10,000 (2) | — | >10,000 (2) |
ND, not determined.
Found to have negligible or no modulation of [3H]methyl (1R,2S,3S)-3-(4-fluorophenyl)-8-methyl-8-azabicyclo[3.2.1]octane-2-carboxylate binding at the hDAT by the PDSP (see Supplemental Material; Supplemental Table 1).
Modulation of [125I]RTI-55 binding (inhibition, enhancement, or undetermined effect). Values are expressed as the mean ± S.E.M. of n values (in parentheses). For radioligand binding enhancement, a maximal % is given.
Inhibition of DA uptake in HEK cells expressing the DAT. Values are expressed as the mean ± S.E.M. of n values (in parentheses). AR affinities are given for reported compounds in Supplemental Table 2 (see Supplemental Material).
Found to inhibit [3H]methyl (1R,2S,3S)-3-(4-fluorophenyl)-8-methyl-8-azabicyclo[3.2.1]octane-2-carboxylate binding at the hDAT by the PDSP (see Supplemental Material; Supplemental Table 1).
Found to enhance [3H]methyl (1R,2S,3S)-3-(4-fluorophenyl)-8-methyl-8-azabicyclo[3.2.1]octane-2-carboxylate binding at the hDAT by the PDSP (see Supplemental Material; Supplemental Table 1).
The enlarged set of nucleoside analogs can be categorized according to the substitution at the C2 position as follows: simple C2 derivatives (H, compound 21, or Cl, compounds 1 and 2); arylalkyne derivatives (5′-methylamides 3–20 and 4′-truncated compounds 22–24); and triazole derivatives (compounds 25–31). The truncated derivatives were included because 4′-truncation tends to convert selective A3AR agonists into selective antagonists (Tosh et al., 2012c). The triazole linker was explored as a substitute for the ethynyl group that would maintain key interactions with the A3AR agonists (Tosh et al., 2015b). The expanded set of compounds was initially tested for modulation of binding of [125I]RTI-55 binding to HEK cell membranes expressing the hDAT, hNET, or hSERT.
Many of the nucleosides, particularly those bearing an extended C2 substituent potently modulated DAT binding (Fig. 2, A and B; Supplemental Fig. 1). Compounds containing both a C2-fluorophenylalkyne and a N6-(3-chlorobenzyl) group (compounds 3 and 4) were weakly inhibiting or inactive at the DAT. Only one related compound, 2-chlorophenyl analog 5, displayed a measurable Ki value in [125I]RTI-55 binding inhibition of 2.65 µM. Binding inhibition at the DAT by N6-(3-chlorobenzyl) derivatives, i.e., the truncated nucleoside (compound 22) and triazole (compound 31), was found using the radioligand [125I]RTI-55. The 2-arylalkynyl-N6-methyl derivatives included numerous analogs that interacted with the DAT. However, inhibition by the N6-methyl derivative (compound 17) was not observed using [125I]RTI-55. Furthermore, numerous analogs greatly enhanced the binding of [125I]RTI-55 (40-80 pM) to the DAT: 2-arylalkyne 5′-methylamides (compounds 6–9, 11–13, 16, and 19); truncated derivatives (compounds 23 and 24); and triazole derivative (compound 26). The most potent enhancers (all N6-methyl or ethyl) were (EC50, in nM): 5-bromothien-2-yl 16 (9.1) > 5-chlorothien-2-yl 9 (35.1) > 3,4-difluorophenyl 6 (70) > 5-chlorothien-2-yl 11 (300) > thien-2-yl 8 (446) derivatives. Arranged according to maximal enhancement of binding, the order was as follows (percentage of enhancement): compound 11 (760), compound 6 (690), compound 9 (550), compound 8 (387), compound 12 (268), compound 13 (251), and compound 16 (217). Other compounds that enhanced were weaker in potency (>1 µM) and in maximal enhancement (<200%).
Fig. 2.

Drug-induced enhancement or inhibition of [125I]RTI-55 binding and [3H]mazindol binding to HEK-hDAT cell membranes. Assays were conducted as described in Materials and Methods. A total of n = 3–9 independent experiments were conducted in duplicate, except for compounds with no effect, where n = 2 independent experiments were conducted. (A) [125I]RTI-55 binding of compounds 2, 6, 8, 9, and 22; (B) [125I]RTI-55 binding of compounds 4, 5, 7, 16, 17, and 19; (C) [3H]mazindol binding of compounds 2, 6, 8, 9, and 22; (D) [3H]mazindol binding of compounds 4, 5, 7, 16, 17, and 19 (n = 3–8 independent experiments, except for drugs with no effect, where n = 2).
