

Abstract
We have previously reported that mice lacking the efflux transporter Mrp3 had significant intestinal injury after toxic diclofenac (DCF) challenge, and proposed that diclofenac acyl glucuronide (DCF-AG), as a substrate of Mrp3, played a part in mediating injury. Since both humans and mice express the uptake transporter OATP2B1 in the intestines, OATP2B1 was characterized for DCF-AG uptake. In vitro assays using human embryonic kidney (HEK)-OATP2B1 cells demonstrated that DCF-AG was a substrate with a maximal velocity (Vmax) and Km of 17.6 ± 1.5 pmol/min per milligram and 14.3 ± 0.1 μM, respectively. Another key finding from our in vitro assays was that DCF-AG was more cytotoxic compared with DCF, and toxicity occurred within 1–3 hours of exposure. We also report that 1 mM DCF-AG caused a 6-fold increase in reactive oxygen species (ROS) by 3 hours. Investigation of oxidative stress through inhibition of superoxide dismutase (SOD) revealed that DCF-AG had 100% inhibition of SOD at the highest tested dose of 1 mM. The SOD and ROS results strongly suggest DCF-AG induced oxidative stress in vitro. Lastly, DCF-AG was screened for pharmacologic activity against COX-1 and COX-2 and was found to have IC50 values of 0.620 ± 0.105 and 2.91 ± 0.36 μM, respectively, which represents a novel finding. Since cyclooxygenase (COX) inhibition can lead to intestinal ulceration, it is plausible that DCF-AG can also contribute to enteropathy via COX inhibition. Taken in context, the work presented herein demonstrated the multifactorial pathways by which DCF-AG can act as a direct contributor to toxicity following DCF administration.
Introduction
Prior work on diclofenac (DCF) and its glucuronide metabolite, diclofenac acyl glucuronide (DCF-AG), focused on their disposition and the contribution of efflux transporters in modulating toxic outcomes. Mutant Wistar rats lacking the efflux transporter Mrp2 (Abcc2) exhibited resistance to intestinal injury after DCF administration compared with nonmutant rats (Seitz and Boelsterli, 1998). It was also determined that DCF-AG administration led to increased gastrointestinal (GI) ulceration compared with DCF administration. Since DCF is glucuronidated to DCF-AG, a known Mrp2 substrate (Seitz et al., 1998), the intestinal toxicity in nonmutant rats was probably attributable to DCF-AG. A recent study reported that a single 50-mg oral DCF dose to humans resulted in a DCF-AG plasma exposure that was comparable to DCF exposure, indicating DCF-AG formation constitutes a significant metabolic pathway (Zhang et al., 2015). Our work with efflux transporters demonstrated Bcrp (Abcg2) and Mrp3 (Abcc3) have functional roles with respect to DCF and DCF-AG disposition (submission pending). Toxicodynamic studies conducted in Mrp3 knockout (KO) mice showed conclusively that KO mice developed increased intestinal damage compared with wild-type (WT) (Scialis et al., 2015). Despite the susceptibility differences between genotypes, the mechanisms for the increased injury remain unclear.
DCF administration leads to the covalent adduct formation along the intestinal epithelia, with adduction occurring at the brush border and within enterocytes (Ware et al., 1998; Atchison et al., 2000). Adduction on the extracellular surface of plasma membrane was probably followed by internalization of the adducted protein (Boelsterli, 2003). Another pathway may involve DCF-AG uptake by intestinal transporters followed by intracellular adduction. Indeed, the GI tract expresses a number of transporters that have been quantified by liquid chromatography–tandem mass spectrometry (LC–MS/MS; Groer et al., 2013). One of the major uptake transporters is OATP2B1 (SCLO2B1), a known carrier for conjugated substances (Gao et al., 2012).
There is a distinct possibility that DCF-AG uptake by organic anion–transporting polypeptide (OATP) was contributory to the enteropathy observed in our animal models. Evidence for transporter uptake causing injury has gained recent attention. Stably transfected human embryonic kidney (HEK)-OATP cells were used to explore toxicity resulting from transporter-mediated uptake of statins wherein active uptake by OATP1B1 (SCLO1B1) caused a left-shift in cytotoxicity potency compared with the toxicity observed in HEK-WT cells (Zhang et al., 2013). Since OATP2B1 intestinal expression and activity have been demonstrated (Sai et al., 2006), understanding the relationship between intestinal uptake and toxicity should be pursued.
Intestinal enteropathy is probably not mediated through covalent adduct formation by reactive DCF metabolites. The argument against covalent adduction as an injury mechanism was supported by our observation that covalent adducts were observed in the liver without apparent toxicity or morphologic changes following DCF administration (Scialis et al., 2015). A possible causal factor for enteropathy is oxidative stress through inhibition of superoxide dismutase (SOD), which is a critical cellular defense mechanism for coping with reactive oxygen species (ROS) (Fridovich, 1978). Precedent of SOD inhibition by xenobiotic metabolites comes from a study by Chiou et al. (1999), in which suprofen acyl glucuronide inhibited SOD. In addition to SOD perturbation, DCF-AG may induce oxidative stress by generation of ROS just as DCF has been shown to incite (Lim et al., 2006).
Gastrointestinal injury after DCF administration occurs through inhibition of cyclooxygenase (COX) enzymes that metabolize arachidonic acid into physiologically important cytoprotective derivatives such as prostaglandin E2 (PGE2) (Kotani et al., 2006). DCF, a nonselective inhibitor of COX-1 and COX-2, promotes GI injury though decreased PGE2 signaling. Inhibition of both COX isoforms is necessary for injury to develop, and selective COX inhibitors fail to show significant GI damage (Tanaka et al., 2001). DCF’s ability to provoke injury makes clear that promiscuity for general COX inhibition, rather than potency against a single COX isoform, is pivotal. Since hydroxylated DCF metabolites have shown pharmacological activity against COX, the inhibition of COX by DCF-AG should be explored. Though conjugated metabolites are normally thought to be pharmacologically inactive, glucuronide metabolites with pharmacologic potency equivalent to their parent compound have been reported (Osborne et al., 1992; Schutz et al., 1999). In light of this, DCF-AG may possess enough COX inhibition pharmacology to promote injury.
Our hypothesis is that DCF-AG, as a major metabolite observed in animal models and human subjects, can directly mediate intestinal toxicity through pathways that occur in parallel to or independent from DCF. Thus the goal of the current work was to identify the various mechanisms that DCF-AG can promote injury. This was accomplished through analysis of OATP2B1-mediated uptake of DCF-AG, and by conducting in vitro assays to quantify the extent of cytotoxicity as a result of ROS generation or inhibition of COX and SOD enzymes.
Materials and Methods
Chemicals and Reagents.