Various N6-substituted derivatives in the 2-arylalkyne 5′-methylamide series contained a common 5-chlorothien-2-ylethynyl group, which allowed comparison of the effects of various N6 groups. Di-methyl substitution of the N6 amine in compound 10 in the 5-chlorothien-2-yl series eliminated the potent interaction with the DAT observed with the corresponding monomethyl derivative (compound 9). Enlargement of the N6-methyl group to ethyl in compound 11 maintained enhancement of binding (EC50 value of 300 ± 150 nM); however, this EC50 was 9-fold weaker in comparison with compound 9. The N6-propyl 12 and N6-cyclobutyl (compound 13) analogs were less efficacious in enhancing DAT binding at 10 µM; further enlargement of the N6 group in cyclopropylmethyl (compound 14) and cyclobutylmethyl (compound 15) analogs successively reduced and eliminated DAT interaction. Therefore, in the alkyne series enlargement of the N6 group progressively disfavored interaction with the DAT. The 5-bromothien-2-yl-N6-methyl analog (compound 16) enhanced DAT binding with 4-fold higher potency (EC50 value of 9.1 ± 1.7 nM) and less than half of the percent enhancement in comparison with the corresponding 5-chlorothien-2-yl analog (compound 9). Substitution of the terminal aryl group in the 2-arylalkynyl-N6-methyl series with N-heterocycles in compounds 17–19 greatly decreased the degree of interaction with the DAT, with pyrazine (compound 17) and pyrimidine (compound 18) being essentially inactive. Replacement of the N1-nitrogen in the 1-deaza analog (compound 20) eliminated DAT interaction, which is a major difference compared with the corresponding adenine derivative (compound 9). 4′-Truncation in compounds 21–24 resulted in maintaining weak interaction with the DAT (µM inhibition or enhancement), but only when a 2-arylalkynyl group was present. The 2-triazole derivatives (compounds 25–32) were either weak or inactive in modulating DAT binding. With N6-methyl, the ability of the triazole derivatives to interact with the DAT depended on the terminal aryl group, with 3,4-difluorophenyl (compound 26) and fur-2-yl (compound 27) being weakly permissive, but with 5-chlorothien-2-yl (compound 28) being nonpermissive toward inhibition of DAT binding. Upon enlargement of the N6 group in the 5-chlorothien-2-yl triazole series (compounds 29–31) weak binding inhibition was observed only with the largest group, i.e., N6-3-chlorobenzyl (compound 31).
Six compounds (6, 7, 8, 9, 16, and 19) that enhanced [125I]RTI-55 binding were also evaluated in [3H]mazindol binding to the DAT, and a substantial binding enhancement was found (Fig. 2, C and D). This tetracyclic radioligand is not of the same structural class as the cocaine-related [125I]RTI-55, but it binds in the central substrate binding site (Severinsen et al., 2014).
Functional activity at the DAT was determined using [3H]DA uptake (Fig. 6A). In general, both the inhibitors and the enhancers of radioligand binding at the DAT inhibited the uptake of DA, while those compounds that lacked activity in the binding assay had no functional effect on DA uptake. The most potent inhibitors of DA uptake (all binding enhancers) were the following (IC50, in nM): compound 6 (92), compound 9 (253), compound 11 (206), and compound 16 (229) (Figs. 1 and 6A). All drugs completely inhibited [3H]DA uptake. All four compounds shared N6-methyl or ethyl substitution in the 2-arylalkyne 5′-methylamide series and differed mainly in the terminal aryl substitution, and all enhanced radioligand binding. Three of these four binding enhancers contained a 5-halothien-2-yl group. In the same functional assay cocaine and mazindol had IC50 values of 250 ± 35 and 13.9 ± 1.6 nM, respectively. The DA uptake inhibition among DAT binding inhibitors, such as compound 5, was weak at best and the IC50 values could not be determined.
Fig. 6.

Effect of compounds on HEK-hDAT functional assays: [3H]DA uptake and [3H]DA release. (A) Effect of compounds 6, 9, 11, and 16 on [3H]DA uptake into HEK-hDAT cells (n = 3–5). (B) Effect of compound 9 (1 nM to 1 µM) on cocaine-induced inhibition of [3H]DA uptake into HEK-hDAT cells (n = 2–5 for each concentration of 9). (C) Lack of effect of compound 2 (0.3 and 10 µM) and compound 8 (0.3 and 10 µM), compared with methamphetamine (METH) (1 and 10 µM), on preloaded [3H]DA release from HEK-hDAT cells. Data are from a representative experiment that was repeated with similar results.
Binding activity of the nucleoside derivatives was also measured at the hNET (Fig. 3) and hSERT (Fig. 4). There was mostly negligible activity at the SERT in the [125I]RTI-55 binding (Fig. 4, A and B) and [3H]mazindol binding (Fig. 4, C and D) assays, but the truncated 2-chlorophenyl derivative (compound 23) inhibited SERT binding using [125I]RTI-55 with a Ki value of 5.30 ± 0.92 µM (n = 6) (Fig. 4A) and serotonin uptake with an IC50 value of 5.5 ± 1.9 µM (n = 3). In the same uptake assay cocaine and mazindol had IC50 values of 307 ± 49 and 40.4 ± 9.1 nM, respectively. Compound 5 inhibited SERT [125I]RTI-55 binding with a Ki value of 6.71 ± 0.55 µM (n = 5) (Fig. 4B), but the effect on serotonin uptake was insignificant.
Fig. 3.

Drug-induced enhancement of [125I]RTI-55 binding to HEK-hNET cell membranes is not observed with [3H]mazindol binding to HEK-hNET cell membranes. A total of n = 2–6 independent experiments were conducted in duplicate, except for compounds with no effect, where n = 2 independent experiments were conducted. (A) [125I]RTI-55 binding of compounds 2, 6, 8, 9, and 22; (B) [125I]RTI-55 binding of compounds 4, 5, 7, 16, 17, and 19; (C) [3H]mazindol binding of compounds 2, 6, 8, 9, and 22; (D) [3H]mazindol binding of compounds 4, 5, 7, 16, 17, and 19 (n = 3–10, except for drugs with no effect, where n = 2).
Fig. 4.

Lack of drug-induced enhancement of [125I]RTI-55 binding and [3H]mazindol binding to HEK-hSERT cell membranes. (A) [125I]RTI-55 binding of compounds 2, 6, 8, 9, and 22; (B) [125I]RTI-55 binding of compounds 4, 5, 7, 16, 17, and 19; (C) [3H]mazindol binding of compounds 2, 6, 8, 9, and 22; (D) [3H]mazindol binding of compounds 4, 5, 7, 16, 17, and 19 (n = 3–7 independent experiments, except for drugs with no effect, where n = 2).
There was more widespread interaction of the nucleoside derivatives at the NET (Fig. 3) than at the SERT (Fig. 4), with several compounds modulating binding. The N6-methyl derivatives that enhanced binding of [125I]RTI-55 at the hNET were the following (EC50 in µM, percentage of control, n = 5 to 6): compound 6 (1.76 ± 0.64, 371% ± 67%), compound 9 (1.18 ± 0.36, 386% ± 73%), and compound 16 (0.670 ± 0.200, 285% ± 22%), while compound 5 inhibited binding (IC50 2.45 ± 0.59 µM). Some derivatives with N6 substituents larger than methyl enhanced binding of [125I]RTI-55 at the hNET but with small maximal enhancement (EC50 in µM, percentage of control, n = 3): compound 12 (0.90 ± 0.32, 159% ± 1%) and compound 13 (0.40 ± 0.10, 134% ± 13%). The N6-ethyl derivative (compound 11) weakly enhanced binding at the hNET, with an EC50 value of 4.1 ± 1.2 µM, up to 373% ± 15% of control (n = 6). Curiously, enhancement of NET binding by compounds 6, 9, and 16 was not observed using [3H]mazindol as the radioligand (Fig. 3, C and D). The greatest functional inhibition at the NET was seen with the 5-bromothien-2-yl derivative (compound 16) (IC50 value of 6.11 ± 0.57 µM). This was the only compound that modulated NET binding and also had a significant functional effect on [3H]norepinephrine uptake at 10 µM. In the same functional assay cocaine and mazindol had IC50 values of 230 ± 22 and 1.31 ± 0.12 nM, respectively. Compounds 2, 4, 7, 8, 17, 19, 20, 22–26, 31, and 32 at 10 µM were determined to have no significant effect on [125I]RTI-55 binding at the hNET or hSERT. Furthermore, compounds 1, 3, 10, 14, 15, 18, 21, and 27–30 at 10 µM were inactive in screening at the DAT, NET, and SERT in the PDSP screening.
To determine potential mechanisms for enhancement of [125I]RTI-55 binding to the hDAT, we conducted saturation isotherm binding in the presence of increasing concentrations of compounds 9 and 16. The data in Table 2 and Fig. 5 indicate that compounds 9 and 16 increase the affinity of the hDAT for the radioligand (one-way analysis of variance followed by Dunnett’s multiple comparisons test). However, no significant change in the Bmax value was induced by either compound. Thus, the substantial increase in binding at a fixed, low concentration of the radioligand can be explained as an allosteric enhancement of the affinity of the radioligand.
TABLE 2.