DCF, DCFDA, Dulbecco’s modified Eagle’s medium (DMEM), formic acid, hydrogen peroxide, HEPES, indomethacin (used as the internal standard), MOPS, and SC-560 were purchased from Sigma-Aldrich (MilliporeSigma, St. Louis, MO). DCF-AG was purchased from Toronto Research Chemicals Incorporated (Toronto, Canada). Arachidonic acid (AA), COX-1, COX-2, Dup-697, and SOD were purchased from Cayman Chemical (Ann Arbor, MI). Calcein-AM was purchased from Thermo Fisher Scientific, Inc. (Waltham, MA). Ethidium homodimer-1 was purchased from Biotium Inc. (Hayward, CA). Stably-transfected HEK-OATP2B1 cells were created at Pfizer Inc. (Sandwich, UK) and were previously described (Kalgutkar et al., 2013). All LC–MS/MS solvents were of high analytical grade and were purchased from Burdick and Jackson (Muskegon, MI).
In Vitro Transport.
HEK-WT and HEK-OATP2B1 cells were seeded at 60,000 and 90,000 cells/well, respectively, in 96-well poly-d-lysine coated microplates. Cells were grown in DMEM containing 10% heat-inactivated fetal bovine serum and 5 mM sodium pyruvate. Plates were kept in an incubator set to 95% relative humidity, 5% CO2, and 37°C. Upon reaching confluency 48 hours after seeding, the growth media was discarded, and cells were washed three times with 100 μl of uptake buffer at 37°C. Two types of buffer were used for the studies: 1) Hanks’ balanced salt solution (HBSS) supplemented with 20 mM HEPES and titrated to pH 7.4, and 2) HBSS fortified with 20 mM MOPS and titrated to pH 6.0. The last wash was left of the cells for 15 minutes to equilibrate the cells before dosing commenced. After the equilibration period had passed, the last wash was removed and cells were dosed with 50 μl of uptake buffer containing increasing DCF-AG concentrations. Plates were quickly placed on a shaker set to 150 rpm and 37°C and incubated for 0.1, 0.5, 1, and 2 minutes. Termination of uptake was performed by quickly removing the uptake buffer, and the cells were washed four times with 200 μl of ice-cold uptake buffer. Intracellular DCF-AG was extracted by adding the internal standard (IS) in 100 μl of ice-cold methanol and shaking the cell plates for 15 minutes at 4°C. Cellular extracts were transferred into a clean 1-ml deep-well microplate and mixed with 100 μl of solvent A. Sample plates were vigorously vortex-mixed and injected onto the LC–MS/MS. A DCF-AG standard curve was prepared using naïve cells and extracted using the same process described for samples. The injection volume for all sample types was 10 μl. Total DCF-AG uptake was quantified using the standard curve, and the data were reported as picomoles. Cell counts and cell size were measured using a Scepter device (EMD Millipore, Billerica, MA), and total protein was determined using a Pierce BCA kit following the manufacturer’s recommendations (Thermo Fisher Scientific Incorporated, Waltham, MA). Concentrations were then normalized to picomoles per microliter of total cell volume in each well.
Kinetic Modeling.
The DCF-AG uptake data were subsequently analyzed using a two-compartment model previously described for OATP-mediated uptake (Poirier et al., 2008, 2009). Essentially, the model is governed by three processes: 1) Pdif,in defined as passive diffusion from the media into the cell; 2) Kactive, which is active transport from the media into the cell; and 3) Pdif,out representing passive diffusion from the cell into the media. A schematic of the model is shown in Fig. 1. Each process is described using the following equations:
| (1) |
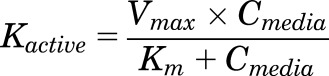 |
(2) |
| (3) |
where Pdif was a passive diffusion constant (μl/min), Cmedia was the DCF-AG concentration of the media (μM), Kactive is the saturable OATP2B1-mediated active transport that follows Michaelis-Menten kinetics, Vmax is the maximal transport velocity (pmol/min), Km is the substrate concentration at half the maximal velocity, and Ccell is the intracellular DCF-AG concentration (μM). The Pdif and Vmax parameters noted above were normalized by the total cellular protein per well to yield units of μl/min per milligram and pmol/min per milligram, respectively. The amount of DCF-AG in each compartment was defined by:
| (4) |
| (5) |
where Amedia and Acell were the DCF-AG amounts (pmol), Vmedia was the volume of the dosing buffer (50 μl), and Vcell was the total cellular volume per well (μl) that was determined during the cell counting procedure using total cell counts and cell diameter. Utilizing the previously described relationships, the temporal uptake of DCF-AG into cells was derived from the following differential equation:
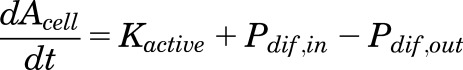 |
(6) |
The concentrations from the uptake studies were imported into Berkeley Madonna software v8.3.18 (Macey et al., 2000). A fourth order Runge-Kutta integration algorithm was implemented using a proportional error model in which the time course data were log transformed. The software was used to fit the data and determine values for Km, Pdif, and Vmax. Each modeling run consisted of six concentrations, four time points in duplicate, and two cell types (HEK-WT and HEK-OATP2B1) giving 96 total data points. Model performance in Berkeley Madonna was assessed by selection of initial guesses and parameter ranges that resulted in the lowest root-mean-square error.
Fig. 1.

Visual representation of a mechanistic two-compartmental transporter model. The movement of DCF-AG in the proposed model is described by three vectorial processes: 1) Pdif,in, passive diffusion from the buffer into the cell; 2) Kactive, active transport from the buffer into the cell; and 3) Pdif,out, passive diffusion from the cell into the buffer. Active transport for this model applies for the OATP2B1-mediated uptake of DCF-AG. Using differential equations within Berkeley Madonna software, the mass transfer of DCF-AG into and out of each compartment is modeled over time using in vitro time-course data to derive kinetic parameters such as Km, Pdif, and Vmax. Pdif reflects passive diffusion while Km and Vmax are used to characterize active uptake that adheres to Michaelis-Menten kinetics. Kinetic parameterization was performed for uptake at pH 6.0 and 7.4 to reflect the physiologic conditions of the intestine and liver, respectively.
Cytotoxicity Assay.