Compounds 9 and 16 increase affinity and mazindol decreases affinity of the hDAT for [125I]RTI-55
None of the treatment conditions had a significant effect on the Bmax values. All Bmax P values are >0.05; one-way analysis of variance; Dunnett’s multiple comparisons test; n = 3–12.
| Treatment |
|||
|---|---|---|---|
| Compound |
Concentration |
Ki (95% Confidence Interval) |
Bmax ± S.E.M. |
| nM | fmol/mg protein | ||
| Control | 1.31 (0.92–1.89) | 7040 ± 590 | |
| 9 | 10 nM | 0.902 (0.58–1.39) | 7820 ± 1170 |
| 9 | 30 nM | 0.60 (0.29–1.23)* | 6960 ± 520 |
| 9 | 100 nM | 0.39 (0.15–1.01)** | 7280 ± 540 |
| 9 | 300 nM | 0.53 (0.42–0.67)* | 7730 ± 880 |
| Control | 1.20 (0.96–1.50) | 6460 ± 420 | |
| 16 | 30 nM | 1.09 (0.50–2.38) | 7240 ± 1560 |
| 16 | 100 nM | 0.94 (0.51–1.72) | 6700 ± 410 |
| 16 | 300 nM | 0.60 (0.38–0.95)* | 7390 ± 770 |
| 16 | 1 µM | 0.58 (0.33–1.00)*** | 7140 ± 580 |
| Control | 1.24 (0.99–1.54) | 6510 ± 350 | |
| Mazindol | 10 nM | 2.30 (1.66–3.18)* | 6310 ± 1150 |
| Mazindol | 30 nM | 2.77 (0.67–1.14)*** | 6360 ± 480 |
P < 0.05, **P < 0.001, *** P < 0.01.
Fig. 5.

Scatchard plots of [125I]RTI-55 binding in the presence of compound 9 or 16. Saturation [125I]RTI-55 binding experiments were conducted in the absence or presence of varying concentrations of compound 9 (A) or compound 16 (B), as described in Materials and Methods. Data shown are from a representative experiment conducted in triplicate, which was replicated at least two times with similar results. In the Scatchard analysis, the (−) reciprocal of the slope of the line is an estimate of the Kd value for the radioligand. The steeper slope in the presence of higher concentrations of drugs indicates a lower Kd value and increased affinity.
The effects of the nucleoside derivative (compound 9) on inhibition of [3H]DA uptake by cocaine are shown in Fig. 6B. A leftward shift of the cocaine concentration-response curve is observed in the presence of increasing, fixed concentrations of compound 9 (1 nM to 1 µM). The average cocaine IC50 values (nM, mean ± S.E.M.) in the presence of 0, 1 nM, 10 nM, 100 nM, 300 nM, and 1 µM of compound 9 were 507 ± 57, 479 ± 78, 614 ± 82, 460 ± 150, 182 ± 73 (P < 0.05, one-way analysis of variance followed by Dunnett’s multiple comparisons test), and 35 ± 11 (P < 0.001), respectively. Furthermore the maximal uptake is decreased in the presence of high nanomolar concentrations of compound 9. There is no significant enhancement of DA uptake or a rightward shift of the cocaine concentration-response curve by compound 9.
The inability of the nucleoside derivatives (compounds 2 and 8) to induce release of preloaded [3H]DA is shown in Fig. 6C. Although these two derivatives had differing effects on binding and uptake—wherein compound 2 had no effect on binding or uptake, while compound 8 enhanced [125I]RTI-55 binding and inhibited [3H]DA uptake—neither compound induced release. The ability to induce release is strong evidence that a drug is a substrate for the hDAT, as shown by the ability of methamphetamine to induce robust release (Fig. 6C).
All of the nucleosides tested (compounds 2, 4–9, 11, 16, 17, 19, 20, 22–26, 31, and 32) were shown to have no effect on [3H]DHTB or [3H]ketanserin binding to VMAT2 (SLC18A2) expressed in HEK cells (Eshleman et al., 2013) (Supplemental Fig. 2). In the [3H]DHTB binding assay 2-hydroxy-2-ethyl-3-isobutyl-9,10-dimethoxy-1,2,3,4,5,6,7-hexahydrobenzo[a]chinolizine, a potent VMAT2 inhibitor, had a Ki value of 50.4 ± 1.8 nM and in the [3H]ketanserin binding assay ketanserin had a Ki value of 13.9 ± 3.8 nM.
Discussion
Multiple binding sites on the DAT have already been identified (Schmitt et al., 2013). In some cases DAT ligands of diverse structure are overlapping in their protein binding sites. The sites of either benztropine inhibitors or cocaine overlap with the DA site (Beuming et al., 2008; Bisgaard et al., 2011). Allosteric ligands of the DAT have been detected by pharmacological methods (Rothman et al., 2009). Photoaffinity labeling of the DAT by a cocaine analog and molecular modeling have established its binding site location (Dahal et al., 2014). The combination of binding enhancement and functional inhibition by the nucleoside derivatives suggests possible allosteric binding with respect to the tropanes. The finding that these compounds enhance the affinity of the hDAT for RTI-55 further supports an allosteric mechanism for the interaction of the nucleoside derivatives with the hDAT. Furthermore, the enhancement of binding applies to two distinct probe molecules, RTI-55 and mazindol. Furthermore, the enhancement of binding by some drugs suggests that they are interacting with different transporter residues compared with residues involved in drug-induced transporter inhibition. Additionally, drug-induced conformational changes that increase binding but decrease uptake cannot be ruled out.
The interaction of nucleoside derivatives at the DAT and NET proteins is novel. None of the previously reported structural classes of small molecule modulators of these proteins resembles nucleosides. The phenomenon of enhanced binding at the hDAT of radioligands that are derived from the structure of cocaine is unprecedented (for a review, see Reith et al., 2015). This phenomenon was observed using radiolabeled tropanes, which bind with high affinity to the DAT, NET, and SERT, and radiolabeled mazindol, which like the nucleoside analogs, binds to the hDAT and hNET with greater affinity than to the hSERT (Eshleman et al., 1999; Severinsen et al., 2014). At the same time, these rigid nucleosides inhibited DA uptake in a similar fashion to cocaine, suggesting an allosteric interaction with respect to the cocaine binding site. Furthermore, those related nucleoside derivatives that inhibit radioligand binding at the DAT are likely to bind to the same site on the DAT as the binding enhancers, based on the close similarity of structure. Therefore, it is likely that even the inhibitors of DAT binding in this structural class are allosteric with respect to cocaine. It is also likely that the enhancement of NET binding observed for a few compounds is the result of nucleoside binding at a similar site on this transporter. The NET and DAT are on the same subfamily of SLC transporters, and there is a high degree of homology between them. Modeling has demonstrated commonality of binding of the same ligand families at different transporters (Koldsø et al., 2013). Recently, four diverse classes of antidepressant drugs were crystallized in complex with a bacterial homologue of biogenic amine transporters, which serves as a model for various transport proteins. Common binding regions were located in association with transmembrane helices 1, 3, 6, and 8, and a mechanism for how this impedes function of the transporters by conformationally locking the helices with respect to each other was proposed (Wang et al., 2013). It will be interesting to see how the nucleoside ligands fit in the DAT, NET, and SERT proteins to inhibit transport and to modulate binding affinity in a complex manner.
The SAR of the nucleoside derivatives at the DAT and other transporters is summarized in Fig. 7. This SAR of adenosine derivatives at the DAT diverges greatly from the SAR of the same compound set at ARs. For example, potent A3AR agonist N6-(3-iodobenzyl)adenosine-5′-N-methylcarboxamide does not interact with the DAT (<10% inhibition at 10 µM; PDSP). Because the (N)-methanocarba nucleoside analogs found here to interact with the DAT are generally also potent AR ligands, we have not yet identified a nucleoside derivative with selectivity for the DAT over various receptor sites. That will be the objective of future exploration of this phenomenon, by further defining the SAR at the DAT. The dimethyl analog (compound 10) was initially intended for this purpose because it is known that disubstitution of the exocyclic amine of adenosine derivatives is generally not tolerated in AR binding (Jacobson and Gao, 2006). However, this compound proved to be inactive at the DAT. Therefore, the requirements for adenosine derivatives to interact with the DAT include at least one amine hydrogen, and if substituted on the N6 group, with small alkyl monosubstitution being most favorable. We have identified the 1-deaza modification and bulky N6 substitution as means of eliminating DAT interaction as an undesirable off-target effect in potent A3AR agonists. However, there might be situations where inhibition of neurotransmitter uptake and modulation of the A3AR agonist might be synergistic, for example, in neuropsychiatric disorders. The SERT and A3AR agonist are colocalized in midbrain serotonergic neurons (Zhu et al., 2011). We do not know if adenosine derivatives containing a native ribose ring rather than the rigid methanocarba bicyclic system will interact similarly at the DAT. The rigid ring system may eventually aid in the structural analysis of protein binding in a systematic fashion, as was done with off-target receptors (Paoletta et al., 2014).
Fig. 7.

Summary of the SAR of the nucleoside derivatives at DAT and other SLC transporters. The structure of the 3,4-difluorophenyl derivative (compound 6) is shown with substitutions leading to thienyl derivatives (compounds 9 and 16), and when truncated to the 2-chlorophenyl derivative (compound 23). Colored regions correspond to structural features on the most potent modulators, which both enhance the binding of tropane radioligands and inhibit DA uptake. The colors correspond to 5′ (blue), N6 (green), and C2-terminal thienyl (yellow) substituents.