HEK-OATP2B1 cells were seeded at 100,000 cells/well in 96-well Optilux microplates (BD Gentest, Waltham, MA). Cells were grown in DMEM containing 10% heat-inactivated fetal bovine serum and 5 mM sodium pyruvate. Plates were kept overnight in an incubator set to 95% relative humidity, 5% CO2, and 37°C. The following day, the growth media was discarded, and cells were washed twice with 100 μl of uptake buffer at 37°C (HBSS, 20 mM HEPES, pH 7.4). After washing, cells were dosed in triplicate with 100 μl of vehicle (uptake buffer with organic solvent), DCF, or DCF-AG and incubated for 3, 6, or 12 hours at 37°C. Thirty minutes before the incubation period ended, the cells were briefly removed from the incubator, overlaid with 100 μl of a cocktail containing 2 μM calcein acetoxymethyl ester (CAM) and 4 μM ethidium homodimer-1 (EthD-1), and placed back in the incubator for a final 30 minutes. Plates were then removed, and fluorescence was measured on a SpectraMax Gemini XPS reader (Molecular Devices, Sunnyvale, CA). The excitation/emission wavelengths for CAM were 490/520 nm and the excitation/emission wavelengths for EthD-1 were 530/620. Responses for CAM and EthD-1 were normalized against the vehicle control cells.
ROS Assay.
HEK-OATP2B1 cells were seeded, grown, and washed as detailed in the preceding section. After washing, the cells were dosed in triplicate with 100 μl of vehicle or increasing concentrations of hydrogen peroxide, DCF, or DCF-AG and incubated for 1, 2, or 3 hours at 37°C. At the end of the incubation period, the cells were removed from the incubator, the dosing media carefully aspirated, and the cells were gently washed once with 100 μl of uptake buffer. Cells were overlaid with 100 μl of 1 μM CAM or 20 μM DCFDA and were placed back in the incubator for an additional 30 minutes at 37°C. At the end of the incubation period, plates were removed, and fluorescence of the cells was measured on a SpectraMax Gemini XPS reader (Molecular Devices, Sunnyvale, CA). The excitation/emission wavelengths for both CAM and DCFDA were 490/530 nm. Responses for CAM and DCFDA were normalized against the vehicle control cells.
SOD Inhibition Assay.
The SOD assay was conducted using a Cayman Chemical SOD kit with minor modifications to the recommended protocol. Briefly, SOD incubations consisted of 198 μl of radical detector, 10 μl of copper/zinc SOD, and 2.3 μl of vehicle or increasing concentration of inhibitor. Reactions commenced upon the addition of 20 μl of xanthine oxidase, and the mixtures were shaken at 60 rpm in the dark for 30 minutes at room temperature. Samples were then read on a BioTek UV/Vis microplate spectrophotometer (BioTek Instruments Incorporated, Winooski, VT) at a wavelength of 450 nm. The SOD activity in all samples was compared against a SOD standard curve. Absorption responses were background subtracted, and the percentage SOD inhibition was determined by normalizing the inhibitor response to vehicle controls.
COX Inhibition Assay.
The COX assay was performed using a Cayman Chemical COX kit with minor modifications to the recommended protocol. Briefly, COX incubations consisted of 194 μl of reaction buffer, 2 μl of heme, 2 μl of COX (1 or 2) enzyme, 2 μl of AA, and 4 μl of vehicle or increasing concentration of inhibitor. Reactions were conducted for 2 minutes in duplicate at 37°C and were quenched by the addition of 10 μl of 1 M HCl. A 200-μl aliquot of the quenched reaction was transferred into a 1-ml deep-well microplate and diluted with 200 μl of ice-cold methanol. From the diluted mixture, a 100-μl aliquot was mixed with 100 μl of IS in ice-cold methanol, vigorously vortex-mixed, and injected onto the LC–MS/MS. Methodology for detecting analytes of interest were modified from Shinde et al. (2012). The injection volume for all sample types was 10 μl. Rather than merely follow AA depletion, COX inhibition was determined by monitoring the appearance of PGE2 (PGE2/IS peak area ratio) in all samples and normalizing the inhibitor response to vehicle controls.
LC–MS/MS Method.
Chromatographic separation of analytes was performed on a Kinetex XB-C18 30 2.6 μm × 2 mm column (Phenomenex Incorporated, Torrance, CA). The system front end consisted of a HTC PAL Autosampler (LEAP Technologies, Carrboro, NC), a CBM-20A system controller, two LC20ADvp pumps, and a DGU-14A degasser (Shimadzu Scientific Instruments, Columbia, MD). Analytes of interest were eluted using one of two gradient profiles: method 1 (for DCF-AG) began with 10% solvent B for the first 0.5 minutes, which was then increased to 90% solvent B at 1.25 minutes using a linear gradient and held at this mixture for 0.25 minutes before reverting back to initial solvent conditions for 0.5 minutes to re-equilibrate the column, and method 2 (for PGE2) began with 10% solvent B for the first 0.5 minutes, which was then increased to 95% solvent B at 2.00 minutes using a linear gradient and held at this mixture for 0.5 minutes before reverting back to initial solvent conditions for 0.5 minutes to re-equilibrate the column. The flow rate for both methods was 0.3 ml/min, and the column effluent was directed to waste for the initial 0.5 minutes before being switched to the mass spectrometer. Analytes were detected using an AB Sciex API 4000 LC–MS/MS triple quad mass-spectrometer with a TurboIonSpray probe and Analyst version 1.5.2 software (AB Sciex, Framingham, MA) that was operated in multiple reaction monitoring mode. Ion spray voltage was –4250 V, and the source temperature was set to 400°C. The mass transitions in negative ion mode for monitoring AA, DCF-AG, PGE2, and indomethacin were mass-to-charge ratio (m/z) 303.3→259.0, 470.1→192.9, 351.1→271.0, and 356.0→311.8, respectively. The retention times of AA, DCF-AG, PGE2, and indomethacin were 2.46, 1.32, 1.55, and 1.47 (1.85 for method 2) minutes, respectively. Concentrations of analytes in the samples were determined by comparing the peak area ratios (analyte/IS) to those in the standard curve using a linear regression model. The criterion of acceptance for standards was defined to be ±20% of nominal concentration.
Statistical Analysis.
Data are expressed as mean ± standard error of the mean. P values ≤0.05 were considered statistically significant. Statistical analysis of data were performed using R version 3.2.1 (R Core Team, 2015). Two groups were compared by Student’s t test, and multiple groups were compared by an analysis of variance followed by Tukey’s post hoc test. GraphPad Prism version 6.0 (GraphPad Software Incorporated, La Jolla, CA) was used to calculate the IC50 for COX inhibition assays.
Results
In Vitro Transport.
The uptake of DCF-AG by a major intestinal transporter was measured using stably transfected HEK-OATP2B1 cells. DCF-AG kinetic uptake by OATP2B1 was measured at pH 6.0 and 7.4 to reflect the physiologic conditions that OATP2B1 is exposed to in the intestine and liver, respectively (Fallingborg, 1999). DCF-AG was incubated at six concentrations ranging from 1 to 300 μM with time points taken at 0.1, 0.5, 1, and 2 minutes. The uptake of DCF-AG was both time- and concentration-dependent (Fig. 2). Concentration-versus-time data were then analyzed using a two-compartment model as shown in Fig. 1. The model was used to estimate transporter kinetic parameters that are summarized in Table 1. The kinetic parameters of Vmax, Km, and Pdif at pH 6.0 were 27.8 ± 4.1 pmol/min per milligram, 15.2 ± 0.8 μM, and 0.0522 ± 0.0120 pmol/min per milligram, respectively. It was determined that at pH 7.4, DCF-AG has Vmax, Km, and Pdif values of 17.6 ± 1.5 pmol/min per milligram, 14.3 ± 0.1 μM, and 0.0245 ± 0.0030 pmol/min per milligram, respectively. The higher Vmax and Pdif values at pH 6.0 were found to be statistically significant compared with the parameter estimates at pH 7.4.
Fig. 2.

Concentration-versus-time profiles of DCF-AG uptake by OATP2B1. HEK-WT and HEK-OATP2B1 cells were seeded for 48 hours and incubated with increasing concentrations of DCF-AG at multiple time points in buffer titrated to either pH 6.0 or 7.4 at 37°C. Intracellular concentrations were determined by LC–MS/MS, and the concentrations were modeled in Berkeley Madonna. (A) and (B) show the uptake of DCF-AG by HEK-OATP2B1 cells at pH 6.0 and 7.4, respectively. Fitted lines from a two-compartment model represent the best fit. Data are the individual replicates from a typical study.
TABLE 1.
Summary of DCF-AG uptake kinetics mediated by OATP2B1
Each value represents the mean ± standard error of the mean from three studies (n = 2 replicates per study). *P < 0.05 for pH 6.0 versus pH 7.4.
| Media pH | Vmax | Km | Uptake CLint | Pdif |
|---|---|---|---|---|
| pmol/min per milligram | μM | μl/min per milligram | μl/min per milligram | |
| 6.0 | 27.8 ± 4.1* | 15.2 ± 0.8 | 1.82 ± 0.19 | 0.0522 ± 0.0120* |
| 7.4 | 17.6 ± 1.5 | 14.3 ± 0.1 | 1.23 ± 0.11 | 0.0245 ± 0.0030 |
CLint, intrinsic uptake clearance.
Cytotoxicity Assay.
Having established a role for OATP2B1-mediated uptake of DCF-AG, HEK-OATP2B1 cells were incubated with DCF or DCF-AG and monitored over time. Cell viability was assessed using two fluorescent dyes that offer different modalities and are complementary to each other. Cell viability measured by CAM, which detects intact cells through esterase activity, indicated that as early as 3 hours, cell death by DCF was apparent at the 1 mM dose and by 12 hours 29% of all cells were dead (Fig. 3A). The cell viability was confirmed by EthD-1, which detects cell death by binding to DNA once the nuclear envelope becomes compromised, and EthD-1 response increased by a maximum of 1.3-fold for the 12-hour 1 mM incubation (Fig. 3C). Compared with DCF, DCF-AG induced greater cell death. After 3 hours, approximately 61% of cells were dead at the 1 mM dose, and the number of dead cells increased to 85% by 12 hours for the 1 mM condition (Fig. 3B). EthD-1 in DCF-AG incubations corroborated the CAM results. The EthD-1 response for a 12-hour incubation with 1 mM DCF increased over 2-fold compared with vehicle control (Fig. 3D).
Fig. 3.

Cytotoxicity of DCF and DCF-AG using HEK cells. HEK-OATP2B1 cells were incubated in the absence or presence of increasing concentrations of compound. Incubations were conducted for 3, 6, or 12 hours at 37°C. Cytotoxicity was assessed using calcein AM to determine live cells and ethidium homodimer-1 as an indicator of dead cells. The CAM and EthD-1 responses indicate concentration-dependent cell death by DCF, and the cytotoxicity was more pronounced for DCF-AG. *P < 0.05, **P < 0.01; ***P < 0.001 compound versus its time-matched vehicle incubation. RFU, relative fluorescence units.
ROS Assay.
Taking into account that DCF had been shown to promote generation of reactive oxygen species, we investigated if DCF-AG had a similar potential. As for the cytotoxicity assay, HEK-OATP2B1 were exposed to DCF or DCF-AG and observed for ROS generation. Since cytotoxicity was evident by 3 hours, the ROS assay was conducted from 1 to 3 hours to determine if ROS preceded cell death. ROS production was measured with the fluorescent probe DCFDA. Hydrogen peroxide, a known ROS producer, was used as positive control and exhibited an increase in DCFDA signal in a dose-dependent manner (Fig. 4A). All hydrogen peroxide incubations achieved statistical significance compared with vehicle controls. ROS production by DCF was relatively modest by comparison, resulting in a statistically significant 1- to 2-fold increase at 3 hours for the 1 mM dose (Fig. 4B). DCF ROS induction appeared to indicate a dose response at 3 hours. DCF-AG demonstrated a clearer time-dependent ROS production culminating with a 5.5-fold increase at 3 hours for the 1 mM dose (Fig. 4C). The 500 μM DCF-AG showed trends of increased ROS generation, though only for the 3-hour incubation, which yielded a 1.8-fold increase versus vehicle that was statistically significant.
Fig. 4.

Generation of reactive oxygen species by in HEK cells. HEK-OATP2B1 cells were incubated in the absence or presence of increasing concentrations of compound. Incubations were conducted for 1, 2, or 3 hours at 37°C, after which ROS were detected with the use of DCFDA, which becomes oxidized by ROS to a fluorogenic form. Fluorescence was normalized to vehicle controls and expressed as percentage change from vehicle. Each bar represents the mean ± the standard deviation of n = 3 measurements per condition. *P < 0.05, **P < 0.01; ***P < 0.001 compound versus its time-matched vehicle incubation. RFU, relative fluorescence units.
SOD Inhibition Assay.
The SOD inhibition assay was used to explore whether DCF or DCF-AG could inhibit SOD enzymatic activity. SOD incubations were noncell-based and consisted of a mixture of substrate, enzymes, inhibitor, and detection reagent. Rather than inhibiting SOD activity over the concentration range, DCF caused an apparent stimulation, with the stimulus becoming statistically significant at the 250 μM dose and beyond (Fig. 5). DCF-AG had an opposite effect and was associated with a dose-dependent decrease in SOD activity such that there was a striking 100% inhibition of SOD at the 1 mM dose (Fig. 5). Furthermore, a statistically significant inhibition of 27% was observed at the 250 μM DCF-AG dose.
Fig. 5.

Inhibition of superoxide dismutase as an indication of oxidative stress. Copper/zinc SOD was incubated in the absence or presence of increasing concentrations of DCF (□) or DCF-AG (▪) for 30 minutes at room temperature (∼25°C). Vehicle shows assay performance in the presence of the same level of organic solvent (dimethyl sulfoxide) used in the inhibitor incubations. Each bar reflects the mean ± standard error of the mean from three separate studies (n = 2 replicates per study). **P < 0.01, ***P < 0.001 inhibitor versus its vehicle incubation. #P < 0.05, ###P < 0.001 for DCF versus DCF-AG.