In conclusion, we have identified a new class of ligands, i.e., rigidified adenosine derivatives containing a (N)-methanocarba ribose substitution and a 2-arylethynyl group, which appear to be moderately selective modulators of the DAT within the neurotransmitter sodium symporter family. They alternately enhanced or inhibited binding of two diverse radioligands at the hDAT. The combination of binding enhancement and inhibition of DA uptake suggests possible allosteric binding with respect to cocaine analogs. Given the structural similarities within this family it is likely that there is a conserved binding site for this nucleoside series. The SAR of this novel class of DAT ligands was explored; the adenine N1 was essential and terminal aryl groups that promote DAT interaction include thien-2-yl and 3,4-difluorophenyl, but not nitrogen heterocycles. Several compounds also inhibited or enhanced binding at the NET and SERT and inhibited function in the µM range. Truncation at the 4′-position in compound 23 allowed for weak inhibition of the SERT. Studies aimed at determining the mode of binding of these atypical ligands to the family of neurotransmitter sodium symporters, and additional structural modification will be performed in follow-up studies (see the (Supplemental Material of the PSDP screening results, which include off-target activities other than transporters, and (Supplemental Table 2 of the AR affinities of previously reported nucleosides).
Supplementary Material
Acknowledgments
The authors thank Robert Johnson, Katherine Wolfrum, John Reed, and Sunyoung Kim for technical assistance; the Addiction Treatment Discovery Program (ATDP) of the National Institute on Drug Abuse, Dr. Bryan L. Roth (University of North Carolina at Chapel Hill), and the National Institute of Mental Health’s Psychoactive Drug Screening Program for screening data.
Abbreviations
- AR
adenosine receptor
- DA
dopamine
- DAT
dopamine transporter
- DHTB
dihydrotetrabenezine
- hDAT
human dopamine transporter
- HEK
human embryonic kidney
- hNET
human norepinephrine transporter
- hSERT
human serotonin transporter
- NET
norepinephrine transporter
- (N)-methanocarba
North-methanocarba
- PDSP
Psychoactive Drug Screening Program
- RTI-55
methyl (1R,2S,3S)-3-(4-iodophenyl)-8-methyl-8-azabicyclo[3.2.1]octane-2-carboxylate
- SAR
structure-activity relationship
- SERT
serotonin transporter
- SLC
solute carrier
- VMAT2
vesicular monoamine transporter 2
Authorship Contributions
Participated in research design: Janowsky, Jacobson.
Conducted experiments: Eshleman, Tosh.
Contributed new reagents or analytic tools: Jacobson, Tosh.
Performed data analysis: Eshleman, Janowsky.
Wrote or contributed to writing of the manuscript: Janowsky, Eshleman, Jacobson.
Footnotes
This work was supported by the Intramural Research Program of the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grant ZIA DK031117] and the National Institute of Mental Health’s Psychoactive Drug Screening Program [Contract HHSN-271-2008-00025-C]. This work was also supported by National Institutes of Health National Institute on Drug Abuse/Veterans Association Interagency Agreement ADA12013, the Methamphetamine Abuse Research Center [Grant P50 DA018165-06], and the Department of Veterans Affairs Research Career Scientist Program.