COX Inhibition Assay.
The pharmacology of DCF for COX enzymes has been thoroughly evaluated, yet the pharmacology for DCF-AG has not been characterized. Hence, the final aspect of our work was to explore DCF-AG inhibition of COX enzymes. COX inhibition assays were noncell-based and used isolated COX enzymes that were mixed with substrate, cofactor, and inhibitor. Selective inhibitors of COX-1 and COX-2 were used as controls. Inhibition was assessed by measuring the decreased synthesis of PGE2 from AA. A summary of the findings is listed in Table 2. SC-560 and DuP-691, as selective COX-1 and COX-2 inhibitors, had IC50 values of 0.00166 ± 0.00022 μM and 0.00714 ± 0.0007 μM, respectively (Fig. 6A and Fig. 7A). DCF, as a nonselective COX inhibitor had COX-1 and COX-2 IC50 concentrations of 0.0206 ± 0.0037 μM and 0.103 ± 0.005 μM, respectively (Fig. 6B and Fig. 7B). OH-DCF was more potent against COX-1 than COX-2 and reached an apparent plateau for COX-2 inhibition. The OH-DCF IC50 values were estimated to be 0.375 ± 0.075 μM and 21.2 ± 0.3 μM for COX-1 and COX-2, respectively (Fig. 6C and Fig. 7C). Last, DCF-AG was demonstrated to inhibit PGE2 synthesis, with COX-1 and COX-2 IC50 estimates of 0.620 ± 0.105 μM and 2.91 ± 0.36 μM, respectively (Fig. 6D and Fig. 7D). COX-1 inhibition potency of DCF-AG was weaker compared with OH-DCF; however, COX-2 inhibition potency by DCF-AG was more intermediate with respect to DCF and OH-DCF.
TABLE 2.
Summary of in vitro COX inhibition assays
IC50 values reflect the mean ± standard error of the mean from three experiments (n = 2 replicates per experiment).
| Compound | COX-1 IC50 | COX-2 IC50 |
|---|---|---|
| μM | μM | |
| SC-560 (COX-1 selective) | 0.00166 ± 0.00022 | N.D. |
| DuP-697 (COX-2 selective) | N.D. | 0.00714 ± 0.0007 |
| DCF | 0.0206 ± 0.0037 | 0.103 ± 0.005 |
| OH-DCF | 0.375 ± 0.075 | 21.2 ± 0.3 |
| DCF-AG | 0.620 ± 0.105 | 2.91 ± 0.36 |
N.D., not determined.
Fig. 6.

COX-1 inhibition profiles of DCF, OH-DCF, and DCF-AG. Recombinant COX-1 was incubated in the presence of inhibitors for 2 minutes at 37°C, and the formation of PGE2 from AA was monitored via LC–MS/MS. Data reflect the mean ± standard error of the mean from three separate studies (n = 2 replicates per study). Inhibition data were fit with a three-parameter model to calculate IC50. Dotted lines reflect the 95% confidence intervals of the fit.
Fig. 7.

COX-2 inhibition profiles of DCF, OH-DCF, and DCF-AG. Recombinant COX-2 was incubated in the presence of inhibitors for 2 minutes at 37°C, and the formation of PGE2 from arachidonic acid was monitored via LC–MS/MS. Data reflect the mean ± standard error of the mean from three separate studies (n = 2 replicates per study). Inhibition data were fit with a three parameter model to calculate IC50. Dotted lines reflect the 95% confidence intervals of the fit.
Discussion
Our interest in exploring the toxicity attributed to DCF-AG was born through our initial work of characterizing the role that efflux transporters have in modulating toxicity following acute DCF administration. We observed that mice lacking the efflux transporter Mrp3 were more susceptible to intestinal injury compared with WT (Scialis et al., 2015). Though the intestinal toxicity differences between WT and KO were unequivocal, the mechanisms behind those differences were uncertain. Immunohistochemical staining of adducts within the intestinal villi led us to theorize that DCF-AG, as a causative agent to the covalent adducts, was subjected to an active transport process.
The intestines are host to an array of transporters some of which may be operative with respect to DCF-AG transport (Drozdzik et al., 2014). One of the probable candidates for DCF-AG uptake would be OATP2B1, which is known to mediate uptake of glucuronide conjugates (Gao et al., 2012; Grosser et al., 2014). Thus, OATP2B1 was investigated for DCF-AG affinity using a stably transfected cell system. The HEK-OATP2B1 cells were not used as an intestinal model, but served as an in vitro tool to test the uptake of DCF-AG by OATP without concomitant transport by multidrug resistance–associated protein 3 (MRP3) expression of which in HEK cells is quite low compared with other transporters (Ahlin et al., 2009). Using a compartmental model (Poirier et al., 2008), we defined the kinetic parameters describing DCF-AG uptake by OATP2B1. Preliminary studies showed that OATPs increased intracellular DCF-AG concentrations to levels exceeding the initial buffer conditions (data not shown). On the basis of those observations, we investigated the activity of OATP2B1 as a concentrative force for DCF-AG uptake.
We report that OATP2B1 acts an uptake transporter for DCF-AG, and that its affinity for DCF-AG varies with extracellular pH. OATP2B1 transporter clearance for DCF-AG was 1.82 μl/min per milligram at pH 6.0, whereas the transport clearance at pH 7.4 was 1.23 μl/min per milligram (Table 1). These findings suggest OATP2B1 activity is greater at pH 6.0, which is a typical pH in the intestines (Evans et al., 1988). Modeling estimates of DCF-AG passive diffusion indicated passive clearance was nearly 2.1-fold greater at pH 6.0 compared with pH 7.4 (0.0522 versus 0.0242 μl/min per milligram, respectively). Despite the increased passive diffusion at pH 6.0, active uptake of DCF-AG would be dominant. DCF, in contrast, as a smaller molecule with high passive uptake, has no such limitations and can more freely enter cells (Huang et al., 2010). Overall, the OATP2B1 studies provide insight into how intestinal uptake of DCF-AG can occur.
Our next studies indicated that DCF-AG is indeed cytotoxic, more so than DCF, and that cytotoxicity can manifest within 3 hours of exposure (Fig. 3B). The cytotoxic DCF-AG concentrations are physiologically relevant as they reflect the biliary concentrations detected in mice after an administration of 75 mg/kg of DCF (Scialis et al., 2015). Importantly, the upper concentration of 1 mM in the cytotoxicity assay reflects the lowest DCF-AG biliary concentration observed in our previous toxicokinetic mouse study. Furthermore, the cytotoxicity caused by DCF-AG was greater in HEK-OATP2B1 cells than observed for HEK wild-type in our preliminary assays (Supplemental Fig. 1). Cell death by DCF reached a maximum of 29% by 12 hours at 1 mM compared with nearly 62% cell death for DCF-AG by 3 hours. These data substantiate the findings reported in transport-deficient (TR–) rats, in which DCF-AG administration elicited greater intestinal injury compared with an equimolar dose of DCF (Seitz and Boelsterli, 1998). OATP2B1 has a mouse homolog, Oatp2b1, which is expressed in the intestinal tract (Cheng et al., 2005). It is conceivable that Oatp-mediated uptake of DCF-AG, along with the cytotoxicity potential of DCF-AG, may have contributed to the increased enteropathy in Mrp3 KO mice since those mice had reduced capacity to eliminate DCF-AG compared with WT.
Oxidative stress, through generation of reactive oxygen species, may overwhelm a cell’s ability to maintain homeostatic redox levels. The pesticide rotenone is a known inducer of ROS, and exposure to rotenone results in cell death (Tamilselvam et al., 2013). We conducted ROS assays with DCF and DCF-AG to determine their ROS activity. DCF ROS induction was relatively weak, yet ROS production was significantly increased after 2 hours of exposure. The modest ROS increase by DCF in our assay was less than a nearly 6-fold increase in ROS caused by DCF after a 30-hour exposure in a hepatocyte model of cytotoxicity (Lim et al., 2006). DCF-AG instigated a dose-dependent increase in ROS with a maximal 5.5-fold increase after 3 hours. Given that ROS generation occurred from 1 to 3 hours with cytotoxicity detected from 3 to 12 hours, it is plausible that DCF-AG ROS production contributed to cell death in our assays. Possible pathways of ROS-induced cell death are inhibition of ATPases, oxidation of DNA, perturbation of the mitochondrial permeability transition pore, or protein inactivation (Cantoni et al., 1989; Martinez-Reyes and Cuezva, 2014).
Oxidative stress can occur through inhibition of superoxide dismutase, an important part of the cell’s antioxidant response system (Fukui and Zhu, 2010). Therefore, we conducted noncell-based SOD inhibition assays. DCF failed to yield any inhibition and surprisingly appeared to stimulate SOD activity (Fig. 5). A striking observation was the clear dose-dependent inhibition of SOD by DCF-AG, with 1 mM DCF-AG reducing SOD activity to nondetectable levels (Fig. 5). Prior evidence for SOD inhibition by a glucuronide was reported by Chiou and colleagues (1999). In their study, 5 mM suprofen acyl glucuronide reduced SOD activity by 88% after a 14-day treatment using a noncell-based in vitro model, whereas 5 mM suprofen decreased SOD activity by only 5%. Our data show that DCF-AG can elicit complete inhibition at lower doses and within a shorter time scale. Taken in context, the SOD inhibition and ROS generation by DCF-AG may lead to sustained oxidative stress and cell death.
DCF was developed as a treatment of inflammation and pain via inhibition of COX enzymes. Hydroxylated metabolites of DCF have been tested for COX inhibition, but there has been no investigation of DCF-AG’s pharmacology. Hence, we sought to identify if DCF-AG possessed COX inhibitory activity. Our inhibition assays with COX-1 and COX-2 led to the novel finding that DCF-AG had apparent COX-1 and COX-2 IC50 values of 0.620 ± 0.105 μM and 2.91 ± 0.36 μM, respectively (Table 2). The COX inhibition potency of DCF-AG was notably weaker compared with DCF, for which we determined IC50 values of 0.0206 ± 0.0037 μM and 0.103 ± 0.005 μM for COX-1 and COX-2, respectively, values that are in line with published findings (Johnson et al., 1995).
The COX inhibition assays monitored formation of PGE2, which has several important physiologic functions such as protection of the GI mucosa, and PGE2-signaling deficiencies were associated with increased GI injury (Takeuchi, 2014). Mere inhibition of COX-1 or COX-2 is not sufficient to cause gastrointestinal injury. Administration to rats of a selective COX-1 or COX-2 inhibitor did not induce GI injury; however, coadministration of selective inhibitors caused extensive damage, implying that inhibition of both COX isoforms is essential to injury formation (Tanaka et al., 2001). Studies with COX-1 or COX-2 KO mice demonstrated that COX-1 activity promotes intestinal intactness, and COX-2 activity is necessary for healing of ulcers (Schmassmann et al., 2006). That DCF-AG can inhibit both COX enzymes represents a novel finding and offers a possible cause for the enteropathy associated with DCF-AG exposure.
In Fig. 8 we propose a toxicity model that captures our collective investigations. The model is predicated on the transfer of DCF into the liver, whereupon it undergoes metabolism. Hepatic DCF-AG can be excreted into the blood via MRP3 or transported into the bile by breast cancer resistance protein (BCRP) or MRP2. Residual hepatic DCF-AG may adduct albumin or be sequestered by glutathione (Grillo et al., 2003). Biliary DCF-AG enters the small intestine at the proximal duodenal region, after which it may undergo conversion back to DCF by bacterial β-glucuronidase present in the gut microflora (Louis et al., 2014). Free DCF-AG may covalently adduct to the brush border or undergo active uptake by OATP2B1. Intracellular DCF-AG can induce cytotoxicity through ROS generation or SOD inhibition causing oxidative stress. The acidic environment of the intestinal lumen stabilizes DCF-AG by decreasing the rate of nonenzymatic hydrolysis. DCF-AG may be cleared from enterocytes if MRP3 is present and functional, whereas lack of MRP3 may result in greater DCF-AG retention and injury. COX inhibition by DCF-AG may potentiate enteropathy via loss of PGE2 protection as well as interference of COX-2-directed wound healing. Finally, DCF is expected to contribute to intestinal injury in conjunction to DCF-AG via COX inhibition, oxidative stress, and ROS generation.
Fig. 8.

Proposed pathways on the disposition and mechanisms of toxicity for DCF-AG. DCF-AG is generated in the liver after DCF (intraperitoneal or by mouth) administration, where it undergoes excretion into bile or blood via ABC transporters. In the intestinal lumen DCF-AG is taken up by OATPs and exerts various effects that lead to intestinal injury. The absence of MRP3 exacerbates GI injury from DCF-AG owing to the loss of a clearance mechanism from enterocytes. Solid arrows indicate vectorial transport via uptake or efflux transporters, or via blood flow. Dotted arrows signify transport by passive processes. Dashed lines demonstrate possible enzymatic reactions such as inhibition of COX, generation of ROS, or cleavage by bacterial β-glucuronidases (β-gluc). The question mark denotes uncertainty regarding the efflux pathways for the indicated metabolites.