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Axelrod J, Whitby LG, Hertting G. (1961) Effect of psychotropic drugs on the uptake of H3-norepinephrine by tissues. Science 133:383–384. [DOI] [PubMed] [Google Scholar]
- Besnard J, Ruda GF, Setola V, Abecassis K, Rodriguiz RM, Huang XP, Norval S, Sassano MF, Shin AI, Webster LA, et al. (2012) Automated design of ligands to polypharmacological profiles. Nature 492:215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuming T, Kniazeff J, Bergmann ML, Shi L, Gracia L, Raniszewska K, Newman AH, Javitch JA, Weinstein H, Gether U, et al. (2008) The binding sites for cocaine and dopamine in the dopamine transporter overlap. Nat Neurosci 11:780–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisgaard H, Larsen MA, Mazier S, Beuming T, Newman AH, Weinstein H, Shi L, Loland CJ, Gether U. (2011) The binding sites for benztropines and dopamine in the dopamine transporter overlap. Neuropharmacology 60:182–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borea PA, Varani K, Vincenzi F, Baraldi PG, Tabrizi MA, Merighi S, Gessi S. (2015) The A3 adenosine receptor: history and perspectives. Pharmacol Rev 67:74–102. [DOI] [PubMed] [Google Scholar]
- Carroll FI, Runyon SP, Abraham P, Navarro H, Kuhar MJ, Pollard GT, Howard JL. (2004) Monoamine transporter binding, locomotor activity, and drug discrimination properties of 3-(4-substituted-phenyl)tropane-2-carboxylic acid methyl ester isomers. J Med Chem 47:6401–6409. [DOI] [PubMed] [Google Scholar]
- Chen Z, Janes K, Chen C, Doyle T, Bryant L, Tosh DK, Jacobson KA, Salvemini D. (2012) Controlling murine and rat chronic pain through A3 adenosine receptor activation. FASEB J 26:1855–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 22:3099–3108. [DOI] [PubMed] [Google Scholar]
- Dahal RA, Pramod AB, Sharma B, Krout D, Foster JD, Cha JH, Cao J, Newman AH, Lever JR, Vaughan RA, et al. (2014) Computational and biochemical docking of the irreversible cocaine analog RTI 82 directly demonstrates ligand positioning in the dopamine transporter central substrate-binding site. J Biol Chem 289:29712–29727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshleman AJ, Carmolli M, Cumbay M, Martens CR, Neve KA, Janowsky A. (1999) Characteristics of drug interactions with recombinant biogenic amine transporters expressed in the same cell type. J Pharmacol Exp Ther 289:877–885. [PubMed] [Google Scholar]
- Eshleman AJ, Wolfrum KM, Hatfield MG, Johnson RA, Murphy KV, Janowsky A. (2013) Substituted methcathinones differ in transporter and receptor interactions. Biochem Pharmacol 85:1803–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felts B, Pramod AB, Sandtner W, Burbach N, Bulling S, Sitte HH, Henry LK. (2014) The two Na+ sites in the human serotonin transporter play distinct roles in the ion coupling and electrogenicity of transport. J Biol Chem 289:1825–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen FH, Skjørringe T, Yasmeen S, Arends NV, Sahai MA, Erreger K, Andreassen TF, Holy M, Hamilton PJ, Neergheen V, et al. (2014) Missense dopamine transporter mutations associate with adult parkinsonism and ADHD. J Clin Invest 124:3107–3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Gao ZG. (2006) Adenosine receptors as therapeutic targets. Nature Rev Drug Discovery 5:247–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilty JE, Lorang D, Amara SG. (1991) Cloning and expression of a cocaine-sensitive rat dopamine transporter. Science 254:578–579. [DOI] [PubMed] [Google Scholar]
- Koldsø H, Christiansen AB, Sinning S, Schiøtt B. (2013) Comparative modeling of the human monoamine transporters: similarities in substrate binding. ACS Chem Neurosci 4:295–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laasko A, Vilkman H, Alakare B, Haaparanta M, Bergman J, Solin O, Peurasaari J, Räkköläinen V, Syvälahti E, Hietala J. (2000) Striatal dopamine transporter binding in neuroleptic-naive patients with schizophrenia studied with positron emission tomography. Am J Psychiatry 157:269–271. [DOI] [PubMed] [Google Scholar]
- Little JW, Ford A, Symons-Liguori AM, Chen Z, Janes K, Doyle T, Xie J, Luongo L, Tosh DK, Maione S, et al. (2015) Endogenous adenosine A3 receptor activation selectively alleviates persistent pain states. Brain 138:28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little KY, Kirkman JA, Carroll FI, Breese GR, Duncan GE. (1993) [125I]RTI-55 binding to cocaine-sensitive dopaminergic and serotonergic uptake sites in the human brain. J Neurochem 61:1996–2006. [DOI] [PubMed] [Google Scholar]