It may not be clear which of the proposed mechanisms are dominant or minor. Nonetheless, our results lay the foundation of a novel multifactorial process of intestinal injury attributable directly to DCF-AG. The in vitro assays detailed in the current work offer potential explanations and insight that have not been previously explored. In conclusion, we believe we have made a rational argument for the potential for DCF-AG to directly mediate toxicity and have provided credible mechanistic leads that could explain this toxicity.
Supplementary Material
Acknowledgments
The authors thank Drs. Chester Costales and A. David Rodrigues for the use of transfected HEK cells.
Abbreviations
- AA
arachidonic acid
- CAM
calcein acetomethoxyl ester
- COX
cyclooxygenase
- DCF
diclofenac
- DCF-AG
diclofenac acyl glucuronide
- DCFDA
5-(and-6)-carboxy-2′,7′-dichlorofluorescein diacetate
- DMEM
Dulbecco’s modified Eagle’s medium
- Dup-697
5-bromo-2-(4-fluorophenyl)-3-[4-(methylsulfonyl)phenyl]-thiophene
- EthD-1
ethidium homodimer 1
- GI
gastrointestinal
- HBSS
Hanks’ balanced salt solution
- HEK
human embryonic kidney
- IS
internal standard
- Km
substrate concentration at half the maximal velocity
- KO
knockout
- LC–MS/MS
liquid chromatography–tandem mass spectrometry
- MOPS
3-(N-morpholino)propanesulfonic acid
- MRP
multidrug resistance–associated protein
- OATP
organic anion–transporting polypeptide
- OH-DCF
4′-hydroxy diclofenac
- Pdif
passive diffusion
- PGE2
prostaglandin E2
- ROS
reactive oxygen species
- SC-560
5-(4-chlorophenyl)-1-(4-methoxyphenyl)-3-(trifluoromethyl)-1H-pyrazole
- SOD
superoxide dismutase
- Vmax
maximal transport velocity
- WT
wild type
Authorship Contributions
Participated in research design: Manautou, Scialis.
Conducted experiments: Scialis.
Performed data analysis: Scialis.
Wrote or contributed to the writing of the manuscript: Manautou, Scialis.
Footnotes
This work was supported by the National Institutes of Health [Grant DK069557].
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Ahlin G, Hilgendorf C, Karlsson J, Szigyarto CA, Uhlén M, Artursson P. (2009) Endogenous gene and protein expression of drug-transporting proteins in cell lines routinely used in drug discovery programs. Drug Metab Dispos 37:2275–2283. [DOI] [PubMed] [Google Scholar]
- Atchison CR, West AB, Balakumaran A, Hargus SJ, Pohl LR, Daiker DH, Aronson JF, Hoffmann WE, Shipp BK, Treinen-Moslen M. (2000) Drug enterocyte adducts: possible causal factor for diclofenac enteropathy in rats. Gastroenterology 119:1537–1547. [DOI] [PubMed] [Google Scholar]
- Boelsterli UA. (2003) Diclofenac-induced liver injury: a paradigm of idiosyncratic drug toxicity. Toxicol Appl Pharmacol 192:307–322. [DOI] [PubMed] [Google Scholar]
- Cantoni O, Brandi G, Salvaggio L, Cattabeni F. (1989) Molecular mechanisms of hydrogen peroxide cytotoxicity. Ann Ist Super Sanita 25:69–73. [PubMed] [Google Scholar]
- Cheng X, Maher J, Chen C, Klaassen CD. (2005) Tissue distribution and ontogeny of mouse organic anion transporting polypeptides (Oatps). Drug Metab Dispos 33:1062–1073. [DOI] [PubMed] [Google Scholar]
- Chiou YJ, Tomer KB, Smith PC. (1999) Effect of nonenzymatic glycation of albumin and superoxide dismutase by glucuronic acid and suprofen acyl glucuronide on their functions in vitro. Chem Biol Interact 121:141–159. [DOI] [PubMed] [Google Scholar]
- Drozdzik M, Gröer C, Penski J, Lapczuk J, Ostrowski M, Lai Y, Prasad B, Unadkat JD, Siegmund W, Oswald S. (2014) Protein abundance of clinically relevant multidrug transporters along the entire length of the human intestine. Mol Pharm 11:3547–3555. [DOI] [PubMed] [Google Scholar]
- Evans DF, Pye G, Bramley R, Clark AG, Dyson TJ, Hardcastle JD. (1988) Measurement of gastrointestinal pH profiles in normal ambulant human subjects. Gut 29:1035–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallingborg J. (1999) Intraluminal pH of the human gastrointestinal tract. Dan Med Bull 46:183–196. [PubMed] [Google Scholar]
- Fridovich I. (1978) The biology of oxygen radicals. Science 201:875–880. [DOI] [PubMed] [Google Scholar]
- Fukui M, Zhu BT. (2010) Mitochondrial superoxide dismutase SOD2, but not cytosolic SOD1, plays a critical role in protection against glutamate-induced oxidative stress and cell death in HT22 neuronal cells. Free Radic Biol Med 48:821–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C, Zhang H, Guo Z, You T, Chen X, Zhong D. (2012) Mechanistic studies on the absorption and disposition of scutellarin in humans: selective OATP2B1-mediated hepatic uptake is a likely key determinant for its unique pharmacokinetic characteristics. Drug Metab Dispos 40:2009–2020. [DOI] [PubMed] [Google Scholar]
- Grillo MP, Knutson CG, Sanders PE, Waldon DJ, Hua F, Ware JA. (2003) Studies on the chemical reactivity of diclofenac acyl glucuronide with glutathione: identification of diclofenac-S-acyl-glutathione in rat bile. Drug Metab Dispos 31:1327–1336. [DOI] [PubMed] [Google Scholar]
- Gröer C, Brück S, Lai Y, Paulick A, Busemann A, Heidecke CD, Siegmund W, Oswald S. (2013) LC-MS/MS-based quantification of clinically relevant intestinal uptake and efflux transporter proteins. J Pharm Biomed Anal 85:253–261. [DOI] [PubMed] [Google Scholar]
- Grosser G, Doring B, Ugele B, Geyer J, Kulling SE, Soukup ST. (2015) Transport of the soy isoflavone daidzein and its conjugative metabolites by the carriers SOAT, NTCP, OAT4, and OATP2B1. Arch Toxicol 89:2253–2263. [DOI] [PubMed] [Google Scholar]
- Huang L, Berry L, Ganga S, Janosky B, Chen A, Roberts J, Colletti AE, Lin MH. (2010) Relationship between passive permeability, efflux, and predictability of clearance from in vitro metabolic intrinsic clearance. Drug Metab Dispos 38:223–231. [DOI] [PubMed] [Google Scholar]
- Johnson JL, Wimsatt J, Buckel SD, Dyer RD, Maddipati KR. (1995) Purification and characterization of prostaglandin H synthase-2 from sheep placental cotyledons. Arch Biochem Biophys 324:26–34. [DOI] [PubMed] [Google Scholar]
- Kalgutkar AS, Chen D, Varma MV, Feng B, Terra SG, Scialis RJ, Rotter CJ, Frederick KS, West MA, Goosen TC, et al. (2013) Elucidation of the biochemical basis for a clinical drug-drug interaction between atorvastatin and 5-(N-(4-((4-ethylbenzyl)thio)phenyl)sulfamoyl)-2-methyl benzoic acid (CP-778875), a subtype selective agonist of the peroxisome proliferator-activated receptor alpha. Xenobiotica 43:963–972. [DOI] [PubMed] [Google Scholar]
- Kotani T, Kobata A, Nakamura E, Amagase K, Takeuchi K. (2006) Roles of cyclooxygenase-2 and prostacyclin/IP receptors in mucosal defense against ischemia/reperfusion injury in mouse stomach. J Pharmacol Exp Ther 316:547–555. [DOI] [PubMed] [Google Scholar]
- Lim MS, Lim PL, Gupta R, Boelsterli UA. (2006) Critical role of free cytosolic calcium, but not uncoupling, in mitochondrial permeability transition and cell death induced by diclofenac oxidative metabolites in immortalized human hepatocytes. Toxicol Appl Pharmacol 217:322–331. [DOI] [PubMed] [Google Scholar]
- Louis P, Hold GL, Flint HJ. (2014) The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol 12:661–672. [DOI] [PubMed] [Google Scholar]
- Macey R, Oster G, Zahley T. (2000) Berkeley Madonna, Berkely, CA. [Google Scholar]
- Martínez-Reyes I, Cuezva JM. (2014) The H(+)-ATP synthase: a gate to ROS-mediated cell death or cell survival. Biochim Biophys Acta 1837:1099–1112. [DOI] [PubMed] [Google Scholar]
- Osborne R, Thompson P, Joel S, Trew D, Patel N, Slevin M. (1992) The analgesic activity of morphine-6-glucuronide. Br J Clin Pharmacol 34:130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier A, Lavé T, Portmann R, Brun ME, Senner F, Kansy M, Grimm HP, Funk C. (2008) Design, data analysis, and simulation of in vitro drug transport kinetic experiments using a mechanistic in vitro model. Drug Metab Dispos 36:2434–2444. [DOI] [PubMed] [Google Scholar]
- Poirier A, Cascais AC, Funk C, Lavé T. (2009) Prediction of pharmacokinetic profile of valsartan in human based on in vitro uptake transport data. J Pharmacokinet Pharmacodyn 36:585–611. [DOI] [PubMed] [Google Scholar]
- R Core Team (2015) R: A language and environment for statistical computing, R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- Sai Y, Kaneko Y, Ito S, Mitsuoka K, Kato Y, Tamai I, Artursson P, Tsuji A. (2006) Predominant contribution of organic anion transporting polypeptide OATP-B (OATP2B1) to apical uptake of estrone-3-sulfate by human intestinal Caco-2 cells. Drug Metab Dispos 34:1423–1431. [DOI] [PubMed] [Google Scholar]
- Schmassmann A, Zoidl G, Peskar BM, Waser B, Schmassmann-Suhijar D, Gebbers JO, Reubi JC. (2006) Role of the different isoforms of cyclooxygenase and nitric oxide synthase during gastric ulcer healing in cyclooxygenase-1 and -2 knockout mice. Am J Physiol Gastrointest Liver Physiol 290:G747–G756. [DOI] [PubMed] [Google Scholar]
- Schütz E, Shipkova M, Armstrong VW, Wieland E, Oellerich M. (1999) Identification of a pharmacologically active metabolite of mycophenolic acid in plasma of transplant recipients treated with mycophenolate mofetil. Clin Chem 45:419–422. [PubMed] [Google Scholar]
- Scialis RJ, Csanaky IL, Goedken MJ, Manautou JE. (2015) Multidrug Resistance-Associated Protein 3 Plays an Important Role in Protection against Acute Toxicity of Diclofenac. Drug Metab Dispos 43:944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz S, Boelsterli UA. (1998) Diclofenac acyl glucuronide, a major biliary metabolite, is directly involved in small intestinal injury in rats. Gastroenterology 115:1476–1482. [DOI] [PubMed] [Google Scholar]
- Seitz S, Kretz-Rommel A, Oude Elferink RP, Boelsterli UA. (1998) Selective protein adduct formation of diclofenac glucuronide is critically dependent on the rat canalicular conjugate export pump (Mrp2). Chem Res Toxicol 11:513–519. [DOI] [PubMed] [Google Scholar]
- Shinde DD, Kim KB, Oh KS, Abdalla N, Liu KH, Bae SK, Shon JH, Kim HS, Kim DH, Shin JG. (2012) LC-MS/MS for the simultaneous analysis of arachidonic acid and 32 related metabolites in human plasma: Basal plasma concentrations and aspirin-induced changes of eicosanoids. J Chromatogr B Analyt Technol Biomed Life Sci 911:113–121. [DOI] [PubMed] [Google Scholar]
- Takeuchi K. (2014) Gastric cytoprotection by prostaglandin E₂ and prostacyclin: relationship to EP1 and IP receptors. J Physiol Pharmacol 65:3–14. [PubMed] [Google Scholar]
- Tamilselvam K, Braidy N, Manivasagam T, Essa MM, Prasad NR, Karthikeyan S, Thenmozhi AJ, Selvaraju S, Guillemin GJ. (2013) Neuroprotective effects of hesperidin, a plant flavanone, on rotenone-induced oxidative stress and apoptosis in a cellular model for Parkinson’s disease. Oxid Med Cell Longev 2013:102741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A, Araki H, Komoike Y, Hase S, Takeuchi K. (2001) Inhibition of both COX-1 and COX-2 is required for development of gastric damage in response to nonsteroidal antiinflammatory drugs. J Physiol Paris 95:21–27. [DOI] [PubMed] [Google Scholar]
- Ware JA, Graf ML, Martin BM, Lustberg LR, Pohl LR. (1998) Immunochemical detection and identification of protein adducts of diclofenac in the small intestine of rats: possible role in allergic reactions. Chem Res Toxicol 11:164–171. [DOI] [PubMed] [Google Scholar]
- Zhang X, Scialis RJ, Feng B, Leach K. (2013) Detection of statin cytotoxicity is increased in cells expressing the OATP1B1 transporter. Toxicol Sci 134:73–82. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Han YH, Putluru SP, Matta MK, Kole P, Mandlekar S, Furlong MT, Liu T, Iyer RA, Marathe P, et al. (2016) Diclofenac and Its Acyl Glucuronide: Determination of In Vivo Exposure in Human Subjects and Characterization as Human Drug Transporter Substrates In Vitro. Drug Metab Dispos 44:320–328. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.