- Paoletta S, Tosh DK, Salvemini D, Jacobson KA. (2014) Structural probing of off-target G protein-coupled receptor activities within a series of adenosine/adenine congeners. PLoS One 9:e97858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penmatsa A, Wang KH, Gouaux E. (2013) X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature 503:85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reith ME, Blough BE, Hong WC, Jones KT, Schmitt KC, Baumann MH, Partilla JS, Rothman RB, Katz JL. (2015) Behavioral, biological, and chemical perspectives on atypical agents targeting the dopamine transporter. Drug Alcohol Depend 147:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice ME, Cragg SJ. (2008) Dopamine spillover after quantal release: rethinking dopamine transmission in the nigrostriatal pathway. Brain research reviews 58(2):303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman RB, Dersch CM, Ananthan S, Partilla JS. (2009) Studies of the biogenic amine transporters. 13. Identification of “agonist” and “antagonist” allosteric modulators of amphetamine-induced dopamine release. J Pharmacol Exp Ther 329:718–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt KC, Rothman RB, Reith ME. (2013) Nonclassical pharmacology of the dopamine transporter: atypical inhibitors, allosteric modulators, and partial substrates. J Pharmacol Exp Ther 346:2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine Y, Minabe Y, Ouchi Y, Takei N, Iyo M, Nakamura K, Suzuki K, Tsukada H, Okada H, Yoshikawa E, et al. (2003) Association of dopamine transporter loss in the orbitofrontal and dorsolateral prefrontal cortices with methamphetamine-related psychiatric symptoms. Am J Psychiatry 160:1699–1701. [DOI] [PubMed] [Google Scholar]
- Severinsen K, Koldsø H, Thorup KA, Schjøth-Eskesen C, Møller PT, Wiborg O, Jensen HH, Sinning S, Schiøtt B. (2014) Binding of mazindol and analogs to the human serotonin and dopamine transporters. Mol Pharmacol 85:208–217. [DOI] [PubMed] [Google Scholar]
- Stoilov RM, Licheva RN, Mihaylova MK, Reitblat T, Dimitrov EA, Shimbova KM, Bhatia G, Pispati A, Gurman-Balbir A, Bagaria BR, et al. (2014) Therapeutic effect of oral CF101 in patients with rheumatoid arthritis: a randomized, double-blind, placebo-controlled phase II study. Immunome Res 11:1000087. [Google Scholar]
- Torres GE, Gainetdinov RR, Caron MG. (2003) Plasma membrane monoamine transporters: structure, regulation and function. Nat Rev Neurosci 4:13–25. [DOI] [PubMed] [Google Scholar]
- Tosh DK, Crane S, Chen Z, Paoletta S, Gao ZG, Gizewski E, Auchampach JA, Salvemini D, Jacobson KA. (2015a) Rigidified A3 adenosine receptor agonists: 1-deaza modification maintains high in vivo efficacy. ACS Med Chem Lett 6:804–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosh DK, Deflorian F, Phan K, Gao ZG, Wan TC, Gizewski E, Auchampach JA, Jacobson KA. (2012a) Structure-guided design of A3 adenosine receptor-selective nucleosides: combination of 2-arylethynyl and bicyclo[3.1.0]hexane substitutions. J Med Chem 55:4847–4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosh DK, Finley A, Paoletta S, Moss SM, Gao ZG, Gizewski ET, Auchampach JA, Salvemini D, Jacobson KA. (2014) In vivo phenotypic screening for treating chronic neuropathic pain: modification of C2-arylethynyl group of conformationally constrained A3 adenosine receptor agonists. J Med Chem 57:9901–9914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosh DK, Paoletta S, Chen Z, Crane S, Lloyd J, Gao ZG, Gizewski ET, Auchampach JA, Salvemini D, Jacobson KA. (2015b) Structure-based design, synthesis by click chemistry and in vivo activity of highly selective A3 adenosine receptor agonists. MedChemComm 6:555–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosh DK, Paoletta S, Deflorian F, Phan K, Moss SM, Gao ZG, Jiang X, Jacobson KA. (2012b) Structural sweet spot for A1 adenosine receptor activation by truncated (N)-methanocarba nucleosides: receptor docking and potent anticonvulsant activity. J Med Chem 55:8075–8090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosh DK, Paoletta S, Phan K, Gao ZG, Jacobson KA. (2012c) Truncated nucleosides as A3 adenosine receptor ligands: combined 2-arylethynyl and bicyclohexane substitutions. ACS Med Chem Lett 3:596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan RA, Kuhar MJ. (1996) Dopamine transporter ligand binding domains. Structural and functional properties revealed by limited proteolysis. J Biol Chem 271:21672–21680. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fischman MW, Foltin RW, Fowler JS, Abumrad NN, Vitkun S, Logan J, Gatley SJ, Pappas N, et al. (1997) Relationship between subjective effects of cocaine and dopamine transporter occupancy. Nature 386:827–830. [DOI] [PubMed] [Google Scholar]
- Wang CIA, Shaikh NH, Ramu S, Lewis RJ. (2012) A second extracellular site is required for norepinephrine transport by the human norepinephrine transporter. Mol Pharmacol 82:898–909. [DOI] [PubMed] [Google Scholar]
- Wang H, Goehring A, Wang KH, Penmatsa A, Ressler R, Gouaux E. (2013) Structural basis for action by diverse antidepressants on biogenic amine transporters. Nature 503:141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KH, Penmatsa A, Gouaux E. (2015) Neurotransmitter and psychostimulant recognition by the dopamine transporter. Nature 521:322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu CB, Lindler KM, Campbell NG, Sutcliffe JS, Hewlett WA, Blakely RD. (2011) Colocalization and regulated physical association of presynaptic serotonin transporters with A₃ adenosine receptors. Mol Pharmacol 80:458–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.